







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Hormonalna terapia zastępcza w leczeniu objawów niedoboru estrogenów u kobiet po menopauzie (naturalnej lub wywołanej chirurgicznie) z zachowaną macicą.
Profilaktyka osteoporozy u kobiet po menopauzie, u których występuje zwiększone ryzyko złamań kości
w przyszłości, a które nie tolerują lub, dla których przeciwwskazane są inne leki stosowane
w profilaktyce osteoporozy.
Produkt Estalis nie może być stosowany w celu profilaktyki osteoporozy, jako lek pierwszego rzutu.
Doświadczenie w leczeniu kobiet po 65 roku życia jest ograniczone.
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w
punkcie 6.1,
Rozpoznany nowotwór piersi, nowotwór piersi w wywiadzie lub podejrzenie tej choroby,
Rozpoznanie lub podejrzenie zależnych od estrogenów nowotworów złośliwych (np. rak endometrium),
Niezdiagnozowane krwawienie z dróg rodnych,
Nieleczony rozrost endometrium,
Podawana w wywiadzie lub aktualnie występująca żylna choroba zakrzepowo-zatorowa (zakrzepica żył głębokich, zatorowość płucna),
Znane zaburzenia świadczące o zwiększonej skłonności do powstawania zakrzepów (np. niedobór białka C, białka S lub antytrombiny, patrz punkt 4.4),
Czynna lub niedawno przebyta choroba zakrzepowo-zatorowa tętnic (np. dusznica bolesna, zawał mięśnia sercowego),
Ostra choroba wątroby lub choroba wątroby w wywiadzie, jeżeli wyniki testów czynności wątroby nie powróciły do normy,
Porfiria,
Rozpoznanie lub podejrzenie ciąży,
Karmienie piersią.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Mięśniaki (włókniaki macicy) lub endometrioza
Czynniki ryzyka zaburzeń zakrzepowo-zatorowych (patrz niżej)
Czynniki ryzyka wystąpienia nowotworów hormonozależnych np. nowotwór piersi u bliskich z
pokrewieństwem 1-szego stopnia
Nadciśnienie tętnicze
Choroby wątroby (np. gruczolak wątroby)
Cukrzyca z zajęciem naczyń lub bez zajęcia naczyń
Kamica żółciowa
Migrena lub ciężkie bóle głowy
Toczeń rumieniowaty układowy
Rozrost endometrium w wywiadzie (patrz niżej)
Padaczka
Astma
Otoskleroza
Wskazania do natychmiastowego przerwania leczenia
Leczenie należy przerwać w przypadku stwierdzenia przeciwwskazań do jego stosowania oraz
w następujących sytuacjach:
Żółtaczka lub pogorszenie czynności wątroby
Istotne podwyższenie ciśnienia tętniczego krwi
Wystąpienie bólów typu migrenowego u osoby, która dotychczas nie miała takich dolegliwości
Ciąża
U kobiet z zachowaną macicą ryzyko rozrostu i raka endometrium ulega zwiększeniu, gdy estrogeny stosuje się w monoterapii przez dłuższy czas. Zaobserwowano, że u kobiet stosujących leczenie samymi estrogenami, wzrost ryzyka wystąpienia raka endometrium jest od 2 do 12 razy większy w porównaniu z ryzykiem u kobiet niestosujących tych preparatów, w zależności od czasu trwania leczenia i dawki estrogenów (patrz punkt 4.8). Po przerwaniu leczenia zwiększone ryzyko może utrzymywać się jeszcze, przez co najmniej 10 lat.
Cykliczne stosowanie dodatkowego progestagenu, przez co najmniej 12 dni w miesiącu przy 28-dniowym cyklu leczenia lub stosowanie skojarzenia estrogenu z progestagenem w sposób ciągły u kobiet z zachowaną macicą zapobiega wzrostowi ryzyka związanego ze stosowaniem HTZ samymi estrogenami.
W pierwszych miesiącach leczenia może dojść do krwawienia lub plamienia śródcyklicznego. Gdy tego typu objawy wystąpią po pewnym czasie od rozpoczęcia terapii lub będą się utrzymywać po jej zakończeniu, należy przeprowadzić badania ukierunkowane na wykrycie ich przyczyny. Do badań tych może należeć biopsja endometrium w celu wykluczenia nowotworu złośliwego endometrium.
Dane zbiorcze potwierdzają wzrost ryzyka wystąpienia nowotworu piersi u kobiet stosujących złożone produkty estrogenu z progestagenem lub terapię HTZ opartą na podawaniu samego estrogenu, w zależności od czasu trwania terapii HTZ.
Terapia skojarzona estrogenami z progestagenem
Randomizowane, kontrolowane badanie kliniczne z użyciem placebo – Inicjatywa na rzecz Zdrowia Kobiet (ang. Women’s Health Initiative study, WHI) oraz metaanaliza prospektywnych badań epidemiologicznych przyniosły podobne wyniki, potwierdzające wzrost ryzyka wystąpienia nowotworu piersi u kobiet stosujących HTZ estrogenami w skojarzeniu z progestagenem. Wzrost ten jest widoczny po około 3 (1-4) latach (patrz punkt 4.8).
W badaniu WHI nie stwierdzono wzrostu ryzyka wystąpienia nowotworu piersi u kobiet z usuniętą macicą stosujących HTZ samymi estrogenami. W badaniach obserwacyjnych zgłaszano na ogół niewielki wzrost ryzyka rozpoznania nowotworu piersi, które było mniejsze niż ryzyko u kobiet stosujących leczenie skojarzone estrogenami z progestagenem (patrz punkt 4.8).
Wyniki szeroko zakrojonej metaanalizy wykazały, że po zaprzestaniu leczenia dodatkowe ryzyko z czasem maleje, a czas powrotu do poziomu początkowego zależy od czasu trwania HTZ. Jeśli HTZ trwała ponad 5 lat, ryzyko może się utrzymywać przez 10 lat lub dłużej.
Hormonalna terapia zastępcza, a w szczególności złożona terapia estrogenowo-progestagenowa, zwiększa gęstość radiologicznych obrazów mammograficznych, co może utrudniać radiologiczne wykrywanie nowotworu piersi.
Nowotwór jajnika występuje znacznie rzadziej niż nowotwór piersi. Z danych epidemiologicznych z dużej metaanalizy wynika nieznacznie zwiększone ryzyko wystąpienia nowotworu jajnika, które uwidacznia się w ciągu 5 lat stosowania i zmniejsza się w czasie po odstawieniu tych środków, u kobiet przyjmujących HTZ w postaci samych estrogenów lub skojarzenia estrogenów i progestagenów. Z niektórych innych badań, w tym badania WHI, wynika, że stosowanie skojarzonej HTZ może wiązać się z podobnym lub nieznacznie mniejszym ryzykiem wystąpienia nowotworu jajnika (patrz punkt 4.8).
Stosowanie HTZ wiąże się z 1,3-3-krotnym wzrostem ryzyka wystąpienia żylnej choroby zakrzepowo-zatorowej (ŻChZZ), tj. zakrzepicy żył głębokich lub zatorowości płucnej. Wystąpienie przypadków zakrzepicy żylnej jest bardziej prawdopodobne w pierwszym roku stosowania HTZ, niż w latach późniejszych (patrz punkt 4.8).
U pacjentek ze skłonnością do zakrzepicy żył ryzyko wystąpienia ŻChZZ jest większe, a HTZ może to ryzyko dodatkowo zwiększyć. Z tego względu stosowanie HTZ u tych pacjentek jest przeciwwskazane (patrz punkt 4.3).
Powszechnie uznane czynniki ryzyka wystąpienia ŻChZZ obejmują: stosowanie estrogenów, starszy wiek, rozległy zabieg operacyjny, dłuższe unieruchomienie, otyłość (wskaźnik masy ciała BMI >
30 kg/m), ciążę/okres poporodowy, toczeń rumieniowaty układowy (SLE) oraz nowotwór złośliwy. Badacze nie są zgodni, co do roli żylaków w występowaniu ŻChZZ.
Jak w przypadku wszystkich pacjentów poddanych operacji należy rozważyć profilaktykę, aby zapobiegać wystąpieniu ŻChZZ po zabiegu chirurgicznym. W przypadku dłuższego unieruchomienia pacjentki w związku z planowaną operacją, zaleca się tymczasowe przerwanie HTZ na 4 do 6 tygodni wcześniej. Nie należy rozpoczynać leczenia zanim pacjentka nie osiągnie pełnej aktywności ruchowej.
Kobietom bez ŻChZZ w wywiadzie, których krewny pierwszego stopnia doznał zakrzepicy w młodym wieku można zaproponować badania przesiewowe informując je wcześniej o ograniczeniach takich badań (jedynie część zaburzeń świadczących o skłonnościach do zakrzepicy jest wykrywana w badaniach przesiewowych). Jeśli badania wykażą zaburzenia świadczące o skłonności do zakrzepicy u kobiet z zakrzepicą w wywiadzie rodzinnym, lub jeśli zaburzenia te są ciężkie (niedobór antytrombiny, białka S lub białka C bądź współwystępowanie tych niedoborów), stosowanie HTZ jest przeciwwskazane.
U kobiet przewlekle stosujących leki przeciwzakrzepowe należy dokładnie rozważyć, czy ryzyko związane ze stosowaniem HTZ nie przewyższa oczekiwanych korzyści.
Jeśli ŻChZZ rozwinie się po rozpoczęciu terapii, produkt należy odstawić. Pacjentki należy poinformować, że jeśli stwierdzą u siebie wystąpienie ewentualnych objawów zakrzepicy (np. bolesny obrzęk kończyny dolnej, nagły ból w klatce piersiowej, duszność), powinny bezzwłocznie zgłosić się do lekarza.
Wyniki randomizowanych, kontrolowanych badań klinicznych nie wykazały działania zapobiegającego zawałowi mięśnia sercowego u kobiet z chorobą niedokrwienną serca lub bez tej
choroby, które otrzymywały leczenie skojarzone estrogenami z progestagenem lub HTZ samymi estrogenami.
Leczenie skojarzone estrogenami z progestagenem
Względne ryzyko wystąpienia choroby niedokrwiennej serca podczas stosowania skojarzonej HTZ estrogenami z progestagenami jest nieznacznie zwiększone. Ponieważ wyjściowe bezwzględne ryzyko wystąpienia choroby niedokrwiennej serca w dużym stopniu zależy od wieku, u zdrowych kobiet w wieku okołomenopauzalnym liczba dodatkowych przypadków choroby niedokrwiennej serca spowodowanych stosowaniem estrogenów w skojarzeniu z progestagenem jest bardzo niewielka, natomiast wzrasta ona wraz z wiekiem pacjentki.
W randomizowanych, kontrolowanych badaniach nie stwierdzono wzrostu ryzyka wystąpienia
choroby niedokrwiennej serca u kobiet po histerektomii stosujących terapię samymi estrogenami.
Terapia estrogenami w skojarzeniu z progestagenem oraz terapia samymi estrogenami związana jest z maksymalnie 1,5-krotnym wzrostem ryzyka wystąpienia udaru niedokrwiennego. Ryzyko względne nie zmienia się wraz z wiekiem lub w miarę upływu czasu od menopauzy. Ponieważ jednak wyjściowe ryzyko udaru jest w dużym stopniu zależne od wieku, całkowite ryzyko udaru u kobiet stosujących HTZ będzie wzrastać wraz z wiekiem (patrz punkt 4.8).
Pacjentki wymagające stosowania terapii zastępczej hormonami tarczycy powinny regularnie kontrolować czynność tarczycy podczas przyjmowania HTZ, aby upewnić się, że stężenie hormonów tarczycy mieści się w dopuszczalnym zakresie wartości.
Estrogeny mogą wywoływać lub zaostrzać objawy obrzęku naczynioruchowego, w szczególności u kobiet z dziedzicznym obrzękiem naczynioruchowym.
Ciężkie reakcje anafilaktyczne/ rzekomoanafilaktyczne
Po wprowadzeniu leku do obrotu zgłaszano przypadki reakcji anafilaktycznych/ rzekomoanafilaktycznych występujących na różnych etapach leczenia estradiolem i wymagających nagłej pomocy medycznej. Reakcje te obejmowały skórę (pokrzywka, świąd, obrzęk twarzy, gardła, warg, języka, skóry i obrzęk okostnej oczodołu) oraz układ oddechowy (zaburzenia oddychania) lub układ pokarmowy (ból brzucha, wymioty).
Stosowanie estrogenów może prowadzić do zatrzymania płynów, w związku z czym pacjentki z zaburzeniami czynności serca lub nerek należy starannie obserwować.
Kobiety z wcześniej istniejącą hipertrójglicerydemią muszą być dokładnie obserwowane w trakcie estrogenowej terapii zastępczej lub hormonalnej terapii zastępczej, gdyż stwierdzono rzadkie przypadki znacznego zwiększenia stężenia trójglicerydów w osoczu, prowadzące do zapalenia trzustki podczas doustnej terapii estrogenowej u kobiet z tym zaburzeniem.
Estrogeny zwiększają stężenia globuliny wiążącej tyroksynę (ang. TBG), co prowadzi do zwiększenia stężenia całkowitej ilości tyroksyny krążącej, mierzonej poprzez obliczenie stężenia jodu związanego z białkami (ang. PBI), stężeniem T4 (metodą kolumnową lub radioimmunologiczną) lub T3 (metodą
radioimmunologiczną). Dochodzi do spadku wychwytu T3 na żywicy, co jest konsekwencją zwiększonego stężenia TBG. Nie stwierdza się zmian stężenia wolnej T4 i T3. Może dochodzić do zwiększenia stężenia w osoczu innych białek wiążących, tj. globuliny wiążącej kortykosteroidy (ang. CBG), globuliny wiążącej hormony płciowe (ang. SHBG), co prowadzi odpowiednio do zwiększenia stężenia krążących kortykosteroidów i hormonów płciowych. Nie stwierdza się zmian stężenia hormonów w postaci niezwiązanej lub biologicznie czynnej. Może dochodzić do zwiększenia stężenia innych białek w osoczu (angiotensynogenu/substratu reniny, alfa-1-antytrypsyny, ceruloplazminy)
Stosowanie HTZ nie poprawia funkcji poznawczych. Istnieją pewne dowody na temat zwiększonego ryzyka wystąpienia otępienia u kobiet, które rozpoczęły stosowanie ciągłej terapii skojarzonej lub HTZ samymi estrogenami po 65 roku życia.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Obserwuje się maksymalnie 2-krotny wzrost ryzyka rozpoznania nowotworu piersi u kobiet stosujących
leczenie skojarzone estrogenami z progestagenem przez ponad 5 lat.
Wszelki wzrost ryzyka u pacjentek stosujących terapię samymi estrogenami jest mniejszy niż ryzyko obserwowane u pacjentek otrzymujących leczenie skojarzone estrogenami z progestagenem.
Wielkość ryzyka zależy od czasu stosowania leku (patrz punkt 4.4).
Ryzyko całkowite oszacowane na podstawie wyników największego randomizowanego badania kontrolowanego placebo (badania WHI) oraz największej metaanalizy prospektywnych badań epidemiologicznych przedstawiono poniżej.
Ryzyko wystąpienia choroby niedokrwiennej serca jest nieznacznie zwiększone u pacjentek stosujących HTZ opartą na leczeniu skojarzonym estrogenami z progestagenem w wieku powyżej 60 lat (patrz punkt 4.4).
Stosowanie leczenia samymi estrogenami oraz leczenia skojarzonego estrogenami z progestagenem wiąże sie z maksymalnie 1,5-krotnym wzrostem względnego ryzyka wystąpienia udaru niedokrwiennego. Ryzyko wystąpienia udaru krwotocznego nie ulega zwiększeniu podczas stosowania HTZ.
To względne ryzyko nie zależy od wieku pacjentki ani od czasu trwania leczenia, ponieważ jednak wyjściowe ryzyko jest silnie zależne od wieku, całkowite ryzyko udaru u kobiet stosujących HTZ wzrasta wraz z wiekiem, patrz punkt 4.4.
Połączone dane z badań WHI – Dodatkowy wzrost ryzyka udaru niedokrwiennego* w okresie 5 lat stosowania leczenia
Zakres
Częstość występowania na
Współczynnik ryzyka i
Liczba dodatkowych przypadków na
wiekowy (lata)
1000 kobiet w grupie placebo
w okresie 5 lat
95% CI
1000 pacjentek stosujących HTZ przez
okres 5 lat
50-59
8
1,3 (1,1- 1,6)
3 (1-5)
* Nie wprowadzono rozróżnienia pomiędzy udarem niedokrwiennym a udarem krwotocznym
Inne działania niepożądane odnotowane w związku z terapią estrogenowo-progestagenową:
Choroba pęcherzyka żółciowego
Zaburzenia skóry i tkanki podskórnej: ostuda, rumień wielopostaciowy, rumień guzowaty, plamica
naczyniowa
Prawdopodobne otępienie po 65 roku życia (patrz punkt 4.4)
Zespół suchego oka
Zmiana składu filmu łzowego
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
ESTALIS 0,5 mg; 50 μg/24h + 4,8 mg; 250 μg/24h, system transdermalny
ESTALIS 0,6 mg; 50 μg/24h + 2,7 mg; 140 μg/24h, system transdermalny
Substancje czynne: estradiol półwodny i noretysteronu octan.
Jeden system transdermalny Estalis zawiera 0,5 mg estradiolu półwodnego (Estradiolum hemihydratum)
i 4,8 mg noretysteronu octanu (Norethisteroni acetas), co zapewnia dostarczenie 50 µg estradiolu i 250 µg noretysteronu octanu na dobę z powierzchni 16 cm2 lub jeden system transdermalny zawiera 0,6 mg estradiolu półwodnego (Estradiolum hemihydratum) i 2,7 mg noretysteronu octanu (Norethisteroni acetas) co zapewnia dostarczenie 50 µg estradiolu i 140 µg noretysteronu octanu na dobę z powierzchni 9 cm2.
Nazwa | Nominalna szybkość uwalniania mikrogramy/dobę estradiol/ noretysteronu octan | Zawartość estradiolu półwodnego (mg)* | Zawartość noretysteronu octanu (mg) | Powierzchnia (cm2) | Kształt |
Estalis | 50/250 | 0,5 | 4,8 | 16 | okrągły |
Estalis | 50/140 | 0,6 | 2,7 | 9 | okrągły |
* 1 mg estradiolu półwodnego odpowiada 0,968 mg estradiolu.
Substancje czynne są uwalniane przez 4 dni.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Dorośli i pacjenci w podeszłym wieku
We wszystkich wskazaniach stosuje się najmniejszą skuteczną dawkę.
Hormonalną terapię zastępczą (HTZ), w tym podawanie samych estrogenów lub podawanie estrogenów i progestagenu w leczeniu skojarzonym, należy stosować, dopóki w każdym indywidualnym przypadku korzyści wynikające z leczenia przewyższają ryzyko.
Rozpoczęcie leczenia
U pacjentek po menopauzie, nie stosujących produktów estrogenowo-progestagenowych, leczenie produktem Estalis można rozpocząć w dowolnym czasie.
Pacjentki „przechodzące” z ciągłej sekwencyjnej terapii estrogenowo-progestagenowej powinny rozpocząć stosowanie produktu Estalis po zakończeniu dotychczasowego cyklu leczenia. Po zakończeniu cyklu leczenia w terapii ciągłej sekwencyjnej, często występuje krwawienie z odstawienia. Pierwszy dzień krwawienia zwykle jest dniem rozpoczęcia nowego cyklu leczenia produktem Estalis.
Schemat leczenia
Zarówno na początku leczenia jak i w czasie trwania leczenia należy stosować najmniejszą skuteczną dawkę, przez jak najkrótszy czas. Estalis jest stosowany w sposób ciągły (stosowany nieprzerwanie). W czterotygodniowym cyklu nakłada się na skórę jeden system transdermalny, co 3-4 dni. Należy poinformować pacjentki, że przez kilka pierwszych miesięcy leczenia mogą wystąpić nieregularne krwawienia z macicy, zazwyczaj w okresie zanim miesiączkowanie trwale zaniknie.
Systemy transdermalne Estalis należy stosować starannie, nalepiając na skórę czystą, suchą, bez otarć
i podrażnień (nie należy stosować żadnych kremów nawilżających, mleczek ani olejków kosmetycznych). System transdermalny umieszcza się na gładkiej powierzchni skóry brzucha lub pośladków (gdy tylko to możliwe), w miejscu pozbawionym fałdów skórnych. Nie należy nalepiać systemu transdermalnego
w okolicy talii, gdyż ściśle przylegające tam ubranie może spowodować jego odlepienie. Produktu Estalis nie wolno naklejać na skórę piersi lub ich okolice. System transdermalny należy zmieniać, co 3 lub 4 dni, umieszczając go każdorazowo w innym miejscu. Należy zachować, co najmniej 1 tydzień przerwy między przylepieniem kolejnego systemu transdermalnego w tym samym miejscu.
Po otwarciu saszetki zawierającej system transdermalny należy usunąć połowę folii ochronnej nie dotykając
przy tym przylepnej powierzchni systemu transdermalnego.

System transdermalny należy nakleić na skórę jak najszybciej. Następnie należy usunąć drugą połowę folii
ochronnej.
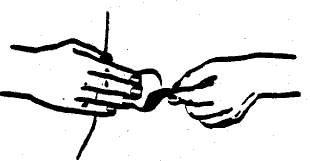
Przyciskając system transdermalny dłonią do skóry przez ok. 10 sekund, należy ostrożnie wygładzić jego brzegi.
Należy uważać, aby system transdermalny nie odlepił się w czasie kąpieli lub podczas wykonywania innych czynności. Wzmożona aktywność fizyczna, silne pocenie się lub tarcie ubrania mogą spowodować odklejenie systemu transdermalnego. Można wtedy ten sam system transdermalny nakleić w inne miejsce na skórze. W razie konieczności, można nakleić nowy system transdermalny kontynuując pierwotny schemat leczenia. Należy unikać długotrwałego działania światła słonecznego na system transdermalny przyklejony na skórę.
System transdermalny należy odklejać ostrożnie i powoli, aby uniknąć podrażnienia skóry. Jeśli na skórze pozostanie substancja klejąca, miejsce należy wysuszyć przez 15 minut, a następnie można ją zmyć tłustym kremem lub mleczkiem kosmetycznym.
W przypadku, gdy pacjentka zapomni nakleić system transdermalny, powinna to uczynić najszybciej jak to tylko możliwe według stosowanego schematu leczenia. Przerwa w leczeniu mogłaby zwiększyć prawdopodobieństwo wystąpienia nawrotów objawów choroby, krwawienia śródcyklicznego i plamienia.
Szczególne populacje pacjentów
Pacjenci z zaburzeniami czynności nerek i (lub) wątroby
Nie przeprowadzono badań z udziałem pacjentek z zaburzeniami czynności nerek i wątroby. Wszystkie produkty estrogenowe są przeciwwskazane u pacjentek z ostrą chorobą wątroby lub chorobą wątroby w wywiadzie, jeżeli wyniki prób wątrobowych nie powróciły do normy (patrz punkt 4.3).
Dzieci i młodzież
Produktu leczniczego Estalis nie należy stosować u dzieci i młodzieży.
Hormonalną terapię zastępczą (HTZ) można rozpocząć tylko w przypadku występowania objawów menopauzy niekorzystnie wpływających na jakość życia pacjentki. We wszystkich przypadkach, przynajmniej raz w roku należy przeprowadzić dokładną ocenę ryzyka i korzyści wynikających ze stosowania HTZ i leczenie kontynuować tak długo, jak korzyści przewyższają ryzyko związane z jego stosowaniem.
Dane dotyczące ryzyka związanego ze stosowaniem HTZ u kobiet z przedwczesną menopauzą są ograniczone. Jednak z uwagi na niskie ryzyko bezwzględne u młodszych kobiet, stosunek korzyści do ryzyka u tych pacjentek może być wyższy niż u pacjentek starszych.
Przed rozpoczęciem lub ponownym wdrożeniem HTZ należy przeprowadzić pełny wywiad osobisty
i rodzinny. Badanie przedmiotowe (włącznie z badaniem ginekologicznym i badaniem piersi) powinno być przeprowadzane i obejmować dane uzyskane z wywiadu oraz uwzględniać treść punktu 4.3 Przeciwwskazania i punktu 4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania. W trakcie leczenia zaleca się przeprowadzanie okresowych badań kontrolnych, których częstotliwość i charakter powinny być dostosowywane indywidualnie. Pacjentki należy poinformować o tym, że wszelkie zmiany w obrębie piersi powinny zgłaszać lekarzowi lub pielęgniarce (patrz rozdział Nowotwór piersi poniżej).
Badania dodatkowe, w tym odpowiednie techniki obrazowania, np. mammografię, należy przeprowadzać zgodnie z aktualnie przyjętymi metodami wykonywania badań przesiewowych, w sposób zmodyfikowany zależnie od potrzeb klinicznych danej osoby.
Stany, które wymagają obserwacji
Pacjentkę należy poddać starannej obserwacji w razie obecności któregokolwiek z wymienionych poniżej stanów w danym momencie lub w przeszłości i (lub) zaostrzenia ich w ciąży lub w trakcie poprzedniego leczenia hormonalnego. Należy uwzględnić fakt, że mogą one nawracać lub nasilać się w trakcie stosowania estradiolu/octanu noretysteronu w dawce 50 g/250 g/24 godziny. Chodzi tu szczególnie o następujące stany:
Metabolizm estrogenów i progestagenów może zostać nasilony w przypadku równoczesnego stosowania substancji, które są znane jako aktywatory enzymów uczestniczących w metabolizmie leku, zwłaszcza enzymów cytochromu P450 – takich jak środki przeciwdrgawkowe (np. fenobarbital, fenytoina, karbamazepina) oraz leki przeciwzakaźne (np. ryfampicyna, ryfabutyna, newirapina, efawirenz).
Estradiol jest metabolizowany głównie przez CYP3A4; dlatego jednoczesne podawanie inhibitorów CYP3A4 takich jak ketokonazol, erytromycyna lub ritonawir może spowodować zwiększenie pola AUC estradiolu o około 50%.
Rytonawir, telaprewir i nelfinawir, o których wiadomo, że są silnymi inhibitorami enzymów cytochromu P450, kiedy są stosowane równocześnie z hormonami steroidowymi wykazują właściwości indukujące. Produkty lecznicze roślinne zawierające ziele dziurawca zwyczajnego (Hypericum perforatum) mogą nasilać metabolizm estrogenów (i progestagenów).
Podczas stosowania systemów transdermalnych nie występuje efekt pierwszego przejścia przez wątrobę, w związku z czym na estrogeny i progestageny podawane przezskórnie mniejszy wpływ wywierają substancje indukujące działanie enzymów niż ma to miejsce w przypadku tych samych hormonów podawanych doustnie.
Pod względem klinicznym, zwiększony metabolizm estrogenów i progestagenów może zmniejszać ich działanie oraz prowadzić do nieregularnych krwawień z macicy.
Zastosowanie estrogenów może wpłynąć na zmianę niektórych wyników badań laboratoryjnych, takich jak
test tolerancji glukozy lub badań monitorujących czynność tarczycy.
Stosowanie estradiolu/octanu noretysteronu w dawce 50 g/250 g/24 godziny jest przeciwwskazane w czasie ciąży. W przypadku zajścia w ciążę podczas stosowania estradiolu/octanu noretysteronu w dawce 50 g/250 g/24 godziny, leczenie należy natychmiast przerwać.
Dane kliniczne z udziałem ograniczonej liczby ciężarnych pacjentek wskazują na brak działań niepożądanych octanu noretysteronu na płód. W przypadku przyjmowania wyższych dawek niż zazwyczaj stosowane w terapii doustnymi środkami antykoncepcyjnymi oraz w preparatach HTZ obserwowano maskulinizację płodów żeńskich.
Wyniki większości dotychczas przeprowadzonych badań epidemiologicznych odnoszących się do niezamierzonej ekspozycji płodu na skojarzenie estrogenów z progestagenami wskazują na brak teratogennych lub toksycznych działań na płód.
Stosowanie estradiolu/octanu noretysteronu w dawce 50 g/250 g/24 godziny w czasie karmienia piersią
jest przeciwwskazane.
Płodność
Nie dotyczy.
Estalis nie ma wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Można oczekiwać, że działania niepożądane wystąpią u około jednej trzeciej kobiet leczonych estradiolem/octanem noretysteronu w dawce 50 g/250 g/24 godziny. Do najczęściej zgłaszanych działań niepożądanych należą: napięcie i bolesność piersi (31%), reakcje w miejscu stosowania (20% stanowi rumień o nasileniu przeważnie łagodnym), bolesne miesiączkowanie (19%), nieregularne krwawienie (12,7%) oraz ból głowy (10%).
Tabela 1:
W obrębie każdej grupy o określonej częstości występowania objawy niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem. Bardzo często (≥1/10); Często (≥1/100 do <1/10); Niezbyt często
(≥1/1 000 do <1/100); Rzadko (≥1/10 000 do <1/1 000); Bardzo rzadko (<1/10 000); Częstość nieznana (nie
może być oszacowana na podstawie dostępnych danych).
Bardzo często | Często | Niezbyt | Rzadko | Bardzo | Częstość | |
(≥1/10) | (≥1/100 do | często | (≥1/10 000 do | rzadko | nieznana** | |
<1/10) | (≥1/1 000 do <1/100) | <1/1 000) | (<1/10 000) | (niemożliwa do oszacowania na podstawie | ||
dostępnych | ||||||
danych) | ||||||
Zaburzenia | Nadwrażliwość | Reakcje | ||||
układu | anafilaktycz- | |||||
immunologicz- | ne, reakcje | |||||
nego | rzekomoana- | |||||
filaktyczne | ||||||
Zaburzenia | Depresja*, | Zaburzenia libido | ||||
psychiczne | nerwowość*, | |||||
chwiejność | ||||||
emocjonalna | ||||||
Zaburzenia układu nerwowego | Ból głowy* | Zawroty głowy*, bezsenność* | Migrena, zawroty głowy | Parestezje | ||
Zaburzenia naczyniowe | Nadciśnienie, żylaki | Zator żylny | ||||
Zaburzenia | Nudności, | Wymioty | ||||
żołądka i jelit | wzdęcie | |||||
brzucha*, | ||||||
biegunka*, |
niestrawność*, ból brzucha | ||||||
Zaburzenia wątroby i dróg żółciowych | Choroba pęcherzyka żółciowego, kamica żółciowa | Żółtaczka cholestatyczna | ||||
Zaburzenia skóry i tkanki podskórnej | Trądzik*, wysypka, świąd*, suchość skóry | Przebarwienie skóry | Łysienie, kontaktowe zapalenie skóry | |||
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Ból pleców*, ból kończyn* | Osłabienie mięśni | ||||
Zaburzenia układu rozrodczego i piersi | Ból piersi*, tkliwość piersi, bolesne miesiączkowa- nie*, zaburzenia miesiączkowa- nia* | Powiększenie piersi*, krwotok miesiączkowy*, wydzielina z dróg rodnych*, nieregularne krwawienia z dróg rodnych, skurcze macicy, zakażenie pochwy, hiperplazja endometrium | Nowotwór piersi | Mięśniaki gładkokomórko- we macicy, torbiele jajowodu, polipy wewnątrzszyj- kowe macicy | ||
Zaburzenia ogólne i stany w miejscu podania | Reakcje w miejscu stosowania*** | Ból, osłabienie, obrzęki obwodowe*, przyrost masy ciała* | ||||
Badania diagnostyczne | Wzrost aktywności transaminaz |
(*) Działania niepożądane związane z zastosowaniem estrogenu i progestagenu względnie rzadziej występowały po zastosowaniu produktu leczniczego w najmniejszych dawkach.
(**) Zgłaszane po wprowadzeniu produktu leczniczego do obrotu.
(***) Do reakcji w miejscu stosowania leku należy miejscowe krwawienie, zasinienie, pieczenie, uczucie dyskomfortu, suchość, wyprysk, obrzęk, rumień, zapalenie, podrażnienie, ból, grudki, parestezje, świąd, wysypka, przebarwienia skóry, pigmentacja skóry, opuchlizna, pokrzywka i powstawanie pęcherzy.
Największa metaanaliza prospektywnych badań epidemiologicznych
Oszacowane dodatkowe ryzyko raka piersi po 5 latach leczenia u kobiet z BMI równym 27 (kg/m2)
Wiek na początku HTZ (lata) | Zapadalność na 1000 kobiet, w ogóle niestosujących HTZ przez okres 5 lat (50-54 lata)* | Współczynnik ryzyka | Dodatkowe przypadki na 1000 kobiet stosujących HTZ po 5 latach |
HTZ samymi estrogenami | |||
50 | 13,3 | 1,2 | 2,7 |
Leczenie skojarzone estrogenami z progestagenem | |||
50 | 13,3 | 1,6 | 8,0 |
Uwaga: Ponieważ częstość występowania nowotworu piersi jest różna w różnych krajach UE, liczba dodatkowych przypadków nowotworu piersi będzie się także proporcjonalnie zmieniać. | |||
*Na podstawie wyjściowej zapadalności w Anglii w 2015 r. u kobiet z BMI równym 27 (kg/m2).
Oszacowane dodatkowe ryzyko raka piersi po 10 latach leczenia u kobiet z BMI równym 27 (kg/m2)
Wiek na początku HTZ (lata) | Zapadalność na 1000 kobiet, w ogóle niestosujących HTZ przez okres 10 lat (50-59 lat)* | Współczynnik ryzyka | Dodatkowe przypadki na 1000 kobiet stosujących HTZ po 10 latach |
HTZ samymi estrogenami | |||
50 | 26,6 | 1,3 | 7,1 |
Leczenie skojarzone estrogenami z progestagenem | |||
50 | 26,6 | 1,8 | 20,8 |
Uwaga: Ponieważ częstość występowania nowotworu piersi jest różna w różnych krajach UE, liczba dodatkowych przypadków nowotworu piersi będzie się także proporcjonalnie zmieniać. | |||
*Na podstawie wyjściowej zapadalności w Anglii w 2015 r. u kobiet z BMI równym 27 (kg/m2).
Amerykańskie badania WHI – dodatkowy wzrost ryzyka wystąpienia nowotworu piersi po 5 latach stosowania leczenia
Zakres wiekowy (lata) | Częstość występowania na 1000 kobiet w grupie placebo przez 5 lat stosowania | Współczynnik ryzyka i 95% CI | Liczba dodatkowych przypadków na 1000 pacjentek stosujących HTZ przez 5 lat (95% CI) |
Terapia samymi skoniugowanymi estrogenami końskimi (CEE) | |||
50-79 | 21 | 0,8 (0,7 – 1,0) | -4 (-6 – 0)* |
Terapia estrogenami w skojarzeniu z progestagenem CEE+MPA ‡ | |||
50-79 | 17 | 1,2 (1,0 – 1,5) | +4 (0 – 9) |
‡Po tym, jak analizę danych ograniczono do kobiet niestosujących HTZ przed badaniem, nie stwierdzono wzrostu
ryzyka w ciągu pierwszych 5 lat leczenia; po upływie 5 lat ryzyko to było większe niż u kobiet niestosujących terapii.
*Badanie WHI u kobiet po usunięciu macicy, w którym nie obserwowano wzrostu ryzyka wystąpienia nowotworu
piersi.
Kobiety po menopauzie z zachowaną macicą
Ryzyko wystąpienia raka endometrium wynosi około 5 na każde 1000 kobiet z zachowaną macicą niestosujących HTZ.
U kobiet z zachowaną macicą nie zaleca się stosowania HTZ samymi estrogenami z uwagi na wzrost ryzyka
raka endometrium (patrz punkt 4.4).
W zależności od czasu trwania terapii samymi estrogenami i dawki estrogenów wzrost ryzyka wystąpienia raka endometrium w badaniach epidemiologicznych wahał się od 5 do 55 dodatkowych przypadków na każde 1000 kobiet w wieku od 50 do 65 lat.
Dodanie progestagenu do terapii samymi estrogenami, przez co najmniej 12 dni każdego cyklu leczenia może zapobiegać temu wzrostowi ryzyka. W badaniu MWS stosowanie przez 5 lat HTZ opartej na skojarzeniu estrogenów z progestagenami (w terapii sekwencyjnej lub ciągłej) nie spowodowało wzrostu ryzyka raka endometrium (współczynnik ryzyka 1,0 (0,8 – 1,2)).
Stosowanie HTZ obejmującej jedynie estrogeny lub skojarzenie estrogenów z progestagenami wiąże się z
nieznacznie zwiększonym ryzykiem rozpoznania nowotworu jajnika (patrz punkt 4.4).
Metaanaliza 52 badań epidemiologicznych wykazała zwiększone ryzyko wystąpienia nowotworu jajnika u kobiet aktualnie stosujących HTZ w porównaniu do kobiet, które nigdy nie stosowały HTZ (RW 1,43%, 95% PU 1,31-1,56). U kobiet w wieku od 50 do 54 lat stosowanie HTZ przez 5 lat może spowodować 1 dodatkowe rozpoznanie na 2000 stosujących. Wśród kobiet w wieku od 50 do 54 lat, które nie stosują HTZ, nowotwór jajnika zostanie rozpoznany w okresie 5 lat u 2 na 2000 kobiet.
Ryzyko żylnej choroby zakrzepowo-zatorowej
HTZ jest związana z 1,3-3-krotnym wzrostem względnego ryzyka wystąpienia żylnej choroby zakrzepowo- zatorowej (ŻChZZ), tzn. zakrzepicy żył głębokich lub zatorowości płucnej. Wystąpienie tych działań niepożądanych jest bardziej prawdopodobne w pierwszym roku stosowania terapii hormonalnej (patrz punkt 4.4). Poniżej przedstawiono wyniki badań WHI:
Badania WHI – Dodatkowe ryzyko ŻChZZ w okresie 5 lat stosowania leczenia
Zakres wiekowy (lata) | Częstość występowania na 1000 kobiet w grupie placebo w okresie 5 lat | Współczynnik ryzyka i 95% CI | Liczba dodatkowych przypadków na 1000 pacjentek stosujących HTZ |
Doustne leczenie samymi estrogenami* | |||
50-59 | 7 | 1,2 (0,6-2,4) | 1 (-3 – 10) |
Doustne leczenie skojarzone estrogenami z progestagenem | |||
50-59 | 4 | 2,3 (1,2 – 4,3) | 5 (1 - 13) |
*Badanie prowadzone z udziałem kobiet z usuniętą macicą
Ryzyko wystąpienia choroby niedokrwiennej serca
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem:
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Ze względu na drogę podania, przedawkowanie estradiolu lub octanu noretysteronu jest mało prawdopodobne. Jeśli jednak objawy przedawkowania wystąpią, należy odlepić system transdermalny zawierający estradiol/octan noretysteronu w dawce 50 g/250 g/24 godziny. Do objawów przedawkowania estrogenów doustnych należą tkliwość piersi, nudności, wymioty i (lub) krwotok maciczny.
Przedawkowanie progestagenów może również prowadzić do obniżenia nastroju, zmęczenia, wystąpienia trądziku oraz nadmiernego owłosienia.
Kod ATC: G03FA01
Estradiol
Jak w przypadku wszystkich hormonów steroidowych działania metaboliczne estrogenów zachodzą
w komórkach. Estrogeny wchodzą w interakcje ze swoistymi receptorami w komórkach narządów docelowych, tworząc kompleks modulujący transkrypcję genową i następującą po niej syntezę białka. Takie receptory zidentyfikowano w różnych narządach, np. w podwzgórzu, przysadce, pochwie, cewce moczowej, macicy, sutkach i wątrobie oraz w osteoblastach.
Estradiol, który od pierwszej miesiączki aż do menopauzy jest wytwarzany głównie przez pęcherzyki jajnikowe, należy do najbardziej aktywnych estrogenów. Jest on w dużej mierze odpowiedzialny za rozwój i prawidłowe funkcjonowanie układu moczowo-płciowego kobiet oraz za drugorzędowe cechy płciowe.
Po menopauzie, gdy czynność jajników ustaje, wytwarzane są jedynie niewielkie ilości estradiolu, poprzez aromatyzację androstendionu i, w mniejszym stopniu, testosteronu przez enzym aromatazę, w wyniku czego powstają odpowiednio: estron i estradiol. Estron jest dalej przekształcany do estradiolu pod wpływem enzymu dehydrogenazy 17beta-hydroksysteroidowej. Oba enzymy występują w tkance tłuszczowej, mięśniowej i w wątrobie.
U wielu kobiet zakończenie wytwarzania estradiolu przez jajniki wiąże się z występowaniem objawów naczynioruchowych (uderzenia gorąca), zaburzeniami snu i postępującą atrofią układu moczowo-płciowego. Zaburzenia te można w dużym stopniu wyeliminować stosując estrogenową terapię zastępczą. Wykazano również, że HTZ lub stosowanie estrogenów jest skuteczne w zapobieganiu scieńczaniu skóry obserwowanym po menopauzie.
Według dobrze ugruntowanego poglądu estrogenowa terapia zastępcza zapobiega pomenopauzalnej utracie tkanki kostnej, zwłaszcza, jeśli rozpoczyna się ją na wczesnym etapie menopauzy.
Noretysteronu octan
Noretysteronu octan (NETA) jest silnym progestagenem imitującym biologiczne działanie endogennego progesteronu. Jest on hydrolizowany w skórze do noretysteronu (NET), czynnego hormonu obecnego w krwiobiegu.
Progesteron zmniejsza liczbę receptorów estradiolowych w narządach docelowych oraz pobudza działanie dehydrogenazy 17beta-hydroksysteroidowej, która miejscowo utlenia estradiol do słabszego estrogennego metabolitu, estronu.
Jednym z głównych narządów docelowych dla progestagenów jest macica. U kobiet przed menopauzą oraz
u kobiet po menopauzie otrzymujących cykliczną HTZ progestageny wywołują przemianę endometrium
w fazie wydzielniczej, po czym endometrium ulega złuszczeniu.
U większości kobiet octan noretysteronu podawany przezskórnie jest skuteczny w dawkach mniejszych niż
w przypadku doustnej drogi podania z uwagi na brak metabolizmu pierwszego przejścia.
Skojarzenie estradiolu i NETA
Stosowanie samych estrogenów zwiększa częstość występowania rozrostu endometrium oraz ryzyko raka endometrium. W badaniach stwierdzono, że dodatkowe podanie progestagenu, przez co najmniej 10 dni cyklu stosowania estrogenu znacznie zmniejsza częstość występowania rozrostu endometrium, a przez to również częstość występowania nieregularnych krwawień i raka endometrium w porównaniu z leczeniem samym estrogenem. Natomiast zastosowanie tego cyklicznego schematu leczenia skutkuje wystąpieniem regularnego złuszczania stymulowanego estrogenem endometrium (comiesięczne krwawienia), podanie produktu Estalis, w sposób ciągły/złożony estradiol/progestagen, skutkuje zanikiem endometrium i brakiem miesiączki.
Dalsze informacje o badaniach klinicznych z produktem Estalis
W badaniach klinicznych prowadzonych z udziałem kobiet po menopauzie, trwających od trzech miesięcy do jednego roku, Estalis zmniejszał liczbę oraz nasilenie przypadków uderzeń gorąca i potów. Miał on również pozytywny wpływ na inne wskaźniki jakości życia takie jak zaburzenia snu i czynności seksualne. Nie obserwowano żadnych działań niepożądanych dotyczących wartości ciśnienia krwi i wyników testów krzepnięcia.
Po podaniu produktu Estalis 50/140, Estalis 50/250 µ/dobę odnotowano zmniejszenie stężenia cholesterolu całkowitego, LDL-cholesterolu, apoproteiny B, lipoproteiny (a) i trójglicerydów w porównaniu ze stanem wyjściowym. Obserwowano również spadek stężenia HDL-cholesterolu. Wszystkie lipoproteiny osocza
pozostawały w zakresie wartości prawidłowych. Ponadto, stosunek całkowitego cholesterolu do HDL- cholesterolu oraz LDL-cholesterolu do HDL cholesterolu pozostawał niezmieniony względem wartości wyjściowych w okresie do jednego roku.
Estradiol
Po naklejeniu systemu transdermalnego Estalis stężenia estradiolu w surowicy oraz stosunek estronu do estradiolu mieszczą się w tych samych zakresach wartości jak w przypadku kobiet przed menopauzą we wczesnej fazie pęcherzykowej lub w środku tej fazy (wartości estradiolu > 40 pg/ml). Stężenia te utrzymują się przez cały 84 lub 96-godzinny okres stosowania leku.
Wielokrotne stosowanie systemów transdermalnych Estalis (50/250 µg/dobę, 50/140 µg/dobę) dało maksymalne stężenia estradiolu w surowicy (Cmax) w stanie równowagi dynamicznej wynoszące odpowiednio 71 i 73 pg/ml. Średnie stężenia estradiolu w surowicy wynosiły 52 i 46 pg/ml. Pod koniec okresu stosowania średnie stężenie estradiolu w surowicy wynosiło odpowiednio 46 i 30 pg/ml.
Noretysteronu octan
Wielokrotne stosowanie systemów transdermalnych Estalis (50/250 µg/dobę, 50/140 µg/dobę) dało maksymalne stężenia noretysteronu w surowicy (Cmax) w stanie równowagi dynamicznej wynoszące odpowiednio 1060 i 638 pg/ml. Średnie stężenia noretysteronu w surowicy w stanie stacjonarnym wynosiły odpowiednio 832 i 492 pg/ml. Pod koniec okresu stosowania średnie stężenie noretysteronu w surowicy wynosiło odpowiednio 681 i 393 pg/ml. Stężenia noretysteronu w osoczu wzrastały liniowo wraz ze zwiększeniem dawek NETA.
Minimalne wahania w stężeniach estradiolu i noretysteronu w osoczu wskazują na równomierną dystrybucję substancji przez cały okres stosowania leku. Po wielokrotnym zastosowaniu systemu transdermalnego nie dochodzi do kumulacji estradiolu i noretysteronu w krążeniu. Estradiol jest obecny w krwiobiegu głównie w postaci związanej z globuliną wiążącą hormony płciowe (SHBG) i z albuminą. W osoczu noretyndron wiąże się z SHBG i albuminą w około 90%.
Estradiol
Estradiol, po zastosowaniu na skórę, jest metabolizowany jedynie w niewielkim stopniu przez skórę. Nie
występuje również efekt pierwszego przejścia obserwowany po doustnym podaniu estrogenów. Terapeutyczne stężenia estradiolu w surowicy przy niższych stężeniach estronu i estronu skoniugowanego uzyskuje się po podaniu na skórę mniejszych dawek (dobowych i całkowitych) w porównaniu do terapii doustnej, osiągając stężenia bardziej zbliżone do tych obserwowanych u kobiet przed menopauzą.
Estradiol podany przezskórnie podlega takim samym przemianom jak hormon endogenny. Estradiol jest metabolizowany do estronu, a następnie – głównie w wątrobie – do estriolu, epiestriolu i estrogenów katecholowych, które następnie zostają skoniugowane z siarczanami i glukuronidami.
Noretysteronu octan
Noretyndron przechodzi intensywną redukcję pierścienia A, w wyniku, której powstają metabolity dihydro- i
tetrahydronoretyndron podlegające sprzęganiu. Eliminacja
Estradiol
Estradiol charakteryzuje się krótkim okresem półtrwania, wynoszącym ok. 2-3 godzin i dlatego po usunięciu systemu transdermalnego obserwuje się szybkie zmniejszenie stężenia w surowicy. Po odklejeniu systemu transdermalnego, w ciągu 4-8 godzin stężenie estradiolu w surowicy powraca do wartości obserwowanych u nieleczonych kobiet po menopauzie (<20 pg/ml).
Octan noretysteronu
Okres półtrwania eliminacji noretysteronu wynosi od 6 do 8 godzin. Po usunięciu systemu transdermalnego
Estalis stężenie noretysteronu w surowicy szybko zmniejsza się i w ciągu 48 godzin wynosi mniej niż
50 pg/ml.
Profil toksycznego działania estradiolu i noretysteronu jest dobrze udokumentowany. Długotrwałe, ciągłe podawanie estrogenów naturalnych lub sztucznych pewnym gatunkom zwierząt zwiększa częstość występowania nowotworu piersi, macicy, szyjki macicy, pochwy, jąder i wątroby. Długotrwałe, ciągłe podawanie noretysteronu pewnym gatunkom zwierząt zwiększa częstość występowania guzów przysadki i jajników u samic oraz wątroby i piersi u samców.
Silikonowa warstwa przylepna, akrylowa warstwa przylepna, powidon, kwas olejowy, glikol dipropylenowy, poliestrowa folia laminowana, fluoropolimer (warstwa ochronna).
30 miesięcy (24 miesiące w temperaturze 2-8°C i 6 miesięcy w temperaturze do 25°C).
Przechowywać w temperaturze 2-8°C przez okres 24 miesięcy i w temperaturze do 25°C nie dłużej niż
6 miesięcy.
8 lub 24 systemy transdermalne w tekturowym pudełku. Systemy transdermalne umieszczone w zgrzewanym opakowaniu z folii papier/polietylen /AL/polietylen.
Patrz punkt 4.2.
Zużyty system transdermalny należy złożyć klejącymi powierzchniami do siebie i wyrzucić tak, aby był niedostępny dla dzieci.
OBROTU
Novartis Poland Sp. z o.o. ul. Marynarska 15
02-674 Warszawa
Estalis, system transdermalny, 0,5 mg; 50 µg/24 h + 4,8 mg; 250 µg/24 h - Pozwolenie nr 8165 Estalis, system transdermalny, 0,6 mg; 50 µg/24 h + 2,7 mg; 140 µg/24 h - Pozwolenie nr 8166
DATA PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 21 czerwca 2000
Data ostatniego przedłużenia pozwolenia: 2 października 2013
PRODUKTU LECZNICZEGO
09/2020







