







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Wprowadzenie ogólne
Bromek tiotropiowy jest czwartorzędowym związkiem amoniowym, nie wykazującym izomerii optycznej, bardzo słabo rozpuszczalnym w wodzie. Bromek tiotropiowy stosowany jest w postaci proszku do inhalacji. Na ogół po podaniu wziewnym większość dostarczonej dawki osadza się
w przewodzie pokarmowym, w mniejszym zaś stopniu w narządzie docelowym, czyli w płucach. Wiele z opisanych poniżej danych farmakokinetycznych uzyskano po podaniu dawek większych niż zalecane.
Ogólna charakterystyka substancji czynnej po podaniu produktu leczniczego
Wchłanianie:
Po podaniu produktu w postaci proszku do inhalacji u młodych, zdrowych ochotników, całkowita biodostępność wynosi 19,5%, co sugeruje dużą biodostępność frakcji trafiającej do płuc.
Całkowita biodostępność doustnych roztworów tiotropium wynosi 2-3%.
Maksymalne stężenie tiotropium w osoczu występuje po upływie 5-7 minut od inhalacji. W stanie stacjonarnym najwyższe stężenia tiotropium w osoczu u pacjentów z POChP wynosiły 12,9 pg/ml i szybko zmniejszały się, co jest charakterystyczne dla dystrybucji wielokompartmentowej. Najniższe stężenia w stanie stacjonarnym wynosiły 1,71 pg/ml.
Dystrybucja:
Tiotropium wiąże się w 72% z białkami osocza i wykazuje objętość dystrybucji 32 l/kg. Miejscowe stężenie leku w płucach nie jest znane, ale sposób podawania sugeruje, że jest ono znacznie większe niż w osoczu. Badania na szczurach wykazały, że stopień przenikania bromku tiotropiowego przez barierę krew-mózg nie jest znaczący.
Metabolizm:
Stopień biotransformacji jest mały. Wykazano, że u młodych, zdrowych ochotników 74% dawki podanej dożylnie wydala się w postaci niezmienionej z moczem. Ester bromku tiotropiowego ulega nieenzymatycznemu rozkładowi do alkoholu (N-metyloskopina) i kwasu ditienyloglikolowego, które nie mają powinowactwa do receptorów muskarynowych. Badania in vitro z użyciem mikrosomów wątroby ludzkiej i hepatocytów ludzkich wskazują, że pozostała część produktu (< 20% dawki podanej dożylnie) jest metabolizowana na drodze oksydacji zależnej od cytochromu P-450,
a następnie sprzęgana z glutationem, co prowadzi do powstania różnych metabolitów II fazy. Badania in vitro z użyciem mikrosomów wątroby wskazują na możliwość zablokowania tego szlaku enzymatycznego przez inhibitory CYP 2D6 (i 3A4), chinidynę, ketokonazol i gestoden. Tak więc CYP 2D6 i 3A4 są włączone w szlak metaboliczny odpowiedzialny za przemianę mniejszej części podanej dawki. Bromek tiotropiowy, nawet w stężeniach większych niż lecznicze, nie hamuje aktywności CYP 1A1, 1A2, 2B6, 2C9, 2C19, 2D6, 2E1 lub 3A w mikrosomach wątroby ludzkiej.
Eliminacja:
Skuteczny okres półtrwania tiotropium u pacjentów z POChP wynosi 27-45 godzin. Całkowity klirens po podaniu dożylnym u młodych, zdrowych ochotników wynosił 880 ml/min. Po podaniu dożylnym tiotropium jest wydalane głównie w postaci niezmienionej z moczem (74%). Po podaniu leku w postaci proszku do inhalacji u pacjentów z POChP do osiągnięcia stanu stacjonarnego 7% (1,3 µg) niezmienionego leku wydala się z moczem w ciągu 24 godzin, pozostała część dawki, głównie niewchłonięta w jelitach, wydala się z kałem.
Klirens nerkowy tiotropium jest większy od klirensu kreatyniny, co wskazuje na aktywne wydzielanie leku do moczu. Podczas długotrwałego, wziewnego stosowania produktu raz na dobę u pacjentów z POChP, stan stacjonarny uzyskiwano po7 dniach, bez późniejszej kumulacji.
Liniowość lub nieliniowość:
Tiotropium wykazuje liniową farmakokinetykę w zakresie dawek terapeutycznych niezależnie od postaci farmaceutycznej.
Charakterystyka poszczególnych grup pacjentów
Pacjenci w podeszłym wieku: Zgodnie z oczekiwaniem, podobnie jak w przypadku innych leków wydalanych głównie przez nerki, zaawansowanie wieku pacjenta wiąże się ze zmniejszeniem klirensu nerkowego tiotropium (365 ml/min u pacjentów w wieku < 65 lat z POChP do 271 ml/min u pacjentów w wieku ≥ 65 lat z POChP). Nie skutkowało to wzrostem wartości AUC0-6,ss lub Cmax,ss.
Pacjenci z zaburzeniami czynności nerek: Po podaniu raz na dobę tiotropium w postaci wziewnej do stanu stacjonarnego pacjentom z POChP, łagodne zaburzenia czynności nerek (Clkr 50-80 ml/min) skutkowały nieznacznym zwiększeniem AUC0-6,ss (od 1,8 do 30% wyższe) i podobnymi wartościami Cmax,ss w porównaniu do pacjentów z niezaburzoną czynnością nerek (Clkr >80 ml/min).
U pacjentów z POChP z umiarkowanymi do ciężkich zaburzeniami czynności nerek
(Clkr < 50 ml/min), dożylne podanie tiotropium spowodowało dwukrotne zwiększenie ekspozycji na lek (82% większe AUC0-4 godz i 52% większe Cmax) w porównaniu do pacjentów z POChP z niezaburzoną czynnością nerek, co zostało potwierdzone po podaniu suchego proszku do inhalacji.
Pacjenci z zaburzeniami czynności wątroby: Niewydolność wątroby nie powinna mieć znaczącego wpływu na właściwości farmakokinetyczne tiotropium. Tiotropium jest wydalane przede wszystkim przez nerki (74% dawki u młodych, zdrowych ochotników) i ulega prostej, nieenzymatycznej reakcji hydrolizy estru z wytworzeniem farmakologicznie nieaktywnych produktów.
Dzieci i młodzież: Patrz punkt 4.2.
Zależności farmakokinetyczno-farmakodynamiczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Tekturowe pudełko zawierające 30 kapsułek (3 blistry po 10 sztuk)
Tekturowe pudełko zawierające aparat do inhalacji HandiHaler i 30 kapsułek (3 blistry po 10 sztuk)
Tekturowe pudełko zawierające 90 kapsułek (9 blistrów po 10 sztuk) Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Spiriva, 18 mikrogramów/dawkę inhalacyjną, proszek do inhalacji w kapsułkach twardych
Każda kapsułka zawiera 18 mikrogramów tiotropium (Tiotropium) w postaci bromku tiotropiowego jednowodnego mikronizowanego - 22,5 mikrograma.
Dawka dostarczona (uwalniana z ustnika aparatu do inhalacji HandiHaler) zawiera 10 mikrogramów tiotropium.
Substancja pomocnicza: laktoza jednowodna.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek do inhalacji w kapsułkach twardych.
Jasnozielone kapsułki twarde. Na każdej kapsułce nadrukowany jest kod leku TI 01 i logo firmy.
Produkt leczniczy Spiriva jest wskazany jako lek rozszerzający oskrzela w leczeniu podtrzymującym w celu złagodzenia objawów u pacjentów z przewlekłą obturacyjną chorobą płuc (POChP).
Dawkowanie
Ten produkt leczniczy jest przeznaczony jedynie do stosowania wziewnego.
Zaleca się inhalację zawartości jednej kapsułki raz na dobę, o tej samej porze każdego dnia, za pomocą aparatu do inhalacji HandiHaler.
Nie należy stosować dawki większej niż zalecana.
Kapsułki Spiriva mogą być stosowane tylko wziewnie. Nie stosować doustnie. Kapsułek Spiriva nie należy połykać.
Kapsułki Spiriva mogą być inhalowane wyłącznie za pomocą aparatu do inhalacji HandiHaler.
Szczególne grupy pacjentów
Pacjenci w podeszłym wieku mogą stosować bromek tiotropiowy w zalecanej dawce.
Pacjenci z zaburzeniami czynności nerek mogą stosować bromek tiotropiowy w zalecanej dawce. Pacjenci z umiarkowanymi do ciężkich zaburzeniami czynności nerek (klirens kreatyniny
50 ml/min), patrz punkty: 4.4 i 5.2.
Pacjenci z zaburzeniami czynności wątroby mogą stosować bromek tiotropiowy w zalecanej dawce (patrz punkt 5.2).
Dzieci i młodzież
Produktu leczniczego Spiriva nie należy stosować u osób w wieku poniżej 18 lat. Nie ustalono skuteczności i bezpieczeństwa stosowania tego produktu u dzieci i młodzieży.
POChP
Produkt leczniczy nie ma zastosowania u dzieci i młodzieży we wskazaniu podanym w punkcie 4.1.
Mukowiscydoza
Nie określono bezpieczeństwa stosowania i skuteczności produktu leczniczego Spiriva u dzieci i młodzieży. Brak dostępnych danych.
Sposób podawania
Aparat do inhalacji HandiHaler jest dostosowany tylko do kapsułek leku Spiriva. Nie należy go używać do podawania innych leków. Aparat do inhalacji HandiHaler jest przewidziany jedynie dla jednego pacjenta i jest przeznaczony do wielokrotnego użycia. W celu zapewnienia właściwego sposobu podawania produktu leczniczego, lekarz lub inny wykwalifikowany pracownik służby zdrowia powinien przeszkolić pacjenta w zakresie prawidłowego stosowania inhalatora.
Instrukcja użycia i obsługi aparatu do inhalacji HandiHaler
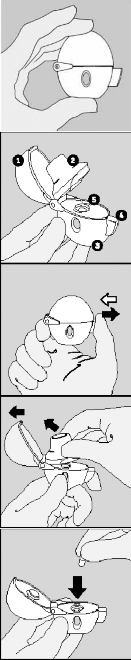
Pacjenci powinni postępować zgodnie z poniższymi punktami podczas korzystania z aparatu do inhalacji HandiHaler.
Należy dokładnie przestrzegać zaleceń lekarza dotyczących stosowania leku Spiriva. Po pierwszym użyciu, aparat do inhalacji HandiHaler można używać przez okres jednego roku w celu podawania produktu leczniczego. | |
HandiHaler | |
1. Aby otworzyć osłonę górną, należy wcisnąć całkowicie przycisk przekłuwający, a następnie go puścić. | |
2. Należy otworzyć osłonę górną aparatu do inhalacji odciągając ją do góry. Następnie należy podnieść do góry ustnik. | |
3. Bezpośrednio przed użyciem należy wyjąć kapsułkę produktu leczniczego Spiriva z blistra (patrz punkt „Wyjmowanie kapsułki z blistra”) i umieścić ją w komorze centralnej (5) w sposób pokazany na rysunku. Nie ma znaczenia, którym końcem kapsułka zostanie wprowadzona do komory. |
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
Nadwrażliwość na bromek tiotropiowy, atropinę lub jej pochodne, takie jak ipratropium lub oksytropium, bądź też na substancję pomocniczą, laktozę jednowodną, która zawiera białka mleka.
Bromku tiotropiowego jako leku rozszerzającego oskrzela przeznaczonego do stosowania raz na dobę w terapii podtrzymującej, nie należy stosować jako doraźnego leczenia ostrych napadów skurczu oskrzeli.
Po zastosowaniu bromku tiotropiowego w postaci proszku do inhalacji, mogą wystąpić natychmiastowe reakcje nadwrażliwości.
Podobnie jak w przypadku innych leków przeciwcholinergicznych, należy zachować ostrożność podczas stosowania bromku tiotropiowego u pacjentów z jaskrą z wąskim kątem przesączania, rozrostem gruczołu krokowego lub niedrożnością szyi pęcherza moczowego (patrz punkt 4.8).
Leki stosowane wziewnie mogą powodować skurcz oskrzeli wywołany inhalacją.
Ponieważ stężenie leku w osoczu krwi zwiększa się wraz ze zmniejszaniem się czynności nerek, u pacjentów z umiarkowanymi do ciężkich zaburzeniami czynności nerek (klirens kreatyniny
≤ 50 ml/min), bromek tiotropiowy należy stosować jedynie w przypadku, gdy spodziewana korzyść
dla pacjenta przewyższa potencjalne ryzyko. Brak długoterminowego doświadczenia dotyczącego stosowania bromku tiotropiowego u pacjentów z ciężkimi zaburzeniami czynności nerek (patrz punkt 5.2).
Nie wolno dopuścić, aby podczas inhalacji bromek tiotropiowy dostał się do oczu. Pacjenta należy uprzedzić, że może to spowodować wystąpienie lub zaostrzenie objawów jaskry z wąskim kątem przesączania, ból oka lub dyskomfort, przemijające niewyraźne widzenie, widzenie tęczowej obwódki wokół źródła światła lub zmienione widzenie kolorów, jednocześnie z zaczerwienieniem oczu wywołanym przekrwieniem spojówek i obrzękiem rogówki. W przypadku wystąpienia któregokolwiek z tych objawów, pacjent powinien przerwać stosowanie bromku tiotropiowego
i niezwłocznie skonsultować się z lekarzem specjalistą.
Suchość błony śluzowej jamy ustnej obserwowana w trakcie leczenia przeciwcholinergicznego, może po dłuższym czasie powodować próchnicę zębów.
Bromku tiotropiowego nie należy stosować częściej niż raz na dobę (patrz punkt 4.9). Dzieci i młodzież
Produktu leczniczego Spiriva nie należy stosować u osób w wieku poniżej 18 lat. Nie ustalono skuteczności i bezpieczeństwa stosowania tego produktu u dzieci i młodzieży.
Kapsułka produktu Spiriva zawiera 5,5 mg laktozy jednowodnej.
Chociaż nie przeprowadzono badań dotyczących interakcji leku, podawanie bromku tiotropiowego
w postaci proszku do inhalacji równocześnie z innymi lekami często stosowanymi w leczeniu POChP, takimi jak: sympatykomimetyki rozszerzające oskrzela, metyloksantyny, steroidy doustne i wziewne, nie powodowało wystąpienia klinicznie istotnych interakcji lekowych.
Nie stwierdzono wpływu często stosowanych przez pacjentów z POChP leków (LABA, ICS i ich połączenia) na ekspozycję na tiotropium.
Jednoczesne podawanie bromku tiotropiowego i innych leków zawierających substancje o działaniu przeciwcholinergicznym nie było badane i w związku z tym nie jest zalecane.
Ciąża
Istnieją tylko ograniczone dane dotyczące stosowania tiotropium u kobiet w okresie ciąży. Badania na zwierzętach nie wykazały bezpośredniego lub pośredniego szkodliwego wpływu na reprodukcję w zakresie dawek klinicznych (patrz punkt 5.3). W celu zachowania ostrożności zaleca się unikanie stosowania produktu Spiriva w okresie ciąży.
Karmienie piersią
Nie wiadomo, czy bromek tiotropiowy przenika do mleka kobiecego. Pomimo, że wyniki badań przeprowadzonych w okresie laktacji u gryzoni wskazują, że tylko niewielkie ilości bromku tiotropiowego przenikają do mleka, stosowanie produktu leczniczego Spiriva w okresie karmienia piersią nie jest zalecane. Bromek tiotropiowy jest substancją o długim działaniu. Decyzję
o kontynuowaniu lub przerwaniu karmienia piersią, bądź też o kontynuowaniu lub przerwaniu stosowania produktu leczniczego Spiriva należy podjąć, biorąc pod uwagę korzyści dla dziecka, wynikające z karmienia piersią oraz korzyści dla matki wynikające ze stosowania produktu leczniczego Spiriva.
Płodność
Brak danych klinicznych dla tiotropium dotyczących płodności. Badanie przedkliniczne z zastosowaniem tiotropium nie wykazało negatywnego wpływu na płodność (patrz punkt 5.3).
Nie przeprowadzono badań nad wpływem produktu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Jeżeli wystąpią zawroty głowy, niewyraźne widzenie lub ból głowy, nie należy prowadzić pojazdów ani obsługiwać maszyn.
Podsumowanie profilu bezpieczeństwa
Większość wymienionych działań niepożądanych związana jest z przeciwcholinergicznym działaniem produktu Spiriva.
Tabelaryczne zestawienie działań niepożądanych
Częstość wymienionych poniżej działań niepożądanych została oszacowana na podstawie częstości działań niepożądanych zaobserwowanych w grupie 9647 pacjentów przyjmujących tiotropium
w trakcie 28 łącznie analizowanych badań klinicznych, kontrolowanych placebo, z okresem leczenia od czterech tygodni do czterech lat.
Częstość występowania została określona na podstawie następującej konwencji:
Bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1 000 do <1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000); częstość nieznana (częstość nie może być określona na podstawie dostępnych danych).
Klasyfikacja układów i narządów (wg terminologii MedDRA) | Częstość występowania |
Zaburzenia metabolizmu i odżywiania | |
Odwodnienie | Częstość nieznana |
Zaburzenia układu nerwowego | |
Zawroty głowy | Niezbyt często |
Ból głowy | Niezbyt często |
Zaburzenia smaku | Niezbyt często |
Bezsenność | Rzadko |
Zaburzenia oka | |
Niewyraźne widzenie | Niezbyt często |
Jaskra | Rzadko |
Zwiększone ciśnienie wewnątrzgałkowe | Rzadko |
Zaburzenia serca | |
Migotanie przedsionków | Niezbyt często |
Częstoskurcz nadkomorowy | Rzadko |
Tachykardia | Rzadko |
Kołatanie serca | Rzadko |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Zapalenie gardła | Niezbyt często |
Dysfonia | Niezbyt często |
Kaszel | Niezbyt często |
Skurcz oskrzeli | Rzadko |
Krwawienie z nosa | Rzadko |
Zapalenie krtani | Rzadko |
Zapalenie zatok | Rzadko |
Zaburzenia żołądka i jelit | |
Suchość błony śluzowej jamy ustnej | Często |
Choroba refluksowa przełyku | Niezbyt często |
Zaparcia | Niezbyt często |
Kandydoza jamy ustnej i gardła | Niezbyt często |
Niedrożność jelit, w tym porażenna niedrożność jelit | Rzadko |
Zapalenie dziąseł | Rzadko |
Zapalenie języka | Rzadko |
Dysfagia (utrudnione przełykanie) | Rzadko |
Zapalenie jamy ustnej | Rzadko |
Nudności | Rzadko |
Próchnica zębów | Częstość nieznana |
Zaburzenia skóry i tkanki podskórnej, zaburzenia układu immunologicznego | |
Wysypka | Niezbyt często |
Pokrzywka | Rzadko |
Świąd | Rzadko |
Nadwrażliwość (w tym reakcje natychmiastowe) | Rzadko |
Obrzęk naczynioruchowy | Rzadko |
Reakcje anafilaktyczne | Częstość nieznana |
Zakażenie skórne, owrzodzenie skóry | Częstość nieznana |
Suchość skóry | Częstość nieznana |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Obrzęk stawów | Częstość nieznana |
Zaburzenia nerek i dróg moczowych | |
Bolesne lub utrudnione oddawanie moczu | Niezbyt często |
Zatrzymanie moczu | Niezbyt często |
Zakażenie dróg moczowych | Rzadko |
Opis wybranych działań niepożądanych
Najczęściej obserwowanymi w kontrolowanych badaniach klinicznych działaniami niepożądanymi były działania związane z przeciwcholinergicznym działaniem produktu, takie jak suchość błony śluzowej jamy ustnej, która pojawiła się u około 4% pacjentów.
W 28 badaniach klinicznych suchość błony śluzowej jamy ustnej była przyczyną przerwania leczenia u 18 z 9647 pacjentów przyjmujących tiotropium (0,2%).
Do ciężkich działań niepożądanych związanych z przeciwcholinergicznym działaniem produktu należą: jaskra, zaparcia, niedrożność jelit, w tym porażenna niedrożność jelit oraz zatrzymanie moczu.
Inne szczególne grupy pacjentów
Częstość występowania objawów związanych z przeciwcholinergicznym działaniem produktu może zwiększać się z wiekiem.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C, 02-222 Warszawa,
tel.: + 48 22 49-21-301
faks: +48 22 49-21-309
strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu lub przedstawicielowi podmiotu odpowiedzialnego.
Podawanie dużych dawek bromku tiotropiowego może prowadzić do wystąpienia przedmiotowych i podmiotowych objawów związanych z przeciwcholinergicznym działaniem leku.
Jednakże, po podaniu zdrowym ochotnikom do 340 mikrogramów bromku tiotropiowego w pojedynczej dawce wziewnej nie zaobserwowano działań niepożądanych związanych z ogólnoustrojowym działaniem przeciwcholinergicznym. Poza tym, po zastosowaniu u zdrowych ochotników bromku tiotropiowego w dawce do 170 mikrogramów na dobę przez 7 dni, nie zanotowano żadnych istotnych działań niepożądanych, z wyjątkiem suchości błony śluzowej jamy ustnej. W badaniu z zastosowaniem wielokrotnych dawek u pacjentów z POChP, przy maksymalnej dawce dobowej wynoszącej 43 mikrogramy bromku tiotropiowego, stosowanej przez cztery tygodnie, nie zanotowano znaczących działań niepożądanych.
Ostre zatrucie po nieumyślnym spożyciu doustnym bromku tiotropiowego w kapsułkach jest mało prawdopodobne ze względu na małą biodostępność po podaniu doustnym.
Grupa farmakoterapeutyczna: Inne leki stosowane w chorobach obturacyjnych dróg oddechowych podawane drogą wziewną, leki przeciwcholinergiczne, kod ATC: R03B B04
Mechanizm działania
Bromek tiotropiowy jest długo działającym, wybiórczym antagonistą receptorów muskarynowych, w medycynie klinicznej określanym jako lek przeciwcholinergiczny. Poprzez wiązanie się
z receptorami muskarynowymi w mięśniówce gładkiej oskrzeli, bromek tiotropiowy hamuje cholinergiczne (zwężające oskrzela) działanie acetylocholiny wydzielanej na zakończeniach nerwów przywspółczulnych. Wykazuje on podobne powinowactwo do poszczególnych podtypów receptora muskarynowego (od M1 do M5). Bromek tiotropiowy, działając jako kompetycyjny i odwracalny antagonista receptorów M3 w drogach oddechowych, powoduje rozszerzenie oskrzeli. Działanie to jest zależne od dawki i utrzymuje się ponad 24 godziny. Długotrwałe działanie jest prawdopodobnie spowodowane bardzo powolną dysocjacją cząsteczki produktu od receptora M3, przy czym okres połowicznej dysocjacji jest znacznie dłuższy w porównaniu z ipratropium. Jako lek przeciwcholinergiczny o strukturze N-czwartorzędowej, bromek tiotropiowy stosowany wziewnie odznacza się wybiórczym, miejscowym działaniem na oskrzela. Stężenia terapeutyczne, przy których nie występują ogólnoustrojowe objawy działania przeciwcholinergicznego mieszczą się
w dopuszczalnym zakresie.
Działanie farmakodynamiczne
Rozszerzenie oskrzeli jest wynikiem działania miejscowego (w drogach oddechowych), a nie ogólnoustrojowego. Dysocjacja tiotropium od receptora M2 jest szybsza niż w przypadku receptora M3, czego przejawem w czynnościowych badaniach in vitro była (kinetycznie zależna) selektywność podtypu receptora M3 względem M2. Duża siła działania i wolna dysocjacja od receptora znalazły swoje kliniczne odzwierciedlenie w postaci znaczącego i długotrwałego działania rozszerzającego oskrzela u pacjentów z POChP.
Elektrofizjologia serca
Elektrofizjologia: W badaniach dotyczących wpływu na odstęp QT z udziałem 53 zdrowych ochotników, produkt Spiriva podawany w dawkach 18 µg oraz 54 µg (tj. dawka trzykrotnie większa
od dawki terapeutycznej), przez 12 dni, nie powodował znaczącego wydłużenia odstępu QT w zapisie EKG.
Skuteczność kliniczna i bezpieczeństwo stosowania w POChP
Zasadniczy program fazy klinicznej obejmował cztery roczne i dwa półroczne badania z randomizacją i podwójnie ślepą próbą, z udziałem 2663 pacjentów (1308 otrzymywało bromek tiotropiowy).
Program roczny obejmował dwa badania z kontrolą placebo i dwa z kontrolą aktywną (ipratropium). Dwa badania trwające 6 miesięcy przeprowadzono z kontrolą placebo i salmeterolu. W badaniach tych oceniano czynność płuc i istotne dla zdrowia wskaźniki, takie jak: duszność, występowanie zaostrzeń choroby i jakość życia związaną ze stanem zdrowia.
Czynność płuc
Zastosowanie bromku tiotropiowego raz na dobę pozwalało uzyskać znaczącą poprawę czynności płuc (natężona objętość wydechowa pierwszosekundowa FEV1 i natężona pojemność życiowa FVC) w ciągu 30 minut po podaniu pierwszej dawki. Działanie to utrzymywało się przez 24 godziny.
Farmakodynamiczny stan stacjonarny uzyskiwano w ciągu jednego tygodnia, przy czym najsilniejsze działanie rozszerzające oskrzela obserwowano trzeciego dnia. Podanie bromku tiotropiowego powodowało znaczącą poprawę porannych i wieczornych wyników pomiaru PEFR (szczytowy przepływ wydechowy), ustaloną na podstawie codziennych zapisów pacjenta. Działanie rozszerzające oskrzela bromku tiotropiowego utrzymywało się przez okres jednego roku podawania leku, bez objawów tachyfilaksji.
Badanie kliniczne z randomizacją, kontrolowane placebo przeprowadzone u 105 pacjentów z POChP wykazało, że, w porównaniu do placebo, działanie rozszerzające oskrzela utrzymywało się przez
24 godziny (okres pomiędzy kolejnymi dawkami), niezależnie od tego, czy lek był podany rano, czy wieczorem.
Badania kliniczne (trwające do 12 miesięcy)
Duszność, tolerancja wysiłku
Bromek tiotropiowy wykazywał znaczącą poprawę u pacjentów z dusznością (jak oceniono z zastosowaniem Transition Dyspnoea Index). Poprawa ta utrzymywała się przez cały okres leczenia.
W dwóch badaniach klinicznych z randomizacją i podwójnie ślepą próbą, kontrolowanych placebo z udziałem 433 pacjentów z umiarkowaną do ciężkiej postacią POChP badano stopień zmniejszenia uczucia duszności powysiłkowej. W badaniach tych wykazano, że przyjmowanie produktu Spiriva
przez 6 tygodni znacząco poprawia ograniczony, z powodu choroby, czas tolerancji wysiłku. W czasie ergometrii rowerowej przy 75% maksymalnym obciążeniu osiągnięto wynik 19,7% (badanie A) i 28,3% (badanie B) w porównaniu do placebo.
Jakość życia związana ze stanem zdrowia
W trwającym dziewięć miesięcy podwójnie ślepym badaniu klinicznym z randomizacją oraz kontrolą placebo z udziałem 492 pacjentów, produkt leczniczy Spiriva poprawił jakość życia związaną ze stanem zdrowia, jak wykazano za pomocą kwestionariusza St. George’s Respiratory Questionnaire (SGRQ) na podstawie punktacji ogólnej. Odsetek pacjentów przyjmujących produkt leczniczy Spiriva, którzy uzyskali znaczącą poprawę w zakresie ogólnej punktacji SGRQ (to znaczy więcej niż 4 jednostki) wyniósł 10,9% w porównaniu z placebo (59,1% w grupie stosującej produkt Spiriva w porównaniu z 48,2% w grupie placebo (p=0,029)). Średnia różnica między grupami wyniosła 4,19 jednostki (p=0,001; przedział ufności: 1,69-6,68). Poprawa poddomen w punktacji SGRQ wyniosła 8,19 jednostek dla poddomeny „objawy”, 3,91 jednostek dla poddomeny „aktywność” oraz 3,61 jednostek dla poddomeny „wpływ na życie codzienne”. Poprawa w każdej z wymienionych poddomen była znamienna statystycznie.
Zaostrzenia POChP
W badaniu klinicznym z randomizacją i podwójnie ślepą próbą, kontrolowanym placebo, z udziałem 1829 pacjentów z umiarkowaną do bardzo ciężkiej postacią POChP wykazano, że bromek tiotropiowy statystycznie znamiennie zmniejsza odsetek pacjentów, u których wystąpiły zaostrzenia POChP
(32,2% do 27,8%) oraz statystycznie znamiennie zmniejsza liczbę zaostrzeń do 19% (1,05 do 0,85 zdarzeń w przeliczeniu na pacjenta na rok stosowania produktu leczniczego). Ponadto, 7% pacjentów przyjmujących bromek tiotropiowy i 9,5% pacjentów z grupy placebo było hospitalizowanych z powodu zaostrzeń POChP (p=0,056). Liczba hospitalizacji z powodu POChP zmniejszyła się o 30% (0,25 do 0,18 zdarzeń w przeliczeniu na pacjenta na rok stosowania produktu leczniczego).
Roczne randomizowane podwójnie zaślepione i podwójnie maskowane, prowadzone w równoległych grupach badanie porównało wpływ leczenia produktem Spiriva 18 mikrogramów raz na dobę do wpływu leczenia salmeterolem 50 mikrogramów HFA pMDI dwa razy na dobę na częstość występowania umiarkowanych i ciężkich zaostrzeń u 7376 pacjentów z POChP i zaostrzeniami przebytymi w okresie ostatniego roku.
Tabela 1: Zestawienie punktów końcowych zaostrzeń
Punkt końcowy | Spiriva 18 mikrogramów (HandiHaler) N = 3707 | Salmeterol 50 mikrogramów (HFA pMDI) N = 3669 | Współczynnik (95% CI) | Wartość p |
Czas [dni] do pierwszego zaostrzenia† | 187 | 145 | 0,83 (0,77 – 0,90) | <0,001 |
Czas do pierwszego ciężkiego zaostrzenia (hospitalizacja)§ | - | - | 0,72 (0,61 – 0,85) | <0,001 |
Pacjenci, u których wystąpiło ≥1 zaostrzenie, n (%)* | 1277 (34,4) | 1414 (38,5) | 0,90 (0,85 – 0,95) | <0,001 |
Pacjenci, u których wystąpiło ≥1 ciężkie zaostrzenie (hospitalizacja), n (%)* | 262 (7,1) | 336 (9,2) | 0,77 (0,66 – 0,89) | <0,001 |
† Czas [dni] odnosi się do 1. kwartyla pacjentów. Analizę czasu do zdarzenia przeprowadzono za pomocą modelu proporcjonalnego hazardu Coxa z uwzględnieniem danych z połączonych ośrodków i leczenia jako współzmiennych; współczynnik odnosi się do współczynnika ryzyka.
§ Analizę czasu do zdarzenia przeprowadzono za pomocą modelu proporcjonalnego hazardu Coxa z uwzględnieniem danych z połączonych ośrodków i leczenia jako współzmiennych; współczynnik odnosi się do współczynnika ryzyka. Obliczenie czasu [dni] dla 1. kwartyla pacjentów jest niemożliwe, ponieważ odsetek pacjentów z ciężkimi zaostrzeniami jest za mały.
* Liczba pacjentów, u których wystąpiło zdarzenie, została przeanalizowana za pomocą testu Cochrana-Mantela-Haenszela ze stratyfikacją wg danych z połączonych ośrodków; współczynnik odnosi się do współczynnika ryzyka.
W porównaniu do salmeterolu, produkt Spiriva spowodował wydłużenie czasu do pierwszego zaostrzenia (187 dni w porównaniu do 145 dni) i zmniejszenie ryzyka o 17% (współczynnik ryzyka 0,83; 95% przedział ufności [CI], 0,77 do 0,90; P<0,001). Produkt Spiriva spowodował również wydłużenie czasu do pierwszego ciężkiego zaostrzenia (hospitalizacja) (współczynnik ryzyka 0,72; 95% CI, 0,61 do 0,85; P<0,001).
Długoterminowe badania kliniczne (trwające od roku do czterech lat)
W trwającym cztery lata podwójnie zaślepionym badaniu klinicznym z randomizacją oraz kontrolą placebo z udziałem 5993 pacjentów (3006 pacjentów otrzymywało placebo, a 2987 otrzymywało produkt Spiriva), poprawa FEV1 w wyniku stosowania produktu leczniczego Spiriva, w porównaniu do placebo, utrzymywała się przez czteroletni okres obserwacji. W grupie pacjentów leczonych
produktem Spiriva, w porównaniu do grupy placebo, odnotowano wyższy odsetek pacjentów, którzy ukończyli co najmniej 45-miesięczny okres leczenia (63,8% w porównaniu do 55,4%, p<0,001).
Roczne tempo spadku FEV1 było porównywalne dla produktu Spiriva oraz placebo. Podczas leczenia ryzyko zgonu zostało zmniejszone o 16%. Częstość występowania przypadków zgonu wynosiła 4,79 na 100 pacjentów na rok w grupie placebo w porównaniu z 4,10 na 100 pacjentów na rok w grupie tiotropium (hazard względny HR tiotropium/placebo wyniosło 0,84, 95% Cl = 0,73, 0,97). Leczenie tiotropium zmniejszyło ryzyko niewydolności oddechowej (jak odnotowano na podstawie zgłaszanych działań niepożądanych) o 19% (2,09 w porównaniu do 1,68 przypadków na 100 pacjentów na rok, ryzyko względne tiotropium/placebo wyniosło 0,81, 95% Cl = 0,65, 0,999).
Badania tiotropium z aktywną kontrolą
Długoterminowe, aktywnie kontrolowane badanie kliniczne przeprowadzone metodą podwójnie ślepej próby z randomizacją i okresem obserwacji do 3 lat, zostało dokonane celem porównania skuteczności i bezpieczeństwa produktów Spiriva Handihaler oraz Spiriva Respimat (5 694pacjentów otrzymujących Spiriva Handihaler, 5711 pacjentów otrzymujących Spiriva Respimat).
Pierwszorzędowymi punktami końcowymi były czas do pierwszego zaostrzenia POChP, czas do zgonu z jakiejkolwiek przyczyny oraz w badaniu cząstkowym (906 pacjentów) najniższa wartość FEV1 (przed podaniem).
Czas do pierwszego zaostrzenia POChP był liczbowo podobny zarówno dla Spiriva HandiHaler i Spiriva Respimat (wskaźnik ryzyka (Spiriva HandiHaler /Spiriva Respimat) 1,02 przy 95% CI: 0,97 do 1,08). Mediana dla liczby dni do pierwszego zaostrzenia POChP wynosiła 719 dni dla produktu Spiriva HandiHaler i 719 dla Spiriva Respimat.
Działanie rozkurczające oskrzela Spiriva HandiHaler utrzymywało się w ciągu 120 tygodni i było podobne do Spiriva Respimat. Średnia różnica wartości FEV1 pomiędzy Spiriva HandiHaler a Spiriva Respimat wynosiła -0,010 l (95% CI: 0,018 do 0,038 l).
W prowadzonym po dopuszczeniu do obrotu badaniu TioSpir, porównującym produkty Spiriva Respimat i Spiriva HandiHaler, śmiertelność z jakiejkolwiek przyczyny (łącznie z obserwacją statusu życiowego) była podobna dla Spiriva HandiHaler i Spiriva Respimat (iloraz ryzyka (Spiriva HandiHaler /Spiriva Respimat) 1,04 z 95% CI: 0,91 do 1,19).
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań produktu leczniczego Spiriva we wszystkich podgrupach populacji dzieci i młodzieży w POChP oraz mukowiscydozie (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Parametry farmakokinetyczne produktu nie wpływają bezpośrednio na jego właściwości farmakodynamiczne.
Wiele działań leku obserwowanych w konwencjonalnych badaniach farmakologicznych dotyczących bezpieczeństwa stosowania, toksyczności po podaniu wielokrotnym i toksycznego wpływu na reprodukcję, można wyjaśnić przeciwcholinergicznymi właściwościami bromku tiotropiowego.
Typowe zaobserwowane u zwierząt objawy to: zmniejszenie łaknienia, zmniejszenie przyrostu masy ciała, suchość w jamie ustnej i nosie, zmniejszenie wydzielania płynu łzowego i śliny, rozszerzenie źrenic i zwiększona częstość akcji serca. Inne istotne objawy zanotowane w badaniach toksyczności po podaniu wielokrotnym to: łagodne podrażnienie dróg oddechowych u szczurów i myszy przejawiające się zapaleniem błony śluzowej nosa i zmianami w nabłonku jamy nosowej i krtani, zapaleniem gruczołu krokowego, złogami białkowymi i kamicą pęcherza moczowego u szczurów.
Szkodliwy wpływ na przebieg ciąży, rozwój zarodka i płodu, poród lub rozwój pourodzeniowy, mógł być wykazany tylko po zastosowaniu dawek toksycznych dla matki. Bromek tiotropiowy nie wykazywał działania teratogennego ani u szczurów, ani u królików. W badaniu wpływu na ogólną rozrodczość i płodność szczurów, nie wykazano żadnego działania niepożądanego na płodność lub cykle płciowe zarówno badanych rodziców, jak i ich potomstwa po żadnej z zastosowanych dawek leku.
Po miejscowym lub ogólnoustrojowym narażeniu pięciokrotnie przekraczającym narażenie po zastosowaniu dawek terapeutycznych obserwowano zmiany w układzie oddechowym (podrażnienie)
i moczowo-płciowym (zapalenie gruczołu krokowego) oraz toksyczny wpływ na reprodukcję. Badania genotoksyczności i potencjalnego działania rakotwórczego nie ujawniły występowania szczególnego zagrożenia dla ludzi.
Laktoza jednowodna mikronizowana Laktoza jednowodna 200 M
Skład kapsułki żelatynowej: Żelatyna
Makrogol 3350
Tytanu dwutlenek (E171) Żelaza tlenek żółty (E172) Indygotyna (E 132)
Nie dotyczy.
2 lata.
Po pierwszym otwarciu blistra: 9 dni.
Aparat do inhalacji HandiHaler należy wyrzucić po upływie 12 miesięcy od pierwszego użycia.
Przechowywać w temperaturze poniżej 25ºC. Nie zamrażać.
Blistry Al/PVC/Al umieszczone w tekturowym pudełku oraz blistry Al/PVC/Al wraz z aparatem do inhalacji HandiHaler umieszczone w tekturowym pudełku.
HandiHaler - aparat do inhalacji pojedynczych dawek produktu, wykonany z tworzywa sztucznego akrylonitryl-butadien-styren (ABS) i stali nierdzewnej. Komora kapsułki wykonana jest z tworzywa sztucznego metakrylan-metylu-akrylonitryl-butadien-styren (MABS) lub poliwęglanu (PC).
Wielkość opakowań:
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Boehringer Ingelheim International GmbH Binger Strasse 173
55216 Ingelheim/Rhein Niemcy
Pozwolenie nr 9851
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 30 marca 2003 Data ostatniego przedłużenia pozwolenia: 27 marca 2013
18 marca 2023







