Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- NUMERY POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Zapobieganie zakrzepicy żył głębokich oraz komplikacjom zakrzepowo-zatorowym w sytuacjach klinicznych o podwyższonym ryzyku (szczególnie podczas zabiegów chirurgicznych i okresie okołoporodowym) w połączeniu z heparyną, o ile istnieją wskazania do jej podawania.
Zapobieganie rozwoju zakrzepicy żył głębokich lub komplikacjom zakrzepowo-zatorowym w połączeniu z heparyną jeśli jest to wskazane.
Nabyty niedobór antytrombiny.
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
dokonywanie właściwej selekcji dawców na podstawie wywiadu lekarskiego oraz badań przesiewowych pojedynczych donacji i pul osocza w kierunku obecności HBsAg oraz przeciwciał przeciw antygenom wirusów HIV i HCV.
badanie puli osocza w kierunku obecności materiału genetycznego wirusa HCV
zastosowania podczas produkcji zwalidowanych przy użyciu wirusów modelowych metod inaktywacji/usuwania wirusów. Procesy te uważa się za skuteczne wobec wirusów HIV, HCV, HBV oraz bezotoczkowego wirusa HAV.
W celu dostosowania dawki heparyny w taki sposób aby zapobiec nadmiernemu zmniejszeniu krzepliwości, należy kontrolować regularnie i w krótkich okresach czasu parametry układu krzepnięcia (APPT, i odpowiednio aktywność anty-FXa). Dotyczy to szczególnie pierwszych minut i godzin stosowania antytrombiny.
Należy codziennie dokonywać oznaczeń poziomu antytrombiny w celu ustalenia pojedynczej dawki ze względu na możliwość znacznego obniżenia poziomu antytrombiny wskutek jednoczesnego długotrwałego stosowania niefrakcjonowanej heparyny,
Interakcje z innymi lekami i inne rodzaje interakcji
Ciąża i laktacja
Wpływ na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności przy przechowywaniu
Rodzaj i zawartość opakowania
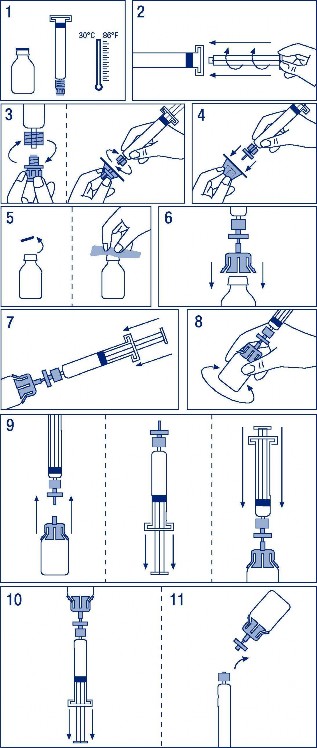
Szczególne środki ostrożności dotyczące usuwania i przygotowania leku do stosowania.
Anbinex, 50 j.m./ml, proszek i rozpuszczalnik do sporządzania roztworu do infuzji,
Ludzka antytrombina III
Anbinex, występuje w postaci liofilizowanego proszku w fiolkach zawierających nominalnie 500 j.m. lub 1000 j.m. antytrombiny pochodzącej z ludzkiego osocza
Po odtworzeniu w 10 ml (500 j.m.) lub 20 ml (1000 j.m.) wody do wstrzykiwań produkt zawiera około 50 j.m./ml antytrombiny ludzkiej.
Moc leku (w j.m.) określana jest przy użyciu metody chromogennej zgodnej z Farmakopeą Europejską. Aktywność swoista leku Anbinex wynosi co najmniej 5 j.m./mg białka.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Fiolka zawiera białą, hygroskopijną, kruchą substancję stałą lub proszek, ampułko-strzykawka zawiera wodę do wstrzykiwań (rozpuszczalnik).
Pacjenci z wrodzonym niedoborem antytrombiny:
Rozpoczęcie leczenia powinno odbywać się pod nadzorem lekarza posiadającego doświadczenie w leczeniu pacjentów z niedoborem antytrombiny
Dawkowanie
We wrodzonym niedoborze antytrombiny dawkowanie i czas trwania leczenia powinno być dostosowane indywidualnie dla każdego pacjenta w zależności od wyniku wywiadu rodzinnego uwzględniającego incydenty zakrzepowo-zatorowe, aktualnie występujące czynniki ryzyka klinicznego oraz wyniki badań laboratoryjnych.
Dawkowanie i czas trwania leczenia substytucyjnego w nabytym niedoborze antytrombiny zależy od poziomu antytrombiny w osoczu, obecności objawów wzrastającego zużycia, choroby podstawowej
oraz ciężkości objawów klinicznych. Wielkość dawek i częstotliwość ich podawania należy zawsze dostosować indywidualnie dla każdego pacjenta , zgodnie z wynikami badań lekarskich i laboratoryjnych.
Podawaną dawkę antytrombiny wyraża się w jednostkach międzynarodowych (j.m.) zgodnie z aktualnymi normami WHO. Aktywność antytrombiny w osoczu może być wyrażona w % (w stosunku do aktywności w normalnym osoczu) lub w jednostkach międzynarodowych (zgodnie z międzynarodowym standardem dla antytrombiny osoczowej).
Jedna jednostka międzynarodowa (j.m.) antytrombiny odpowiada przeciętnej ilości antytrombiny w jednym ml normalnego osocza ludzkiego. Obliczanie potrzebnej dawki antytrombiny, opiera się na obserwcji empirycznej, że podanie 1 j.m. antytrombiny na kg masy ciała powoduje wzrost aktywności antytrombiny w osoczu o około 1,1 % do 1,6 %.
Dawkę początkową oblicza się na podstawie następującego wzoru:
Wymagana liczba jednostek = masa ciała (kg) x (100 – wyjściowa aktywność antytrombiny (w procentach) x 0,8
W początkowej fazie leczenia należy ustalić pożądany poziom aktywności antytrombiny w zależności od sytuacji klinicznej. Po ustaleniu wskazań do stosowania antytrombiny należy podać taką dawkę aby uzyskać pożądany poziom aktywności antytrombiny, a następnie podtrzymywać poziom zapewniający skuteczność leczenia. Dawka powinna być obliczana i monitorowana laboratoryjnymi oznaczeniami aktywności antytrombiny osoczu. Oznaczenia powinny być przeprowadzane co najmniej dwa razy dziennie, a gdy stan pacjenta stabilizuje się – raz dziennie; zawsze tuż przed kolejnym podaniem leku. Należy pamiętać o tym, że w przypadku ciężkich stanów klinicznych, takich jak zespół rozsianego krzepnięcia śródnaczyniowego, okres półtrwania antytrombiny może być znacznie skrócony. Korekcje wielkości dawki należy dokonywać biorąc pod uwagę zarówno szybkość zużycia antytrombiny określaną na podstawie oznaczeń laboratoryjnych, jak i w oparciu o przebieg kliniczny. Aktywność antytrombiny należy utrzymywać powyżej 80% normy przez cały okres leczenia lub odpowiednio dostosować, gdy objawy kliniczne wskazują, że inny poziom może być bardziej skuteczny.
W leczeniu wrodzonego niedoboru początkowa dawka wynosi 30 – 50 j.m/kg masy ciała.
Następnie, wielkość dawki, częstotliwość podawania i długość okresu leczenia zależy od wyników badań i stanu klinicznego pacjenta
Stosowanie u dzieci
Nie zaleca się stosowania leku u dzieci poniżej 6 roku życia, gdyż doświadczenie dotyczące skuteczności i bezpieczeństwa stosowania w tej grupie wiekowej jest ograniczone.
Sposób podawania
Rozpuścić preparat zgodnie z zaleceniami (patrz punkt 6.6). Produkt podawać powoli dożylnie. Szybkość podawania nie powinna przekraczać 0,08 ml/kg/min.
Zaleca się zachowanie ostrożności u chorych o znanej nadwrażliwości na substancję czynną lub substancje pomocnicze.
Tak jak w przypadku innych produktów podawanych dożylnie, możliwe jest wystąpienie reakcji nadwrażliwości typu alergicznego. Podczas infuzji pacjent powinien być ściśle monitorowany z powodu ryzyka wystąpienia objawów niepożądanych. Pacjent powinien być poinformowany o wczesnych objawach reakcji nadwrażliwości, włączając w to wysypkę, uogólnioną pokrzywkę, uczucie ucisku w klatce piersiowej, świstów, podciśnienia i objawów anafilaksji. W przypadku wystąpienia tych objawów, należy powiadomić o tym lekarza.
W przypadku wystąpienia wstrząsu należy zastosować aktualnie zalecane leczenie przeciwwstrząsowe.
Nie można całkowicie wykluczyć prawdopodobieństwa przeniesienia czynników zakaźnych przez podanie preparatów otrzymywanych z krwi lub osocza ludzkiego. Dotyczy to również czynników zakaźnych i patogenów dotychczas nieznanych.
Ryzyko przeniesienia czynników zakaźnych może być ograniczone przez:
Skuteczność metod unieczynniania i usuwania wirusów bezotoczkowych jak np. parwowirus B19 oraz innych czynników zakaźnych może być ograniczona. Zakażenie parwowirusem B19 może być szczególnie groźne u kobiet w ciąży (zakażenie płodu) oraz u osób z obniżoną odpornością lub zwiększoną erytropoezą (np. w anemii hemolitycznej).
U pacjentów otrzymujących regularnie antytrombinę ludzką, należy rozważyć zastosowanie odpowiednich szczepień (przeciwko wirusowi zapalenia wątroby A i B).
Dla dobra pacjentów zdecydowanie zaleca się, aby przy każdym podaniu pacjentowi leku Anbinex odnotować nazwisko pacjenta i numer serii produktu, aby móc powiązać pacjenta z serią leku.
Kliniczne i biologiczne metody monitorowania leczenia, gdy antytronbinę stosuje się w skojarzeniu z heparyną;
Dzieci i młodzież
Dane z badań klinicznych i z systematycznych przeglądów dotyczących stosowania antytrombiny u wcześniaków w niezatwierdzonym wskazaniu: leczenie zespołu niewydolności oddechowej noworodków (IRDS, ang. Infant Respiratory Distress Syndrome), świadczą o zwiększonym ryzyku krwawienia śródczaszkowego i zgonu, bez wykazywania korzystnego działania w tej grupie pacjentów.
Heparyna: Podawanie antytrombiny równocześnie z terapeutycznymi dawkami heparyny zwiększa ryzyko krwawień. Efekt stosowania antytrombiny jest znacznie potęgowany przez heparynę. Równoczesne podawanie heparyny może znacznie zwiększyć zużycie i skrócić okres półtrwania antytrombiny. Jednoczesne podawanie heparyny pacjentom ze zwiększonym ryzykiem krwawień powinno być starannie monitorowane pod względem klinicznym i biologicznym.
Doświadczenie dotyczące bezpieczeństwa stosowania preparatów zawierających antytrombinę ludzką podczas ciąży jest ograniczone.
Lek Anbinex powinien być stosowany podczas ciąży i w okresie laktacji tylko wtedy, gdy jest to jednoznacznie wskazane. Decyzję należy podjąć po uwzględnieniu faktu, że podczas ciąży istnieje zwiększone ryzyko wystąpienia incydentów zakrzepowo-zatorowych.
Anbinex nie ma istotnego wpływu na zdolność do prowadzenia pojazdów i obsługiwania urządzeń mechanicznych w ruchu.
Rzadko obserwowano nadwrażliwość lub reakcje alergiczne (obrzęk naczynioruchowy, pieczenie lub uczucie kłucia w miejscu podania, dreszcze, zaczerwienienie twarzy, uogólniona pokrzywka, ból głowy, wysypka, spadek ciśnienia krwi, letarg, nudności, niepokój, tachykardia, uczucie ucisku w klatce piersiowej, świąd, wymioty, świszczący oddech), które w niektórych przypadkach prowadziły do rozwoju ciężkiej anafilaksji (włączając w to wstrząs).
W rzadkich przypadkach obserwowano gorączkę.
Informacje dotyczące zabezpieczeń przed przeniesieniem czynników zakaźnych (patrz punkt 4.4).
Nie obserwowano objawów przedawkowania.
Grupa farmakoterapeutyczna: Leki przeciwzakrzepowe, kod ATC: B01A B02
Antytrombina to składająca się z 432 aminokwasów glikoproteina o m.cz. 58 kD, należąca do rodziny serpin (białek inaktywujących proteazy serynowe). Jest jednym z głównych naturalnych inhibitorów układu krzepnięcia krwi. Przede wszystkim, hamuje czynność trombiny i czynnika Xa, a także czynników kontaktu, system wewnątrzpochodny oraz kompleks czynnik VIIa/czynnik tkankowy. Aktywność antytrombiny jest znacznie potęgowana przez heparynę, której działanie zależy od obecności antytrombiny.
Antytrombina posiada dwie ważne dla jej funkcji domeny.
W pierwszej dochodzi do inaktywacji trombiny przez antytrombinę co następuje przez utworzenie nieaktywnego kompleksu tych cząsteczek. Druga, stanowi miejsce łączenia się z
glikozaminoglikanami oraz współdziałania z heparyną i substancjami pokrewnymi biorącymi udział w potęgowaniu szybkości inaktywacji trombiny. Powstałe kompleksy enzymów inhibitorów krzepnięcia są następnie usuwane przez układ siateczkowo-śródbłonkowy.
Aktywność antytrombiny we krwi dorosłych wynosi od 80% do 120%, a u noworodków 40 – 60%.
Badania właściwości farmakokinetycznych antytrombiny wykazały, że średni biologiczny okres półtrwania wynosi około 3 dni. Może on ulec zmniejszeniu do około 1,5 dnia podczas jednoczesnego stosowania heparyny. Okres półtrwania może zostać skrócony do kilku godzin w przypadkach wzmożonego zużycia.
W badaniach klinicznych preparatu Anbinex u pacjentów z wrodzonym niedoborem antytrombiny z zastosowaniem niezależnej analizy do oceny wyników, otrzymano następujące wartości parametrów farmakokinetycznych:
Narastający odzysk (Incremental Recovery) 1,3 ± 0,2 w granicach od 1,1 do 1,6% w oparciu o analizę funkcji.
Powierzchnia pod krzywą (AUC) = 66461 ± 15445 j.m. h/l.
Końcowy okres półtrwania = 98,1 ± 45,0 h w oparciu o analizę funkcji.
Średni czas przebywania (MRT) = 121,7 ± 52,1 h. Klirens = 0,931 ± 0,214 ml/h/kg.
Antytrombina jest normalnym składnikiem ludzkiego osocza.
Badanie toksyczności po jednorazowej dawce ma niewielkie znaczenie i nie umożliwia oceny toksycznej lub letalnej dawki oraz zależności pomiędzy dawką a osiągniętym efektem.
Badanie toksyczności przewlekłej u zwierząt nie jest możliwe ze względu na tworzenie się przeciwciał.
Nie opisywano objawów toksyczności ostrej u badanych zwierząt.
Proszek:
D-Mannitol Sodu cytrynian Sodu chlorek. Rozpuszczalnik:
Woda do wstrzykiwań
Ze względu na brak wyników badań dotyczących niezgodności farmaceutycznych, leku nie należy mieszać z innymi lekami.
3 lata
Po odtworzeniu, chemiczne i fizyczne badania stabilności wskazują na okres trwałości do 12 godzin w temperaturze 25°C. Z punktu widzenia mikrobiologicznego, produkt powinien zostać zużyty natychmiast. Jeśli po odtworzeniu produkt nie został zużyty, może być przechowywany nie dłużej niż
24 godziny w temperaturze 2oC - 8oC, ale tylko wtedy, gdy odpowiedzialność za to weźmie użytkownik a przygotowanie roztworu odbyło się zgodnie z zasadami jałowości.
Nie przechowywać w temperaturze powyżej 30oC. Nie zamrażać.
Proszek w fiolce ze szkła typu II z korkiem z gumy bromobutylowej, po 500 j.m. lub 1000 j.m. antytrombiny ludzkiej , woda do wstrzykiwań (rozpuszczalnik) w ampułko-strzykawce ze szkła typu I z tłokiem z gumy bromobutylowej, po 10 ml (500 j.m.) lub 20 ml (1000 j.m.)
Zestaw do sporządzania roztworu: łącznik mocujący do fiolki i mikrofiltr.
Każde opakowanie zawiera:
1 fiolka z proszkiem.
1 ampułko-strzykawka z rozpuszczalnikiem. Zestaw do sporządzania roztworu.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
INSTITUTO GRIFOLS, S.A.
Poligono Levante, c/Can Guasch, 2
08150 Parets del Vallés, Barcelona, Hiszpania.
Anbinex 50 j.m./ml; 500 j.m. - 9405
Anbinex 50 j.m./ml; 1000 j.m. - 9406
26.06.2002
09.12.2016