







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
odmierzona dawka (rozpylenie) zawiera 21 mikrogramów ipratropiowego bromku (Ipratropii bromidum), co odpowiada 20 mikrogramom ipratropiowego bromku bezwodnego.
Substancja pomocnicza o znanym działaniu: jedna dawka odmierzona zawiera 8,415 mg etanolu bezwodnego.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
odmierzone dawki (rozpylenia) 4 razy na dobę.
200 rozpyleń. Po wykorzystaniu tej liczby dawek (zazwyczaj po 3 tygodniach stosowania zgodnie
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Atrovent N, 20 mikrogramów/dawkę inhalacyjną, aerozol inhalacyjny, roztwór
Aerozol inhalacyjny, roztwór
Atrovent N, aerozol inhalacyjny, roztwór jest wskazany jako środek rozszerzający oskrzela w leczeniu podtrzymującym stanów skurczowych oskrzeli w przewlekłej obturacyjnej chorobie płuc (POChP), obejmującej przewlekłe zapalenie oskrzeli i rozedmę płuc oraz w astmie oskrzelowej.
Dawkowanie
Dawkowanie należy dostosować do indywidualnych potrzeb pacjenta. W trakcie leczenia pacjenci
powinni być pod opieką lekarza. Nie należy przekraczać zalecanej dobowej dawki.
Pacjenta należy poinformować o konieczności skontaktowania się z lekarzem w celu ustalenia nowego planu leczenia, jeśli leczenie nie przynosi znaczącej poprawy lub jeśli stan pacjenta ulega pogorszeniu oraz o konieczności bezzwłocznego skonsultowania się z lekarzem w przypadku nagłej lub gwałtownie nasilającej się duszności (trudności w oddychaniu).
Zaleca się następujące dawkowanie:
Dorośli i dzieci w wieku powyżej 6 lat:
Potrzeba zwiększania dawek sugeruje konieczność włączenia dodatkowych produktów leczniczych, nie należy więc przekraczać dawki całkowitej 12 rozpyleń w ciągu doby.
Ze względu na niewystarczającą ilość informacji na temat stosowania produktu u dzieci, Atrovent N aerozol inhalacyjny, roztwór powinien być stosowany tylko po przepisaniu produktu przez lekarza
i pod kontrolą osoby dorosłej.
Sposób podawania
Należy uważnie przeczytać instrukcję użycia, aby mieć pewność właściwego dawkowania. Właściwe stosowanie ma istotne znaczenie dla skuteczności terapii.
Przed pierwszym użyciem inhalatora należy dwukrotnie nacisnąć zawór dozujący.
Podczas każdego użycia produktu leczniczego należy przestrzegać następujących zasad:
z zalecanym dawkowaniem), w pojemniku może nadal znajdować się niewielka ilość roztworu. Należy jednak wymienić inhalator na nowy, by mieć pewność, że każde rozpylenie dostarcza odpowiednią dawkę leku.
Ustnik inhalatora należy myć przynajmniej raz w tygodniu. Ważne jest utrzymywać go w czystości, aby mieć pewność, że lek nie odkłada się na ściankach ustnika i nie zablokuje on użycia inhalatora.
Aby umyć ustnik inhalatora należy najpierw zdjąć kapturek ochronny i usunąć pojemnik z lekiem
z ustnika. Przepłukać ustnik inhalatora ciepłą wodą aż do momentu usunięcia wszystkich widocznych zanieczyszczeń.
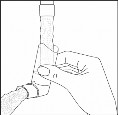
(rys. 2)
Po umyciu wytrząsnąć ustnik i pozostawić na powietrzu do wyschnięcia. Nie używać jakichkolwiek urządzeń suszących. Kiedy ustnik inhalatora jest suchy, należy z powrotem umieścić w ustniku pojemnik z lekiem i kapturek ochronny.
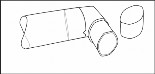
(rys. 3)
UWAGA:
Plastikowy ustnik zaprojektowano specjalnie do użytku z aerozolem inhalacyjnym Atrovent N, by zapewnić dostarczanie każdorazowo odpowiedniej dawki produktu leczniczego. Ustnika nigdy nie wolno wykorzystywać z jakimkolwiek innym aerozolem inhalacyjnym i odwrotnie, Atrovent N aerozol inhalacyjny nie może być stosowany z ustnikiem innym niż dołączony do produktu.
Pojemnik znajduje się pod ciśnieniem i pod żadnym pozorem nie należy używać siły przy jego otwieraniu ani wystawiać go na działanie temperatury wyższej niż 50°C.
Nadwrażliwość na atropinę lub jej pochodne (takie jak substancja czynna ipratropiowy bromek) lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Nadwrażliwość
Po podaniu produktu leczniczego mogą wystąpić reakcje nadwrażliwości typu natychmiastowego, co potwierdziły rzadkie przypadki wysypki, pokrzywki, obrzęku naczynioruchowego, obrzęku błony śluzowej jamy ustnej i gardła, skurczu oskrzeli oraz anafilaksji.
Paradoksalny skurcz oskrzeli
Jak w przypadku innych leków podawanych wziewnie, po podaniu produktu Atrovent N może wystąpić paradoksalny skurcz oskrzeli mogący stanowić zagrożenie dla życia. W przypadku wystąpienia paradoksalnego skurczu oskrzeli, należy natychmiast przerwać stosowanie produktu Atrovent N i zastosować inne leczenie.
Powikłania dotyczące narządu wzroku
Zaleca się zachowanie ostrożności w czasie stosowania produktu leczniczego u pacjentów ze skłonnością do rozwoju jaskry z wąskim kątem przesączania.
Istnieją nieliczne doniesienia o wystąpieniu powikłań dotyczących narządu wzroku (np. rozszerzenie źrenic, zwiększenie ciśnienia wewnątrzgałkowego, jaskra z wąskim kątem przesączania, ból oczu)
w następstwie kontaktu z oczami aerozolu zawierającego sam bromek ipratropiowy lub bromek ipratropiowy w połączeniu z agonistą receptorów beta2-adrenergicznych.
Ból oka lub dyskomfort, niewyraźne widzenie, widzenie tęczowej obwódki wokół źródła światła lub zmienione widzenie kolorów współistniejące z zaczerwienieniem oczu w następstwie przekrwienia spojówki i obrzęku rogówki mogą być objawami ostrej jaskry z wąskim kątem przesączania. W razie wystąpienia któregokolwiek z wymienionych objawów, w dowolnym połączeniu, należy rozpocząć leczenie kroplami zwężającymi źrenicę oka i natychmiast zasięgnąć porady lekarza specjalisty.
Należy poinstruować pacjentów odnośnie prawidłowego stosowania produktu Atrovent N.
Należy zachować ostrożność tak, aby mgiełka powstająca podczas stosowania produktu nie dostała się do oczu. Ponieważ aerozol inhalacyjny jest stosowany z ustnikiem i kontrolowany manualnie, ryzyko przedostania się mgiełki leku do oczu jest ograniczone.
Wpływ na nerki i układ moczowy
Zaleca się zachowanie ostrożności w czasie stosowania produktu leczniczego u pacjentów ze zwężeniem drogi odpływu moczu (np. rozrostem gruczołu krokowego lub zwężeniem szyi pęcherza moczowego).
Zaburzenia motoryki przewodu pokarmowego
Pacjenci z mukowiscydozą mogą być bardziej podatni na zaburzenia motoryki przewodu pokarmowego.
Informacje dotyczące substancji pomocniczej o znanym działaniu
Ten produkt leczniczy zawiera około 8,415 mg alkoholu (etanolu) w każdym rozpyleniu (w każdej dawce odmierzonej). Ilość alkoholu w każdym rozpyleniu tego produktu leczniczego jest równoważna mniej niż 1 ml piwa lub 1 ml wina.
Mała ilość alkoholu w tym produkcie leczniczym nie będzie powodowała zauważalnych skutków.
Długotrwałe podawanie produktu Atrovent N z innymi lekami przeciwcholinergicznymi nie było badane. W związku z tym długotrwałe podawanie produktu Atrovent N z tymi lekami nie jest zalecane.
Produkty lecznicze oddziałujące na receptory beta-adrenergiczne i produkty ksantynowe mogą nasilać działanie rozszerzające oskrzela.
Ciąża
Bezpieczeństwo stosowania produktu leczniczego Atrovent N w okresie ciąży nie zostało ustalone.
Należy rozważyć, czy korzyści terapeutyczne wynikające ze stosowania produktu leczniczego w czasie potwierdzonej ciąży lub w przypadku podejrzenia ciąży przeważają nad potencjalnym ryzykiem dla płodu. Badania przedkliniczne nie wykazały embriotoksycznego ani teratogennego
wpływu produktu podanego w postaci wziewnej lub donosowo w dawkach znacznie przekraczających zalecane do stosowania u ludzi.
Karmienie piersią
Nie wiadomo, czy produkt leczniczy przenika do mleka kobiecego. Wydaje się mało prawdopodobne, by produkt leczniczy – zwłaszcza po zastosowaniu w postaci wziewnej - mógł przedostać się do organizmu dziecka w znaczącej ilości. Jednakże należy zachować ostrożność stosując produkt leczniczy Atrovent N u kobiet karmiących piersią.
Płodność
Dane kliniczne dotyczące płodności nie są dostępne dla bromku ipratropiowego. Badania niekliniczne przeprowadzone z zastosowaniem bromku ipratropiowego nie wykazały negatywnego wpływu na płodność (patrz punkt 5.3).
Nie przeprowadzono badań dotyczących wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn. Niemniej jednak, pacjentów należy poinformować, że w trakcie leczenia lekiem Atrovent N mogą wystąpić takie działania niepożądane, jak zawroty głowy, zaburzenia akomodacji, rozszerzenie źrenic i zaburzenia widzenia. Dlatego zaleca się zachowanie ostrożności podczas prowadzenia samochodu lub obsługiwania maszyn.
Wiele z wymienionych działań niepożądanych można przypisać właściwościom przeciwcholinergicznym produktu Atrovent N. Tak jak wszystkie leki wziewne Atrovent N może wywołać objawy miejscowego podrażnienia.
Najczęstszymi działaniami niepożądanymi zgłaszanymi w badaniach klinicznych były: ból głowy, podrażnienie gardła, kaszel, suchość błony śluzowej jamy ustnej, zaburzenia motoryki przewodu pokarmowego (w tym zaparcia, biegunka i wymioty), nudności i zawroty głowy.
Następujące działania niepożądane odnotowano w czasie stosowania produktu leczniczego Atrovent N w badaniach klinicznych oraz w ramach monitorowania bezpieczeństwa stosowania po wprowadzeniu do obrotu.
Częstość występowania działań niepożądanych została podana zgodnie z następującą klasyfikacją
MedDRA:
Bardzo często (≥ 1/10) Często (≥ 1/100 do < 1/10)
Niezbyt często (≥ 1/1 000 do < 1/100) Rzadko (≥ 1/10 000 do < 1/1 000) Bardzo rzadko (< 1/10 000)
Częstość nieznana (częstość nie może być określona na podstawie dostępnych danych).
Zaburzenia układu immunologicznego
Niebyt często: Nadwrażliwość, reakcje anafilaktyczne
Zaburzenia układu nerwowego
Często: Ból głowy, zawroty głowy
Zaburzenia oka
Niezbyt często: Niewyraźne widzenie, rozszerzenie źrenic, zwiększone ciśnienie
wewnątrzgałkowe, jaskra, ból oka, aureola wzrokowa, przekrwienie spojówki, obrzęk rogówki
Rzadko: Zaburzenia akomodacji
Zaburzenia serca
Niezbyt często: Kołatanie serca, częstoskurcz nadkomorowy
Rzadko: Migotanie przedsionków, przyspieszenie czynności serca
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia Często: Podrażnienie gardła, kaszel
Niezbyt często: Skurcz oskrzeli, paradoksalny skurcz oskrzeli, skurcz krtani, obrzęk
błony śluzowej gardła, suchość w gardle
Zaburzenia żołądka i jelit
Często: Suchość błony śluzowej jamy ustnej, nudności, zaburzenia motoryki
przewodu pokarmowego
Niezbyt często: Biegunka, zaparcia, wymioty, zapalenie jamy ustnej, obrzęk jamy
ustnej
Zaburzenia skóry i tkanki podskórnej
Niezbyt często: Wysypka, świąd, obrzęk naczynioruchowy
Rzadko: Pokrzywka
Zaburzenia nerek i dróg moczowych
Niezbyt często: Zatrzymanie moczu
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, Al. Jerozolimskie 181C, 02-222 Warszawa,
tel: +48 22 492 13 01, faks: +48 22 492 13 09, strona internetowa: https://smz.ezdrowie.gov.pl. Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu lub przedstawicielowi podmiotu odpowiedzialnego.
Nie stwierdzono objawów typowych dla przedawkowania. Biorąc pod uwagę szeroki zakres terapeutyczny i miejscowe podawanie produktu leczniczego, nie należy spodziewać się żadnych poważnych objawów działania przeciwcholinergicznego. Mogą wystąpić łagodne ogólnoustrojowe objawy działania przeciwcholinergicznego produktu, takie jak: suchość błony śluzowej jamy ustnej, zaburzenia akomodacji oraz przyspieszenie czynności serca.
Grupa farmakoterapeutyczna: Inne leki stosowane w chorobach obturacyjnych dróg oddechowych podawane wziewnie, leki przeciwcholinergiczne; kod ATC: R03BB01
Atrovent N jest produktem leczniczym zawierającym czwartorzędową pochodną amonową
o właściwościach przeciwcholinergicznych (parasympatykolitycznych). Z badań przedklinicznych wynika, że jako antagonista acetylocholiny, neuroprzekaźnika uwalnianego z nerwu błędnego, hamuje odruchy przewodzone drogą nerwu błędnego. Leki przeciwcholinergiczne zapobiegają zwiększeniu wewnątrzkomórkowego stężenia jonów wapnia Ca++. Efekt działania acetylocholiny na receptory muskarynowe w mięśniach gładkich oskrzeli osiągany jest za pośrednictwem układu drugiego przekaźnika, który składa się z trójfosforanu inozytolu (IP3) oraz diacyloglicerolu (DAG).
Rozszerzenie oskrzeli w następstwie inhalacji produktu leczniczego Atrovent N (bromek
ipratropiowy) jest wywołane miejscowym, a nie ogólnoustrojowym działaniem produktu leczniczego.
Badania niekliniczne i kliniczne nie wykazały ujemnego wpływu produktu leczniczego Atrovent N na
wydzielanie śluzu w drogach oddechowych, oczyszczanie rzęskowe czy wymianę gazową.
Badania kliniczne
Badania, w których leczono przez okres do trzech miesięcy pacjentów dorosłych z astmą i przewlekłą obturacyjną chorobą płuc (POChP) oraz dzieci z astmą oskrzelową, porównujące Atrovent N zawierający w składzie HFA (hydrofluoroalkan) z produktem Atrovent N zawierającym CFC (chlorofluorowęglowodór), wykazały, że obydwa produkty lecznicze są równoważne pod względem terapeutycznym.
W kontrolowanych 90-dniowych badaniach u pacjentów ze skurczem oskrzeli w przebiegu przewlekłej obturacyjnej choroby płuc (przewlekłe zapalenie oskrzeli i rozedma płuc) znacząca poprawa czynności płuc wystąpiła w ciągu 15 minut, a maksymalna poprawa nastąpiła po 1-2 godzinach. U większości pacjentów działanie utrzymywało się przez okres do 4 - 6 godzin.
W kontrolowanych 90-dniowych badaniach u pacjentów ze skurczem oskrzeli w przebiegu astmy oskrzelowej uzyskano znaczącą poprawę czynności płuc (zwiększenie wartości FEV1 o 15%) u 51% pacjentów. Poprawa wartości FEV1 była odnotowana w czasie do 5 godzin po podaniu leku.
Wchłanianie
Działanie terapeutyczne produktu leczniczego jest wynikiem jego miejscowego działania w drogach oddechowych. Czas występowania działania terapeutycznego - rozszerzenia oskrzeli jest niezależny od farmakokinetyki ogólnoustrojowej.
W następstwie podania produktu leczniczego drogą wziewną, 10-30% dawki, w zależności od składu i techniki inhalacji, gromadzi się głównie w drogach oddechowych. Większa część dawki jest połykana i przechodzi przez przewód pokarmowy.
Część dawki dostająca się do płuc bardzo szybko przedostaje się do krwiobiegu (w ciągu kilku minut).
Łączne wydalanie przez nerki (0-24 godzin) niezmienionej substancji czynnej oszacowano na 46% dawki po podaniu dożylnym, poniżej 1% po podaniu doustnym i około 3 do 13% po podaniu wziewnym. Na podstawie tych danych oceniono, że całkowita ogólnoustrojowa biodostępność bromku ipratropiowego po podaniu doustnym i inhalacji wynosiła odpowiednio 2% i od 7 do 28%. Biorąc to pod uwagę połknięta część dawki nie ma istotnego wpływu na stężenie substancji czynnej w osoczu.
Dystrybucja
Parametry farmakokinetyczne opisujące rozmieszczenie ipratropium wyliczono na podstawie stężeń
w osoczu krwi po podaniu dożylnym. Obserwuje się szybkie dwufazowe zmniejszenie stężenia
w osoczu krwi. Pozorna objętość dystrybucji w stanie równowagi (Vdss) wynosi około 176 l (ok. 2,4 l/kg). Wiązanie produktu z białkami osocza jest niewielkie (poniżej 20%). Dane niekliniczne wskazują, że czwartorzędowa amina ipratropium nie przenika przez barierę łożyskową i barierę krew- mózg.
Metabolizm
Po podaniu dożylnym około 60% dawki jest metabolizowane, prawdopodobnie głównie w wątrobie na drodze utleniania.
Zidentyfikowane metabolity powstałe na drodze hydrolizy, odwodnienia lub eliminacji grupy hydroksymetylowej w obrębie kwasu tropowego wykazują słabe powinowactwo do receptora muskarynowego i uważane są za nieczynne.
Eliminacja
Okres półtrwania w końcowej fazie eliminacji wynosi około 1,6 godziny. Klirens całkowity ipratropium wynosi 2,3 l/min., a klirens nerkowy 0,9 l/min.
W badaniu bilansu wydalania z użyciem produktu leczniczego znakowanego radioaktywnie stwierdzono, że w ciągu 6 dni 72,1%, 9,3% i 3,2% dawki znakowanego produktu (zarówno w postaci niezmienionej jak i w postaci metabolitów) odpowiednio po podaniu dożylnym, doustnym
i wziewnym wydalane jest z moczem. Odzysk w kale wynosił 6,3% po podaniu dożylnym, 88,5% po podaniu doustnym i 69,4% po podaniu wziewnym. Wydalanie produktu leczniczego po podaniu dożylnym oznaczane jako odzysk radioaktywności odbywa się głównie przez nerki. Okres półtrwania dla eliminacji mierzonej z użyciem znakowanego izotopowo związku (pomiar dla związku w postaci niezmienionej, jak i w postaci metabolitów) wynosi 3,6 godziny.
Badanie tolerancji miejscowej i ogólnoustrojowej bromku ipratropiowego badano na kilku gatunkach zwierząt z wykorzystaniem różnych dróg podania.
Toksyczność po podaniu jednorazowym
Toksyczność ostra po podaniu wziewnym, doustnym i dożylnym była oceniana na wielu gatunkach
gryzoni i innych zwierząt.
Po podaniu wziewnym minimalna dawka śmiertelna u samców świnek morskich wynosiła 199 mg/kg. U szczurów nie zanotowano śmiertelności nawet po największych, technicznie możliwych do podania dawkach (np. 0,05 mg/kg po 4 godzinach podawania lub 160 rozpyleniach bromku ipratropiowego, 0,02 mg/rozpylenie).
Wartości LD50 dla myszy, szczurów i królików wynosiły odpowiednio 1585, 1925 i 1920 mg/kg po podaniu doustnym. Po podaniu dożylnym LD50 dla myszy, szczurów i psów wynosił odpowiednio 13,6, 15,8 i około 18,2 mg/kg. Objawy kliniczne obejmują: rozszerzenie źrenic, suchość błony śluzowej jamy ustnej, duszność, drżenie, skurcze i (lub) tachykardię.
Toksyczność po podaniu wielokrotnym
Badania toksyczności po podaniu wielokrotnym zostały przeprowadzone u szczurów, królików, psów i małp gatunku Rhesus. Badania dotyczące podawania wziewnego trwające do 6 miesięcy u szczurów, psów i małp gatunku Rhesus, wykazały, że dawki wynoszące odpowiednio 0,38 mg/kg/dobę,
0,18 mg/kg/dobę i 0,8 mg/kg/dobę, stanowiły poziom dawkowania, przy którym nie obserwowano działań niepożądanych (NOAEL, ang. no-observed adverse effect level). U psów zaobserwowano suchość błony śluzowej jamy ustnej oraz tachykardię. Nie zaobserwowano żadnych histopatologicznych zmian związanych z podawaniem substancji w obrębie układu oskrzelowo- płucnego oraz w żadnych innych organach. U szczurów wartość NOAEL po podaniu doustnym po 18 miesiącach stosowania wyniosła 0,5 mg/kg/dobę.
Badania toksyczności po podaniu wielokrotnym dawki wziewnej u szczurów przez okres do 6 miesięcy i u psów przez okres do 3 miesięcy prowadzone z wykorzystaniem leków w innej postaci (postać donosowa, alternatywny propelent HFA 134a lub proszek do inhalacji z laktozą), nie dostarczyły nowych danych dotyczących ogólnego profilu toksyczności bromku ipratropiowego. Podczas podawania psom produktu donosowo przez okres do 6 miesięcy wykazano, że nie wywiera on toksycznego działania w dawkach większych niż 0,20 mg/kg/dobę i tym samym potwierdzono wyniki wcześniejszych badań, w których produkt podawany był donosowo przez okres do 13 tygodni.
Badania toksyczności po podaniu wielokrotnym bromku ipratropiowego wykazały, że profil toksykologiczny leku zawierającego w składzie HFA oraz tradycyjnego zawierającego w składzie CFC jest podobny.
Tolerancja miejscowa
Wodny roztwór bromku ipratropiowego (0,05 mg/kg) był dobrze tolerowany miejscowo po podaniu wziewnym szczurom (pojedyncze podanie przez ponad 4 godziny). W badaniach toksyczności po podaniu wielokrotnym bromek ipratropiowy był dobrze tolerowany miejscowo.
Immunogenność
Nie wykazano żadnych czynnych ani biernych skórnych reakcji anafilaktycznych u świnek morskich.
Genotoksyczność i rakotwórczość
Nie wykazano genotoksycznego działania w badaniach in vitro (test Amesa) oraz in vivo (test mikrojądrowy, test dominującego czynnika letalnego u myszy, cytogenny na komórkach szpiku kostnego chomika chińskiego).
Długotrwałe badania na myszach i szczurach nie wykazały działania guzotwórczego ani rakotwórczego.
Toksyczny wpływ na reprodukcję i rozwój
Przeprowadzono badania dotyczące ewentualnego wpływu bromku ipratropiowego na płodność, badania toksyczności w odniesieniu do zarodka i płodu oraz dotyczące rozwoju okołoporodowego i poporodowego u myszy, szczurów i królików. Największe zastosowane doustnie dawki, tj.
1000 mg/kg/dobę u szczurów i 125 mg/kg/dobę u królików były toksyczne dla matek obydwu gatunków i toksyczne dla zarodka i płodu u szczurów, u których stwierdzono zmniejszenie masy płodu. Nie stwierdzono wad rozwojowych związanych z leczeniem. Największe, technicznie możliwe
do podania wziewnego dawki aerozolu inhalacyjnego wynoszące 1,5 mg/kg/dobę u szczurów
i 1,8 mg/kg/dobę u królików, nie wykazały niepożądanego wpływu na rozmnażanie.
1,1,1,2-czterofluoroetan (HFA 134a) Kwas cytrynowy bezwodny
Etanol bezwodny Woda oczyszczona
3 lata
Przechowywać w temperaturze poniżej 25°C. Nie zamrażać.
Chronić przed bezpośrednim działaniem światła słonecznego.
Pojemnik ze stali nierdzewnej, z zastawką dozującą i ustnikiem w tekturowym pudełku.
1 pojemnik zawierający 10 ml roztworu (200 dawek)
stosowania
Patrz punkt 4.2.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami.
DOPUSZCZENIE DO OBROTU
Boehringer Ingelheim International GmbH
Binger Strasse 173
D-55216 Ingelheim/Rhein Niemcy
Pozwolenie nr 9990
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 11 września 2003 r.
Data ostatniego przedłużenia pozwolenia: 12 marca 2014 r.







