







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
zespołach pierwotnego niedoboru odporności (PID) z upośledzeniem wytwarzania przeciwciał;
zespołach wtórnego niedoboru odporności (SID) u pacjentów, u których występują ciężkie lub nawracające zakażenia i u których leczenie środkami przeciwdrobnoustrojowymi jest nieskuteczne oraz występuje potwierdzone niepowodzenie wytworzenia swoistych przeciwciał (PSAF)* lub stężenie IgG w surowicy < 4 g/l.
*PSAF = niepowodzenie uzyskania co najmniej dwukrotnego wzrostu miana przeciwciał IgG w odpowiedzi na szczepionki przeciwko pneumokokom zawierające antygeny polisacharydowe i polipeptydowe.
Leczenie immunomodulujące u dorosłych oraz dzieci i młodzieży (0-18 lat) w:
pierwotnej małopłytkowości immunologicznej (ITP) u pacjentów z dużym ryzykiem krwawień lub w celu skorygowania liczby płytek krwi przed zabiegiem chirurgicznym;.
zespole Guillaina-Barrégo;
chorobie Kawasakiego (w skojarzeniu z kwasem acetylosalicylowym; patrz punkt 4.2);
przewlekłej demielinizacyjnej polineuropatii zapalnej (CIDP);
wieloogniskowej neuropatii ruchowej (MMN).
Leczenie immunomodulujące u dorosłych powyżej18 roku życia w:
- - ciężkim zaostrzeniu miastenii.
Dawkowanie i sposób podawania
0,8–1 g/kg podawane w dniu pierwszym; dawkę tę można powtórzyć jeden raz w ciągu trzech dni
0,4 g/kg podawane codziennie przez dwa do pięciu dni.
g/kg przez 1–2 kolejnych dni co 3 tygodnie.
Wynik leczenia należy oceniać po każdym cyklu; w przypadku braku widocznego efektu leczenia po upływie 6 miesięcy leczenie należy przerwać.
Jeśli leczenie jest skuteczne, decyzja w sprawie jego długotrwałego stosowania należy do lekarza
i powinna być oparta na odpowiedzi pacjenta na leczenie, w tym na dawki podtrzymujące. Może istnieć konieczność dostosowania odstępów dawkowania do indywidualnego przebiegu choroby.
Wieloogniskowa neuropatia ruchowa (MMN)
Dawka początkowa: 2 g/kg podzielone na 2–5 kolejnych dni.
Dawka podtrzymująca: 1 g/kg co 2–4 tygodni lub 2 g/kg co 4–8 tygodni.
Wynik leczenia należy oceniać po każdym cyklu; w przypadku braku widocznego efektu leczenia po upływie 6 miesięcy leczenie należy przerwać.
Jeśli leczenie jest skuteczne, decyzja w sprawie jego długotrwałego stosowania należy do lekarza
i powinna być oparta na odpowiedzi pacjenta na leczenie, w tym na dawki podtrzymujące. Może istnieć konieczność dostosowania odstępów dawkowania do indywidualnego przebiegu choroby.
Ciężkie zaostrzenia miastenii:
g/kg w dawkach podzielonych przez 2 dni (w dawkach 1 g/kg dziennie)
Nie przeprowadzono badań klinicznych nad zastosowaniem Gamunex 10% u wystarczającej liczby chorych w wieku 65 lat lub powyżej, aby można było określić precyzyjnie wynik leczenia.
Zalecane dawkowanie podsumowano w poniższej tabeli:
Wskazanie
Dawka
Częstość wstrzyknięć
Leczenie substytucyjne
Zespoły pierwotnego niedoboru odporności
Dawka początkowa: 0,4–0,8 g/kg
Dawka podtrzymująca: 0,2–0,8 g/kg
co 3–4 tygodnie
Wtórne niedobory odporności (wg definicji w punkcie 4.1)
0,2–0,4 g/kg
co 3–4 tygodnie
Leczenie immunomodulacyjne:
Pierwotna małopłytkowość immunologiczna
0,8–1 g/kg
lub
w dniu 1, z możliwością powtórzenia jeden raz w ciągu 3 dni
0,4 g/kg/d
przez 2–5 dni
Zespół Guillaina-Barrégo
0,4 g/kg/d
przez 5 dni
Wskazanie
Dawka
Częstość wstrzyknięć
Choroba Kawasakiego
2 g/kg
w jednej dawce w leczeniu skojarzonym
z kwasem acetylosalicylowym
Przewlekła demielinizacyjna polineuropatia zapalna (CIDP)
Dawka początkowa: 2 g/kg
w dawkach podzielonych przez 2–5 dni
Dawka podtrzymująca: 1 g/kg
co 3 tygodnie przez 1–2 dni
Wieloogniskowa neuropatia ruchowa (MMN)
Dawka nasycająca: 2 g/kg
w dawkach podzielonych przez 2– 5 kolejnych dni
Dawka podtrzymująca: 1 g/kg
lub
co 2–4 tygodnie lub
2 g/kg
co 4–8 tygodni w dawkach podzielonych przez 2–5 dni
Ciężkie zaostrzenia miastenii
2 g/kg
W dawkach podzielonych przez 2 dni (w dawkach 1 g/kg dziennie)
Dzieci i młodzież
Dawkowanie w przypadku dzieci i młodzieży (0-18 lat) nie różni się od dawkowania u dorosłych, ponieważ przy każdym wskazaniu dawkowanie jest podawane w przeliczeniu na masę ciała i korygowane w zależności od wyniku postępowania klinicznego w powyższych chorobach.
Zaburzenia czynności wątroby
Nie są dostępne dowody przemawiające za koniecznością dostosowywania dawki.
Zaburzenia czynności nerek
Nie należy dostosowywać dawki, chyba że jest to uzasadnione ze względów klinicznych (patrz punkt 4.4).
Osoby w podeszłym wieku
Nie należy dostosowywać dawki, chyba że jest to uzasadnione ze względów klinicznych (patrz punkt 4.4). Sposób podawania
Do podawania dożylnego.
Immunoglobulinę ludzką normalną należy podawać dożylnie z początkową szybkością infuzji wynoszącą 0,6-1,2 ml/kg/godz. przez pół godziny. Patrz punkt 4.4. W przypadku wystąpienia reakcji niepożądanych należy zmniejszyć szybkość podawania lub przerwać infuzję. Jeżeli infuzja jest dobrze tolerowana, szybkość infuzji można stopniowo zwiększać do maksymalnej szybkości wynoszącej 4,8–8,4 ml/kg/godz.
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
nie są nadwrażliwi na ludzką normalną immunoglobulinę, rozpoczynając wstrzykiwanie produktu powoli (0,1 ml/kg/godzinę),
są uważnie monitorowani w trakcie infuzji, ze zwróceniem uwagi na jakiekolwiek objawy działań niepożądanych. Szczególnej uwagi wymagają pacjenci, którzy wcześniej nie otrzymywali ludzkiej immunoglobuliny, pacjenci, którym zmieniono podawany wcześniej alternatywny produkt IVIg oraz pacjenci, w przypadku których od poprzedniej infuzji upłynął długi okres. Tę grupę pacjentów należy obserwować w szpitalu podczas pierwszej infuzji i przez pierwszą godzinę po pierwszej infuzji w celu zauważenia oznak ewentualnych działań niepożądanych. Wszystkich pozostałych pacjentów należy obserwować przez co najmniej 20 minut po podaniu produktu.
U wszystkich pacjentów podawanie IVIg wymaga:
właściwego nawodnienia przed rozpoczęciem infuzji IVIg,
monitorowania wydalania moczu,
monitorowania stężenia kreatyniny w surowicy,
unikania równoczesnego stosowania diuretyków pętlowych (patrz punkt 4.5).
W przypadku wystąpienia reakcji niepożądanych należy zmniejszyć szybkość podawania lub przerwać infuzję. Wymagane leczenie zależy od charakteru i stopnia ciężkości reakcji niepożądanej.
Reakcje na infuzję
Niektóre działania niepożądane (np. ból głowy, uderzenia gorąca z zaczerwienieniem twarzy, dreszcze, bóle mięśni, świszczący oddech, częstoskurcz, bóle w okolicy lędźwiowej, nudności i niedociśnienie tętnicze) mogą być związane z szybkością infuzji. Należy ściśle przestrzegać zalecanej szybkości infuzji (podanej w punkcie 4.2). Przez cały okres infuzji należy ściśle monitorować i uważnie obserwować pacjenta pod kątem występowania jakichkolwiek objawów.
Działania niepożądane mogą występować częściej:
u pacjentów, którym immunoglobulinę ludzką normalną podaje się po raz pierwszy, w rzadkich przypadkach u pacjentów, którym zmieniono podawany wcześniej preparat immunoglobuliny ludzkiej normalnej na inny, oraz w sytuacjach, gdy od poprzedniej infuzji upłynął długi okres;
u pacjentów z nieleczonym zakażeniem lub podstawowym przewlekłym stanem zapalnym.
Nadwrażliwość
Reakcje nadwrażliwości są rzadkie.
Anafilaksja może wystąpić u pacjentów:
z niewykrywalnym stężeniem IgA, u których występują przeciwciała przeciw IgA;
którzy tolerowali wcześniejsze leczenie ludzką normalną immunoglobuliną.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
dreszcze, ból głowy, zawroty głowy, gorączka, wymioty, reakcje alergiczne, nudności, ból stawów, niskie ciśnienie krwi oraz bóle w okolicy lędźwiowej o umiarkowanym nasileniu;
odwracalne reakcje hemolityczne, szczególnie u pacjentów z grupą krwi A, B oraz AB i (rzadko) niedokrwistość hemolityczna wymagająca przetoczenia;
(rzadko) nagły spadek ciśnienia krwi, a w odosobnionych przypadkach wstrząs anafilaktyczny, nawet u pacjentów, którzy przy wcześniejszym podawaniu preparatu nie wykazywali reakcji nadwrażliwości;
(rzadko) przejściowe reakcje skórne (w tym toczeń rumieniowaty skórny – częstość nieznana);
(bardzo rzadko) incydenty zakrzepowo-zatorowe, takie jak zawał mięśnia sercowego, udar mózgu, zator płucny oraz zakrzepica żył głębokich;
przypadki odwracalnego jałowego zapalenia opon mózgowo-rdzeniowych;
przypadki zwiększenia stężenia kreatyniny w surowicy i/lub występowanie ostrej niewydolności nerek;
przypadki ostrego poprzetoczeniowego uszkodzenia płuc (TRALI).
Działania niepożądane podsumowane w formie tabeli
W poniższej tabeli przedstawiono działania niepożądane według klasyfikacji narządów i układów MeDRA (klasyfikacja układów i narządów oraz zalecane terminy). Częstości oceniono według następującej konwencji: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1000), bardzo rzadko (<1/10 000), nieznana (częstość nie może być określona na podstawie dostępnych danych).
W obrębie każdej grupy częstości występowania działań niepożądanych wymieniono w kolejności malejącej ciężkości.
Źródło bazy danych dotyczących bezpieczeństwa: badania kliniczne prowadzone łącznie u 703 pacjentów leczonych produktem leczniczym Gamunex 10% (ogółem 4378 infuzji)
Klasyfikacja układów
i narządów (SOC) MedDRA
Działanie niepożądane
Częstość na pacjenta
Częstość na infuzję
Zakażenia i zarażenia pasożytnicze
Zapalenie gardła
Niezbyt często
Niezbyt często
Zapalenie zatok, zapalenie cewki
moczowej, wirusowe zakażenie górnych dróg oddechowych
Niezbyt często
Rzadko
Zaburzenia krwi i układu
chłonnego
Niedokrwistość hemolityczna,
limfocytoza
Niezbyt
często
Rzadko
Klasyfikacja układów
i narządów (SOC) MedDRA
Działanie niepożądane
Częstość na pacjenta
Częstość na infuzję
Zaburzenia układu
immunologicznego
Nadwrażliwość
Niezbyt
często
Rzadko
Zaburzenia psychiczne
Niepokój
Niezbyt
często
Rzadko
Zaburzenia układu nerwowego
Ból głowy
Bardzo często
Często
Zawroty głowy
Niezbyt
często
Niezbyt
często
Bezgłos
Niezbyt
często
Rzadko
Zaburzenia oka
Światłowstręt
Niezbyt
często
Rzadko
Zaburzenia naczyniowe
Nadciśnienie tętnicze
Często
Niezbyt
często
Przełom nadciśnieniowy, niedociśnienie tętnicze, uderzenia
gorąca, przekrwienie
Niezbyt często
Rzadko
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia
Świszczący oddech, kaszel,
przekrwienie nosa
Niezbyt
często
Niezbyt
często
Duszność
Niezbyt
często
Rzadko
Zaburzenia żołądka i jelit
Nudności, wymioty
Często
Niezbyt często
Ból brzucha, biegunka, niestrawność
Niezbyt
często
Rzadko
Zaburzenia skóry i tkanki podskórnej
Wysypka, świąd, pokrzywka
Często
Niezbyt
często
Złuszczanie skóry, zapalenie skóry, kontaktowe zapalenie skóry, rumień
dłoni
Niezbyt często
Rzadko
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej
Bóle stawów, bóle pleców
Często
Niezbyt często
Bóle mięśni
Niezbyt często
Niezbyt często
Ból mięśniowo-szkieletowy, sztywność mięśni szkieletowych, ból
szyi
Niezbyt często
Rzadko
Zaburzenia nerek i dróg
moczowych
Hemoglobinuria
Niezbyt
często
Rzadko
Zaburzenia ogólne i stany w miejscu podania
Gorączka
Często
Często
Objawy grypopodobne, dreszcze,
zmęczenie
Często
Niezbyt
często
Osłabienie
Niezbyt
często
Niezbyt
często
Ból w klatce piersiowej, reakcje
w miejscu wstrzyknięcia, złe samopoczucie
Niezbyt często
Rzadko
Badania diagnostyczne
Podwyższone ciśnienie tętnicze, zmniejszenie liczby białych krwinek, zmniejszenie stężenia hemoglobiny, obecność wolnej hemoglobiny, zwiększenie szybkości opadania
krwinek czerwonych
Niezbyt często
Rzadko
Urazy, zatrucia i powikłania po
zabiegach
Stłuczenie
Niezbyt
często
Rzadko
Dzieci i młodzież
Przypuszczalnie częstotliwość, rodzaj i nasilenie działań niepożądanych u dzieci będą takie same jak u dorosłych.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych,
Al. Jerozolimskie 181C, 02-222 Warszawa,
Tel.: + 48 22 49 21 301,
Faks: + 48 22 49 21 309,
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
lata
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Gamunex 10%,
100 mg/ml roztwór do infuzji
Immunoglobulina ludzka normalna (IVIg) Jeden ml zawiera:
immunoglobulina ludzka normalna 100 mg
(o czystości co najmniej 98% IgG)
Każda fiolka po 10 ml zawiera: 1 g immunoglobuliny ludzkiej normalnej Każda fiolka po 50 ml zawiera: 5 g immunoglobuliny ludzkiej normalnej Każda fiolka po 100 ml zawiera: 10 g immunoglobuliny ludzkiej normalnej Każda fiolka po 200 ml zawiera: 20 g immunoglobuliny ludzkiej normalnej Każda fiolka po 400 ml zawiera: 40 g immunoglobuliny ludzkiej normalnej
Rozkład podklas IgG (wartości przybliżone):
IgG1. 62,8%
IgG2. 29,7%
IgG3. 4,8%
IgG4 2,7%
Maksymalna zawartość IgA to 84 mikrogramów/ml. Wytwarzana z osocza pozyskanego od ludzkich dawców.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na pojedynczą dawkę (maksymalnie 2 g/kg), to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
Roztwór do infuzji
Roztwór jest przezroczysty lub lekko opalizujący, bezbarwny lub jasnożółty.
Leczenie substytucyjne u dorosłych oraz dzieci i młodzieży (0-18 lat) w:
Leczenie substytucyjne należy rozpoczynać i monitorować pod nadzorem lekarza posiadającego doświadczenie w zakresie leczenia niedoborów odporności.
Dawkowanie:
Dawka i schemat dawkowania zależy od wskazań.
Może być konieczne dostosowanie dawek, indywidualnie dla każdego pacjenta w zależności od odpowiedzi klinicznej. Dawka oparta na masie ciała może wymagać dostosowania u pacjentów z niedowagą lub nadwagą.
Poniższe schematy dawkowania podano jako wytyczne.
Leczenie substytucyjne w zespołach pierwotnego niedoboru odporności
Dawkowanie powinno pozwolić na uzyskanie minimalnego stężenia IgG (mierzonego przed podaniem następnej infuzji), wynoszącego co najmniej 6 g/l lub mieszczącego się w zakresie prawidłowym odpowiednim dla wieku. Uzyskanie równowagi (stężenie IgG w stanie stacjonarnym) wymaga trzech do sześciu miesięcy od rozpoczęcia leczenia. Zalecana dawka początkowa wynosi 0,4-0,8 g/kg i jest podawana jednorazowo, a następnie co 3–4 tygodnie podaje się co najmniej 0,2 g/kg.
Dawka wymagana do osiągnięcia minimalnego stężenia IgG 6 g/l wynosi 0,2–0,8 g/kg/miesiąc. Odstęp czasowy pomiędzy dawkami po uzyskaniu stabilnego stanu waha się od 3 do 4 tygodni. Stężenia minimalne IgG należy mierzyć i oceniać w połączeniu z częstością występowania zakażeń. W celu zmniejszenia częstości zakażeń bakteryjnych konieczne może być zwiększenie dawkowania i dążenie do uzyskania większych stężeń minimalnych.
Wtórne niedobory odporności (wg definicji w punkcie 4.1)
Zalecana dawka wynosi 0,2–0,4 g/kg co 3–4 tygodnie.
Należy oznaczać stężenia minimalne IgG w celu dostosowania dawek w zależności od częstości występowania zakażeń. W razie konieczności należy dostosować dawkę, aby uzyskać optymalną ochronę przed zakażeniami. Może być konieczne zwiększenie dawki u pacjentów z utrzymującym się zakażeniem. Zmniejszenie dawki można rozważyć, gdy pacjent pozostaje wolny od zakażeń.
Pierwotna małopłytkowość immunologiczna
Istnieją dwa alternatywne schematy leczenia:
Leczenie można powtórzyć w razie wystąpienia nawrotu.
Zespół Guillaina-Barrégo
0,4 g/kg/dobę przez 5 dni (możliwe powtórzenie dawki w przypadku nawrotu).
Choroba Kawasakiego
2,0 g/kg należy podać w pojedynczej dawce. Pacjenci powinni jednocześnie otrzymywać leczenie kwasem acetylosalicylowym.
Przewlekła demielinizacyjna polineuropatia zapalna (CIDP)
Dawka początkowa: 2 g/kg podzielone na 2–5 kolejnych dni Dawki podtrzymujące:
Nadwrażliwość na substancję czynną (immunoglobuliny ludzkie) lub na którąkolwiek substancję pomocniczą (patrz punkty 4.4 i 6.1).
Pacjenci z selektywnym niedoborem IgA, u których pojawiły się przeciwciała przeciw IgA, gdyż podawanie produktu zawierającego IgA może doprowadzić do anafilaksji.
Wszystkich pacjentów należy uważnie monitorować przy stosowaniu wysokich szybkości infuzji (8,4 ml/kg/godzinę). U dzieci lub pacjentów zagrożonych niewydolnością nerek maksymalna szybkość infuzji nie powinna przekraczać 4,8 ml/kg/godzinę.
Produktu leczniczego Gamunex10% nie wolno mieszać z innymi roztworami do infuzji (np. solą fizjologiczną) i innymi produktami leczniczymi. Jeżeli przed infuzją konieczne jest rozcieńczenie preparatu, można do tego celu użyć roztworu glukozy 50 mg/ml. Jednakże w przypadku pacjentów z cukrzycą utajoną (może pojawić się przejściowy cukromocz), cukrzycą lub u pacjentów będących na diecie o obniżonej zawartości cukru zastosowanie 50 mg/ml roztworu glukozy powinno być uważnie monitorowane. Patrz także ostrzeżenie o ostrej niewydolności nerek poniżej.
Należy unikać jednoczesnego podawania produktu leczniczego Gamunex 10% oraz heparyny poprzez jedno urządzenie podające.
Identyfikowalność
W celu poprawy identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę handlową i numer serii podawanego produktu.
Środki ostrożności dotyczące stosowania
Potencjalnych powikłań często można uniknąć upewniając się, że pacjenci:
W przypadku wystąpienia wstrząsu należy wdrożyć standardowe leczenie wstrząsu. Incydenty zakrzepowo-zatorowe
Istnieją kliniczne dowody istnienia związku pomiędzy podawaniem dożylnym IVIg a incydentami zakrzepowo-zatorowymi, takimi jak zawał mięśnia sercowego, naczyniowe zdarzenie mózgowe (w tym udar mózgu), zatorowość płucna oraz zakrzepica żył głębokich, co do których uważa się, że są związane ze względnym wzrostem lepkości krwi po podaniu dużej ilości immunoglobulin pacjentom z grup ryzyka. Należy zachować ostrożność przy przepisywaniu oraz stosowaniu IVIg u pacjentów otyłych oraz u których istnieje ryzyko wystąpienia stanów zakrzepowych (takich jak podeszły wiek, nadciśnienie tętnicze, cukrzyca oraz choroby naczyniowe lub stany zakrzepowe w wywiadzie; pacjentów z nabytymi lub wrodzonymi zaburzeniami zakrzepowymi, pacjentów długotrwale unieruchomionych, pacjentów z ciężką hipowolemią, pacjentów z chorobami objawiającymi się wzrostem lepkości krwi).
W przypadku pacjentów zagrożonych zakrzepowo-zatorowymi działaniami niepożądanymi produkty IVIg należy podawać z minimalną szybkością infuzji, w minimalnej skutecznej dawce.
Ostra niewydolność nerek
Istnieją doniesienia o przypadkach ostrej niewydolności nerek u pacjentów otrzymujących leczenie IVIg. W większości przypadków zidentyfikowano czynniki ryzyka, takie jak wcześniej istniejąca niewydolność nerek, cukrzyca, hipowolemia, nadwaga; jednoczesne leczenie produktami leczniczymi o właściwościach nefrotoksycznych, lub wiek powyżej 65. roku życia.
Przed infuzją IVIg należy dokonać oceny parametrów nerkowych, zwłaszcza u pacjentów, u których stwierdzono potencjalnie zwiększone ryzyko wystąpienia ostrej niewydolności nerek, a ocenę tę należy powtarzać w odpowiednich odstępach czasu. U pacjentów z ryzykiem ostrej niewydolności nerek preparaty IVIg należy podawać z minimalną szybkością infuzji, w minimalnej skutecznej dawce. W razie wystąpienia zaburzenia czynności nerek należy rozważyć przerwanie leczenia IVIg.
Mimo iż doniesienia o zaburzeniu funkcji nerek i ostrej niewydolność nerek dotyczyły wielu dopuszczonych do obrotu preparatów IVIg, to za wystąpienie większości powikłań w głównej mierze odpowiedzialne były produkty zawierające różne substancje pomocnicze, takie jak sacharoza, glukoza i maltoza, preparaty zawierające sacharozę jako stabilizator. U pacjentów grupy ryzyka można więc rozważyć zastosowanie produktów IVIg niezawierających tych substancji pomocniczych. Gamunex10% nie zawiera sacharozy, maltozy ani glukozy.
Zespół jałowego zapalenia opon mózgowo-rdzeniowych (AMS)
Istnieją doniesienia o występowaniu zespołu jałowego zapalenia opon mózgowych w związku z leczeniem IVIg. Zespół ten zazwyczaj zaczyna się w ciągu od kilku godzin do dwóch dni po leczeniu IVIg. Badania płynu mózgowo-rdzeniowego dają zazwyczaj wynik dodatni, a pleocytoza wynosi do kilku tysięcy komórek na mm3, szczególnie granulocytów. Stężenie białka jest podwyższone i wynosi do kilkuset mg/dl. AMS może występować częściej przy leczeniu wysokimi dawkami IVIg (2 g/kg).
Pacjentów, u których występują takie objawy przedmiotowe i podmiotowe, należy poddać dokładnemu badaniu neurologicznemu, w tym badaniom płynu mózgowo-rdzeniowego, aby wykluczyć inne przyczyny zapalenia opon mózgowo-rdzeniowych.
Przerwanie leczenia IVIg powodowało wystąpienie remisji AMS w ciągu kilku dni bez żadnych następstw.
Niedokrwistość hemolityczna
Produkty IVIg mogą zawierać przeciwciała przeciw grupom krwi, które mogą działać jako hemolizyny i powodować in vivo opłaszczanie czerwonych krwinek immunoglobulinami, powodując dodatni wynik bezpośredniego odczynu antyglobulinowego (odczynu Coombsa) i, w rzadszych przypadkach, hemolizę. Niedokrwistość hemolityczna może wystąpić po leczeniu IVIg z powodu nasilonej sekwestracji czerwonych krwinek (RBC). Osoby otrzymujące IVIg powinny być monitorowane pod kątem klinicznych przedmiotowych i podmiotowych objawów hemolizy (patrz punkt 4.8).
Z rozwojem hemolizy związane są następujące czynniki ryzyka: duże dawki, niezależnie od tego, czy zostają podane jako dawka pojedyncza, czy też podzielona na kilka dni, grupa krwi inna niż 0 czy też toczący się stan zapalny. Większą uwagę należy poświęcić pacjentom z grupą krwi inną niż 0, otrzymujących duże dawki we wskazaniu niezwiązanym z pierwotnym niedoborem odporności. Hemoliza występuje rzadko u pacjentów otrzymujących terapię zastępczą w pierwotnym niedoborze odporności.
Odnotowano pojedyncze przypadki dysfunkcji nerek/niewydolności nerek ze skutkiem śmiertelnym związane z hemolizą.
Neutropenia/leukopenia
Po leczeniu z zastosowaniem IVIg zgłaszano przejściowe zmniejszenie liczby neutrofilów i/lub epizody neutropenii, niekiedy ciężkie. Zwykle dochodzi do nich w ciągu kilku godzin lub dni po podaniu IVIg, a epizody ustępują samoistnie w ciągu 7–14 dni.
Ostre poprzetoczeniowe uszkodzenie płuc (TRALI)
Wśród pacjentów otrzymujących IVIg zgłaszano przypadki ostrego niekardiogennego obrzęku płuc (ostrego poprzetoczeniowego uszkodzenia płuc [TRALI]). TRALI charakteryzuje się ciężką hipoksją, dusznością, przyspieszonym oddechem, sinicą, gorączką i niedociśnieniem tętniczym. Objawy TRALI zwykle występują w czasie przetoczenia lub w ciągu 6 godzin po jego zakończeniu – często w ciągu 1– 2 godzin. Dlatego w przypadku wystąpienia działań niepożądanych ze strony płuc osoby otrzymujące IVIg należy monitorować, a infuzję IVIg natychmiast przerwać. TRALI jest stanem potencjalnie zagrażającym życiu, który wymaga natychmiastowego leczenia na oddziale intensywnej opieki medycznej.
Interakcja z testami serologicznymi
Po podaniu immunoglobuliny przejściowy wzrost różnych przekazywanych biernie przeciwciał we krwi pacjenta może doprowadzić do fałszywie dodatnich wyników badań serologicznych.
Bierne przenoszenie przeciwciał przeciwko antygenom erytrocytów, np. A, B, D może zakłócać niektóre testy serologiczne pod kątem przeciwciał przeciw czerwonym krwinkom, na przykład bezpośredni odczyn antyglobulinowy (DAT, bezpośredni odczyn Coombsa).
Ryzyko przeniesienia zakażeń
Standardowe środki ostrożności stosowane w celu uniknięcia potencjalnych zakażeń wynikających z podawania produktów krwiopochodnych obejmują selekcję dawców, badania przesiewowe indywidualnych donacji krwi i puli osocza w kierunku swoistych markerów chorób zakaźnych oraz stosowanie skutecznych procedur inaktywacji/usuwania wirusów w procesie produkcji. Pomimo zastosowania powyższych środków ostrożności, kiedy podawane są produkty krwiopochodne nie można całkowicie wykluczyć ryzyka przeniesienia czynników zakaźnych, w tym nieznanych dotychczas wirusów lub innych patogenów.
Uważa się, że podejmowane działania zapobiegawcze są skuteczne wobec wirusów otoczkowych, takich jak ludzki wirus niedoboru odporności (HIV), wirus zapalenia wątroby typu B (HBV) i wirus zapalenia wątroby typu C (HCV). Usuwanie/inaktywacja wirusów bezotoczkowych takich jak HAV i parwowirus B19 może mieć ograniczoną skuteczność.
Z dotychczasowego doświadczenia klinicznego wynika, że preparaty immunoglobulin nie przenoszą wirusa zapalenia wątroby typu A ani parwowirusa B19.
Przyjmuje się, że obecność przeciwciał odgrywa istotną rolę zabezpieczającą przed zakażeniami wirusowymi.
Zdecydowanie zaleca się, aby przy każdym podaniu Gamunex 10% zapisać nazwę i numer serii preparatu, tak aby można było w przyszłości ustalić, jaką serię preparatu otrzymał pacjent.
Dzieci i młodzież
Pomimo, iż dostępne dane są ograniczone, można się spodziewać, że te same ostrzeżenia, środki ostrożności i czynniki ryzyka dotyczyć będą również populacji pediatrycznej. Z raportów po wprowadzeniu do obrotu wynika, że jest wysokie dawki IVIg stosowane u dzieci, zwłaszcza w chorobie Kawasakiego, powodują zwiększenie szybkości reakcji hemolitycznej w porównaniu do innych wskazań do stosowania immunoglobulin u dzieci.
Lekarze muszą zdecydowanie rozważyć monitorowanie stężenia hemoglobiny 24 do 48 godzin po zakończeniu stosowania IVIg, jeśli występuje podejrzenie wystąpienia hemolizy. Jeśli wymagane jest ponowne leczenie zaleca się monitorowanie stężenia hemoglobiny w tydzień po kolejnym podaniu IVIg, jeśli podejrzewane jest wystąpienie hemolizy. Rodziny powinny być zobowiązana do powrotu do lekarza, jeśli u ich dziecka rozwijają się objawy hemolizy, takie jak: bladość, ospałość, ciemne zabarwienie moczu, duszność i kołatanie serca.
Szczepionki żywe, atenuowane
Leczenie immunoglobulinami może osłabiać skuteczność szczepionek zawierających żywe, atenuowane wirusy (takich jak szczepionka przeciwko śwince, różyczce czy ospie wietrznej) w okresie co najmniej 6 tygodni do 3 miesięcy. Po podaniu tego produktu należy zachować odstęp 3 miesięcy przed podawaniem szczepionek zawierających żywe, atenuowane wirusy. W przypadku szczepienia przeciwko odrze może być ono nieskuteczne nawet do 1 roku. Dlatego u pacjentów otrzymujących szczepienie przeciw odrze należy sprawdzić miano przeciwciał.
Diuretyki pętlowe
Unikanie równoczesnego stosowania diuretyków pętlowych.
Dzieci i młodzież
Choć szczegółowe badania interakcji nie zostały przeprowadzone w populacji pediatrycznej, nie należy spodziewać się różnic pomiędzy dorosłymi i dziećmi.
Ciąża
Bezpieczeństwo stosowania u kobiet w ciąży nie zostało ustalone w kontrolowanych badaniach klinicznych i z tego powodu powinien być on stosowany z zachowaniem ostrożności u kobiet w ciąży lub karmiących piersią. Wykazano, że preparaty IVIg przenikają przez łożysko, zwłaszcza w czasie trzeciego trymestru. Doświadczenie kliniczne dotyczące stosowania immunoglobulin wskazuje, że nie należy spodziewać się wystąpienia szkodliwego działania preparatu na przebieg ciąży, na płód oraz na noworodka.
Karmienie piersią
Immunoglobuliny przenikają do mleka ludzkiego. Nie należy się spodziewać ujemnego wpływu na organizm noworodków/dzieci karmionych piersią.
Płodność
Doświadczenie kliniczne dotyczące stosowania immunoglobulin sugeruje, że nie należy się spodziewać szkodliwego wpływu na płodność.
Niektóre działania niepożądane związane ze stosowaniem produktu Gamunex 10% mogą upośledzać zdolność prowadzenia pojazdów i obsługiwania maszyn. Pacjenci, u których występują działania niepożądane podczas leczenia, powinni przed prowadzeniem pojazdów mechanicznych i obsługiwaniem urządzeń mechanicznych w ruchu odczekać, aż te reakcje ustąpią.
Podsumowanie profilu bezpieczeństwa
Do działań niepożądanych wywoływanych przez ludzkie normalne immunoglobuliny należą (podano zgodnie z malejącą częstością; patrz także punkt 4.4):
Przedawkowanie może spowodować hiperwolemię lub nadmierną lepkość krwi, zwłaszcza u pacjentów w podeszłym wieku oraz u pacjentów z zaburzoną czynnością serca lub nerek (patrz punkt 4.4).
Grupa farmakoterapeutyczna: surowice odpornościowe i immunoglobuliny; immunoglobuliny nieswoiste do podawania donaczyniowego, kod ATC: J06BA02
Ludzka normalna immunoglobulina zawiera głównie immunoglobulinę G (IgG) z szerokim spectrum przeciwciał przeciwko czynnikom zakaźnym.
Immunoglobulina ludzka normalna zawiera przeciwciała klasy IgG występujące w prawidłowej populacji. Przygotowywana jest zwykle z puli osocza pobieranego z nie mniej niż 1000 donacji. Rozkład podklas immunoglobuliny G jest ściśle proporcjonalny do rozkładu w natywnym osoczu ludzkim. Odpowiednie dawki tego produktu leczniczego mogą przywrócić normalnie niskie poziomy immunoglobuliny G do normalnego zakresu. Mechanizm działania we wskazaniach innych niż leczenie substytucyjne nie został w pełni wyjaśniony.
Produkt leczniczy Gamunex 10% doprowadza się do lekko kwaśnego odczynu pH. Ze względu na swoje słabe właściwości buforujące, Gamunex 10% podczas infuzji dożylnej jest szybko zobojętniany przez krew. Nawet po podaniu dużych dawek preparatu Gamunex 10% nie stwierdzano zmiany pH krwi.
Osmolalość roztworu wynosi 258 mOsmol/kg, a więc jest zbliżona do prawidłowego zakresu (285– 295 mOsmol/kg).
Badania kliniczne przeprowadzone z udziałem pacjentów z przewlekłą demielinizacyjną polineuropatią zapalną (CIDP):
Badanie IVIG-C CIDP skuteczności (badanie ICE) – prowadzone metodą podwójnie ślepej próby, randomizowane, kontrolowane placebo badanie skuteczności i bezpieczeństwa Gamunex 10% w leczeniu przewlekłej demielinizacyjnej polineuropatii zapalnej. 117 pacjentów z przewlekłą demielinizacyjną polineuropatią zapalną przydzielono w sposób randomizowany (losowy) do grup otrzymujących Gamunex 10% lub placebo co trzy tygodnie. Dawka nasycająca wynosiła 2 g/kg masy ciała, dawka podtrzymująca wynosiła 1 g/kg masy ciała.
Odsetek uczestników z pożądaną reakcją na leczenie (poprawa o co najmniej 1 punkt w skali niepełnosprawności INCAT utrzymująca się przez cały okres 24 tygodni badania skuteczności) był znacząco większy w grupie uczestników otrzymujących Gamunex 10% (54%) w porównaniu do grupy
otrzymującej placebo (21%, p=0,0002).Siła mięśni mierzona przy użyciu skali MRC i siła chwytu, jak również odczuwanie bodźców mierzone przy użyciu skali ISS uległy znacząco większej poprawie w grupie otrzymującej Gamunex 10% w porównaniu do grupy otrzymującej placebo.
Badania kliniczne produktu leczniczego nie obejmowały wystarczającej liczby uczestników w wieku 65 lat lub powyżej, aby stwierdzić pożądany wynik leczenia w tej grupie w odniesieniu do skali INCAT. Dla siły chwytu wykazano w badaniach statystycznie znaczący efekt leczenia dla Gamunex 10%.
Wśród uczestników z pożądaną reakcją na leczenie u mniej niż połowy pożądana reakcja wystąpiła do 3 tygodni od dawki nasycającej, a większość odpowiedziała na leczenie po drugiej dawce do 6. tygodnia. Uczestnicy, u których nie wystąpiła pożądana reakcja na leczenie zostali poddani innemu leczeniu przez kolejny okres 24 tygodni.
Wszyscy uczestnicy badania, którzy zareagowali na leczenie, byli ponownie randomizowani w fazie rozszerzenia na kolejny 6-miesięczny okres leczenia podtrzymującego z użyciem Gamunex 10% lub placebo. Spośród wcześniej przyjmujących Gamunex10% rzeczywisty wskaźnik nawrotów był istotnie większy u pacjentów z randomizacją do grupy otrzymującej placebo (42%) niż u pacjentów z Gamunex 10% (13%, p = 0,012).
Badanie ICE wykazało krótkoterminową i długoterminową skuteczność Gamunex 10% w leczeniu przewlekłej demielinizacyjnej polineuropatii zapalnej. Wyniki podsumowano w tabeli poniżej.
Pierwszorzędowy punkt końcowy i inne wyniki badania ICE
Gamunex 10% | Placebo | p | |
Odsetek uczestników z pożądaną reakcją na leczenie w czasie badania skuteczności (pierwszorzędowy punkt końcowy) | 54% | 21% | 0,0002 |
Prawdopodobieństwo nawrotu choroby w fazie przedłużenia badań | 13% | 45% | 0,013 |
Siła chwytu (kPA)1 (zmiana w porównaniu do poziomu wyjściowego) | |||
Ręka dominująca | 13,2 | 1,5 | 0,0008 |
Ręka niedominująca | 13,3 | 4,3 | 0,005 |
Siła mięśni (wynik końcowy wg skali MRCa)1 (zmiana w porównaniu do poziomu wyjściowego) | 3,3 | 0,2 | 0,001 |
Odczuwanie bodźców (skala ISSb)2 (zmiana w porównaniu do poziomu wyjściowego) | -1,2 | 0,2 | 0,021 |
1Poprawa wyrażona przez liczbę dodatnią 2Poprawa wyrażona przez liczbę ujemną aMRC: Medical Research Council
bISS: INCAT Sensory Sum Score
Badanie kliniczne z zastosowaniem Gamunex 10% w leczeniu pacjentów z zaostrzeniem miastenii:
Badanie Zinman et. al. (2007) prowadzone w sposób randomizowany, z podwójnie ślepą próbą, kontorolowane placebo w celu oceny skuteczności leczenia Gamunex 10% u 51 pacjentów z zaostrzeniem miastenii w dawce 2 g/kg przez dwa dni. Za pierwotny punkt końcowy badania przyjęto zmianę wyniku QMG między ocena przed leczeniem i w dniu 14-tym. W dniu 14-tym średnia zmiana wyniku QMG wynosiła -2,54 (p = 0,047).
Klinicznie istotny wpływ na przebieg zaostrzenia miastenii obserwowano jedymie w grupie badawczej z umiarkowanymi do ciężkich objawów miastenii z wynikiem QMG > 10,5 przed leczeniem, ze średnią zmianą wyniku -3,39 (p = 0,010).
Dodatkowe potwierdzenie wyników otrzymano z wieloośrodkowego, prospektywnego, otwartego niekontrolowanego badania klinicznego skuteczności i bezpieczeństwa Gamunex 10% w leczeniu zaostrzeń miastenii.
Do badania włączono 49 pacjentów, którym podawano Gamunex 10% w ilości 2 g/kg w dawkach podzielonych po 1 g/kg dziennie. Z badania wykluczono chorych z dodatnim wynikiem badań w kierunku przeciwciał MuSK.
Pierwotny punkt końcowy badania stanowiła zmiana wyniku Quantitative Myastenia Gravis (QMG) od początku leczenia do dnia 14-tego. Średnia zmiana wyniku QMG wyniosła -6,4 w grupie badanej skuteczności a w grupie badanego bezpieczeństwa -6,7.
Analiza wyników z punktów wtórnych i końcowych badań skuteczności (metodą QMG, MG-ADL oraz MG Composite) potwierdziła wyniki z pierwotnych punktów końcowych.
Immunoglobulina ludzka normalna po podaniu dożylnym jest w pełni i niezwłocznie biodostępna. Jej dystrybucja pomiędzy osoczem, a płynem zewnątrznaczyniowym zachodzi stosunkowo szybko; po około 3–5 dniach osiągana jest równowaga pomiędzy przestrzenią wewnątrz- i zewnątrznaczyniową.
Okres półtrwania immunoglobuliny ludzkiej normalnej oznaczany u pacjentów z pierwotnym zespołem niedoboru przeciwciał, wynosi 35 dni. Jest zatem dłuższy, niż podaje się w piśmiennictwie dla osób zdrowych (21 dni). Okres półtrwania IgG może być jednak różny u różnych pacjentów, zwłaszcza u tych z pierwotnymi zespołami niedoboru odporności.
Immunoglobuliny i kompleksy IgG są rozkładane w komórkach układu siateczkowo-śródbłonkowego. Dzieci i młodzież
Nie należy spodziewać się różnic w zakresie właściwości farmakokinetycznych w populacji
pediatrycznej.
Immunoglobuliny są normalnymi składnikami organizmu człowieka. Ponieważ w badaniach na zwierzętach podawanie immunoglobulin może prowadzić do powstawania przeciwciał, dlatego liczba danych z badań przedklinicznych dotyczących bezpieczeństwa preparatu jest ograniczona. W przeprowadzonych badaniach toksyczności ostrej i podostrej na zwierzętach, Gamunex 10% nie wykazywał szczególnego ryzyka dla ludzi.
Glicyna, woda do wstrzykiwania.
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, oprócz wymienionych w punkcie 6.6.
Przechowywać w temperaturze 2°C – 8°C (w lodówce). Nie zamrażać. Przechowywać w opakowaniu zewnętrznym.
Produkt może być przechowywany w oryginalnym opakowaniu zewnętrznym przez okres do 6 miesięcy w temperaturze pokojowej (nie powyżej 25ºC). W tym przypadku okres ważności upływa po 6 miesiącach. Nowy termin ważności musi być zapisany na zewnętrznym opakowaniu oraz na etykiecie fiolki. Nowy termin ważności nie może być późniejszy niż wydrukowany termin ważności. Dlatego, produkt należy zużyć lub zniszczyć. Nie wolno produktu ponownie umieszczać w lodówce lub zamrażać.
Roztwór do infuzji w fiolkach z przezroczystego szkła typu I lub typu II z korkiem z gumy chlorobutylowej.
Dostępne opakowania:
Jedno opakowanie po 10 ml zawierające: 1 g immunoglobuliny ludzkiej normalnej Jedno opakowanie po 50 ml zawierające: 5 g immunoglobuliny ludzkiej normalnej Jedno opakowanie po 100 ml zawierające: 10 g immunoglobuliny ludzkiej normalnej Jedno opakowanie po 200 ml zawierające: 20 g immunoglobuliny ludzkiej normalnej Jedno opakowanie po 400 ml zawierające: 40 g immunoglobuliny ludzkiej normalnej Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Przed podaniem produkt leczniczy należy doprowadzić do temperatury pokojowej lub temperatury ciała. Roztwór powinien być przezroczysty lub lekko opalizujący, bezbarwny lub bladożółty. Mętny roztwór, lub zawierający osad, nie powinien być stosowany. Wszelkie resztki niewykorzystanego produktu lub jego odpady należy usunąć zgodnie z lokalnymi przepisami. Po otwarciu opakowania jego zawartość należy natychmiast zużyć. Ze względu na możliwość rozwoju bakterii powtórne przechowywanie preparatu, nawet w lodówce, jest niedozwolone.
Jeżeli konieczne jest rozcieńczenie preparatu przed podaniem, należy użyć do tego celu roztworu glukozy 50 mg/ml. Nie rozcieńczać roztworami soli.
Należy unikać jednoczesnego podawania produktu leczniczego Gamunex 10% oraz heparyny poprzez jedno urządzenie podające.
Linie infuzyjne można przepłukać roztworem glukozy 50 mg/ml lub chlorku sodu (9 mg/ml), ale nie roztworem heparyny.
Korek heparynowy, przez który podawany jest Gamunex 10%, można przepłukać roztworem glukozy 50 mg/ml lub chlorku sodu (9 mg/ml), i nie można przepłukać roztworami heparyny.
Grifols Deutschland GmbH Colmarer Straße 22
60528 Frankfurt Niemcy
Tel.: +49 69-660 593 100
12750
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:05 kwietnia 2007
Data ostatniego przedłużenia pozwolenia: 8 czerwca 2011 roku
30 stycznia 2020
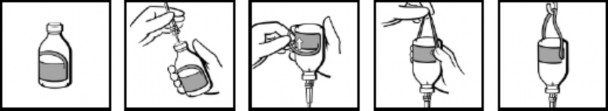
Instrukcja użycia fiolek (dotyczy tylko fiolek 50 ml, 100 ml, 200 ml i 400 ml)
Fiolki są dostarczone z etykietą ze zintegrowanym zawieszeniem (ryc. 1). Po podłączeniu zestawu do infuzji (ryc. 2) odwrócić fiolkę i odgiąć część etykiety w kształcie pętli (ryc. 3). Pociągnąć mocnym, zdecydowanym ruchem za pętlę, mocno przytrzymując ją palcami w miejscu, gdzie łączy się z resztą etykiety (ryc. 4). Zawiesić fiolkę na stojaku za powstałą pętlę (ryc. 5).
ryc. 1 ryc. 2 ryc. 3 ryc. 4 ryc. 5








