







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO\
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
podczas pierwszego trymestru ciąży występuje zwiększone ryzyko pojawienia się wrodzonych wad rozwojowych (w szczególności rozszczep wargi/podniebienia, spodziectwo oraz zaburzenia dotyczące innych układów).
Dane z rejestru ciąż North American Antiepileptic Drug dotyczące monoterapii topiramatem wykazały około 3-krotne zwiększenie częstości występowania dużych wad wrodzonych (4,3%), w porównaniu z grupą referencyjną, w której nie przyjmowano leków przeciwpadaczkowych (1,4%). Ponadto dane z innych badań wskazują, że w porównaniu z monoterapią występuje zwiększone ryzyko działania teratogennego związanego ze stosowaniem leków przeciwpadaczkowych w terapii skojarzonej. Stwierdzone ryzyko było zależne od dawki; działanie stwierdzano po wszystkich dawkach. U leczonych topiramatem kobiet, które mają dziecko z wadą wrodzoną, ryzyko wystąpienia wad wrodzonych jest zwiększone w kolejnych ciążach, jeśli są narażone na topiramat.
zwiększona jest częstość występowania małej urodzeniowej masy ciała (<2500 gramów) w porównaniu z grupą wzorcową
zwiększona jest częstość występowania niedoboru masy ciała w stosunku do wieku ciążowego (SGA, ang. small for gestational age; definiowane jako masa ciała poniżej 10 percentyla skorygowana pod względem wieku ciążowego i stratyfikowana względem płci). Nie można określić długotrwałych następstw SGA.
Wpływ na zdolność prowadzenia i obsługiwania maszyn
Działania niepożądane
zidentyfikowane jako działania niepożądane zgłoszone spontanicznie po wprowadzeniu produktu do obrotu. Częstość ich występowania została obliczona na podstawie danych z badań klinicznych lub zostało obliczone, jeśli zdarzenie nie wystąpiło w badaniach klinicznych.
Wady wrodzone i ograniczenie rozwoju płodu (patrz punkty 4.4 i 4.6). Dzieci i młodzież
Działania niepożądane zgłaszane częściej u dzieci (≥2-krotnie) niż u osób dorosłych podczas badań z
podwójnie ślepą próbą i kontroląplacebo obejmują:
zmniejszony apetyt
zwiększony apetyt
kwasicę hiperchloremiczną
hipokalemię
zaburzenia zachowania
napady agresji
apatię
zaburzenia zasypiania
myśli samobójcze
zaburzenia koncentracji
letarg
zaburzenia rytmu okołodobowego
niską jakość snu
nasilone łzawienie
bradykardię zatokową
samopoczucie odbiegające od normy
zaburzenia chodu.
Działania niepożądane, które odnotowano tylko u dzieci podczas badań z podwójnie ślepą i kontrolą placebo obejmują:
eozynofilię
nadaktywność psychoruchową
zawroty głowy
wymioty
hipertermię
gorączkę
trudności w uczeniu się
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO\
Etopro, 50 mg, tabletki powlekane
Jedna tabletka powlekana zawiera 50 mg topiramatu Substancje pomocnicze o znanym działaniu:
Każda tabletka zawiera 0,4 mg laktozy jednowodnej
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana.
Żółta, okrągła, obustronnie wypukła.
Monoterapia częściowych napadów padaczkowych, z wtórnym uogólnieniem lub bez oraz pierwotnie uogólnionych napadów kloniczno-tonicznych u osób dorosłych, młodzieży i dzieci w wieku powyżej 6 lat.
Terapia uzupełniająca u dzieci (w wieku 2 lat i powyżej), młodzieży i osób dorosłych z częściowymi napadami padaczkowymi z wtórnym uogólnieniem lub bez albo z pierwotnie uogólnionymi napadami padaczkowymi toniczno-klonicznymi oraz w leczeniu napadów padaczkowych związanych z zespołem Lennoxa-Gastauta.
Topiramat jest wskazany w zapobieganiu migrenie u dorosłych po dokładnym rozważeniu innych alternatywnych metod leczenia. Topiramat nie jest zalecany w leczeniu ostrego bólu głowy.
Dawkowanie
Zaleca się rozpoczęcie leczenia od małej dawki, a następnie należy ją stopniowo zwiększać do osiągnięcia dawki skutecznej. Dawka oraz stopniowe jej zwiększanie powinna być dostosowana do odpowiedzi klinicznej na leczenie.
Nie ma konieczności monitorowania stężeń topiramatu w osoczu w celu optymalizacji leczenia lekiem Etopro. W rzadkich przypadkach, dodanie topiramatu do leczenia fenytoiną może wymagać dostosowania dawki fenytoiny dla uzyskania optymalnego wyniku leczenia. Dodanie lub odstawienie fenytoiny lub karbamazepiny jako terapii uzupełniającej do Etopro, może powodować konieczność dostosowania dawki produktu leczniczego Etopro.
U pacjentów z atakami padaczki lub bez w wywiadzie, zakończenie podawania leków przeciwdrgawkowych (LPP), w tym topiramatu, powinno być przeprowadzane stopniowo, aby zminimalizować prawdopodobieństwo wystąpienia, bądź zwiększenia częstości napadów padaczki. W badaniach klinicznych dawki dobowe były zmniejszane w tygodniowych odstępach czasu, o 50-100 mg u osób dorosłych z epilepsją i o 25-50 mg u dorosłych przyjmujących topiramat w dawkach do 100 mg na dobę w profilaktyce migreny. W badaniach klinicznych z udziałem dzieci podawanie topiramatu przerywano stopniowo przez okres 2-8 tygodni.
Monoterapia padaczki
Zalecenia ogólne
W przypadku przerwania leczenia jednocześnie stosowanymi lekami przeciwpadaczkowymi w celu zmiany leczenia na monoterapię topiramatem, należy uwzględnić, jaki może mieć to wpływ na skuteczność kontroli napadów. Zaleca się stopniowe zmniejszanie ich dawki o około jedną trzecią, w odstępach dwutygodniowych, chyba, że względy bezpieczeństwa powodują konieczność natychmiastowego zakończenia podawania (stosowanych jednocześnie) innych leków przeciwpadaczkowych.
Po zakończeniu stosowania leków indukujących enzymy może nastąpić zwiększenie stężenia topiramatu w osoczu. Zmniejszenie dawkowania topiramatu może być wskazane ze względów klinicznych.
Dorośli
Dawkę oraz stopniowe jej zwiększanie należy ustalać na podstawie odpowiedzi klinicznej. Leczenie należy rozpocząć od dawki 25 mg podawanej wieczorem, przez jeden tydzień.
Następnie dawkę należy zwiększać w odstępach 1- lub 2-tygodniowych o 25 lub 50 mg/dobę i podawać w dwóch dawkach podzielonych. Jeśli pacjent nie toleruje powyższego schematu stopniowego zwiększania dawki leku, można ją zwiększać o mniejsze ilości lub w dłuższych odstępach czasu.
Zalecana początkowa dawka docelowa w monoterapii topiramatem u dorosłych wynosi od 100 mg na dobę do 200 mg na dobę w dwóch dawkach podzielonych. Maksymalna zalecana dawka dobowa wynosi 500 mg w dwóch dawkach podzielonych. Niektórzy pacjenci z opornymi na leczenie postaciami padaczki tolerowali topiramat stosowany w monoterapii w dawkach do 1000 mg na dobę. Takie dawkowanie jest zalecane w przypadku wszystkich osób dorosłych, w tym u osób w wieku podeszłym, u których nie występują choroby nerek.
Dzieci i młodzież (dzieci w wieku powyżej 6 lat)
Dawkę oraz stopniowe jej zwiększanie u dzieci należy ustalać na podstawie odpowiedzi klinicznej na leczenie. Leczenie dzieci w wieku powyżej 6 lat należy rozpocząć od dawki od 0,5 do 1 mg/kg masy ciała podawanej na noc przez pierwszy tydzień. Dawkę należy stopniowo zwiększać o 0,5 do 1 mg/kg masy ciała na dobę, w odstępach jedno- lub dwutygodniowych, podając produkt w dwóch dawkach podzielonych. Jeśli dziecko nie toleruje powyższego schematu stopniowego zwiększania dawki, można ją zwiększać o mniejsze ilości lub w dłuższych odstępach czasu.
Zalecana początkowa dawka docelowa w monoterapii topiramatem u dzieci w wieku powyżej 6 lat wynosi 100 mg na dobę w zależności od odpowiedzi klinicznej na leczenie (tj. około 2,0 mg/kg masy ciała na dobę u dzieci w wieku 6-16 lat).
Terapia uzupełniająca w leczeniu padaczki (częściowe napady padaczkowe z lub bez wtórnego uogólnienia, pierwotnie uogólnione napady padaczkowe toniczno-kloniczne lub napady padaczkowe związane z zespołem Lennoxa-Gastauta)
Dorośli
Stosowanie produktu należy rozpocząć od dawki 25 do 50 mg na noc przez jeden tydzień. Odnotowano stosowanie niższych dawek początkowych, ale nie przeprowadzono systematycznych badań z użyciem tego schematu terapeutycznego. Następnie dawkę należy stopniowo zwiększać, w odstępach jedno- lub dwutygodniowych, o 25 do 50 mg na dobę, podając produkt w dwóch dawkach podzielonych.
U niektórych pacjentów można uzyskać skuteczność terapeutyczną przy stosowaniu produktu raz na dobę.
Dawka 200 mg stanowiła najmniejszą skuteczną dawkę leczniczą, jako terapia uzupełniająca w badaniach klinicznych. Typowa dawka dobowa produktu wynosi 200 do 400 mg, podawana w dwóch dawkach podzielonych.
Takie dawkowanie jest zalecane w przypadku wszystkich osób dorosłych, w tym u osób w wieku podeszłym, u których nie występują choroby nerek (patrz rozdział 4.4).
Dzieci i młodzież (w wieku 2 lat i powyżej)
Zalecana całkowita dawka dobowa topiramatu stosowanego w leczeniu uzupełniającym wynosi około 5 do 9 mg/kg mc. na dobę podawana w dwóch dawkach podzielonych. Stopniowe zwiększanie dawki
należy rozpocząć od 25 mg (lub mniej- w zakresie od 1 do 3 mg/kg masy ciała na dobę) podawanych na noc przez pierwszy tydzień. Następnie dawkę należy stopniowo zwiększać w odstępach 1- lub 2- tygodniowych o 1 do 3 mg/kg mc./dobę i podawać w dwóch dawkach podzielonych, aż do uzyskania optymalnej odpowiedzi klinicznej.
Dotychczas przeprowadzono badania dotyczące dawek do 30 mg/kg mc./dobę i dawki te były na ogół dobrze tolerowane.
Migrena
Dorośli
Zalecana całkowita dawka dobowa topiramatu w zapobieganiu migrenie wynosi 100 mg na dobę podawana w dwóch dawkach podzielonych. Stopniowe zwiększanie dawki należy rozpocząć od dawki 25 mg podawanej na noc przez pierwszy tydzień. Następnie dawkę można zwiększać o 25 mg na dobę,
w odstępach jednotygodniowych. Jeśli pacjent nie toleruje powyższego schematu zwiększania dawki, można ją zwiększać w dłuższych odstępach czasu. Niektórzy pacjenci mogą uzyskać korzyść ze stosowania produktu w całkowitej dawce dziennej rzędu 50 mg na dobę. Pacjenci otrzymywali produkt w całkowitej dawce dziennej do 200 mg na dobę. Chociaż u niektórych pacjentów dawka ta może być odpowiednia, zalecane jest, aby zwrócić szczególną uwagę na zwiększone ryzyko wystąpienia działań niepożądanych.
Dzieci i młodzież
Z powodu niewystarczających danych na temat bezpieczeństwa i skuteczności, produkt Etopro nie jest zalecany do stosowania w leczeniu lub zapobieganiu atakom migreny u dzieci.
Ogólne zalecenia dotyczące dawkowania produktu Etopro w szczególnych grupach pacjentów
Zaburzenia czynności nerek
Topiramat należy stosować ostrożnie u pacjentów z zaburzeniami czynności nerek (CLCR ≤ 70 ml/min), ze względu na zmniejszony klirens osoczowy i nerkowy dla topiramatu. Pacjenci ze stwierdzonymi zaburzeniami nerek wymagają dłuższego czasu do osiągnięcia stanu stacjonarnego dla każdej dawki.
Zaleca się zastosowanie połowy zwykle stosowanej dawki początkowej i podtrzymującej (patrz punkt 5.2).
W związku z tym, że u pacjentów z zaburzeniami czynności nerek w stadium końcowym topiramat jest usuwany z osocza podczas zabiegu hemodializy, należy podać dawkę uzupełniającą topiramatu równą w przybliżeniu połowie dawki dobowej w dniu wykonywania zabiegu hemodializy. Uzupełniającą dawkę leku należy podawać w dwóch dawkach podzielonych, na początku i po zakończeniu zabiegu hemodializy. Dawka uzupełniająca może być różna w zależności od charakterystyki stosowanego sprzętu do dializ (patrz punkt 5.2).
Zaburzenia czynności wątroby
Topiramat należy stosować ostrożnie u pacjentów z umiarkowanymi do ciężkich zaburzeniami czynności wątroby, ze względu na zmniejszony klirens topiramatu.
Pacjenci w podeszłym wieku
Nie jest wymagane dostosowywanie dawki u pacjentów w podeszłym wieku z zachowaną prawidłową czynnością nerek.
Sposób podawania
Produkt Etopro jest dostępny w postaci tabletek powlekanych do stosowania doustnego. Tabletek powlekanych nie należy dzielić.
Produkt Etopro może być przyjmowany niezależnie od posiłków.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Zapobieganie migrenie u kobiet w ciąży oraz kobiet w okresie rozrodczym, które nie stosują wysoce skutecznych metod antykoncepcyjnych.
W sytuacjach, w których ze względów medycznych wymagane jest szybkie odstawienie topiramatu, zaleca się właściwą obserwację pacjentów (patrz punkt 4.2).
Podobnie jak w przypadku innych leków przeciwpadaczkowych, po zastosowaniu topiramatu może dojść do zwiększenia częstości napadów padaczkowych lub pojawienia się nowych rodzajów napadów padaczkowych. Może to być wynikiem zastosowania zbyt dużej dawki topiramatu, zmniejszenia stężenia we krwi innych, stosowanych jednocześnie leków przeciwpadaczkowych, postępu choroby lub wystąpienia efektu paradoksalnego.
Podczas stosowania topiramatu bardzo ważne jest zapewnienie odpowiedniego nawodnienia organizmu. Nawodnienie może zmniejszyć ryzyko rozwoju kamicy nerkowej (patrz poniżej). Ponadto prawidłowe nawodnienie przed i w trakcie wysiłku fizycznego czy przebywania w wysokiej temperaturze może zmniejszyć ryzyko wystąpienia działań niepożądanych związanych z przegrzaniem (patrz punkt 4.8).
Oligohydrosis
Zgłaszano przypadki zmniejszonego wydzielania potu po zastosowaniu topiramatu. Zmniejszone wydzielanie potu i hipertermia (podwyższenie temperatury ciała) może wystąpić szczególnie u małych dzieci narażonych na wysokie temperatury otoczenia.
Zaburzenia nastroju/depresja
Podczas stosowania topiramatu obserwowano częstsze występowanie zaburzeń nastroju i depresji.
Próby samobójcze/myśli samobójcze
U pacjentów, u których stosowano leki przeciwpadaczkowe w poszczególnych wskazaniach, odnotowano przypadki myśli i zachowań samobójczych. Meta-analiza randomizowanych, kontrolowanych placebo badań leków przeciwpadaczkowych również wskazuje na niewielkie zwiększenie ryzyka myśli
i zachowań samobójczych. Nie jest znany mechanizm powstawania tego ryzyka, a na podstawie dostępnych danych nie można wykluczyć, że zwiększone ryzyko występuje także podczas stosowania topiramatu.
W badaniach klinicznych przeprowadzonych z podwójnie ślepą próbą, zdarzenia związane
z samobójstwem (myśli samobójcze, próby samobójcze i rzeczywiste samobójstwa) występowały
z częstością 0,5% u pacjentów leczonych topiramatem (u 46 z 8652 leczonych pacjentów), przy czym ich częstość była prawie 3 razy większa niż u pacjentów, którym podawano placebo (0,2%; u 8 z 4045 pacjentów).
W związku z tym należy uważnie obserwować, czy u pacjenta nie występują oznaki myśli i zachowań samobójczych i w razie konieczności rozważyć zastosowanie odpowiedniego leczenia. Pacjentów (oraz ich opiekunów) należy poinformować, że w razie wystąpienia oznak myśli lub zachowań samobójczych należy poradzić się lekarza.
Kamica nerkowa
U niektórych pacjentów, szczególnie z predyspozycją do kamicy nerkowej, może istnieć zwiększone ryzyko powstawania kamieni nerkowych i związanych z tym objawów, takich jak: kolka nerkowa, ból nerki, okolicy lędźwiowej lub w boku.
Czynnikami ryzyka kamicy nerkowej są: wcześniejsze występowanie kamieni, kamica nerkowa w wywiadzie rodzinnym i hiperkalciuria (patrz poniżej – kwasica metaboliczna). Żaden z tych czynników ryzyka nie pozwala jednoznacznie przewidzieć wystąpienia kamicy nerkowej w czasie leczenia topiramatem. Ryzyko to może być również zwiększone u pacjentów, którzy przyjmują inne produkty lecznicze sprzyjające powstawaniu kamieni nerkowych.
Zaburzenia czynności nerek
U pacjentów z zaburzeniami czynności nerek (CLCR ≤ 70 mL/min) topiramat powinien być stosowany z zachowaniem ostrożności, z uwagi na zmniejszony klirens nerkowy i osoczowy topiramatu. Specjalne zalecenia dotyczące dawkowania u pacjentów z upośledzoną czynnością nerek znajdują się w punkcie 4.2.
Zaburzenia czynności wątroby
U pacjentów z zaburzeniami czynności wątroby topiramat należy stosować ostrożnie, ze względu na możliwość zmniejszenia klirensu topiramatu.
Ostra krótkowzroczność z wtórną jaskrą z zamkniętym kątem przesączania
U pacjentów przyjmujących topiramat zgłaszano wystąpienie zespołu składającego się z nagłej krótkowzroczności i wtórnej jaskry z zamkniętym kątem przesączania. Objawami zespołu są: nagłe zmniejszenie ostrości widzenia i (lub) ból gałki ocznej. W badaniach oftalmologicznych stwierdzono
krótkowzroczność, spłycenie przedniej komory oka, przekrwienie gałki ocznej (zaczerwienienie) i podwyższenie ciśnienia wewnątrzgałkowego. Rozszerzenie źrenicy może występować lub nie.
Zespołowi może towarzyszyć wysięk nadrzęskowy (nad ciałkiem rzęskowym) powodujący przesunięcie do przodu soczewki i tęczówki, z wtórnym zamknięciem kąta przesączania. Objawy pojawiają się zazwyczaj w ciągu 1 miesiąca od rozpoczęcia leczenia topiramatem. W przeciwieństwie do pierwotnej jaskry z wąskim kątem przesączania, która rzadko występuje przed 40 rokiem życia, jaskrę wtórną
z zamkniętym kątem przesączania związaną z przyjmowaniem topiramatu obserwowano zarówno u dzieci, jak i u dorosłych. Leczenie obejmuje: najszybsze, jak to tylko możliwe w ocenie lekarza
prowadzącego, przerwanie leczenia topiramatem i postępowanie zmierzające do obniżenia ciśnienia w gałce ocznej. Działania te zwykle prowadzą do obniżenia ciśnienia wewnątrzgałkowego.
Nieleczone podwyższone ciśnienie wewnątrzgałkowe niezależnie od etiologii może prowadzić do poważnych następstw, w tym do całkowitej utraty wzroku.
Należy określić, czy pacjenci z przebytymi zaburzeniami oka mogą być leczeni topiramatem.
Zaburzenia pola widzenia
U pacjentów otrzymujących topiramat zgłaszano zaburzenia pola widzenia niezależnie od zwiększonego ciśnienia śródgałkowego. W badaniach klinicznych, większość tych zdarzeń ustępowała po odstawieniu topiramatu. W razie wystąpienia zaburzeń pola widzenia kiedykolwiek w trakcie terapii topiramatem, należy rozważyć odstawienie leku.
Kwasica metaboliczna
W trakcie przyjmowania topiramatu może wystąpić hiperchloremiczna kwasica metaboliczna
z prawidłową luką anionową (to jest zmniejszenie stężenia wodorowęglanów w surowicy poniżej normy, przy braku zasadowicy oddechowej). To zmniejszenie stężenia wodorowęglanów w surowicy jest spowodowane hamującym działaniem topiramatu na nerkową anhydrazę węglanową. Choć zmniejszenie stężenia wodorowęglanów we krwi może wystąpić na każdym etapie terapii, najczęściej występuje
w początkowej fazie leczenia. Zmniejszenie to jest zwykle niewielkie do umiarkowanego (średnie zmniejszenie stężenia wynosi 4 mmol/1 po dawkach 100 mg/dobę lub większych u dorosłych i około 6 mg/kg mc./dobę u dzieci). Rzadko dochodzi do zmniejszenia stężenia wodorowęglanów poniżej 10 mmol/1. Stany lub terapie sprzyjające powstawaniu kwasicy (takie jak choroby nerek, ciężkie zaburzenia oddychania, stan padaczkowy, biegunka, zabieg chirurgiczny, dieta ketogenna, lub niektóre leki) mogą dodatkowo wpływać na zmniejszenie stężenia wodorowęglanów wywołane przez topiramat.
Przewlekła, nieleczona kwasica metaboliczna zwiększa ryzyko powstawania kamicy nerkowej i nefrokalcynozy i potencjalnie może prowadzić do osteopenii (patrz powyżej – kamica nerkowa).
Przewlekła kwasica metaboliczna u dzieci może powodować zmniejszenie tempa wzrostu. Wpływ topiramatu na wzrastanie i przemiany w tkance kostnej nie były dotąd przedmiotem systematycznych badań ani w populacji dzieci, ani dorosłych pacjentów. Zaleca się, aby podczas terapii topiramatem wykonywane były odpowiednie badania, w tym oznaczenie stężenia wodorowęglanów w surowicy, w zależności od choroby zasadniczej. Jeśli utrzymują się objawy wskazujące na kwasicę metaboliczną (np. pogłębiony oddech Kussmaula, duszność, jadłowstręt, nudności, wymioty, nadmierna męczliwość, tachykardia lub arytmia), zaleca się wykonanie badania stężenia wodorowęglanów w surowicy. W przypadku rozwoju i utrzymywania się kwasicy metabolicznej, należy rozważyć zmniejszenie dawki lub przerwanie leczenia topiramatem z zastosowaniem stopniowego zmniejszania dawki.
Topiramat należy stosować ostrożnie u pacjentów, u których występują choroby lub stosowane jest leczenie mogące stanowić czynnik ryzyka kwasicy metabolicznej.
Zaburzenia funkcji poznawczych
Zaburzenia kognitywne w przebiegu padaczki mają podłoże wieloczynnikowe i mogą być spowodowane przez chorobę zasadniczą, jako skutek samej choroby lub skutek leczenia przeciwpadaczkowego.
Opisywano w piśmiennictwie przypadki zaburzeń funkcji poznawczych u dorosłych leczonych topiramatem, w których konieczne było zmniejszenie dawki lub przerwanie leczenia. Jednakże, badania funkcji kognitywnych u dzieci leczonych topiramatem są niedostateczne i jego działanie w tym zakresie wymaga wyjaśnienia.
Hiperamonemia i encefalopatia
Podczas leczenia topiramatem zgłaszano przypadki hiperamonemii z encefalopatią lub bez (patrz punkt 4.8). Ryzyko hiperamonemii jest zależne od dawki topiramatu. Hiperamonemię jest zgłaszana częściej, gdy topiramat jest stosowany jednocześnie z kwasem walproinowym (patrz punkt 4.5).
U pacjentów, u których wystąpi niewyjaśniony letarg lub zmiany stanu psychicznego związane z monoterapią lub terapią skojarzoną topiramatem, należy rozważyć możliwość wystąpienia encefalopatii związanej hiperamonemią i zaleca się pomiar stężenia amoniaku.
Uzupełnianie substancji odżywczych
U niektórych pacjentów podczas leczenia topiramatem może dojść do utraty masy ciała. Zaleca się monitorowanie pacjentów przyjmujących topiramat w kierunku utraty masy ciała. Pacjentom, u których obniża się waga podczas leczenia topiramatem, można rozważyć zwiększenie ilości spożywanego pokarmu lub stosowanie diety uzupełniającej.
Kobiety w wieku rozrodczym
Topiramat podawany kobietom w ciąży może działać szkodliwie i hamować rozwój płodu (niedobór masy ciała w stosunku do wieku ciążowego i mała urodzeniowa masa ciała). Dane z rejestru ciąż z Ameryki Północnej (NAAED,ang. North American Antiepileptic Drug) dotyczące monoterapii topiramatem wykazały około 3-krotnie większą częstość występowania dużych wad wrodzonych (4,3%), w porównaniu z referencyjną grupą nieotrzymującą leków przeciwpadaczkowych (1,4%). Ponadto dane z innych badań wskazują, że w porównaniu z monoterapią występuje zwiększone ryzyko działania teratogennego związanego ze stosowaniem leków przeciwpadaczkowych w terapii skojarzonej.
Przed rozpoczęciem leczenia topiramatem u kobiety w wieku rozrodczym należy wykonać test ciążowy i zalecić stosowanie wysoce skutecznej metody antykoncepcyjnej (patrz punkt 4.5). Pacjentkę należy szczegółowo poinformować o ryzyku związanym ze stosowaniem topiramatu w czasie ciąży (patrz punkt 4.3 i 4.6).
Nietolerancja laktozy
Produkt Etopro tabletki powlekane 50 mg zawiera laktozę jednowodną. Nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, całkowitym niedoborem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Sód
Lek zawiera mniej niż 1 mmol (23 mg) sodu w jednej tabletce, to znaczy lek uznaje się za „wolny od sodu”.
Wpływ produktu Etopro na inne leki przeciwpadaczkowe
Włączenie topiramatu do leczenia innymi lekami przeciwpadaczkowymi (fenytoiną, karbamazepiną, kwasem walproinowym, fenobarbitalem, prymidonem) nie wpływa na stężenia tych leków w osoczu
w stanie stacjonarnym. Wyjątek stanowią nieliczni pacjenci, u których dodanie topiramatu do leczenia fenytoiną może spowodować zwiększenie stężenia fenytoiny w osoczu. Możliwe, że spowodowane jest to hamowaniem aktywności swoistej izoformy polimorficznego enzymu (CYP2C19). Z związku
z powyższym, u pacjentów leczonych fenytoiną, u których występują objawy toksyczności, zaleca się monitorowanie jej stężenia.
Badania interakcji farmakokinetycznych u pacjentów z padaczką wykazały, że dodanie topiramatu w dawce od 100 do 400 mg/dobę do leczenia lamotryginą nie wpływa na stężenie lamotryginy w stanie stacjonarnym w osoczu. Ponadto, nie stwierdzono zmian stężenia topiramatu w stanie stacjonarnym
w osoczu podczas przerywania leczenia lamotryginą lub po zaprzestaniu jej stosowania (średnia dawka 327 mg/dobę).
Topiramat jest inhibitorem enzymu CYP 2C19, a więc może wpływać na metabolizm innych związków metabolizowanych przez ten enzym (np. diazepamu, imipraminy, moklobemidu, proguanilu, omeprazolu).
Wpływ innych leków przeciwpadaczkowych na produkt Etopro
Fenytoina i karbamazepina zmniejszają stężenie topiramatu w osoczu. Rozpoczynanie lub przerwanie stosowania fenytoiny lub karbamazepiny podczas leczenia topiramatem może wymagać dostosowania dawki topiramatu. Dostosowanie dawki należy prowadzić stopniowo w zależności od efektu klinicznego. Jednoczesne podawanie lub zaprzestanie stosowania kwasu walproinowego nie powoduje klinicznie istotnych zmian stężenia topiramatu w osoczu i w związku z tym nie jest konieczne dostosowanie dawki topiramatu.
Interakcje topiramatu z innymi lekami przeciwpadaczkowymi (LPP) zostały zestawione w poniższej tabeli:
LPP stosowany razem z topiramatem | Stężenie LPP w osoczu | Stężenie topiramatu |
Fenytoina | ↔** |
|
Karbamazepina (CBZ) | ↔ |
|
Kwas walproinowy | ↔ | ↔ |
Lamotrygina | ↔ | ↔ |
Fenobarbital | ↔ | NB |
Primidon | ↔ | NB |
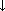
↔ = brak wpływu (zmiana ≤ 15%)
** = zwiększenie stężenia w osoczu u pojedynczych pacjentów
↓ = zmniejszenie stężenia w osoczu NB = nie badano
LPP = lek przeciwpadaczkowy Interakcje z innymi lekami
Digoksyna
Badanie po podaniu pojedynczej dawki topiramatu wykazało zmniejszenie o 12% pola powierzchni pod krzywą obrazującego zmiany stężenia digoksyny w osoczu (AUC). Kliniczne znaczenie tej obserwacji nie zostało ustalone. W przypadku rozpoczynania lub przerwania stosowania topiramatu u pacjentów leczonych digoksyną należy rutynowo monitorować stężenia digoksyny w surowicy.
Leki działające hamująco na ośrodkowy układ nerwowy (OUN)
Jednoczesne przyjmowanie topiramatu i alkoholu lub innych środków działających hamująco na ośrodkowy układ nerwowy (OUN) nie było przedmiotem badań klinicznych. Ze względu na potencjalny hamujący wpływ topiramatu na OUN, niepożądany wpływ na funkcje poznawcze i neuropsychiczne działania niepożądane leku, należy zachować ostrożność, stosując topiramat jednocześnie z alkoholem i innymi środkami działającymi hamująco na OUN.
Dziurawiec zwyczajny (Hypericum perforatum)
Obserwowano zmniejszoną skuteczność topiramatu, będącą wynikiem zmniejszonego jego stężenia we krwi, podczas jednoczesnego stosowania topiramatu z dziurawcem zwyczajnym. Nie przeprowadzono żadnych badań klinicznych oceniających wystąpienie tej interakcji.
Doustne środki antykoncepcyjne
W badaniu interakcji farmakokinetycznych ze złożonymi doustnymi produktami antykoncepcyjnymi u zdrowych ochotniczek użyto złożonego, doustnego środka antykoncepcyjnego zawierającego 1 mg noretyndronu (NET) i 35 μg etynyloestradiolu (EE). Równocześnie stosowany, jako jedyny lek przeciwpadaczkowy, produkt Etopro, w dawce od 50 do 200 mg na dobę nie powodował statystycznie istotnych zmian w średniej ekspozycji (pole pod krzywą –AUC) dla żadnego z komponentów środka antykoncepcyjnego. Jednak w innym badaniu, ekspozycja na EE była istotnie zmniejszona po podaniu topiramatu w dawkach 200, 400 i 800 mg na dobę (odpowiednio o 18%, 21% i 30%) w terapii
wspomagającej u pacjentów z padaczką otrzymujących także kwas walproinowy. W obu wspomnianych badaniach, produkt Etopro (podawany w dawce od 50 do 200 mg na dobę u zdrowych ochotniczek i 200- 800 mg na dobę u pacjentek z padaczką) nie wpływał znamiennie na ekspozycję na NET. Chociaż
w przedziale dawek topiramatu od 200 do 800 mg na dobę (u pacjentek z padaczką) zaobserwowano zależne od dawki zmniejszenie ekspozycji na EE dla dawek od 50 do 200 mg na dobę (u zdrowych ochotniczek). Nie wiadomo jakie jest kliniczne znaczenie obserwowanych zmian. U pacjentek stosujących jednocześnie z produktem Etopro złożone, doustne środki antykoncepcyjne, należy liczyć się ze zmniejszoną skutecznością działania środka antykoncepcyjnego oraz z częstszym występowaniem krwawień międzymiesiączkowych. Pacjentki stosujące środki antykoncepcyjne zawierające estrogen powinny zgłaszać zmiany rytmu krwawień miesiączkowych. Działanie antykoncepcyjne może być osłabione, nawet jeśli nie wystąpią krwawienia międzymiesiączkowe.
Lit
U zdrowych ochotników obserwowano zmniejszenie (AUC o 18%) układowej ekspozycji na lit podczas jednoczesnego stosowania z topiramatem w dawce 200 mg na dobę. U pacjentów z zaburzeniem afektywnym dwubiegunowym podczas leczenia topiramatem w dawce 200 mg na dobę, farmakokinetyka litu była niezmieniona; obserwowano jednak zwiększenie układowej ekspozycji (wartości AUC o 26%) po podaniu topiramatu w dawkach do 600 mg na dobę. W przypadku jednoczesnego stosowania
z topiramatem należy monitorować stężenie litu we krwi.
Rysperydon
Badania interakcji leków prowadzone z zastosowaniem dawki jednorazowej u zdrowych ochotników
i dawek wielokrotnych u pacjentów z zaburzeniem afektywnym dwubiegunowym dały podobne wyniki. Podczas jednoczesnego stosowania rysperydonu (podawanego w dawkach od 1 do 6 mg na dobę)
z topiramatem (stosowanym w narastających dawkach wynoszących 100, 250 i 400 mg na dobę), stwierdzono zmniejszenie układowej ekspozycji na rysperydon (zmniejszenie wartości AUC w stanie stacjonarnym o 16% i 33%, odpowiednio po dawkach 250 i 400 mg na dobę). Jednakże różnice
w wartości AUC dla całej frakcji aktywnej między leczeniem samym rysperydonem oraz leczeniem skojarzonym z topiramatem nie były istotne statystycznie. Obserwowano minimalne zmiany farmakokinetyki całej frakcji aktywnej (rysperydon plus 9-hydroksyrysperydon) i brak zmian farmakokinetyki 9-hydroksyrysperydonu. Nie stwierdzono istotnych zmian układowej ekspozycji na całą frakcję aktywną, ani na topiramat. Po dodaniu topiramatu (250-400 mg na dobę) do trwającego już leczenia rysperydonem (1-6 mg/dobę) znacznie częściej zgłaszano działania niepożądane, niż przed włączeniem topiramatu (odpowiednio 90% i 54%). Najczęściej zgłaszanymi działaniami niepożądanymi po dodaniu topiramatu do leczenia rysperydonem były: senność (27% i 12%), zaburzenia czucia (22%
i 0%) oraz nudności (odpowiednio 18% i 9%).
Hydrochlorotiazyd (HCTZ)
W przeprowadzonym u zdrowych ochotników badaniu interakcji między lekami oceniano farmakokinetykę HCTZ (stosowanego w dawce 25 mg co 24 godziny) i topiramatu (stosowanego w dawce 96 mg co 12 godzin) w stanie stacjonarnym w osoczu, zarówno gdy leki te były stosowane oddzielnie, jak i jednocześnie. Wyniki tego badania wskazują, że po włączeniu HCTZ do leczenia topiramatem maksymalne stężenie topiramatu w osoczu (Cmax) zwiększyło się o 27%, a AUC zwiększyło się o 29%. Znaczenie kliniczne stwierdzonych zmian nie jest znane. Włączenie HCTZ do leczenia topiramatem może wymagać dostosowania dawki topiramatu.
Farmakokinetyka HCTZ w stanie stacjonarnym nie uległa istotnym zmianom podczas jednoczesnego podawania topiramatu. Wyniki badań laboratoryjnych wskazują na zmniejszenie stężenia potasu zarówno po podaniu topiramatu, jak i po podaniu HCTZ. Zmniejszenie to było większe, gdy HCTZ i topiramat były jednocześnie stosowane.
Metformina
W badaniu interakcji między lekami przeprowadzonym u zdrowych ochotników oceniano farmakokinetykę metforminy i topiramatu w osoczu w stanie stacjonarnym po podaniu samej metforminy oraz w sytuacji, gdy metformina i topiramat były stosowane jednocześnie. Wyniki tego badania wskazują, iż średnie wartości Cmax i AUC0-12h dla metforminy zwiększyły się odpowiednio o 18% i 25%, podczas gdy średnia wartość wskaźnika CL/F uległa zmniejszeniu o 20% w przypadku, gdy metformina była podawana jednocześnie z topiramatem. Topiramat nie wpływał na tmax dla metforminy. Kliniczne znaczenie stwierdzonego wpływu topiramatu na farmakokinetykę metforminy jest niejasne. Wydaje się, iż w przypadku jednoczesnego podawania z metformina osoczowy klirens topiramatu po podaniu doustnym jest zmniejszony. Stopień, w jakim zmienia się klirens leku, nie został określony. Kliniczne znaczenie wpływu metforminy na farmakokinetykę topiramatu jest niejasne.
Należy zwrócić szczególną uwagę na właściwe monitorowanie parametrów przebiegu cukrzycy u pacjentów leczonych metforminą, gdy topiramat jest dodawany lub wycofywany z terapii.
Pioglitazon
W badaniu interakcji leków u zdrowych ochotników oceniano farmakokinetykę topiramatu i pioglitazonu w osoczu w stanie stacjonarnym, gdy leki były stosowane osobno, a także gdy były stosowane jednocześnie. Zaobserwowano zmniejszenie o 15% AUC,ss dla pioglitazonu, nie stwierdzono natomiast zmian w Cmax,ss dla tego leku. Wyniki te nie były jednak istotne statystycznie. Ponadto wykazano zmniejszenie wartości Cmax,ss i AUC,ss odpowiednio o 13% i 16% dla aktywnego hydroksy-metabolitu, jak również zmniejszenie o 60% wartości Cmax,ss i AUC,ss dla aktywnego ketometabolitu. Kliniczne znaczenie tych obserwacji nie jest znane.
W przypadku dodania topiramatu do leczenia pioglitazonem lub dodania pioglitazonu do leczenia topiramatem należy zwracać szczególną uwagę na właściwe monitorowanie glikemii i innych parametrów związanych z cukrzycą.
Glibenklamid
W badaniu interakcji leków, przeprowadzonym u pacjentów z cukrzycą typu 2 oceniano parametry farmakokinetyczne glibenklamidu w stanie stacjonarnym (5 mg na dobę) podawanego w monoterapii
i jednocześnie z topiramatem (150 mg na dobę). Podczas podawania topiramatu stwierdzono zmniejszenie wartości AUC24 dla glibenklamidu o 25%. Układowa ekspozycja na aktywne metabolity,
4-trans-hydroksygliburyd (M1) i 3-cis-hydroksygliburyd (M2), także uległa zmniejszeniu – odpowiednio o 13% i 15%. Farmakokinetyka topiramatu w stanie stacjonarnym nie ulegała zmianie podczas jednoczesnego podawania glibenklamidu.
W przypadku dodania topiramatu do terapii glibenklamidem lub glibenklamidu do leczenia topiramatem należy zwrócić szczególną uwagę na rutynowe monitorowanie pacjentów w kierunku odpowiedniej kontroli cukrzycy.
Inne rodzaje interakcji
Środki predysponujące do wystąpienia kamicy nerkowej
Produkt Etopro stosowany jednocześnie z innymi środkami mogącymi wywołać kamicę nerkową może zwiększać ryzyko jej wystąpienia. Podczas stosowania produktu Etopro należy unikać stosowania takich środków, ponieważ mogą one tworzyć fizjologiczne środowisko zwiększające ryzyko powstawania kamieni nerkowych.
Kwas walproinowy
Jednoczesne stosowanie topiramatu z kwasem walproinowym wiązało się ze zwiększeniem stężenia amoniaku we krwi z towarzyszącą encefalopatią lub bez encefalopatii u pacjentów, którzy tolerowali monoterapię każdym z leków. W większości przypadków wystąpienie objawów powodowało przerwanie stosowania jednego z leków (patrz punkty 4.4 i 4.8). To działanie niepożądane nie wynika z interakcji farmakokinetycznych. Zgłaszano przypadki hipotermii, określonej jako niezamierzone obniżenie wewnętrznej temperatury ciała poniżej 35°C, związanej z jednoczesnym stosowaniem topiramatu i kwasu walproinowego, zarówno z towarzyszącą hiperamonemią oraz bez hiperamonemii. To działanie niepożądane występujące u pacjentów leczonych jednocześnie topiramatem i walproinianem może wystąpić po rozpoczęciu stosowania topiramatu lub po zwiększeniu dawki dobowej topiramatu.
Warfaryna
U pacjentów leczonych topiramatem w skojarzeniu z warfaryną stwierdzano zmniejszenie czasu protrombinowego/międzynarodowego współczynnika znormalizowanego (PT/INR). Dlatego należy regularnie badać INR u pacjentów leczonych jednocześnie topiramatem i warfaryną.
Dodatkowe badania interakcji farmakokinetycznych leku
Przeprowadzono badania kliniczne dotyczące oceny potencjalnych interakcji farmakokinetycznych między topiramatem a innymi lekami. Zmiany wartości Cmax i AUC, do których dochodzi w wyniku tych interakcji, zostały podsumowane poniżej. W drugiej kolumnie („Stężenie leku stosowanego z topiramatem”) przedstawiono zmianę wartości stężenia leku, wymienionego w pierwszej kolumnie, po dodaniu topiramatu. Trzecia kolumna („Stężenie topiramatu”) pokazuje wpływ zastosowania leku wymienionego w pierwszej kolumnie na stężenie topiramatu.
Podsumowanie wyników dodatkowych badań klinicznych dotyczących interakcji farmakokinetycznych
Lek stosowany z topiramatem | Stężenie leku stosowanego z topiramatema | Stężenie topiramatu a |
Amitryptylina | ↔ 20% zwiększenie wartości Cmax i AUC dla metabolitu nortryptyliny | NB |
Dihydroergotamina (p.o. i s.c.) | ↔ | ↔ |
Haloperydol | ↔ 31% zwiększenie wartości AUC dla zredukowanego metabolitu | NB |
Propranolol | ↔ 17% zwiększenie wartości Cmax dla 4-OH propranololu (topiramat 50 mg co 12 godzin) | 9% i 16% zwiększenie wartości Cmax 9% i 17% zwiększenie wartości AUC (odpowiednio, propranolol 40 i 80 mg co 12 godzin) |
Sumatryptan (p.o. i s.c.) | ↔ | NB |
Pizotyfen | ↔ | ↔ |
Diltiazem | 25% zmniejszenie wartości AUC dla diltiazemu i 18% zmniejszenie wartości dla DEA oraz ↔ dla DEM* | 20% zwiększenie wartości AUC |
Wenlafaksyna | ↔ | ↔ |
Flunaryzyna | 16% zwiększenie wartości AUC (topiramat 50 mg co 12 godzin)b | ↔ |
a = wartości w % odnoszą się do występujących podczas leczenia zmian w średnich wartościach Cmax lub AUC w odniesieniu do monoterapii
↔ = brak wpływu na wartość Cmax i AUC (≤ 15% zmiany) związku macierzystego NB = nie badano
*DEA = deacetylodiltiazem, DEM = N-demetylodiltiazem
b = Flunaryzyna; zwiększenie wartości AUC o 14% u osób przyjmujących flunaryzynę w monoterapii. Zwiększenie ekspozycji może być spowodowane akumulacją leku podczas osiągania stanu stacjonarnego.
Ciąża
Ogólne ryzyko związane z padaczką i stosowaniem leków przeciwpadaczkowych
Należy udzielić specjalistycznej porady kobietom w wieku rozrodczym. Należy zweryfikować potrzebę stosowania leków przeciwpadaczkowych w przypadku, gdy kobieta planuje zajść w ciążę. U kobiet leczonych na padaczkę należy unikać nagłego odstawiania leków przeciwpadaczkowych, gdyż może to prowadzić do przełomów padaczkowych, które mogą mieć poważne konsekwencje dla matki i nienarodzonego dziecka.
Jeśli to tylko możliwe, preferowana jest monoterapia, ponieważ stosowaniu kilku leków przeciwpadaczkowych towarzyszy większe ryzyko wad wrodzonych niż podczas monoterapii, w zależności od skojarzonych leków.
Ryzyko związane z topiramatem
Topiramat wykazywał działanie teratogenne u myszy, szczurów i królików (patrz punkt 5.3). U szczurów topiramat przenika przez barierę łożyskową.
U ludzi topiramat przenika przez barierę łożyska i stwierdzano podobne stężenia we krwi pępowinowej i krwi matki.
Dane pochodzące z rejestru ciąż wskazują, że u dzieci narażonych na topiramat w monoterapii:
Wskazanie w leczeniu padaczki
U kobiet w wieku rozrodczym zaleca się rozważenie alternatywnych metod leczenia. Zaleca się, aby kobiety w wieku rozrodczym stosowały wysoce skuteczną antykoncepcję (patrz punkt 4.5), a także żeby otrzymały pełną informację o znanym ryzyku jakim jest wystąpienie niekontrolowanych napadów padaczkowych oraz potencjalnym zagrożeniu dla płodu wynikającym ze stosowania produktu leczniczego.
Jeśli kobieta planuje zajść w ciążę, zaleca się odbycie wizyty przed zajściem w ciążę, aby zweryfikować terapię i rozważyć inne opcje leczenia. W przypadku przyjmowania topiramatu podczas pierwszego trymestru ciąży należy prowadzić dokładną obserwację prenatalną.
Wskazanie w zapobieganiu migrenie
Topiramat jest przeciwwskazany do stosowania w ciąży oraz u kobiet w wieku rozrodczym, które nie stosują wysoce skutecznych metod antykoncepcji (patrz punkty 4.3 i 4.5).
Karmienie piersią
Badania na zwierzętach wykazały wydzielanie topiramatu z mlekiem matki. Wydzielanie topiramatu do mleka kobiet nie podlegało ocenie w trakcie kontrolowanych badań. Ograniczone obserwacje u pacjentek wskazują, że topiramat jest w znacznym stopniu wydzielany z mlekiem matki. Objawy obserwowane u noworodków lub niemowląt karmionych piersią przez matki otrzymujące lek to: biegunka, senność, drażliwość i nieprawidłowy przyrost ciała. Dlatego należy podjąć decyzję, czy zaniechać karmienia piersią, czy przerwać terapię topiramatem lub z niej zrezygnować, mając na uwadze korzyści z karmienia piersią dla dziecka i korzyści z leczenia topiramatem dla matki (patrz punkt 4.4).
Płodność
Badania na zwierzętach nie wykazały niekorzystnego wpływu topiramatu na płodność (patrz punkt 5.3). Wpływ topiramatu na płodność u ludzi nie został ustalony.
Etopro wywiera niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Topiramat wywiera wpływ na ośrodkowy układ nerwowy i może powodować senność, zawroty głowy lub inne powiązane zaburzenia. Może także powodować zaburzenia widzenia i(lub) niewyraźne widzenie. Te działania niepożądane mogą być niebezpieczne dla pacjentów kierujących pojazdami mechanicznymi lub obsługujących maszyny, zwłaszcza do czasu ustalenia indywidualnej reakcji pacjenta na produkt.
Bezpieczeństwo stosowania topiramatu określono na podstawie informacji pochodzących z bazy danych badań klinicznych obejmującej 4111 pacjentów (3182 leczonych topiramatem i 929 placebo), którzy wzięli udział w 20 badaniach klinicznych z podwójną ślepą próbą oraz 2847 pacjentów, którzy brali udział w 34 badaniach otwartych, w których topiramat stosowany był w leczeniu uzupełniającym pierwotnie uogólnionych napadów toniczno-klonicznych, częściowych napadów padaczkowych i napadów padaczkowych związanych z zespołem Lennoxa-Gastauta, w monoterapii nowo lub niedawno rozpoznanej padaczki lub profilaktyce migreny. Większość występujących w badaniach klinicznych działań niepożądanych miało nasilenie łagodne do umiarkowanego. Działania niepożądane zidentyfikowane podczas badań klinicznych, oraz po wprowadzeniu leku do obrotu (oznaczone “*”) zostały wymienione wg częstości ich występowania w badaniach klinicznych w tabeli 1.
Uszeregowane są według następującej częstości występowania:
Bardzo często ≥ 1/10
Często ≥ 1/100 do < 1/10 Niezbyt często ≥ 1/1000 do < 1/100 Rzadko ≥ 1/10 000 do <1/1000
Nieznana częstość nie może być określona na podstawie dostępnych danych
Najczęstsze działania niepożądane (o częstości występowania > 5% i większej niż obserwowana w grupie placebo w co najmniej jednym wskazaniu w kontrolowanych badaniach z podwójnie ślepą próbą) obejmują: jadłowstręt, zmniejszenie apetytu, spowolnienie procesów myślowych depresję, zaburzenia ekspresji mowy, bezsenność, zaburzenia koordynacji ruchowej, zaburzenia uwagi, zawroty głowy, zaburzenia mowy i smaku, niedoczulicę, letarg, upośledzenie pamięci, oczopląs, parestezje, senność, drżenia, podwójne widzenie, nieostre widzenie, biegunkę, nudności, zmęczenie, drażliwość i zmniejszenie masy ciała.
Tabela 1. Działania niepożądane topiramatu
Klasyfikacja układów i narządów | Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko |
Zakażenia i zarażenia pasożytnicze | Zapalenie nosa i gardła* | ||||
Zaburzenia krwi i układu chłonnego | Niedokrwistość | Leukopenia, małopłytkowość, | Neutropenia* |
limfadenopatia, eozynofilia | |||||
Zaburzenia układu immunologicznego | Nadwrażliwość | Obrzęk alergiczny*, | |||
Zaburzenia metabolizmu i odżywiania | Jadłowstręt, zmniejszony apetyt | Kwasica metaboliczna, hipokaliemia, zwiększony apetyt, polidypsja | Kwasica hiperchloremiczna, hiperamonemia, encefalopatia hiperamonemiczna | ||
Zaburzenia psychiczne | Depresja | Spowolnienie procesów myślowych, bezsenność, zaburzenia ekspresji mowy, lęk, stan splątania, dezorientacja, agresja, zmieniony nastrój, pobudzenie, chwiejny nastrój, nastrój depresyjny, gniew, nieprawidłowe zachowanie | Myśli samobójcze, próba samobójcza, omamy, zaburzenia psychotyczne, omamy słuchowe, omamy wzrokowe, apatia, brak spontanicznej mowy, zaburzenia snu, chwiejność afektu, zmniejszenie libido, niepokój ruchowy, płacz, zacinanie się w mowie, euforyczny nastrój, paranoja, perseweracja, napad silnego lęku, płaczliwość, trudności z czytaniem, trudności z zaśnięciem, płytki afekt, nieprawidłowe myślenie, utrata libido, zobojętnienie, budzenie się w środku nocy, trudności w skupieniu uwagi, budzenie się wcześnie rano, reakcje paniczne, podwyższony nastrój | Mania, zaburzenia lękowe, uczucie rozpaczy*, hipomania, |
Zaburzenia układu nerwowego | Parestezje, senność, zawroty głowy | Zaburzenia uwagi, upośledzenie pamięci, amnezja, zaburzenia kognitywne, upośledzenie umysłowe, upośledzenie funkcji psychomotorycznych, drgawki, zaburzenia koordynacji ruchowej, drżenie, letarg, niedoczulica, oczopląs, zaburzenia smaku, zaburzenia równowagi, upośledzenie wymowy, drżenie zamiarowe, nadmierne uspokojenie | Zmniejszony poziom świadomości, drgawki typu grand mal, zaburzenia pola widzenia, napady częściowe złożone, zaburzenia mowy, nadmierna aktywność psychomotoryczna, omdlenia, zaburzenia czucia, ślinienie się, nadmierna senność, afazja, powtarzanie słów, hipokinezja, dyskinezja, ortostatyczne zawroty głowy, niska jakość snu, uczucie pieczenia, utrata czucia, węch opaczny, zespół móżdżkowy, dysestezja, upośledzenie smaku, stupor, niezdarność, aura, brak smaku, dysgrafia, dysfazja, neuropatia obwodowa, stany przedomdleniowe, dystonia, mrowienie | Apraksja, zaburzenia rytmu okołodobowego, hiperestezja, osłabienie węchu, brak węchu, drżenie samoistne, akinezja, niereagowanie na bodźce | |
Zaburzenia oka | Nieostre widzenie, podwójne widzenie, zaburzenia widzenia | Zmniejszona ostrość widzenia, mroczki, krótkowzroczność*, nieprawidłowe odczucia w oku*, suchość oka, | Ślepota jednostronna, ślepota przemijająca, jaskra, zaburzenia akomodacji, zmiana | Jaskra z zamkniętym kątem przesączania*, zwyrodnienie plamki żółtej*, zaburzenia |
światłowstręt, kurcz powiek, wzmożone łzawienie, fotopsja, rozszerzenie źrenic, starczowzroczność | postrzegania głębi obrazu, mroczki iskrzące, obrzęk powiek*, ślepota zmierzchowa, niedowidzenie | ruchu gałek ocznych *, obrzęk spojówek*, zapalenie błony naczyniowej | |||
Zaburzenia ucha i błędnika | Zawroty głowy, szumy uszne, ból ucha | Głuchota, głuchota jednostronna, głuchota neurosensoryczna, poczucie dyskomfortu w uchu, upośledzenie słuchu | |||
Zaburzenia serca | Bradykardia, bradykardia zatokowa, kołatanie serca | ||||
Zaburzenia naczyniowe | Niedociśnienie, niedociśnienie ortostatyczne, przejściowe zaczerwienienie twarzy, uderzenia gorąca | Zespół Raynauda | |||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Duszność, krwawienie z nosa, przekrwienie błon śluzowych nosa, wodnisty wyciek z nosa, kaszel* | Duszność wysiłkowa, nadmierne wydzielanie z zatok przynosowych, dysfonia | |||
Zaburzenia żołądka i jelit | Nudności, biegunka | Wymioty, zaparcia, ból w nadbrzuszu, niestrawność, ból brzucha, uczucie suchości w jamie ustnej, uczucie dyskomfortu w żołądku, parestezje okolicy ust, zapalenie błony śluzowej żołądka, dyskomfort brzuszny | Zapalenie trzustki, wzdęcia, choroba refluksowa przełyku, ból w dolnej części brzucha, niedoczulica okolicy ust, krwawienia z dziąseł, wzdęcie brzucha, dyskomfort w nadbrzuszu, tkliwość w obrębie brzucha, nadmierne |
wydzielanie śliny, ból w jamie ustnej, nieprzyjemny zapach z ust, ból języka | |||||
Zaburzenia wątroby i dróg żółciowych | Zapalenie wątroby, niewydolność wątroby | ||||
Zaburzenia skóry i tkanki podskórnej | Łysienie, wysypka, świąd | Brak potu, niedoczulica twarzy, pokrzywka, rumień, świąd uogólniony, wysypka plamkowa, przebarwienia skóry, alergiczne zapalenie skóry, obrzęk twarzy | Zespół Stevensa- Johnsona*, rumień wielopostaciowy*, nieprawidłowy zapach skóry, obrzęk wokół oczu*, miejscowa pokrzywka | Martwica toksyczno- rozpływna naskórka * | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Artralgia, kurcze mięśni, bóle mięśni, drżenie mięśniowe, osłabienie mięśni, ból mięśniowo- szkieletowy w klatce piersiowej | Obrzęk stawów*, sztywność mięśniowo- szkieletowa, ból w boku, męczliwość mięśni | Uczucie dyskomfortu w kończynach* | ||
Zaburzenia nerek i dróg moczowych | Kamica nerkowa, częstomocz, dyzuria, nefrokalcynoza* | Kamienie w drogach moczowych, nietrzymanie moczu, krwiomocz, nagłe uczucie parcia na pęcherz, kolka nerkowa, ból nerki | Kamienie moczowodowe, nerkowa kwasica cewkowa* | ||
Zaburzenia układu rozrodczego i piersi | Zaburzenia erekcji, zaburzenia czynności seksualnych | ||||
Zaburzenia ogólne i stany w miejscu podania | Zmęczenie | Gorączka, astenia, drażliwość, zaburzenia chodu, złe samopoczucie, osłabienie | Hipertermia, pragnienie, objawy grypopodobne*, ospałość, ziębnięcie | Obrzęk twarzy |
obwodowych części ciała, uczucie upojenia alkoholowego, uczucie niepokoju | |||||
Badania diagnostyczne | Zmniejszenie masy ciała | Zwiększenie masy ciała* | Obecność kryształków w moczu, nieprawidłowy test chodzenia z układaniem stóp jedna za drugą (tandem), zmniejszona liczba białych krwinek, zwiększenie aktywności enzymów wątrobowych | Zmniejszenie stężenia wodorowęglanów we krwi | |
Uwarunkowania społeczne | trudności w uczeniu się |
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania do Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181 C, 02-222 Warszawa
Tel.: +48 22 49 21 301, Faks: +48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Objawy przedmiotowe i podmiotowe
Zgłaszano przypadki przedawkowania topiramatu. Do opisanych objawów przedmiotowych
i podmiotowych należały: drgawki, senność, zaburzenia mowy, nieostre widzenie, podwójne widzenie, zaburzenia myślenia, letarg, zaburzenia koordynacji ruchowej, stupor, niedociśnienie, bóle brzucha, pobudzenie, zawroty głowy i depresja. W większości przypadków nie miały one ciężkich następstw klinicznych, zgłaszano jednak zgony związane z zatruciem wielolekowym po przedawkowaniu kilku leków, w tym topiramatu. Przedawkowanie topiramatu może wywołać ciężką kwasicę metaboliczną (patrz punkt 4.4).
Leczenie
W razie ostrego przedawkowania, należy przerwać stosowanie topiramatu i zastosować ogólne leczenie podtrzymujące do czasu zmniejszenia lub ustąpienia toksyczności klinicznej. Pacjent powinien być dobrze nawodniony. Wykazano, iż hemodializa jest skutecznym środkiem eliminacji topiramatu z organizmu. Inne środki zapobiegawcze mogą być również podejmowane według uznania lekarza.
Grupa farmakoterapeutyczna: leki przeciwpadaczkowe, leki przeciwmigrenowe. kod ATC: N03 AX 11
Mechanizm działania
Topiramat jest klasyfikowany jako monosacharyd z podstawioną grupą sulfaminianową.
Dokładny mechanizm działania przeciwpadaczkowego i w profilaktyce migreny topiramatu nie jest znany. W elektrofizjologicznych i biochemicznych badaniach przeprowadzonych na hodowlach neuronów zidentyfikowano trzy właściwości mogące warunkować przeciwpadaczkowe działanie topiramatu.
Działanie farmakodynamiczne
Topiramat blokował w sposób zależny od czasu potencjały czynnościowe wielokrotnie wywoływane przez podtrzymywaną depolaryzację neuronów, co sugeruje, że dochodzi do blokowania zależnych od napięcia kanałów sodowych. Topiramat zwiększa częstość, z jaką kwas γ-aminomasłowy (GABA) aktywował receptory GABAA i zwiększał zdolność GABA do indukowania przepływu jonów chlorkowych do neuronów, co może wskazywać, że topiramat nasila hamujące działanie tego neuroprzekaźnika.
Działanie to nie było blokowane przez flumazenil - antagonistę benzodiazepin. Ponadto topiramat nie wydłużał czasu otwarcia kanałów, co odróżnia go od barbituranów, które wywierają modyfikujący wpływ na receptory GABAA.
Ponieważ profil działania przeciwpadaczkowego topiramatu różni się znacznie od profilu działania przeciwpadaczkowego benzodiazepin, topiramat może działać na niewrażliwy na benzodiazepiny podtyp receptora GABAA.
Topiramat blokuje aktywność kwasu kainowego w stosunku do receptorów glutaminianergiczych typu kainowych i AMPA (alfa-amino-3-hydroksy-5-metyloizoksazolo-4-propionowych), nie wywierając widocznego wpływu na aktywność NMDA (N-metylo-D-asparaginianowych) w stosunku do receptora glutaminergicznego typu NMDA. To działanie topiramatu jest zależne od stężenia w zakresie 1 µM do 200 µM, a najmniejszą aktywność stwierdzano dla stężeń od 1 µM do 10 µM.
Dodatkowo topiramat hamuje niektóre izoenzymy anhydrazy węglanowej, lecz znacznie słabiej niż acetazolamid, znany inhibitor anhydrazy węglanowej. Ten efekt farmakologiczny nie wydaje się stanowić głównej składowej aktywności przeciwpadaczkowej topiramatu.
Skuteczność kliniczna i bezpieczeństwo stosowania
W badaniach na zwierzętach topiramat wykazywał działanie przeciwdrgawkowe u szczurów i u myszy
w testach drgawek wywołanych przez maksymalny wstrząs elektryczny. Wykazywał również skuteczność w modelach padaczki u gryzoni, w tym w napadach tonicznych, i w napadach nieobecności u szczurów ze spontaniczną padaczką oraz w drgawkach tonicznych i klonicznych, wywołanych u szczurów przez pobudzenie ciała migdałowatego lub przez ogólne niedotlenienie. Topiramat tylko nieznacznie blokuje drgawki kloniczne wywołane przez pentetrazol, antagonistę receptora GABAA.
Badania przeprowadzone na myszach wykazały, że jednoczesne stosowania topiramatu i karbamazepiny lub fenobarbitalu powoduje synergistyczne działanie przeciwdrgawkowe, natomiast w skojarzeniu
z fenytoiną stwierdzono działanie addytywne. W kontrolowanych badaniach klinicznych
z wykorzystaniem terapii skojarzonej nie stwierdzono korelacji między stężeniem topiramatu w osoczu a jego kliniczną skutecznością. Nie stwierdzono rozwoju tolerancji na topiramat u ludzi.
Napady typu ”absence”
Przeprowadzono dwa małe jednoramienne badania u dzieci w wieku 4-11 lat (CAPSS-326 i TOPAMAT- ABS-001). Jedno dotyczyło 5 dzieci a drugie 12 dzieci, zanim zostały wcześniej przerwane z powodu braku odpowiedzi na leczenie. Zastosowane dawki wynosiły do około 12 mg/kg w badaniu TOPAMAT- ABS-001i nie więcej niż 9 mg/kg/dobę lub 400 mg/dobę w badaniu CAPSS-326. Badania te nie dostarczyły wystarczających dowodów do wyciągnięcia wniosków dotyczących skuteczności i bezpieczeństwa u dzieci.
Tabletki powlekane są biorównoważne.
Profil farmakokinetyczny topiramatu w porównaniu z innymi lekami przeciwpadaczkowymi charakteryzuje się wydłużonym okresem półtrwania w osoczu, liniową farmakokinetyką, dominacją klirensu nerkowego, niewystępowaniem znaczącego wiązania z białkami i brakiem klinicznie istotnych aktywnych metabolitów.
Topiramat nie jest silnym induktorem enzymów metabolizujących leki, może być podawany niezależnie od posiłków i nie jest konieczne rutynowe monitorowanie stężenia topiramatu w osoczu. W badaniach klinicznych nie zaobserwowano wyraźnej zależności między stężeniem topiramatu w osoczu, a skutecznością terapeutyczną i częstością działań niepożądanych.
Wchłanianie
Topiramat jest szybko i dobrze wchłaniany. U zdrowych ochotników, po doustnym podaniu 100 mg topiramatu średnia wartość maksymalnego stężenia topiramatu w osoczu (Cmax) wynosząca 1,5 µg/ml była osiągana w ciągu 2-3 godzin (Tmax).
Na podstawie pomiaru radioaktywności w moczu ustalono, że średni stopień wchłaniania po podaniu doustnym dawki 100 mg topiramatu znakowanego 14C wynosił co najmniej 81%. Nie wykazano znaczącego wpływu pokarmu na dostępność biologiczną topiramatu.
Dystrybucja
Wiązanie z białkami osocza wynosi 13 do 17%. Stwierdzono występowanie miejsc wiązania topiramatu o niewielkim powinowactwie na/w erytrocytach, które są wysycane, gdy stężenie topiramatu w osoczu jest większe niż 4 µg/ml. Objętość dystrybucji zmienia się w sposób odwrotny do dawki. Dla pojedynczej dawki
w zakresie od 100 do 1200 mg średnia pozorna objętość dystrybucji wynosiła od 0,8 do 0,55 l/kg. Zaobserwowano, że objętość dystrybucji zależy od płci. Wartości tego parametru u kobiet jest o około 50% mniejsza niż u mężczyzn. Jest to związane z większą zawartością tkanki tłuszczowej u kobiet i nie ma następstw klinicznych.
Metabolizm
U zdrowych ochotników topiramat nie jest intensywnie metabolizowany (ok.20%). U pacjentów przyjmujących jednocześnie inne leki przeciwpadaczkowe o działaniu indukującym enzymy metabolizujące leki, topiramat jest metabolizowany do 50%. U ludzi zidentyfikowano i scharakteryzowano 6 metabolitów topiramatu wyizolowanych z osocza, moczu i kału. Metabolity te powstają w wyniku procesów hydroksylacji, hydrolizy i glukuronizacji. Każdy z metabolitów stanowi mniej niż 3% całkowitej dawki wydalonej po podaniu topiramatu znakowanego 14C. Przebadano dwa metabolity o strukturze zbliżonej do topiramatu i stwierdzono, że wykazują one słabe działanie przeciwpadaczkowe lub nie wykazują działania przeciwpadaczkowego.
Eliminacja
U ludzi główną drogą eliminacji topiramatu w postaci niezmienionej i jego metabolitów są nerki (co najmniej 81% dawki). Około 66% topiramatu znakowanego 14C zostało wydalane w postaci niezmienionej z moczem w okresie 4 dni od przyjęcia. Po podaniu topiramatu w dawce 50 mg i 100 mg dwa razy na dobę, średni klirens nerkowy wynosił w przybliżeniu odpowiednio 18 ml/min i 17 ml/min. Badania wykazały reabsorpcję topiramatu w kanalikach nerkowych. Dane te potwierdzone zostały w badaniach na szczurach. Badania te wykazały znaczące zwiększenie nerkowego klirensu topiramatu podczas jednoczesnego stosowania
z probenecydem. Ogólnie, u ludzi, po podaniu doustnym osoczowy klirens topiramatu wynosi około 20 do 30 ml/min.
Liniowość/ nieliniowość
Międzyosobnicze różnice w stężeniu topiramatu w osoczu są niewielkie, dlatego też charakteryzuje się dobrze przewidywalnymi właściwościami farmakokinetycznymi. U zdrowych ochotników farmakokinetyka topiramatu ma przebieg liniowy, klirens osoczowy ma wartość stałą, a pole powierzchni pod krzywą obrazującą zmiany w stężeniu leku po pojedynczej dawce doustnej z przedziału od 100 do 400 mg zwiększa
się proporcjonalnie do dawki. U pacjentów z prawidłową czynnością nerek stężenie leku w osoczu osiąga stan stacjonarny po 4-8 dniach. U zdrowych ochotników średnia wartość Cmax po wielokrotnym podaniu leku doustnie w dawkach 100 mg dwa razy na dobę wynosiła 6,76 µg/ml. Po wielokrotnym podaniu topiramatu
w dawkach 50 mg i 100 mg dwa razy na dobę średni okres półtrwania eliminacji z osocza wynosił w przybliżeniu 21 godzin.
Stosowanie z innymi lekami przeciwpadaczkowymi
Po wielokrotnym podaniu topiramatu w dawkach 100 mg i 400 mg dwa razy na dobę jednocześnie z fenytoiną lub karbamazepiną stwierdzono proporcjonalne do dawki zwiększenie stężenia topiramatu w osoczu.
Zaburzenie czynności nerek
Klirens osoczowy i nerkowy topiramatu zmniejsza się u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności nerek (CLkr ≤ 70 ml/min). W związku z tym u pacjentów z zaburzoną czynnością nerek należy spodziewać się większego stężenia w osoczu w stanie stacjonarnym niż u pacjentów z prawidłową czynnością nerek. Ponadto, pacjenci z zaburzeniami czynności nerek wymagają więcej czasu do uzyskania stanu stacjonarnego dla każdej dawki. U pacjentów z umiarkowanymi i ciężkimi zaburzeniami czynności nerek zaleca się zastosowanie połowy zwykle stosowanej dawki początkowej i podtrzymującej.
Topiramat jest skutecznie usuwany z osocza podczas zabiegu hemodializy. Wydłużony czas trwania hemodializy może skutkować zmniejszeniem stężenia topiramatu poniżej stężenia wymaganego do utrzymania działania przeciwpadaczkowego. Może być konieczne podawanie dodatkowej dawki topiramatu, aby uniknąć szybkich spadków stężenia topiramatu w osoczu podczas hemodializy. Przy dostosowywaniu należy brać pod uwagę: 1) czas trwania dializy, 2) szybkość klirensu systemu dializacyjnego i 3) rzeczywisty klirens nerkowy topiramatu u pacjenta poddawanego dializie.
Zaburzenie czynności wątroby
Klirens osoczowy topiramatu zmniejsza się o około 26% u pacjentów ze średnimi lub ciężkimi zaburzeniami czynności wątroby. Dlatego topiramat należy stosować z ostrożnością u pacjentów z zaburzeniami czynności wątroby.
Osoby w podeszłym wieku
Klirens osoczowy topiramatu nie zmienia się u pacjentów w wieku podeszłym z prawidłową czynnością nerek.
Farmakokinetyka topiramatu u dzieci w wieku do 12 lat
Farmakokinetyka topiramatu u dzieci, podobnie jak u osób dorosłych otrzymujących terapię uzupełniającą, jest liniowa, z klirensem niezależnym od dawki i stężeniem w osoczu w stanie stacjonarnym zwiększającym się proporcjonalnie do dawki. U dzieci klirens jest jednak większy, a okres półtrwania w fazie eliminacji krótszy niż u dorosłych pacjentów. W konsekwencji, po tej samej dawce topiramatu w mg/kg mc. stężenie topiramatu w osoczu może być niższe u dzieci niż u dorosłych. Podobnie, jak u osób dorosłych, leki przeciwpadaczkowe pobudzające aktywność enzymów wątrobowych powodują zmniejszenie stężeń topiramatu w osoczu krwi w stanie stacjonarnym.
W nieklinicznych badaniach wpływu na płodność, pomimo działania toksycznego obserwowanego
u matek i ojców po podaniu niskich dawek 8 mg/kg mc./dobę, nie obserwowano wpływu na płodność samców i samic szczurów nawet przy dawkach osiągających 100 mg/kg mc.
W badaniach przedklinicznych wykazano, że u badanych gatunków zwierząt (myszy, szczury, króliki) topiramat działa teratogennie.
U myszy, topiramat podawany w dawce 500 mg/kg mc./dobę powodował zmniejszenie masy ciała płodu oraz hamował proces kostnienia szkieletu, przy jednoczesnym działaniu toksycznym na organizm matki. Całkowita liczba wad wrodzonych u płodu myszy uległa zwiększeniu we wszystkich grupach, którym podawano topiramat w dawkach: 20, 100 i 500 mg/kg mc,/dobę.
U szczurów, zależne od dawki działanie toksyczne na organizm matki oraz feto- i embriotoksyczność (zmniejszoną masa ciała płodu i (lub) hamowanie procesu kostnienia szkieletu) obserwowano po podaniu dawek do 20 mg/kg mc./dobę, a działanie teratogenne (zniekształcenia kończyn i palców) po zastosowaniu dawek 400 mg/kg mc./dobę lub większych. U królików, zależne od dawki toksyczne działanie leku na matkę obserwowano po dawkach mniejszych niż 10 mg/kg mc./dobę, feto- i embriotoksyczność (zwiększoną umieralność) obserwowano po dawkach mniejszych niż 35 mg/kg mc./dobę, a działanie teratogenne (zniekształcenia żeber i kręgów) po dawkach 120 mg/kg mc./dobę.
Działanie teratogenne obserwowane u szczurów i królików było podobne do tego, jakie obserwowano w związku ze zastosowaniem inhibitorów anhydrazy węglanowej, które nie powodowały wad
rozwojowych u ludzi. O wpływie topiramatu na wzrost świadczyła również niższa waga urodzeniowa oraz niższa urodzeniowa masa ciała i masa w okresie oseskowym u potomstwa samic otrzymujących lek
w dawce 20 lub 100 mg/kg mc./dobę w okresie ciąży i karmienia. U szczurów topiramat przenika przez barierę łożyskową.
U młodych szczurów topiramat podawany doustnie w dawce do 300 mg/kg mc./dobę w okresie rozwoju odpowiadającym okresowi niemowlęcemu, dzieciństwu i okresowi dojrzewania, wywoływał działania toksyczne podobne do tych, jakie obserwowano u dorosłych osobników (zmniejszenie przyjmowania pokarmów skojarzone ze spowolnieniem zwiększania masy ciała, przerost centralnej części zrazików wątrobowych). Nie stwierdzono istotnego wpływu leku na wzrost kości długich (kość piszczelowa), gęstość mineralną kości (kość udowa), przedwczesne odstawienie od piersi oraz rozwój reprodukcyjny, rozwój neurologiczny (w oparciu o ocenę pamięci i uczenia się), parzenie się, płodność oraz parametry histerotomii.
W badaniach nad mutagennością przeprowadzonych zarówno in vitro, jak i in vivo topiramat nie wykazywał działania genotoksycznego.
Rdzeń tabletki:
Celuloza mikrokrystaliczna Mannitol
Karboksymetyloskrobia sodowa (typ A) Skrobia żelowana, kukurydziana Krospowidon
Powidon
Magnezu stearynian Wosk Carnauba
Otoczka:
Opadry II White OY-LS-28908 Hypromeloza 15cP Hypromeloza 3 cP
Hypromeloza 50 cP
Laktoza jednowodna Tytanu dwutlenek (E 171) Makrogol 4000
Opadry Yellow 02H2229
Hypromeloza 5 cP
Tytanu dwutlenek (E 171) Talk
Glikol propylenowy Żółcień chinolinowa (E 104)
Nie dotyczy.
Pojemnik z HDPE: 3 lata
Blister PVC/PE/PVDC/Aluminium: 3 lata
Brak specjalnych zaleceń dotyczących przechowywania.
Blister PVC/PE/PVDC/Aluminium, w tekturowym pudełku. Każde opakowanie zawiera 28 lub 60 tabletek powlekanych.
Pojemnik z HDPE z wieczkiem z PP i środkiem pochłaniającym wilgoć; w tekturowym pudełku. Każde opakowanie zawiera 60 tabletek powlekanych.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Bez specjalnych wymagań.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Bausch Health Ireland Limited 3013 Lake Drive
Citywest Business Campus Dublin 24, D24PPT3 Irlandia
12894
12.12.2007
05.09.2012
07.03.2021








