







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
premedykacja: 1 ml (0,3 mg) domięśniowo na godzinę przed zabiegiem.
jako uzupełniający lek przeciwbólowy: dożylnie od 1 ml do 1½ ml (0,3 mg do 0,45 mg). U pacjentów w podeszłym wieku nie jest konieczna modyfikacja dawkowania.
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowywania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
BUNONDOL, 0,3 mg/ml, roztwór do wstrzykiwań
Każdy ml roztworu zawiera 0,3 mg buprenorfiny (Buprenorphinum) w postaci buprenorfiny chlorowodorku.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Roztwór do wstrzykiwań
Bezbarwny lub prawie bezbarwny, przezroczysty płyn
Bóle różnego pochodzenia umiarkowane do silnych, wymagające zastosowania opioidowego leku przeciwbólowego.
Dawkowanie
W bólach różnego pochodzenia
Dorośli i młodzież powyżej 12 lat
Zwykle 1 do 2 ml (0,3 do 0,6 mg) co 6 do 8 godzin.
Dzieci poniżej 12 lat
3 do 6 μg/kg mc. co 6 do 8 godzin. Nie przekraczać dawki 9 μg/kg mc.
Nie zbadano bezpieczeństwa stosowania buprenorfiny u dzieci poniżej 6. miesiąca życia. W premedykacji u dorosłych lub jako uzupełniający lek przeciwbólowy
Sposób podawania
Produkt podawać domięśniowo lub w powolnym wstrzyknięciu dożylnym.
Nadwrażliwość na substancję czynną, inne opioidowe leki przeciwbólowe lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Szczególna ostrożność jest zalecana w przypadku podawania buprenorfiny pacjentom otrzymującym leki o działaniu hamującym czynność ośrodkowego układu nerwowego, takie jak leki stosowane
w znieczuleniu ogólnym, przeciwhistaminowe, pochodne fenotiazyny, uspokajające, nasenne.
W przypadku stosowania takiego leczenia skojarzonego zaleca się zmniejszenie dawki jednego lub obu stosowanych leków.
Ryzyko związane z jednoczesnym stosowaniem leków uspokajających, np. benzodiazepin lub leków pochodnych:
Jednoczesne stosowanie produktu Bunondol i leków uspokajających, np. benzodiazepin lub leków pochodnych, może prowadzić do wystąpienia nadmiernego uspokojenia, depresji oddechowej, śpiączki lub śmierci. Z tego względu leczenie skojarzone z takimi lekami uspokajającymi należy stosować wyłącznie u pacjentów, u których nie są dostępne alternatywne metody leczenia. Jeśli
zostanie podjęta decyzja o stosowaniu produktu Bunondol w skojarzeniu z lekami uspokajającymi, należy podać możliwie najmniejszą skuteczną dawkę, a czas leczenia powinien być możliwie jak najkrótszy.
Należy uważnie obserwować, czy u pacjenta nie występują objawy związane z depresją oddechową i nadmierne uspokojenie. W tym kontekście zdecydowanie zaleca się informowanie pacjentów i ich opiekunów o możliwości wystąpienia takich objawów (patrz punkt 4.5).
Buprenorfiny chlorowodorek, tak jak inne opioidowe leki przeciwbólowe, może powodować wzrost ciśnienia płynu mózgowo-rdzeniowego i powinien być podawany z zachowaniem ostrożności
pacjentom z urazami głowy, ze zmianami wewnątrzczaszkowymi oraz innymi, w przebiegu których możliwy jest wzrost ciśnienia płynu mózgowo-rdzeniowego.
Buprenorfina może powodować zwężenie źrenic oraz zaburzenia świadomości, które mogą utrudniać ocenę stanu pacjenta.
Buprenorfiny chlorowodorek powinien być podawany z zachowaniem ostrożności pacjentom
w podeszłym wieku, osłabionym, dzieciom oraz osobom z zaburzeniami czynności nerek lub płuc.
Ostrożnie stosować u osób, u których występują niedoczynność tarczycy, niewydolność kory nadnerczy (np. choroba Addisona), myasthenia gravis, zahamowanie czynności ośrodkowego układu nerwowego, psychozy, śpiączka, przerost gruczołu krokowego lub zwężenie cewki moczowej, alkoholizm, delirium tremens lub kifoskolioza.
Badania przeprowadzone u ludzi oraz na zwierzętach wykazały, że buprenorfina ma niższy potencjał uzależniający w porównaniu z tzw. czystymi agonistami receptorów opioidowych.
Wykazano, że u osób uzależnionych od opioidów podanie małych dawek buprenorfiny zapobiegało wystąpieniu zespołu abstynencyjnego. Sporadycznie, u osób uzależnionych po podaniu buprenorfiny obserwowano euforię. Dlatego pacjentom uzależnionym lub podejrzanym o uzależnienie od opioidów, buprenorfinę należy podawać z zachowaniem ostrożności.
Zespół serotoninowy
Jednoczesne podawanie produktu Bunondol i innych leków serotoninergicznych, takich jak inhibitory MAO, selektywne inhibitory wychwytu zwrotnego serotoniny (SSRI), inhibitory wychwytu
zwrotnego serotoniny i noradrenaliny (SNRI) lub trójpierścieniowe leki przeciwdepresyjne, może prowadzić do zespołu serotoninowego, choroby mogącej zagrażać życiu (patrz punkt 4.5).
Jeśli jednoczesne przyjmowanie innych leków serotoninergicznych jest klinicznie uzasadnione, zaleca się uważną obserwację pacjenta, zwłaszcza w początkowej fazie leczenia i podczas zwiększania dawki.
Objawy zespołu serotoninowego mogą obejmować zmiany stanu psychicznego, niestabilność autonomiczną, zaburzenia nerwowo-mięśniowe lub objawy dotyczące układu pokarmowego.
Jeśli podejrzewa się występowanie zespołu serotoninowego, należy rozważyć zmniejszenie dawki lub przerwanie leczenia, w zależności od nasilenia objawów.
Zaburzenia oddychania podczas snu
Opioidy mogą powodować zaburzenia oddychania podczas snu, w tym centralny bezdech senny (CBS) i hipoksemię. Stosowanie opioidów zwiększa ryzyko wystąpienia CBS w sposób zależny od dawki. U pacjentów, u których występuje CBS, należy rozważyć zmniejszenie całkowitej dawki opioidów.
Specjalne grupy pacjentów
Pacjenci z zaburzeniami czynności układu oddechowego
Podobnie jak w przypadku stosowania innych silnie działających opioidów, po podaniu zalecanych dawek buprenorfiny mogą wystąpić zaburzenia oddychania. Dlatego buprenorfinę należy stosować ostrożnie u osób z zaburzeniami czynności układu oddechowego, np. astmą, niewydolnością oddechową, przerostem prawej komory serca, obniżeniem rezerwy oddechowej, niedotlenieniem narządów, hiperkapnią, uprzednio występującym zahamowaniem oddychania.
Mimo, że badania przeprowadzone u zdrowych ochotników wykazały, że antagoniści receptora opioidowego mogą nie odwracać całkowicie działania buprenorfiny, z praktyki klinicznej wynika, że w przeciwdziałaniu depresji oddechowej korzystne jest stosowanie naloksonu. Skuteczne są również leki pobudzające ośrodek oddechowy, takie jak doksapram.
Pacjenci z zaburzeniami czynności wątroby
Ponieważ buprenorfina jest metabolizowana w wątrobie, jej działanie może być nasilone u osób
z zaburzeniami czynności wątroby, dlatego też u tych pacjentów lek ten należy stosować ostrożnie. Buprenorfina, podobnie jak inne opioidy, powoduje wzrost ciśnienia w drogach żółciowych, dlatego powinna być podawana ostrożnie pacjentom z chorobami utrudniającymi odpływ żółci.
Buprenorfiny chlorowodorek nasila działanie leków uspokajających, nasennych, środków działających hamująco na ośrodkowy układ nerwowy.
Leki uspokajające, np. benzodiazepiny lub leki pochodne
Jednoczesne stosowanie opioidowych leków przeciwbólowych i leków uspokajających, np. benzodiazepin lub leków pochodnych, zwiększa ryzyko wystąpienia nadmiernego uspokojenia, depresji oddechowej, śpiączki lub śmierci na skutek addytywnego działania depresyjnego na OUN. Należy ograniczyć dawkę leku i czas trwania leczenia skojarzonego (patrz punkt 4.4).
Alkohol nasila hamujące działanie buprenorfiny na ośrodkowy układ nerwowy.
Produkt Bunondol należy stosować ostrożnie w przypadku jednoczesnego podawania serotoninergicznych produktów leczniczych, takich jak inhibitory MAO, selektywne inhibitory
wychwytu zwrotnego serotoniny (SSRI), inhibitory wychwytu zwrotnego serotoniny i noradrenaliny (SNRI) lub trójpierścieniowe leki przeciwdepresyjne, ponieważ zwiększają one ryzyko zespołu serotoninowego, choroby mogącej zagrażać życiu (patrz punkt 4.4).
Jednoczesne stosowanie pochodnych fenotiazyny i trójpierścieniowych leków przeciwdepresyjnych zwiększa ryzyko wystąpienia zahamowania czynności oddechowej.
Wykazano, że terapeutyczne dawki buprenorfiny nie zmniejszają skuteczności przeciwbólowej standardowych dawek agonistycznych opioidowych leków przeciwbólowych. W przypadku stosowania dawek leczniczych buprenorfiny, standardowe dawki opioidowych leków
przeciwbólowych mogą być podawane pod koniec działania buprenorfiny i nie ma to wpływu na analgezję.
Nie przeprowadzono odpowiednich badań dotyczących poniżej wymienionych interakcji. Jednak ponieważ buprenorfina jest metabolizowana przez CYP3A4, przypuszcza się, że leki takie jak gestoden, troleandomycyna, ketokonazol, norfluoksetyna, rytonawir, indynawir i sakwinawir hamują jej metabolizm. Natomiast fenobarbital, karbamazepina, fenytoina i ryfampicyna mogą powodować indukcję CYP3A4 i zmniejszać stężenie buprenorfiny. Ponieważ znaczenie skutków hamowania lub indukowania nie zostało wyjaśnione, nie zaleca się stosowania buprenorfiny z lekami wpływającymi na aktywność izoenzymu CYP3A4.
Buprenorfina nie wpływa na wyniki badań laboratoryjnych.
Ciąża
Badania przeprowadzone na zwierzętach wskazują na istnienie zagrożenia dla płodu. Odpowiednich badań u kobiet ciężarnych nie wykonano.
Nie zaleca się stosowania buprenorfiny w okresie ciąży. Karmienie piersią
Buprenorfina przenika do mleka kobiecego w niewielkich ilościach.
Badania przeprowadzone na zwierzętach, u których zastosowano dawki buprenorfiny znacznie przewyższające dawki stosowane u ludzi wykazały, że buprenorfina może zmniejszać wydzielanie mleka. Należy o tym pamiętać podczas zalecania tego leku pacjentkom karmiącym piersią.
Pacjentów leczonych ambulatoryjnie należy ostrzec, aby nie prowadzili pojazdów i nie obsługiwali maszyn w przypadku występowania działań niepożądanych.
Zaburzenia układu immunologicznego
Rzadko (≥1/10 000 do <1/1 000): reakcje alergiczne o ciężkim przebiegu (odnotowywano po zastosowaniu jednorazowej dawki).
Odnotowywano przypadki skurczu oskrzeli, obrzęku naczynioruchowego i wstrząsu anafilaktycznego. Wysypki (występowały sporadycznie).
Zaburzenia psychiczne
Omamy i inne działania niepożądane o charakterze psychomimetycznym - stwierdzano rzadziej po zastosowaniu buprenorfiny, niż po zastosowaniu innych agonistów/antagonistów. U pacjentów
w podeszłym wieku jest większe prawdopodobieństwo wystąpienia tego rodzaju działań niepożądanych.
Zaburzenia układu nerwowego
Zawroty głowy, ból głowy, senność (występowały częściej u pacjentów leczonych ambulatoryjnie).
Zaburzenia oka
Zaburzenia widzenia (występowały sporadycznie).
Zaburzenia naczyniowe
Niedociśnienie tętnicze prowadzące do omdleń.
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia Zahamowanie oddychania (występowało sporadycznie).
Zaburzenia żołądka i jelit
Nudności, wymioty (występowały one częściej u pacjentów leczonych ambulatoryjnie).
Zaburzenia nerek i dróg moczowych
Zaburzenia oddawania moczu (występowały sporadycznie).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań
niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania
Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Objawami przedawkowania buprenorfiny chlorowodorku są: senność, nudności, wymioty, znaczne zwężenie źrenic, zahamowanie czynności układu oddechowego.
W przypadku wystąpienia niewydolności oddechowej należy przede wszystkim zapewnić pacjentowi prawidłową wentylację. W przypadku wystąpienia zahamowania czynności układu oddechowego, właściwe jest podanie naloksonu lub leków pobudzających czynność oddechową. Można zastosować
doksapram w dawce 0,5 do 2,0 mg/kg mc. w ciągłym wlewie dożylnym, nie jest to jednak specyficzna odtrutka.
Grupa farmakoterapeutyczna: leki przeciwbólowe, opioidy, pochodne orypawiny, kod ATC: N02AE01
Buprenorfiny chlorowodorek jest silnym lekiem przeciwbólowym, należącym do grupy egzogennych opioidów, z bardzo silną komponentą antagonistyczną. Działa agonistycznie na receptor μ
i antagonistycznie na receptor κ.
Buprenorfina wykazuje silne i długotrwałe działanie przeciwbólowe.
W porównaniu z morfiną działa słabiej kurcząco na mięśnie gładkie. Może powodować zwężenie źrenic.
Buprenorfiny chlorowodorek wykazuje niewielki potencjał uzależniający: zespół abstynencyjny, jeżeli wystąpi, ma łagodny przebieg.
W zawale mięśnia sercowego wykazuje mniejszy wpływ na parametry układu krążenia - nie zmienia lub nieznacznie obniża ciśnienie późnorozkurczowe w prawej komorze, nie zmienia oporu obwodowego, nieznacznie i tylko w dużych dawkach obniża ciśnienie tętnicze, wskaźnik sercowy
i ciśnienie w tętnicy płucnej.
Buprenorfinę w postaci roztworu do wstrzykiwań można podawać dożylnie lub domięśniowo.
Względna dostępność biologiczna leku podanego domięśniowo w stosunku do podanego dożylnie wynosiła 1,07. Maksymalne stężenie w osoczu osiągane jest w ciągu kilku minut. Po 10 minutach od podania domięśniowego stężenie w osoczu nie różniło się znacząco od stężeń stwierdzanych po podaniu dożylnym takich samych dawek leku.
Buprenorfina jest metabolizowana do norbuprenorfiny, która jest agonistą receptora μ, charakteryzującym się słabą wewnętrzną aktywnością.
Eliminacja charakteryzuje się długo trwającą fazą końcową (od 20 do 25 godzin), co wynika
z częściowej reabsorpcji buprenorfiny po hydrolizie jej sprzężonych metabolitów oraz z właściwości lipofilnych cząsteczki.
Buprenorfina jest wydalana głównie w kale (80%), pozostałe ilości wydalane są w moczu.
Nie ma innych danych, niż zamieszczone w poprzednich punktach Charakterystyki Produktu Leczniczego.
Glukoza
Kwas solny 10% (do ustalenia pH) Woda do wstrzykiwań
Produktu nie należy mieszać ani podawać jednocześnie przez ten sam dostęp żylny z roztworami o odczynie zasadowym.
3 lata
Przechowywać ampułki w opakowaniu zewnętrznym w celu ochrony przed światłem. Przechowywać w temperaturze poniżej 25°C. Nie zamrażać.
Ampułki z bezbarwnego szkła w tekturowym pudełku. 5 ampułek po 1 ml
Instrukcja otwierania ampułki
Przed otwarciem ampułki należy upewnić się, że cały roztwór znajduje się w dolnej części ampułki. Można delikatnie potrząsnąć ampułką lub postukać w nią palcem, aby ułatwić spłynięcie roztworu.
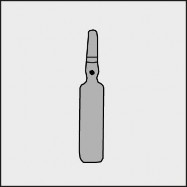
Na każdej ampułce umieszczono kolorową kropkę (patrz rysunek 1.) jako oznaczenie znajdującego się poniżej niej punktu nacięcia.
Rysunek 1.
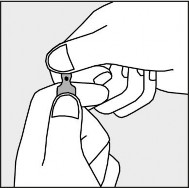
Aby otworzyć ampułkę należy trzymać ją pionowo, w obu dłoniach, kolorową kropką do siebie - patrz rysunek 2. Górną część ampułki należy uchwycić w taki sposób, aby kciuk znajdował się powyżej kolorowej kropki.
Rysunek 2.
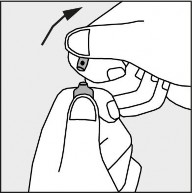
Nacisnąć zgodnie ze strzałką umieszczoną na rysunku 3. Rysunek 3.
Ampułki są przeznaczone wyłącznie do jednorazowego użytku, należy je otwierać bezpośrednio przed użyciem. Wszelkie resztki niewykorzystanego produktu lub jego odpady należy usunąć w sposób zgodny z lokalnymi przepisami.
Warszawskie Zakłady Farmaceutyczne Polfa S.A. ul. Karolkowa 22/24; 01-207 Warszawa
Pozwolenie nr R/3516
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 07.11.1994 r. Data ostatniego przedłużenia pozwolenia: 28.11.2012 r.







