







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Fiolka z proszkiem o pojemności 10 ml z bezbarwnego szkła typu I (Ph. Eur.), zamknięta gumowym korkiem (bezlateksowym) oraz aluminiowo-polipropylenowym karbowanym wieczkiem.
Berinin P 1200 j.m.:
Fiolka z proszkiem o pojemności 17 ml z bezbarwnego szkła typu II (Ph. Eur.), zamknięta gumowym korkiem (bezlateksowym) oraz aluminiowo-polipropylenowym karbowanym wieczkiem.
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Roztwór powinien być przezroczysty lub lekko opalizujący. Po przefiltrowaniu lub pobraniu (patrz poniżej), przed podaniem, produkt po rekonstytucji powinien być oceniony wizualnie w celu wykluczenia obecności cząsteczek i przebarwień. Nie używać roztworów mętnych lub
zawierających pozostałości (osad/cząstki).
Proces rekonstytucji oraz pobierania produktu musi się odbywać w warunkach aseptycznych.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Berinin P 600/1200, 600 j.m./1200 j.m.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań lub infuzji.
Każda fiolka zawiera nominalnie:
600 j.m./1200 j.m. ludzkiego IX czynnika krzepnięcia krwi (FIX).
Berinin P 600/1200 zawiera około 120 j.m./ml IX czynnika po rekonstytucji w 5/10 ml wody do wstrzykiwań.
Aktywność produktu (j.m) jest określana przy użyciu jednostopniowego testu krzepnięcia zgodnego z Farmakopeą Europejską.
Aktywność swoista Berinin P osiąga minimum 50 j.m. IX czynnika na mg białka całkowitego. Produkt jest wytwarzany z ludzkiego osocza.
Substancja pomocnicza o znanym działaniu:
Sód: około 167 mmol/l (3,84 mg/ml)
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań lub infuzji. Biały lub jasnożółty, higroskopijny proszek lub krucha, zestalona masa.
Bezbarwny rozpuszczalnik do sporządzania roztworu do wstrzykiwań lub infuzji.
Leczenie i profilaktyka krwawień u pacjentów z hemofilią typu B (wrodzonym niedoborem IX czynnika krzepnięcia krwi).
Leczenie powinno być prowadzone pod nadzorem lekarza doświadczonego w leczeniu hemofilii.
Monitorowanie leczenia
Podczas leczenia zaleca się właściwe oznaczanie poziomu IX czynnika, w celu ustalenia wielkości podawanej dawki oraz częstotliwości powtarzania wlewów. U poszczególnych pacjentów mogą wystąpić różnice w odpowiedzi na podanie IX czynnika krzepnięcia, co odzwierciedlają różnice
w okresie półtrwania oraz odzysku. Dawka ustalana na podstawie masy ciała może wymagać dostosowania u pacjentów z niedowagą lub nadwagą. Podczas dużych zabiegów chirurgicznych
niezbędne jest precyzyjne monitorowanie terapii substytucyjnej poprzez kontrolę procesu krzepnięcia (poziomu aktywności IX czynnika krzepnięcia w osoczu).
Dawkowanie
Dawkowanie i czas trwania terapii substytucyjnej zależy od stopnia ciężkości niedoboru IX czynnika krzepnięcia krwi, umiejscowienia i intensywności krwawienia oraz od stanu klinicznego pacjenta.
Liczba jednostek podawanego IX czynnika krzepnięcia krwi jest wyrażona w jednostkach międzynarodowych (j.m.), które odnoszą się do aktualnych standardów WHO dla produktów
IX czynnika krzepnięcia. Aktywność IX czynnika krzepnięcia w osoczu jest wyrażana w procentach (w odniesieniu do normalnego osocza ludzkiego) lub w jednostkach międzynarodowych (w odniesieniu do międzynarodowych standardów zawartości IX czynnika krzepnięcia krwi w osoczu).
Jedna jednostka międzynarodowa (j.m.) aktywności IX czynnika jest równa ilości IX czynnika krzepnięcia zawartego w jednym ml normalnego osocza ludzkiego.
Leczenie na żądanie
Obliczenie wymaganej dawki czynnika IX opiera się na doświadczeniach empirycznych dowodzących, że l jednostka międzynarodowa (j.m.) czynnika IX na kg masy ciała podwyższa
aktywność osoczowego czynnika IX o 1, 0% normalnej aktywności. Wymagana dawka jest obliczana przy użyciu następującego wzoru:
Wymagana liczba jednostek = masa ciała [kg] x pożądany wzrost aktywności czynnika IX [% lub j.m./dl] x 1,0
Podawana dawka produktu, częstotliwość oraz czas trwania jego podawania powinny zawsze być indywidualnie dobrane w zależności od efektywności klinicznej u poszczególnych pacjentów.
W razie wystąpienia następujących przypadków krwawień, aktywność czynnika IX nie powinna
zmniejszać się poniżej podanych poziomów aktywności w osoczu (w % normy lub j.m./dl) w danym okresie czasu. Schemat podany w poniższej tabeli może być stosowany w doborze dawkowania
w przypadkach krwawienia lub zabiegów chirurgicznych:
Nasilenie krwawienia / rodzaj | Wymagany poziom czynnika | Częstotliwość dawkowania |
zabiegu chirurgicznego | IX (% lub j.m./dl) | (godziny)/czas trwania leczenia |
(dni) | ||
Krwotok | ||
Wczesne krwawienie do stawów, mięśni lub z jamy ustnej | 20-40 | Powtarzać infuzje, co 24 godziny. Co najmniej |
przez 1 dobę, aż do | ||
ustąpienia bólu | ||
spowodowanego przez | ||
krwawienie lub zagojenia | ||
rany. | ||
Bardziej nasilone krwawienie | 30-60 | Powtarzać infuzję, co 24 |
do stawów, mięśni lub krwiak | godziny przez 3-4 dni lub | |
dłużej aż do ustania bólu i | ||
powrotu sprawności. | ||
Krwawienia zagrażające | 60-100 | Powtarzać infuzję, co 8 do 24 |
życiu | godzin aż do ustąpienia | |
zagrożenia. | ||
Zabiegi chirurgiczne | ||
Małe włącznie z ekstrakcją | 30-60 | Co 24 godziny, co najmniej |
zęba | przez 1 dzień, aż do zagojenia | |
rany. | ||
Duże | 80-100 | Powtarzać infuzję, co 8 do 24 |
(przed- i pooperacyjne) | godzin aż do zagojenia się | |
rany, następnie kontynuować | ||
terapię, przez co najmniej 7 kolejnych dni w celu utrzymania aktywności czynnika IX na poziomie 30% do 60% (j.m./dl) | ||
Profilaktyka
W długotrwałej profilaktyce krwawień u pacjentów z ciężką hemofilią typu B, zwykle podawane są dawki czynnika IX wynoszące 20 do 40 j.m. na kg masy ciała w odstępach od 3 do 4 dni.
U niektórych, zwłaszcza młodszych pacjentów, może być konieczne podanie produktu w krótszych odstępach czasu lub w większych dawkach.
Pacjenci powinni być monitorowani ze względu na możliwość pojawienia się inhibitorów IX czynnika krzepnięcia. Patrz też punkt 4.4.
Dzieci i młodzież
Nie istnieją dane z badań klinicznych pozwalające określić dawkowanie Berinin P u dzieci. Sposób podawania
Do stosowania dożylnego.
Instrukcja dotycząca rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6.
Produkt po rekonstytucji przed podaniem należy ogrzać do temperatury pokojowej lub temperatury ciała. Podawać powoli dożylnie za pomocą wstrzyknięcia lub infuzji, z szybkością dogodną dla pacjenta. Należy zapewnić, aby krew nie dostała się do strzykawki z produktem. Szybkość wstrzyknięć lub infuzji nie powinna przekraczać 2 ml na minutę.
Należy obserwować pacjenta w kierunku jakichkolwiek reakcji natychmiastowych. W
przypadku wystąpienia reakcji, które mogą być związane z podawaniem Berinin P, należy zmniejszyć szybkość infuzji lub wstrzymać podawanie, w zależności od stanu klinicznego pacjenta (patrz też punkt 4.4).
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Nadwrażliwość
Podczas stosowania Berinin P możliwe jest wystąpienie reakcji nadwrażliwości typu alergicznego. W przypadku wystąpienia objawów nadwrażliwości, pacjent powinien natychmiast zaprzestać stosowania produktu i skontaktować się z lekarzem. Pacjenci powinni być poinformowani o wczesnych objawach reakcji nadwrażliwości, do których należą pokrzywka, uogólniona pokrzywka, ucisk w klatce piersiowej, świszczący oddech, spadek ciśnienia tętniczego krwi oraz anafilaksja.
W przypadku wystąpienia wstrząsu należy zastosować odpowiednie postępowanie medyczne w terapii wstrząsu.
Berinin P 600 zawiera do 19,2 mg na fiolkę, Berinin P 1200 zawiera do 38,4 mg sodu na fiolkę. Należy wziąć to pod uwagę u pacjentów będących na kontrolowanej diecie niskosodowej.
Berinin P zawiera heparynę jako substancję pomocniczą. Może ona powodować reakcje alergiczne i obniżony poziom krwinek, przez co może wpływać na układ krzepnięcia. Pacjenci, u których
w wywiadzie występują reakcje alergiczne wywołane przez heparynę powinni unikać produktów leczniczych zawierających heparynę.
Inhibitory
Po wielokrotnym podawaniu produktów ludzkiego czynnika krzepnięcie IX, należy u pacjentów monitorować powstawanie przeciwciał neutralizujących (inhibitorów), które powinny być oznaczane w jednostkach Bethesda (BU, ang. Bethesda Units), za pomocą odpowiednich testów biologicznych.
W literaturze zgłaszano przypadki wskazujące na korelację pomiędzy pojawieniem się inhibitora
czynnika IX, a wystąpieniem reakcji alergicznych. Z tego względu pacjentów, u których wystąpiły reakcje alergiczne należy kontrolować na obecność inhibitora. Należy wziąć pod uwagę, iż u
pacjentów, u których stwierdzono obecność inhibitorów czynnika IX może wystąpić zwiększone ryzyko reakcji anafilaktycznej po kolejnym podaniu czynnika IX.
Z powodu ryzyka wystąpienia reakcji alergicznych związanych z podawaniem koncentratu IX
czynnika, rozpoczęcie jego podawania powinno zgodnie z oceną lekarza prowadzącego odbywać się pod kontrolą lekarską w warunkach, które umożliwiają zastosowanie odpowiedniego leczenia w przypadku pojawienia się reakcji alergicznych.
Choroba zakrzepowo-zatorowa
Ze względu na potencjalne ryzyko wystąpienia powikłań zakrzepowo-zatorowych, podając produkt pacjentom z chorobami wątroby, pacjentom po zabiegach chirurgicznych, noworodkom lub pacjentom z ryzykiem wystąpienia powikłań zakrzepowo-zatorowych lub DIC, należy rozpocząć odpowiednią obserwację kliniczną wystąpienia wczesnych objawów powikłań zakrzepowo-zatorowych i koagulopatii ze zużycia z zastosowaniem odpowiednich testów biologicznych. W każdej z tych sytuacji należy rozważyć korzyści płynące z terapii Berinin P wobec ryzyka wystąpienia
wymienionych powikłań.
Zdarzenia sercowo-naczyniowe
U pacjentów, u których istnieją czynniki ryzyka wystąpienia zaburzeń sercowo-naczyniowych terapia substytucyjna za pomocą FIX może zwiększać to ryzyko.
Ryzyko występowania powikłań przy stosowaniu cewników
W przypadku konieczności użycia kaniuli do cewnikowania żył centralnych (ang. central venous access device, CVAD), należy rozważyć ryzyko związane z ich stosowaniem, takie jak miejscowe zakażenia, bakteriemia oraz zakrzepica w miejscu umieszczenia cewnika.
Bezpieczeństwo przeciwwirusowe
Zastosowano standardowe postępowanie zabezpieczające przed przenoszeniem zakażeń powodowanych zastosowaniem produktów leczniczych otrzymywanych z krwi lub osocza ludzkiego, do którego należą: selekcja dawców, badanie pojedynczych donacji i puli osocza na obecność swoistych markerów zakażenia i zastosowanie w trakcie procesu wytwarzania skutecznych metod inaktywacji/usuwania wirusów. Pomimo zastosowania wspomnianych wyżej metod, nie można
całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych po podaniu produktu
leczniczego pochodzącego z krwi lub osocza ludzkiego. Dotyczy to również nieznanych lub nowo odkrytych wirusów i innych czynników zakaźnych.
Podjęte środki zabezpieczające są uważane za skuteczne w odniesieniu do otoczkowych wirusów, takich jak: wirus ludzkiego niedoboru odporności (HIV), wirus zapalenia wątroby typu B (HBV)
i wirus zapalenia wątroby typu C (HCV) oraz wobec bezotoczkowego wirusa zapalenia wątroby typu A (HAV).
Metody inaktywacji/usuwania wirusów mogą mieć ograniczoną skuteczność wobec wirusów bezotoczkowych takich jak parwowirus B19.
Zakażenie parwowirusem B19 może mieć poważne skutki dla kobiet w ciąży (ze względu na możliwość zakażenia płodu) oraz dla osób z niedoborami odporności lub ze zwiększoną erytropoezą (np. przy anemii hemolitycznej).
Zalecane jest odpowiednie szczepienie przeciw wirusowemu zapaleniu wątroby typu A i B
u pacjentów regularnie/wielokrotnie przyjmujących produkty czynnika IX pochodzącego z osocza.
Stanowczo zaleca się, aby przy każdym podaniu Berinin P odnotować nazwę i serię produktu, w celu umożliwienia powiązania numeru serii produktu z pacjentem.
Dzieci i młodzież
Wymienione ostrzeżenia i środki ostrożności dotyczą zarówno dorosłych, jak i dzieci.
Nie zgłaszano przypadków interakcji produktów ludzkiego IX czynnika krzepnięcia z innymi produktami leczniczymi.
Nie przeprowadzano badań na zwierzętach dotyczących wpływu czynnika IX na reprodukcję. Ciąża i laktacja
Ze względu na rzadkie występowanie hemofilii typu B u kobiet, dane dotyczące stosowania czynnika
IX w okresie ciąży i karmienia piersią nie są dostępne.
Z tego względu w okresie ciąży i laktacji produkty czynnika IX należy stosować tylko w uzasadnionych przypadkach..
Płodność
Nie przeprowadzono badań na zwierzętach dotyczących wpływu IX czynnika krzepnięcia krwi na płodność
Berinin P nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Działania niepożądane odnotowano po wprowadzeniu produktu na rynek oraz na podstawie literatury naukowej.
Podsumowanie profilu bezpieczeństwa
Nadwrażliwość lub reakcje alergiczne, (wśród których występują: obrzęk naczynioruchowy, kłucie i pieczenie w miejscu infuzji, dreszcze, nagłe zaczerwienienie twarzy, uogólniona pokrzywka, ból głowy, pokrzywka, spadek ciśnienia tętniczego krwi, letarg, nudności, niepokój, tachykardia, ucisk w klatce piersiowej, uczucie mrowienia, wymioty, świszczący oddech) były obserwowane rzadko. W niektórych przypadkach wymienione objawy rozwijały się w ciężką anafilaksję (z wstrząsem
włącznie) i były ściśle powiązane czasowo z powstawaniem inhibitorów IX czynnika (patrz też punkt 4.4). Zaobserwowano zespół nerczycowy po próbie indukcji tolerancji immunologicznej u pacjentów z hemofilią B z inhibitorami czynnika IX i reakcją alergiczną w wywiadzie.
Pacjenci z hemofilią typu B mogą wytwarzać przeciwciała neutralizujące (inhibitory) IX czynnika krzepnięcia. Jeżeli pojawią się tego typu inhibitory, stan taki będzie objawiał się niewystarczającą
reakcją na leczenie. W takich przypadkach należy się skontaktować z wyspecjalizowanym ośrodkiem leczenia hemofilii.
Istnieje potencjalne ryzyko powikłań zakrzepowo-zatorowych po stosowaniu produktów zawierających IX czynnik, ze zwiększonym ryzykiem przy stosowaniu produktów
niskooczyszczonych. Stosowanie produktów niskooczyszczonych IX czynnika jest związane z
występowaniem zawału mięśnia sercowego, rozsianego wykrzepiania wewnątrznaczyniowego,
zakrzepicy żylnej i zatorowości płucnej. Stosowanie produktów wysokooczyszczonych IX czynnika rzadko związane jest z takimi działaniami niepożądanymi.
Podczas leczenia produktem Berinin P mogą wystąpić następujące działania niepożądane: reakcje nadwrażliwości lub alergiczne oraz gorączka. Ponadto u pacjentów może dojść do powstania inhibitorów FIX.
Tabelaryczne zestawienie działań niepożądanych po wprowadzeniu produktu na rynek:
Działania niepożądane zostały przedstawione w tabeli poniżej zgodnie z systemem klasyfikacji układów i narządów MedDRA.
Częstość występowania tych działań niepożądanych oceniano według następujących kryteriów: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); niezbyt często (> 1/1 000 do < 1/100); rzadko
(≥ 1/10 000 do < 1/1 000); bardzo rzadko (< 1/10 000); częstość nieznana (nie może być określona na podstawie dostępnych danych).
Klasyfikacja układów i narządów MedDRA | Działanie niepożądane | Częstość występowania |
Zaburzenia nerek i dróg moczowych | Zespół nerczycowy | Nieznana |
Zaburzenia naczyniowe | Epizody zakrzepowo-zatorowe | Nieznana |
Zaburzenia ogólne i stany w miejscu podania | Gorączka | Nieznana |
Zaburzenia układu immunologicznego | Nadwrażliwość (reakcje alergiczne) | Nieznana |
Zaburzenia krwi i układu chłonnego | Inhibicja FIX Trombocytopenia | Nieznana Nieznana |
Opis wybranych reakcji niepożądanych
Zaburzenia krwi i układu limfatycznego:
Zgłaszano przypadki wystąpienia u pacjentów trombocytopenii typu II heparynozależnej (HAT Type II), kiedy liczba płytek krwi znacznie spada poniżej 100 000 na l lub wynosi mniej niż 50% ich początkowej wartości. U pacjentów, u których nadwrażliwość na heparynę dotychczas nie
występowała, do spadku liczby płytek krwi dochodzi zazwyczaj po 6 do 14 dniach od rozpoczęcia leczenia. U pacjentów nadwrażliwych na heparynę do takiego spadku może dojść już w ciągu kilku godzin od jej podania.
Do ciężkiej postaci trombocytopenii może dojść na skutek zakrzepów/zaburzeń zatorowo-
zakrzepowych w tętnicach lub żyłach, koagulopatii ze zużycia, której w niektórych przypadkach może towarzyszyć martwica skóry w miejscu iniekcji, wybroczyny, plamica i smolisty stolec. W takich przypadkach hamujące działanie heparyny na proces krzepnięcia może ulec zmniejszeniu (tolerancja heparyny).
U pacjentów, u których doszło do wystąpienia wyżej wymienionych reakcji alergicznych podawanie Berinin P należy natychmiast przerwać. Pacjenci tacy powinni być poinformowani, że nie wolno im przyjmować w przyszłości żadnych leków zawierających heparynę.
W celu uzyskania informacji dotyczących bezpieczeństwa wirusowego, patrz punkt 4.4.
Dzieci i młodzież
Częstość, rodzaj i nasilenie reakcji niepożądanych u dzieci są porównywalne, jak u dorosłych.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań
niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania
Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie są znane objawy przedawkowania ludzkiego IX czynnika krzepnięcia.
Grupa farmakoterapeutyczna: Leki przeciwkrwotoczne/ IX czynnik krzepnięcia krwi. Kod ATC: B02B D04
IX czynnik jest glikoproteiną o pojedynczym łańcuchu o masie molekularnej wynoszącej około 68 000 Daltonów. Jest czynnikiem krzepnięcia krwi zależnym od witaminy K, syntetyzowanym w
wątrobie. Do aktywacji IX czynnika dochodzi na drodze wewnętrznej przy udziale XIa czynnika oraz przy udziale VII czynnika / tkankowego zespołu czynników na drodze zewnętrznej. Aktywowany IX czynnik, w połączeniu z aktywowanym VIII czynnikiem, powoduje aktywację X czynnika Aktywowany X czynnik powoduje przekształcenie protrombiny w trombinę. Trombina przekształca fibrynogen w fibrynę, co prowadzi do uformowania skrzepu. Hemofilia typu B jest dziedzicznym, sprzężonym z płcią zaburzeniem krzepnięcia krwi, spowodowanym obniżonym poziomem IX
czynnika krzepnięcia, który objawia się obfitymi krwawieniami do stawów, mięśni lub organów wewnętrznych, występującymi samoistnie lub będącymi wynikiem urazów lub zabiegów chirurgicznych. Zastosowanie terapii zastępczej powoduje podwyższenie poziomu IX czynnika w osoczu, umożliwiając czasowe zmniejszenie jego niedoboru i ograniczenie krwawienia.
W badaniach farmakokinetycznych przeprowadzonych na 14 pacjentach średni okres półtrwania wyniósł 23 godziny (10 do 43 godzin), średni IVR wyniósł 1,1 (j.m./dl)/(j.m./kg) ( 0,57 do 1,69 (j.m./dl)/(j.m./kg). Średni MRT wyniósł 27 godzin (18 do 40 godzin), a średnia wartość klirensu wynosi 4,5 ml/godz./kg (2,5 do 12,5 ml/godz./kg).
Dzieci i młodzież
Dane farmakokinetyczne nie są dostępne.
Substancją aktywną Berinin P jest ludzki IX czynnik krzepnięcia, uzyskany z ludzkiego osocza, który działa w ten sam sposób, co endogenny składnik osocza. Podawanie pojedynczych dawek Berinin P różnym gatunkom zwierząt nie ujawniło objawów toksycznych. Przedkliniczne badania po wielokrotnym podaniu produktu (toksyczność przewlekła, kancerogenność, wpływ na toksyczność
reprodukcji) nie mogą być poprawnie przeprowadzone na konwencjonalnych modelach zwierzęcych, ze względu na produkcję przeciwciał skierowanych przeciwko heterologicznym (ludzkim) białkom.
Proszek:
glicyna, antytrombina III, heparyna, wapnia chlorek, sodu chlorek, sodu cytrynian
kwas solny lub sodu wodorotlenek (w małych ilościach do ustalenia pH).
Dołączony rozpuszczalnik:
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, oprócz wymienionych w punkcie 6.1.
3 lata
Stabilność fizykochemiczna produktu po rekonstytucji utrzymuje się przez 16 godzin w temperaturze pokojowej (≤ 25oC).
Z mikrobiologicznego punktu widzenia oraz z powodu, że Berinin P nie zawiera środków konserwujących, produkt powinien być zużyty natychmiast po rekonstytucji. Jeżeli produkt nie zostanie podany natychmiast, okres jego przechowywania nie powinien przekraczać 8 godzin w temperaturze pokojowej (≤ 25oC).
Nie przechowywać w temperaturze powyżej 25oC. Nie zamrażać.
Przechowywać fiolki w opakowaniu zewnętrznym w celu ochrony przed światłem.
Opakowania bezpośrednie
Fiolki z proszkiem:
Berinin P 600 j.m.:
Fiolki z rozpuszczalnikiem (woda do wstrzykiwań):
5 ml dla Berinin P 600 j.m:
- Fiolka z rozpuszczalnikiem o pojemności 6 ml, z bezbarwnego szkła typu I (Ph. Eur.), zamknięta gumowym korkiem (bezlateksowym) oraz aluminiowo-polipropylenowym karbowanym wieczkiem.
10 ml dla Berinin P 1200 j.m.:
- Fiolka z rozpuszczalnikiem o pojemności 10 ml, z bezbarwnego szkła typu I (Ph. Eur.), zamknięta gumowym korkiem (bezlateksowym) oraz aluminiowo-polipropylenowym karbowanym wieczkiem.
Dostępne opakowania:
Berinin P 600:
1 fiolka z proszkiem
1 fiolka z wodą do wstrzykiwań po 5 ml 1 system transferowy z filtrem 20/20
Berinin P 1200:
1 fiolka z proszkiem
1 fiolka z wodą do wstrzykiwań po 10 ml 1 system transferowy z filtrem 20/20
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Informacje ogólne:
Rekonstytucja:
Doprowadzić rozpuszczalnik do temperatury pokojowej. Upewnić się, że nasadki z fiolek produktu
leczniczego i rozpuszczalnika zostały usunięte, przetrzeć powierzchnię gumowych korków roztworem antyseptycznym i pozwolić, by korki wyschły przed otwarciem opakowania Mix2Vial.
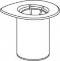
| 1. Otworzyć opakowanie zawierające Mix2Vial usuwając folię zabezpieczające. Nie wyjmować Mix2Vial z opakowania blistrowego! |
| 2. Umieścić fiolkę z rozpuszczalnikiem na czystej i równej powierzchni i mocno przytrzymać. Nie wyjmując z opakowania blistrowego zestawu Mix2Vial nałożyć jego niebieską końcówkę z igłą na korek fiolki z rozpuszczalnikiem i naciskając pionowo w dół przebić korek fiolki z rozpuszczalnikiem. |
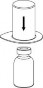
| 3. Przytrzymując krawędź zestawu Mix2Vial ostrożnie zdjąć opakowanie blistrowe pociągając go pionowo do góry. Należy zwrócić uwagę, aby zdjąć jedynie opakowanie blistrowe, a nie cały zestaw Mix2Vial. |
| 4. Umieścić fiolkę z produktem na czystej, równej i twardej powierzchni. Odwrócić do góry dnem fiolkę z rozpuszczalnikiem i dołączonym do niej zestawem Mix2Vial. Naciskając przezroczystą końcówkę z igłą pionowo w dół wbić w korek fiolki z proszkiem. Rozpuszczalnik samoczynnie zostanie przetransferowany do fiolki z proszkiem. |
| 5. Jedną ręką uchwycić fiolkę z produktem przyłączoną do zestawu Mix2Vial, drugą ręką uchwycić fiolkę po rozpuszczalniku także przyłączoną do zestawu Mix2Vial i ostrożnie rozkręcić zestaw na dwie części, przeciwnie do ruchu wskazówek zegara. Fiolkę po rozpuszczalniku wraz z niebieską końcówką zestawu Mix2Vial usunąć. |
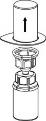
6 | 6. Doprowadzić do pełnego rozpuszczenia produktu delikatnie poruszając ruchem obrotowym fiolkę z produktem z przyłączoną przezroczystą końcówką do zestawu Mix 2Vial. Nie wstrząsać. |
| 7. Nabrać powietrza do pustej, jałowej strzykawki. Trzymając fiolkę z produktem pionowo korkiem do góry, przyłączyć strzykawkę do połączenia Luer Lock zestawu Mix2Vial, przekręcając zgodnie z ruchem wskazówek zegara. Wstrzyknąć powietrze do fiolki z produktem. |
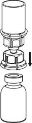
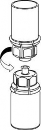


Pobieranie i podawanie
| 8. Przytrzymując tłok strzykawki odwrócić fiolkę wraz ze strzykawką do góry dnem i nabrać roztwór do strzykawki, powoli odciągając tłok. |
| 9. Po napełnieniu strzykawki roztworem, mocno uchwycić cylinder strzykawki (utrzymując strzykawkę tłokiem do dołu) i odłączyć przezroczystą końcówkę zestawu Mix2Vial od strzykawki, przekręcając przeciwnie do ruchu wskazówek zegara. |

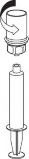
W celu wykonania iniekcji Berinin P należy używać plastikowych jednorazowych strzykawek,
ponieważ roztwory tego typu mają tendencję do przylegania do powierzchni strzykawek wykonanych ze szkła.
Podawać produkt w powoli dożylnie (patrz punkt 4.2.), uważając, aby nie spowodować przedostania się krwi do strzykawki wypełnionej produktem.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
CSL Behring GmbH Emil-von-Behring-Str. 76
35041 Marburg Niemcy
4838; 4839
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 27 marzec 2000 Data ostatniego przedłużenia pozwolenia: 17 luty 2014








