







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- Wykaz substancji pomocniczych
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU 15102; 15103; 21117
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Leczenie i profilaktyka okołooperacyjna krwawień w nabytym niedoborze czynników krzepnięcia zespołu protrombiny, takich jak niedobór wywołany stosowaniem leków z grupy antagonistów witaminy K oraz w przypadku przedawkowania antagonistów witaminy K, kiedy wymagane jest szybkie wyrównanie niedoborów.
Leczenie i profilaktyka okołooperacyjna krwawień we wrodzonych niedoborach któregokolwiek czynnika krzepnięcia zależnego od witaminy K, kiedy zastosowanie preparatu oczyszczonego, swoistego czynnika krzepnięcia nie jest możliwe.
Dawkowanie i sposób podawania Dawkowanie
Krwawienia i profilaktyka okołooperacyjna krwawień podczas leczenia antagonistami witaminy K:
Dawka produktu zależy od wartości wskaźnika INR oznaczonego przed rozpoczęciem leczenia oraz od wartości wskaźnika INR, którą pacjent ma uzyskać. Wskaźnik INR przed leczeniem powinien być mierzony w najkrótszym możliwym czasie przed podaniem leku, aby umożliwić obliczenie odpowiedniej dawki produktu leczniczego Beriplex. Poniższa tabela określa przybliżone dawki (ml/kg masy ciała rekonstytuowanego produktu oraz j.m. czynnika IX/kg masy ciała) tak, aby uzyskać normalizację wskaźnika INR (np. INR≤1,3) przy podanych różnych wyjściowych wartościach wskaźnika INR.
Wartość INR przed leczeniem
2,0 – 3,9
4,0 – 6,0
>6,0
Przybliżona dawka w ml/kg masy ciała
1
1,4
2
Przybliżona dawka w j.m. (czynnik IX) na kilogram masy ciała
25
35
50
Dawkowanie jest ustalane w oparciu o masę ciała, gdy nie przekracza ona 100 kg. W przypadku pacjentów o masie ciała powyżej 100 kg maksymalna pojedyncza dawka (j.m. czynnika IX) nie
powinna przekraczać 2500 j.m. dla wartości wskaźnika INR 2,0 - 3,9; 3500 j.m. dla wartości wskaźnika INR 4,0 – 6,0 oraz 5000 j.m. dla wartości wskaźnika INR >6.0.
Normalizacja zaburzeń hemostazy spowodowanych stosowaniem antagonistów witaminy K jest zwykle osiągana w ciągu około 30 minut po wstrzyknięciu. Równoczesne podawanie witaminy K powinno być rozważone u pacjentów otrzymujących Beriplex w celu pilnego odwrócenia działania antagonistów witaminy K, gdyż zazwyczaj efekt działania witaminy K jest osiągany po 4-6 godzinach. Powtarzane dawkowanie Beriplexu u pacjentów wymagających pilnego odwrócenia działania antagonistów witaminy K nie jest wsparte badaniami klinicznymi i związku z tym nie jest zalecane
Zalecenia te podano w oparciu o badania kliniczne na ograniczonej liczbie osobników. Konieczne jest monitorowanie wartości wskaźnika INR podczas leczenia ponieważ skuteczność i czas działania mogą się różnić u poszczególnych pacjentów.
Krwawienia i profilaktyka okołooperacyjna u pacjentów we wrodzonych niedoborach któregokolwiek czynnika krzepnięcia zależnego od witaminy K, kiedy zastosowanie preparatu swoistego czynnika krzepnięcia nie jest możliwe
Wyliczenie wymaganej dawki koncentratu czynników krzepnięcia zespołu protrombiny zostało ustalone w oparciu o badania kliniczne:
1 j.m. czynnika IX na kg masy ciała powoduje spodziewany wzrost aktywności czynnika IX w osoczu o 1,3% (0,013 j.m./ml) w stosunku do normy,
1 j.m. czynnika VII na kg masy ciała powoduje wzrost aktywności czynnika VII w osoczu o 1,7% w stosunku do normy (0,017 j.m./ml),
1 j.m. czynnika II na kg masy ciała zwiększa aktywność czynnika II w osoczu o 1,9% w stosunku do normy (0,019 j.m./ml),
1 j.m. czynnika X na kg masy ciała zwiększa aktywność czynnika X w osoczu o 1,9% w stosunku do normy (0,019 j.m./ml).
Poniżej podano tylko ogólne zasady dawkowania. Leczenie powinno być rozpoczęte pod kontrolą lekarza doświadczonego w leczeniu zaburzeń krzepnięcia. Dawka i czas leczenia substytucyjnego zależy od wskazania do leczenia, stopnia zaawansowania choroby, umiejscowienia i intensywności krwawienia oraz od stanu klinicznego pacjenta.
Wielkość dawki oraz częstość jej podawania powinny być obliczone dla każdego pacjenta indywidualnie. Odstępy między dawkami powinny być dostosowane do wartości okresu półtrwania poszczególnych czynników krzepnięcia należących do zespołu protrombiny (patrz punkt 5.2). Dawkę indywidualną określa się w oparciu o regularne oznaczanie aktywności w osoczu poszczególnych czynników krzepnięcia lub na podstawie wyników badań laboratoryjnych ogólnie określających działanie zespołu protrombiny (INR, wskaźnik Quicka) oraz poprzez ciągłe monitorowanie stanu klinicznego pacjenta.
W przypadku większych interwencji chirurgicznych konieczne jest dokładne monitorowanie leczenia substytucyjnego poprzez wykonywanie badań układu krzepnięcia (oznaczanie aktywności poszczególnych czynników krzepnięcia i/lub badań określających ogólnie aktywność czynników krzepnięcia zespołu protrombiny).
Dawka poszczególnego czynnika krzepnięcia wyrażana jest w jednostkach międzynarodowych (j.m.), zgodnych z obowiązującym standardem dla każdego czynnika krzepnięcia zatwierdzonym przez Światową Organizację Zdrowia (WHO). Aktywność czynnika krzepnięcia w osoczu jest wyrażana procentowo (w stosunku do prawidłowego osocza ludzkiego) lub w jednostkach międzynarodowych (zgodnie z międzynarodowym standardem dla konkretnego czynnika krzepnięcia w osoczu).
Jedna jednostka międzynarodowa (j.m.) aktywności czynnika krzepnięcia jest równa aktywności tego czynnika zawartego w 1 ml prawidłowego osocza ludzkiego.
Na przykład, obliczenie wymaganej dawki czynnika X opiera się na empirycznym stwierdzeniu, że 1 j.m. czynnika X na kg masy ciała zwiększa aktywność osoczowego czynnika X o 0,019 j.m./ml.
Wymagane dawkowanie obliczane jest przy użyciu następującego wzoru:
Wymagana liczba jednostek = masa ciała [kg] x pożądany wzrost aktywności czynnika X [j.m./ml] x 53
gdzie 53 (ml/kg) jest odwrotnością szacowanej wartości odzysku.
Należy zauważyć, że obliczenia te są oparte na danych dotyczących pacjentów otrzymujących antagonistów witaminy K. Obliczenia oparte na danych dotyczących zdrowych osób dostarczyłyby niższej wartości wymaganej dawki.
Jeśli indywidualna wartość odzysku jest znana powinna być używana do obliczeń.
Dane dotyczące produktu są dostępne na podstawie badań klinicznych prowadzonych na zdrowych ochotnikach (N=15), w odwróceniu działania antagonistów witaminy K w leczeniu ostrych dużych krwawień lub profilaktyki krwawień okołooperacyjnych (N=98, N=43) (patrz punkt 5.2).
Dzieci i młodzież
Bezpieczeństwo i skuteczność produktu leczniczego Beriplex u dzieci i młodzieży nie została do tej pory ustalona w kontrolowanych badaniach klinicznych (patrz punkt 4.4)
Osoby starsze
Dawkowanie i sposób podawania u ludzi starszych (> 65 roku życia) są zgodne z ogólnymi
zaleceniami.
Sposób podawania
Instrukcja dotycząca rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6. Roztwór po rekonstytucji należy podać dożylnie (nie szybciej niż 8 ml/min*).
Roztwór powinien być przezroczysty lub lekko opalizujący.
*W badaniach klinicznych Beriplexu pacjenci ważący poniżej 70 kg mieli zalecane dawkowanie z
maksymalną szybkością infuzji wynoszącą 0.12 ml/kg/min (poniżej 8 ml/min).
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciąże i laktację
Wpływ na zdolność do prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
Beriplex P/N 250, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań. Beriplex P/N 500, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań. Beriplex P/N 1000, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Beriplex jest dostępny jako proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań, zawierający zespół protrombiny ludzkiej. Nominalnie zawiera następujące ilości (j.m.) ludzkich czynników krzepnięcia podane w tabeli poniżej:
Nazwa składnika | Zawartość po rekonstytucji (j.m./ml) | Zawartość w jednej fiolce Beriplex P/N 250 (j.m.) | Zawartość w jednej fiolce Beriplex P/N 500 (j.m.) | Zawartość w jednej fiolce Beriplex P/N 1000 (j.m.) |
Substancje czynne | ||||
Ludzki II czynnik krzepnięcia krwi | 20 - 48 | 200 – 480 | 400 – 960 | 800 – 1920 |
Ludzki VII czynnik krzepnięcia krwi | 10 – 25 | 100 – 250 | 200 -500 | 400 – 1000 |
Ludzki IX czynnik krzepnięcia krwi | 20 - 31 | 200 – 310 | 400 – 620 | 800 – 1240 |
Ludzki X czynnik krzepnięcia krwi | 22 – 60 | 220 – 600 | 440 – 1200 | 880 – 2400 |
Pozostałe substancje czynne | ||||
Białko C | 15 – 45 | 150 -450 | 300 – 900 | 600 – 1800 |
Białko S | 12 – 38 | 120 – 380 | 240 – 760 | 480 - 1520 |
Zawartość białka całkowitego po rekonstytucji wynosi 6 - 14 mg/ml w roztworze. Aktywność swoista czynnika IX wynosi 2,5 j.m. na mg białka całkowitego.
Aktywność wszystkich czynników krzepnięcia jak również białka C i S (antygen) była badana zgodnie z aktualnymi międzynarodowymi standardami WHO.
Substancje pomocnicze o znanym działaniu:
Sód do 343 mg (ok 15 mmol) na 100 ml roztworu. Pełen wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań. Biały lub lekko zabarwiony proszek lub krucha, zestalona masa.
Nadwrażliwość na którąkolwiek substancję czynną lub pomocniczą wymienioną w punkcie 6.1.
W przypadku rozsianego wykrzepiania wewnątrznaczyniowego produkty zawierające kompleks protrombiny można stosować dopiero po zakończeniu fazy nadkrzepliwości.
Znane są incydenty małopłytkowości indukowanej heparyną.
Wskazana jest konsultacja z lekarzem specjalizującym się w leczeniu zaburzeń krzepnięcia. U pacjentów z nabytym niedoborem czynników krzepnięcia zależnych od witaminy K (np.
wywołanych leczeniem antagonistami witaminy K) Beriplex powinien być użyty w przypadku, kiedy konieczne jest szybkie wyrównanie niedoborów czynników krzepnięcia zespołu protrombiny, takich jak duże krwawienia lub pilny zabieg operacyjny. W innych przypadkach zredukowanie dawki leków antagonistów witaminy K i/lub podanie witaminy K jest wystarczające.
U pacjentów otrzymujących leki z grupy antagonistów witaminy K może występować stan nadkrzepliwości, podanie ludzkich czynników krzepnięcia zespołu protrombiny może ten stan zaostrzyć.
U pacjentów z wrodzonym niedoborem któregokolwiek czynnika krzepnięcia z grupy zależnych od witaminy K, swoisty czynnik krzepnięcia powinien być zastosowany, o ile jest dostępny.
W przypadku wystąpienia reakcji alergicznej lub typu anafilaktycznego, podawanie produktu Beriplex powinno być natychmiast przerwane (zaprzestanie wstrzykniecia) i powinno być rozpoczęte odpowiednie leczenie. Działania lecznicze zależą od rodzaju i stopnia ciężkości działania niepożądanego. Należy przestrzegać aktualnych standardów medycznych dotyczących leczenia wstrząsu.
Istnieje ryzyko wystąpienia zakrzepicy lub rozsianego wykrzepiania wewnątrznaczyniowego, zarówno u pacjentów z nabytymi jak i wrodzonymi niedoborami, leczonymi ludzkimi czynnikami krzepnięcia zespołu protrombiny szczególnie powtarzanymi dawkami. Ryzyko to może być większe u chorych leczonych w izolowanym niedoborze czynnika VII, podczas gdy pozostałe zależne od witaminy K czynniki krzepnięcia, z dłuższymi okresami półtrwania, mogą skumulować się do poziomu wyższego niż normalny. Pacjenci podczas leczenia ludzkimi czynnikami krzepnięcia zespołu protrombiny powinni być pod wnikliwą obserwacją w kierunku wystąpienia objawów rozsianego wykrzepiania śródnaczyniowego lub zakrzepicy.
Ponieważ istnieje potencjalne ryzyko powikłań zakrzepowo-zatorowych, należy zachować ostrożność, gdy Beriplex jest stosowany u pacjentów z chorobą wieńcową lub zawałem mięśnia sercowego
w wywiadzie, u pacjentów z chorobami wątroby, w postępowaniu przed i pooperacyjnym,
u noworodków oraz u pacjentów z podwyższonym ryzykiem choroby zakrzepowo-zatorowej lub rozsianego wykrzepiania śródnaczyniowego lub równoczesny niedobór inhibitora. W każdej z tych sytuacji należy rozważyć korzyści płynące z terapii Beriplex wobec ryzyka wystąpienia wymienionych powikłań.
U pacjentów z rozsianym wykrzepianiem wewnątrznaczyniowym może zachodzić konieczność uzupełnienia czynników krzepnięcia kompleksu protrombiny, jednak preparat czynników krzepnięcia można zastosować dopiero po zakończeniu fazy nadkrzepliwości (np. poprzez leczenie choroby podstawowej, trwałej normalizacji poziomu antytrombiny III).
Odwracanie działania antagonistów witaminy K naraża pacjentów na ryzyko zatorowo – zakrzepowe wynikające z choroby zasadniczej. Powrót do leczenia przeciwzakrzepowego powinien być dokładnie rozważony w możliwie najkrótszym czasie.
Wśród działań niepożądanych może wystąpić zależna od heparyny małopłytkowość typu II (HIT Typ II), ze zmniejszeniem liczby płytek poniżej 50% wartości wyjściowej i/lub wystąpieniem nowych lub niewyjaśnionych powikłań zatorowo-zakrzepowych podczas terapii heparyną. Początek typowo po 4- 14 dniach od podania heparyny, choć może pojawić się w ciągu 10 godzin u pacjentów niedawno leczonych heparyną (do 100 dni wstecz).
Zespół nefrotyczny został odnotowany w pojedynczych przypadkach po próbie indukcji immunotolerancji u pacjentów z hemofilią typu B, inhibitorami czynnika IX i historią reakcji alergicznych.
Nie ma dostępnych danych klinicznych dotyczących stosowania produktu Beriplex w przypadku krwawień okołoporodowych z powodu niedoboru witaminy K u noworodków.
Beriplex zawiera do 343 mg sodu (około 15 mmol) w 100 ml. Ilość tę należy uwzględnić w przypadku pacjentów będących na kontrolowanej diecie sodowej.
Bezpieczeństwo przeciwwirusowe
Do standardowych metod zapobiegania zakażeniom związanym z użyciem produktów przygotowanych na bazie krwi lub osocza ludzkiego należą selekcja dawców, skriningowe badanie dawców poszczególnych donacji oraz puli zbiorczej osocza na obecność swoistych markerów zakażeń oraz wdrażanie w procesie wytwarzania skutecznych metod inaktywacji/usuwania wirusów. Pomimo tych zabezpieczeń stosowanie preparatów przygotowanych na bazie krwi lub osocza ludzkiego nie wyklucza całkowicie możliwości przeniesienia czynników zakaźnych. Dotyczy to także nieznanych wirusów i innych patogenów.
Metody uważane za skuteczne dotyczą wirusów otoczkowych takich jak wirus ludzkiego niedoboru odporności (HIV), wirus wirusowego zapalenia wątroby typu B (HBV) i wirus wirusowego zapalenia wątroby typu C (HCV) oraz bezotoczkowego wirusa wirusowego zapalenia wątroby typu A i parwowirusa B19.
Zaleca się rozważenie zastosowania odpowiedniego szczepienia (przeciw wirusowemu zapaleniu wątroby typu A i B), jeżeli pacjent regularnie/wielokrotnie przyjmuje produkty kompleksu protrombiny otrzymywane z ludzkiego osocza.
Zdecydowanie zaleca się przy każdym podaniu produktu Beriplex odnotowanie w dokumentacji medycznej nazwy i numeru serii produktu w celu umożliwienia powiązania numeru serii produktu z pacjentem.
Czynniki krzepnięcia zespołu protrombiny neutralizują efekt leczenia antagonistami witaminy K,
nie są natomiast znane interakcje z innymi produktami leczniczymi.
W przypadku oznaczania testów krzepnięcia wrażliwych na obecność heparyny, u pacjentów otrzymujących wysokie dawki czynników krzepnięcia zespołu protrombiny, w otrzymanych wynikach testów należy uwzględnić dawkę heparyny zawartą w produkcie.
Ciąża i karmienie piersią
Bezpieczeństwo stosowania ludzkiego kompleksu protrombiny podczas ciąży i karmienia piersią nie zostało ustalone. Dotychczasowe wyniki badań na zwierzętach są niewystarczające do oceny wpływu stosowania preparatu na rozrodczość, rozwój zarodka lub płodu, przebieg ciąży oraz rozwój około
i poporodowy.
Dlatego też, u kobiet w okresie ciąży lub karmienia piersią ludzki kompleks protrombiny należy stosować tylko w uzasadnionych przypadkach
Płodność
Brak danych dotyczących wpływu na płodność.
Nie przeprowadzono badań nad wpływem produktu na zdolność prowadzenia pojazdów
i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Reakcje alergiczne lub typu anafilaktycznego były obserwowane niezbyt często, z ciężkimi reakcjami anafilaktycznymi, włącznie (patrz punkt 4.4).
Terapia zastępcza może prowadzić do wytworzenia krążących przeciwciał hamujących jeden lub kilka czynników kompleksu ludzkiej protrombiny. Efektem pojawienia się tych inhibitorów jest słaba odpowiedź na leczenie. W takich przypadkach zaleca się skontaktowanie ze specjalistycznym ośrodkiem leczenia hemofilii w celu uzyskania zaleceń dotyczących postępowania. Reakcje anafilaktyczne były obserwowane u pacjentów z przeciwciałami przeciw czynnikom zawartym
w Beriplexie.
Często obserwowano podwyższenie temperatury ciała.
Istnieje ryzyko rozwinięcia się epizodów zakrzepowo-zatorowych po podaniu ludzkiego kompleksu protrombiny (patrz punkt 4.4).
Tabelaryczne zestawienie działań niepożądanych produktu Beriplex
Poniżej przedstawiono działania niepożądane odnotowane na podstawie badań klinicznych, po wprowadzeniu produktu na rynek jak i w literaturze naukowej.
Poniższa tabela przedstawia działania niepożądane zgodnie z klasyfikacją układów i narządów MedDRA. Częstość występowania oparta na podstawie badań klinicznych przedstawia się w następujący sposób: bardzo często:( ≥ 1/10); często: (≥ 1/100 do< 1/10); niezbyt często:( ≥ 1/1 000 do < 1/100); rzadko:( ≥ 1/10 000 do < 1/1 000); bardzo rzadko:( < 1/10 000) lub nieznana (nie może być określona na podstawie dostępnych danych).
Klasyfikacja układów i narządów MedDRA | Działanie niepożądane | Częstość |
Zaburzenia układu naczyniowego i inne SOC | Zaburzenia zakrzepowo – zatorowe* | Często |
Zaburzenia krwi i układu limfatycznego | Zespół rozsianego wykrzepiania wewnątrznaczyniowego | Nie znana |
Zaburzenia układu immunologicznego | Nadwrażliwość lub reakcje alergiczne | Niezbyt często |
Reakcje anafilaktyczne z wstrząsem włącznie | Nie znana | |
Rozwój przeciwciał | Nie znana | |
Zaburzenia układu nerwowego | Ból głowy | Często |
Zaburzenia ogólne i stany w miejscu podania leku | Wzrost temperatury ciała | Często |
* Z przypadkami śmierci włącznie
Informacje na temat bezpieczeństwa w zakresie przenoszenia czynników zakaźnych, patrz punkt 4.4. Dzieci i młodzież
Brak danych dotyczących stosowania produktu Beriplex u dzieci i młodzieży.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych :
Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W celu uniknięcia przedawkowania wskazana jest regularna kontrola czynników krzepnięcia. Podawanie dużych dawek koncentratu czynników krzepnięcia zespołu protrombiny (przedawkowanie) może być powikłane zawałem mięśnia sercowego, rozsianym wykrzepianiem wewnątrznaczyniowym, zakrzepicą żylną oraz z zatorem tętnicy płucnej. W przypadku przedawkowania ryzyko wystąpienia powikłań zatorowo-zakrzepowych lub rozsianego wykrzepiania wewnątrznaczyniowego znacznie się zwiększa u pacjentów o podwyższonym ryzyku takich powikłań.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Grupa farmakoterapeutyczna: Leki przeciwkrwotoczne, czynniki krzepnięcia krwi II, VII, IX i X
w połączeniu
Kod ATC: B02B D01
Czynniki krzepnięcia II, VII, IX i X, które są syntetyzowane w wątrobie z udziałem witaminy K, są powszechnie nazywane czynnikami krzepnięcia zespołu protrombiny. Poza czynnikami krzepnięcia produkt Beriplex zawiera inhibitory zależnych od witaminy K czynników krzepnięcia białko C
i białko S.
Czynnik VII jest zymogenem aktywnej proteazy serynowej czynnika VIIa, który doprowadza do aktywacji zewnątrzpochodnego toru krzepnięcia. Kompleks tromboplastyna tkankowa – czynnik VIIa aktywuje czynniki krzepnięcia IX i X, czego efektem jest powstanie czynników IXa i Xa. W wyniku dalszej aktywacji kaskady krzepnięcia protrombina (czynnik II) przechodzi w formę aktywną i przekształcana jest w trombinę. Poprzez działanie trombiny, fibrynogen zostaje przekształcony w fibrynę, co doprowadza do uformowania się skrzepu. Prawidłowe wytwarzanie trombiny jest także niezwykle ważne dla prawidłowej funkcji płytek w hemostazie pierwotnej.
Izolowany ciężki niedobór czynnika VII prowadzi do ograniczenia powstawania trombiny i skłonności do krwawienia spowodowanej upośledzonym powstawaniem fibryny i zaburzeniem pierwotnej hemostazy. Izolowany niedobór czynnika IX jest jednym z rodzajów hemofilii (hemofilia B).
Izolowany niedobór czynnika II i czynnika X występuje bardzo rzadko, ale jest przyczyną bardzo poważnych krwawień, podobnych do tych w przebiegu klasycznej hemofilii.
Pozostałe składniki, inhibitory krzepnięcia białko C i białko S, są także syntetyzowane w wątrobie. Biologiczna aktywność białka C jest wymuszona przez kofaktor białko S.
Aktywowane białko C hamuje proces krzepnięcia poprzez inaktywację czynnika Va i VIIIa. Białko S jako kofaktor białka C wspomaga proces hamowania krzepnięcia. Niedobór białka C związany jest ze zwiększonym ryzykiem zakrzepicy.
Objawy nabytego niedoboru zależnych od witaminy K czynników krzepnięcia pojawiają się podczas leczenia antagonistami witaminy K. W przypadku ciężkiego niedoboru, pojawia się skłonność do ciężkich krwawień, szczególnie do przestrzeni zaotrzewnowej lub w postaci krwawień domózgowych, rzadziej w postaci wylewów do mięśni lub stawów. Ciężka postać niewydolności wątroby także objawia się znaczącym obniżeniem poziomów zależnych od witaminy K czynników krzepnięcia, przejawiającym się klinicznie tendencją do krwawień, nie mniej jest to często zespół równolegle przebiegającego wykrzepiania śródnaczyniowego o małym nasileniu, niskiego poziomu płytek krwi, niedoboru inhibitorów krzepnięcia oraz zaburzeń procesu fibrynolizy.
Podanie ludzkich czynników krzepnięcia zespołu protrombiny prowadzi do podwyższenia stężenia w osoczu czynników krzepnięcia zależnych od witaminy K i może chwilowo wyrównać zaburzenia procesu krzepnięcia u pacjentów z niedoborem jednego lub kilku czynników krzepnięcia.
Dane farmakokinetyczne i odzysk in-vivo były uzyskane z badań prowadzonych ze zdrowymi ochotnikami (N=15) i z dwóch badań nad odwracaniem działania antagonistów witaminy K
w leczeniu ostrych dużych krwawień lub w profilaktyce krwawień okołooperacyjnych(N=98, N=43).
Badanie na zdrowych ochotnikach:
15 zdrowych ochotników otrzymywało 50 j.m./kg produktu leczniczego Beriplex. IVR oznacza wzrost mierzalnego poziomu czynnika w osoczu (j.m./ml) jakiego można oczekiwać po infuzji czynników (j.m./kg) podawanych jako dawka produktu Beriplex. Przyrosty IVR dla czynników II, VII, IX, X
i Bałek C i S były ocenione. Maksymalne poziomy wszystkich czynników pojawiały się w przedziale 3 godzin. Średni zakres przyrostów IVR wynosił od 0,016 jednostek/ml dla czynnika IX do 0,028 dla Białka C. Mediana okresów półtrwania w osoczu oraz przyrosty IVR zostały przedstawione poniżej:
Parametr | Mediana okresu półtrwania w osoczu (zakres)/godziny | Przyrost IVR (j.m./ml na j.m./kg m.c.) | |
Średnia geometryczna | 90 % CI† | ||
Czynnik II | 60 (25 – 135) | 0,022 | (0,020 – 0,023) |
Czynnik VII | 4 (2 – 9) | 0,024 | (0,023 – 0,026) |
Czynnik IX | 17 (10 – 127) * | 0,016 | (0,014 – 0,018) |
Czynnik X | 31 (17 – 44) | 0,021 | (0,020 – 0,023) |
Czynnik C | 47 (9 – 122) * | 0,028 | (0,027 – 0,030) |
Czynnik S | 49 (33 – 83) * | 0,020 | (0,018 – 0,021) |
† przedział ufności
* końcowy okres półtrwania, model dwukompartmentowy
Beriplex jest dystrybuowany i metabolizowany w ustroju w ten sam sposób jak endogenne czynniki krzepnięcia II, VII, IX i X.
Podanie dożylne oznacza, że dostępność preparatu jest natychmiastowa; dostępność biologiczna jest
proporcjonalna do podanej dawki.
Badanie odwracania działania antagonistów witaminy K w leczeniu ostrych dużych krwawień: Średnia odzysku in –vivo została obliczona u 98 osób otrzymujących Beriplex w leczeniu krwawień podczas leczenia antagonistami witaminy K. Zakres przyrostowego IVR wynosił pomiędzy 0,016 j.m./ml dla czynnika VII i 0,019 j.m./ml dla białka C.
Badanie odwracania działania antagonistów witaminy K w leczeniu ostrych, dużych krwawień lub profilaktyki krwawień okołooperacyjnych:
Średnia odzysku in –vivo została obliczona u 43 osób otrzymujących Beriplex w leczeniu krwawień lub profilaktyce krwawień okołooperacyjnych w czasie podawania antagonistów witaminy K. Dożylne podawanie 1 j.m./kg produktu leczniczego Beriplex spowodowało wzrost poziomu czynników krzepnięcia zależnych od witaminy K w osoczu w zakresie od 0,013 do 0,023 j.m./ml.
Beriplex jako substancję czynną zawiera czynniki krzepnięcia zespołu protrombiny (czynniki II, VII, IX i X). Są one otrzymywane z ludzkiego osocza i działają w ten sam sposób jak czynniki endogenne występujące w ludzkim osoczu.
Badania nad toksycznością pojedynczej dawki pasteryzowanego lecz nienanofiltrowanego produktu wykazały umiarkowane działanie toksyczne u myszy po podaniu 200 j.m./kg, najwyższej testowanej dawki. Pojedyncza dawka do 100 j.m./kg w infuzji dożylnej pasteryzowanego i nanofiltrowanego produktu była tolerowana u szczurów. Badania przedkliniczne z podawaniem wielokrotnym dawek (przewlekła toksyczność, kancerogenność, wpływ na reprodukcję) nie mogą być przeprowadzone w standardowym modelu zwierzęcym z powodu powstawania przeciwciał po podaniu heterogennych ludzkich białek.
Zbadanie miejscowej tolerancji po podaniu dożylnym produktu Beriplex było przeprowadzone na królikach. Badania nad antygenowością przeprowadzone na królikach nie wykazały powstawania nowych determinant antygenowych wywołanych procesem pasteryzacji.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Proszek Heparyna Albumina ludzka
Ludzka Antytrombina III Sodu chlorek
Sodu cytrynian
HCl lub NaOH (w małych ilościach do ustalenia pH)
Rozpuszczalnik
Woda do wstrzykiwań
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywaniu
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania leku do stosowania Dawkowanie
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, oprócz wymienionych w
punkcie 6.6.
3 lata
Stabilność chemiczna i fizyczna produktu gotowego została wykazana przez 24 godziny
w temperaturze pokojowej (maksymalnie 25ºC). Jednak z mikrobiologicznego punktu widzenia, produkt powinien być zużyty natychmiast.
Nie przechowywać w temperaturze powyżej 25˚C. Nie zamrażać!
Fiolkę przechowywać w opakowaniu zewnętrznym, w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Beriplex P/N 250:
Proszek: fiolka z proszkiem ze szkła bezbarwnego(typ II), zamknięta korkiem (z gumy bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
Rozpuszczalnik: 10ml wody do wstrzykiwań w fiolce ze szkła bezbarwnego (typ I), zamkniętej korkiem (z gumy chlorobutylowej lub bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
1 system transferowy 20/20 z filtrem.
Beriplex P/N 500:
Proszek: fiolka z proszkiem ze szkła bezbarwnego(typ II), zamknięta korkiem (z gumy bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
Rozpuszczalnik: 20ml wody do wstrzykiwań w fiolce ze szkła bezbarwnego (typ I), zamkniętej korkiem (z gumy chlorobutylowej lub bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
1 system transferowy 20/20 z filtrem.
Beriplex P/N 1000:
Proszek: fiolka z proszkiem ze szkła bezbarwnego (typ II), zamknięta korkiem (z gumy bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
Rozpuszczalnik: 40ml wody do wstrzykiwań w fiolce ze szkła bezbarwnego (typ I), zamkniętej korkiem (z gumy chlorobutylowej lub bromobutylowej), aluminiową uszczelką oraz plastikowym wieczkiem typu flip-off.
1 system transferowy 20/20 z filtrem.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Instrukcje ogólne
Roztwór powinien być przezroczysty lub lekko opalizujący. Po przefiltrowaniu/pobraniu roztworu po rekonstytucji (patrz niżej), przed jego podaniem, należy sprawdzić, czy nie zawiera on widocznych gołym okiem cząstek lub odbarwień. Nie należy stosować roztworów mętnych lub zawierających osad lub cząstki.
Rekonstytucja i pobranie z fiolki należy przeprowadzać w warunkach aseptycznych.
Rekonstytucja
Doprowadzić rozpuszczalnik do temperatury pokojowej. Upewnić się, czy wieczka fiolek z proszkiem i z rozpuszczalnikiem są usunięte, przetrzeć gumowe korki płynem antyseptycznym oraz pozwolić by korki wyschły przed otwarciem opakowania Mix2Vial.
| 1. Otworzyć opakowanie zawierające Mix2Vial usuwając folię zabezpieczającą. Nie wyjmować Mix2Vial z blistra. |
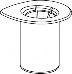
2 | 2. Umieścić fiolkę z rozpuszczalnikiem na czystej i równej powierzchni i mocno przytrzymać. Nie wyjmując z blistra systemu Mix2Vial nałożyć jego niebieską końcówkę z igłą na korek fiolki rozpuszczalnika i naciskając pionowo w dół przebić korek fiolki rozpuszczalnika. |
| 3. Przytrzymując krawędź systemu Mix2Vial ostrożnie zdjąć blister pociągając go pionowo do góry. Należy zwrócić uwagę aby zdjąć jedynie blister a nie cały zestaw Mix2Vial. |
4 | 4. Przytrzymując mocno fiolkę z produktem leczniczym na czystej i gładkiej powierzchni odwrócić fiolkę z rozpuszczalnikiem z systemem Mix2Vial i założyć przezroczystą część łącznika na korek fiolki z produktem. Naciskając pionowo w dół przebić korek fiolki z produktem. Rozpuszczalnik samoczynnie zostanie przeniesiony do fiolki z produktem leczniczym. |
5 | 5. Jedną ręką uchwycić fiolkę z produktem leczniczym przyłączoną do systemu Mix2Vial, drugą ręką uchwycić fiolkę z rozpuszczalnikiem także przyłączoną do systemu Mix2Vial i ruchem przeciwnym do wskazówek zegara ostrożnie rozkręcić zestaw na dwie części. Fiolkę po rozpuszczalniku wraz z niebieską końcówką systemu Mix2Vial usunąć. |
6 | 6. Doprowadzić do pełnego rozpuszczenia produktu delikatnie poruszając ruchem obrotowym fiolkę z produktem z przyłączoną przezroczystą końcówką systemu Mix 2Vial. Nie wstrząsać. |
7 | 7. Nabrać powietrza do pustej jałowej strzykawki. Trzymając fiolkę z produktem leczniczym pionowo korkiem do góry, przyłączyć strzykawkę do połączenia Luer Lock systemu Mix2Vial zgodnie z ruchem wskazówek zegara. Wstrzyknąć powietrze do fiolki z produktem. |
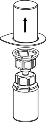

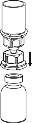

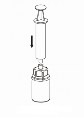
Pobieranie i podawanie
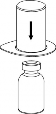
8 | 8. Przytrzymując tłok strzykawki odwrócić fiolkę wraz ze strzykawką do góry dnem i nabrać roztwór do strzykawki, powoli odciągając tłok. |
9 | 9. Po napełnieniu strzykawki roztworem, mocno uchwycić cylinder strzykawki (utrzymując strzykawkę tłokiem do dołu) i odłączyć system Mix2Vial od strzykawki ruchem przeciwnym do ruchu wskazówek zegara. |

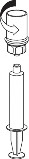
Należy uważać, aby krew nie dostała się do strzykawki wypełnionej produktem. Może to spowodować reakcję czynników krzepnięcia znajdujących się w produkcie i powstania skrzepów fibryny, które mogą być podane pacjentowi.
W przypadku jeżeli wymagane jest podanie więcej niż jednej fiolki produktu Beriplex, istnieje możliwość połączenia kilku fiolek w celu podania jako pojedynczej infuzji przez dostępny na rynku zestaw do podawania.
Roztworu produktu leczniczego Beriplex nie wolno rozcieńczać.
Roztwór po rekonstytucji powinien być podany dożylnie (nie szybciej niż 8 ml/min*).
Wszelkie niewykorzystanego resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami.
*W badaniach klinicznych Beriplexu pacjenci ważący poniżej 70 kg mieli zalecane dawkowanie z
maksymalną szybkością infuzji wynoszącą 0.12 ml/kg/min (poniżej 8 ml/min).
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU 15102; 15103; 21117
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
DOPUSZCZENIE DO OBROTU
CSL Behring GmbH Emil-von-Behring-Str. 76
35041 Marburg Niemcy








