







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Ogniskowej spastyczności stawu skokowego i stopy u dzieci z mózgowym porażeniem dziecięcym, w wieku dwóch lat i starszych jako uzupełnienie terapii rehabilitacyjnej.
Ogniskowej spastyczności nadgarstka i dłoni u pacjentów dorosłych po udarze.
Ogniskowej spastyczności stawu skokowego i stopy u pacjentów dorosłych po udarze
(patrz punkt 4.4).
Kurczu powiek (blefarospazm), połowiczego kurczu twarzy i związane z nim ogniskowe dystonie.
Idiopatycznego kręczu karku (dystonia szyjna).
Profilaktyki bólów głowy u pacjentów dorosłych cierpiących na przewlekłą migrenę (bóle głowy występujące 15 dni w miesiącu lub częściej).
Zaburzenia czynności pęcherza moczowego:
Idiopatyczna nadreaktywność pęcherza moczowego z objawami nietrzymania moczu, parciem naglącym lub częstomoczem, u pacjentów dorosłych, z niewystarczającą odpowiedzią lub
z nadwrażliwością na leki antycholinergiczne.
Nietrzymanie moczu u pacjentów dorosłych z nadreaktywnością mięśnia wypieracza pęcherza moczowego o podłożu neurogennym po stabilnych urazach rdzenia kręgowego poniżej odcinka szyjnego oraz u pacjentów ze stwardnieniem rozsianym.
Zaburzenia skóry i jej przydatków:
Uporczywa, ciężka, pierwotna nadpotliwość pach, przeszkadzająca w codziennych czynnościach i oporna na leczenie miejscowe.
Przejściowa poprawa wyglądu następujących zmarszczek:
zmarszczki pionowe między brwiami o nasileniu umiarkowanym lub ciężkim widoczne przy maksymalnym zmarszczeniu brwi (tzw. zmarszczki gładzizny czoła) i (lub),
zmarszczki w okolicy bocznego kąta oka o nasileniu umiarkowanym lub ciężkim widoczne przy pełnym uśmiechu (zmarszczki typu „kurze łapki”) i (lub),
zmarszczki poziome czoła o nasileniu umiarkowanym lub ciężkim widoczne przy maksymalnym uniesieniu brwi,
u dorosłych osób, gdy ich nasilenie ma psychologiczny wpływ na pacjenta.
Dawkowanie i sposób podawania
Górny margines aktywności mięśnia czołowego: ok. 1 cm powyżej najwyższej zmarszczki na czole.
Dolna linia leczenia: pośrodku pomiędzy górnym marginesem aktywności mięśnia czołowego
a brwiami, co najmniej 2 cm powyżej brwi.
Górna linia leczenia: pośrodku pomiędzy górnym marginesem aktywności mięśnia czołowego a dolną linią leczenia.
Pięć wstrzyknięć należy wykonać w punktach przecięcia poziomych linii leczenia z następującymi pionowymi liniami orientacyjnymi:
Na dolnej linii leczenia, w linii środkowej twarzy oraz 0,5-1,5 cm przyśrodkowo od stwierdzonego palpacyjnie grzebienia skroniowego; powtórzyć po drugiej stronie.
Na górnej linii leczenia, pośrodku pomiędzy bocznym a przyśrodkowym miejscem wstrzyknięcia w dolnej linii leczenia; powtórzyć po drugiej stronie.
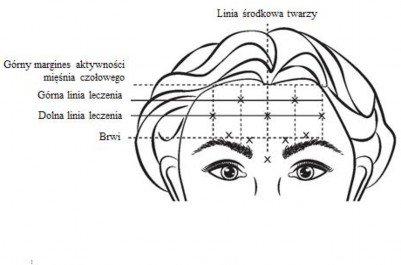
Należy zwrócić szczególną uwagę, aby nie wstrzyknąć produktu leczniczego BOTOX do naczynia krwionośnego podczas wstrzykiwania w zmarszczki poziome czoła widoczne podczas maksymalnego uniesienia brwi (patrz punkt 4.4).
Zalecana dawka: Do każdego z 5 miejsc wstrzyknięcia w mięsień czołowy podaje się po 0,1 ml (4 jednostki) produktu, co łącznie daje dawkę 20 jednostek o całkowitej objętości 0,5 ml (patrz Ryc. 4).
Łączna dawka podawana w leczeniu zmarszczek poziomych czoła (20 jednostek) oraz zmarszczek gładzizny czoła
(20 jednostek) wynosi 40 jednostek/1,0 ml.
W przypadku jednoczesnego leczenia zmarszczek gładzizny czoła i zmarszczek typu „kurze łapki” całkowita dawka wynosi 64 jednostki i obejmuje 20 jednostek na zmarszczki poziome czoła, 20 jednostek na zmarszczki gładzizny czoła (patrz Zalecana dawka na zmarszczki gładzizny czoła
i Ryc. 1) oraz 24 jednostki na zmarszczki typu „kurze łapki” (patrz Zalecana dawka na zmarszczki typu „kurze łapki”
i Ryc. 2 i 3).
Dodatkowe informacje: Przerwy pomiędzy zabiegami nie powinny być krótsze niż
3 miesiące.
Nie zbadano skuteczności i bezpieczeństwa wielokrotnych wstrzyknięć produktu leczniczego BOTOX w leczeniu poziomych zmarszczek czoła trwającym powyżej
12 miesięcy.
UWAGI DOTYCZĄCE WSZYSTKICH WSKAZAŃ
W przypadku niepowodzenia terapeutycznego po pierwszym zabiegu, definiowanego jako brak znamiennej poprawy po miesiącu od podania produktu w porównaniu ze stanem wyjściowym, należy rozważyć następujące strategie postępowania:
Kliniczną weryfikację, która może obejmować badanie elektromiograficzne w celu oceny działania toksyny w mięśniu/mięśniach, po wstrzyknięciu.
Analizę przyczyn niepowodzenia, do których można zaliczyć między innymi:
niewłaściwy wybór mięśni do wstrzyknięcia,
za małą dawkę produktu,
nieprawidłową technikę wstrzyknięć,
występowanie stałego przykurczu,
za słabe mięśnie antagonistyczne,
powstanie przeciwciał neutralizujących toksynę.
Ponowne rozważenie wskazania do leczenia z zastosowaniem toksyny botulinowej typu A.
Jeśli nie wystąpiły jakiekolwiek działania niepożądane po pierwszym zabiegu, należy rozważyć powtórzenie leczenia ze zwróceniem uwagi na następujące problemy:
właściwy dobór dawki produktu w oparciu o analizę wcześniejszego niepowodzenia terapeutycznego,
zastosowanie EMG,
zachowanie trzymiesięcznego odstępu pomiędzy kolejnymi zabiegami.
W przypadku braku lub niezadowalającego skutku leczniczego po drugim cyklu leczenia należy rozważyć zastosowanie alternatywnych metod postępowania.
W leczeniu dorosłych pacjentów, w tym leczenia z powodu wielu wskazań, maksymalna łączna dawka nie powinna przekraczać 400 jednostek, w odstępach 12-tygodniowych.
Przeciwwskazania
u osób o znanej nadwrażliwości na kompleks neurotoksyny Clostridium botulinum typu A lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
jeśli w miejscu, w którym ma być podany, występuje zakażenie.
W przypadku leczenia zaburzeń czynności pęcherza moczowego produkt leczniczy BOTOX jest przeciwwskazany także:
u pacjentów z zakażeniem dróg moczowych w momencie leczenia;
u pacjentów z ostrym zatrzymaniem moczu w momencie terapii, którzy nie są rutynowo poddani cewnikowaniu;
u pacjentów, którzy nie chcą i (lub) nie mogą rozpocząć cewnikowania po terapii, jeśli byłoby to konieczne.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Ogólne ostrzeżenia dotyczące wszystkich wskazań
Produkt leczniczy BOTOX należy stosować ostrożnie w leczeniu ogniskowej spastyczności kończyny górnej (nadgarstka i dłoni) oraz kończyny dolnej (stawu skokowego i stopy) po udarze u pacjentów w podeszłym wieku, z istotnymi chorobami współistniejącymi, a leczenie należy rozpoczynać wyłącznie wtedy, gdy uważa się, że korzyści z niego wynikające przewyższają potencjalne ryzyko.
Produkt leczniczy BOTOX należy stosować wyłącznie w leczeniu poudarowej spastyczności kończyn górnych i (lub) dolnych po ocenie przeprowadzonej przez lekarza specjalistę, mającego doświadczenie w rehabilitacji pacjentów po udarze.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Ogólne
W kontrolowanych badaniach klinicznych działania niepożądane klasyfikowane jako mające związek z podaniem produktu leczniczego BOTOX obserwowano u 35% pacjentów z kurczem powiek (blefarospazm), u 28% pacjentów z dystonią szyjną, u 8% pacjentów ze spastycznością dziecięcą,
u 11% pacjentów z nadpotliwością pach, u 16% pacjentów z ogniskową spastycznością górnej kończyny powiązanej z udarem oraz u 15% pacjentów z ogniskową spastycznością kończyny dolnej powiązanej z udarem. W badaniach klinicznych dotyczących przewlekłej migreny częstość występowania działań niepożądanych wyniosła 26% podczas pierwszej terapii ze spadkiem do 11% podczas drugiej terapii. W badaniach klinicznych nad idiopatyczną nadreaktywnością pęcherza moczowego częstość występowania wyniosła 26% podczas pierwszej terapii ze spadkiem do 22% podczas drugiej terapii. W badaniach klinicznych dotyczących zastosowania tego produktu u dorosłych z neurogenną nadczynnością mięśnia wypieracza działania niepożądane obserwowano
u 32% pacjentów podczas pierwszej terapii ze spadkiem do 18% podczas drugiej terapii.
W przypadku neurogennej nadczynności wypieracza u dzieci i młodzieży częstość występowania wyniosła 6,2% po pierwszym leczeniu.
W pierwszym cyklu leczenia, w badaniach klinicznych dotyczących leczenia zmarszczek poziomych czoła widocznych przy maksymalnym uniesieniu brwi, zdarzenia niepożądane uznane przez badaczy za związane z podaniem produktu leczniczego BOTOX zgłosiło 20,6% pacjentów, którzy otrzymali 40 jednostek (20 jednostek do mięśnia czołowego i 20 jednostek podanych w okolicę gładzizny czoła) i 14,3% pacjentów leczonych dawką 64 jednostek (20 jednostek do mięśnia czołowego, 20 jednostek podanych w okolicę gładzizny czoła i 24 jednostki w okolice kurzych łapek), w porównaniu z 8,9% pacjentów, którzy otrzymali placebo.
W większości przypadków, działania niepożądane występują w ciągu pierwszych kilku dni po wstrzyknięciu i mają charakter przejściowy.
W rzadkich przypadkach działania niepożądane mogą utrzymywać się przez kilka miesięcy lub dłużej. Miejscowe osłabienie mięśni stanowi spodziewane farmakologiczne działanie toksyny botulinowej.
Zgłaszano również występowanie osłabienia sąsiadujących mięśni i (lub) mięśni oddalonych od miejsca wstrzyknięcia.
Tak jak w przypadku każdego wstrzyknięcia, w miejscu wstrzyknięcia mogą wystąpić: miejscowy ból, zapalenie, parestezje, niedoczulica, tkliwość, obrzęk, rumień, miejscowe zakażenie, krwawienie i (lub) zasinienie, działania niepożądane związane z samą procedurą wstrzyknięcia.
Ból i (lub) lęk związany z igłą mogą doprowadzić do wystąpienia reakcji wazowagalnej, obejmującej przemijające objawowe niedociśnienie i omdlenia.
Po podaniu toksyny botulinowej zgłaszano także przypadki gorączki i objawów grypopodobnych.
Działania niepożądane – częstość w zależności od wskazań
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Statystycznie znaczące różnice od placebo (p<0.05)
a Punktacja MAS jest 6-punktową skalą (0 [brak wzrostu napięcia mięśniowego], 1, 1+, 2, 3, i 4 [kończyna sztywna w zgięciu lub wyproście]), która mierzy siłę potrzebną do poruszania kończyną w stawie, przy czym zmniejszenie wyniku oznacza poprawę spastyczności.
b CGI oceniło odpowiedź na leczenie pod kątem tego, jak pacjent radził sobie w życiu, używając 9-stopniowej skali (-4 = bardzo wyraźne pogorszenie do +4 = bardzo wyraźna poprawa).
c GAS to 6-punktowa skala (-3 [gorzej niż na początku], -2 [tak jak na początku], -1 [mniej niż oczekiwano], 0 [oczekiwany cel], +1 [nieco więcej niż oczekiwano], +2 [znacznie więcej niż oczekiwano]).
d Ból oceniano u uczestników, którzy byli w wieku 4 lat i starsi i mieli na początku punktację bólu> 0 za pomocą skali bólu twarzy (FPS: 0 = brak bólu do 10 = bardzo silny ból)
Ogniskowa spastyczność kończyny dolnej u dzieci
Skuteczność i bezpieczeństwo stosowania produktu leczniczego BOTOX w leczeniu spastyczności kończyny dolnej u dzieci w wieku 2 lat i starszych oceniono w randomizowanym, wieloośrodkowym, podwójnie zaślepionym badaniu, kontrolowanym placebo. Badanie obejmowało 384 pacjentów pediatrycznych (128 BOTOX 8 jednostek/kg, 126 BOTOX 4 jednostki/kg i 128 placebo) ze spastycznością kończyny dolnej z powodu porażenia mózgowego (punktacja dla stawu skokowego co najmniej 2 w zmodyfikowanej skali Ashwortha). Całkowitą dawkę 4 jednostki/kg (maksymalnie 150 jednostek) lub 8 jednostek/kg (maksymalnie 300 jednostek) lub placebo wstrzykiwano domięśniowo
i podzielono pomiędzy mięśnie brzuchaty łydki, płaszczkowaty i piszczelowy tylny. Wszyscy pacjenci przeszli fizjoterapię. Aby pomóc właściwie zlokalizować mięśnie do iniekcji wymagane było zastosowanie kontroli elektromiograficznej, stymulacji nerwów lub technik ultrasonografii.
Pierwszorzędowym punktem końcowym była średnia zmiana punktacji dla stawu skokowego w MAS w 4 i 6 tygodniu w stosunku do wartości wyjściowej, a kluczowym drugorzędowym punktem końcowym była średnia wartość CGI w 4 i 6 tygodniu. Skala oceny GAS przez lekarza, dla czynnych i biernych celów czynnościowych była drugorzędowym punktem końcowym w tygodniach 8 i 12.
Chód oceniano skalą Edinburgh Visual Gait (EVG) w tygodniach 8 i 12 w podgrupie pacjentów. Pacjentów obserwowano przez 12 tygodni.
Pacjenci spełniający wymagania mogli zostać włączeni do otwartego badania rozszerzonego,
w którym otrzymywali do 5 sesji terapeutycznych w dawkach do 10 jednostek/kg (maksymalnie 340 jednostek), w przypadku leczenia więcej niż jednej kończyny.
Statystycznie znacząca poprawa w porównaniu z placebo została wykazana u pacjentów leczonych produktem leczniczym BOTOX 4 i 8 jednostek/kg dla pierwszorzędowego punktu końcowego
i w większości punktów czasowych do Tygodnia 12. Poprawa wyniku MAS była podobna w obu grupach leczenia produktem leczniczym BOTOX. Jednak w żadnym momencie różnica w porównaniu z placebo wynosiła ≥ 1 punkt w skali MAS. Patrz tabela poniżej. Efekt leczenia w analizie odpowiedzi na leczenie był mniejszy niż 15% we wszystkich punktach czasowych.
Pierwszorzędowe i drugorzędowe punkty końcowe w ocenie skuteczności
BOTOX 4 jednostki/kg (N=125)
BOTOX 8 jednostek/kg (N=127)
Placebo (N=129)
Średnia zmiana mięśnia zginacza podeszwowego kostki w punktacji MAS
w odniesieniu do wartości wyjścioweja
Średnio 4 i 6 tydzień
-1,01*
-1,06*
-0,80
Średni wynik CGIb
Średnio 4 i 6 tydzień
1,49
1,65*
1,36
Średni wynik GASc
Cele pasywne w tygodniu 8
0,18*
0,19*
-0,26
Cele pasywne w tygodniu 12
0,27
0,40*
0,00
Cele aktywne w tygodniu 8
-0,03*
0,10*
-0,31
Cele aktywne w tygodniu 12
0,09
0,37*
-0,12
Średnia zmiana wyniku EVG w odniesieniu do wartości wyjściowejd
Tydzień 8
-2,11
-3,12*
-0,86
Tydzień 12
-2,07
-2,57
-1,68
Statystycznie znaczące różnice od placebo (p<0,05)
jednostek/kg, zostali przydzieleni do najbliższej możliwej grupy dawkowania; N= 38, N=45
i N=30 odpowiednio dla jednostek BOTOX 50, BOTOX 100 i BOTOX 200. Przed zastosowaniem leczenia, pacjenci otrzymywali znieczulenie w zależności od wieku i miejscowej praktyki. Stu dziewięciu pacjentów (97,3%) otrzymało znieczulenie ogólne lub płytką sedację (wymaganą dla pacjentów < 12 lat), a 3 pacjentów (2,7%) otrzymało znieczulenie miejscowe (dozwolone tylko dla pacjentów ≥ 12 lat).
Wartość wyjściowa i zmiana w stosunku do wartości wyjściowej w dziennej częstości epizodów nietrzymania moczu w ciągu dnia, objętość moczu przy pierwszym porannym cewnikowaniu oraz maksymalne ciśnienie wypieracza podczas fazy magazynowania (cmH2O) w badaniu klinicznym prowadzonym metodą podwójnie ślepej próby w grupach równoległych
BOTOX 200
jednostek (N=30)
BOTOX 100
jednostek (N=45)
BOTOX 50
jednostek (N=38)
Poziom istotności p*
BOTOX 200 vs. 50
jednostek
Poziom istotności p*
BOTOX 100 vs. 50
jednostek
Dzienna częstotliwość epizodów nietrzymania moczu w ciągu dniaa
0,9123
0,9949
Średnia wartość na początku badania (SD)
3,7 (5,06)
3,0 (1,07)
2,8 (1,04)
Średnia zmiana* w
2. tygodniu
-1,1
-1,0
-1,2
Średnia zmiana* w
6. tygodniu** (95% CI)
-1,3 (-1.8, -
0,9)
-1,3 (-1,7,
-0,9)
-1,3 (-1,7, -
0,9)
Średnia zmiana*w
12. tygodniu
-0,9
-1,4
-1,2
Objętość moczu przy pierwszym porannym cewnikowaniu (mL)b
0,0055
0,5117
Średnia wartość na początku badania (SD)
187,7
(135,70)
164,2
(114,48)
203,5
(167,48)
Średnia zmiana* w
2. tygodniu
63,2
29,4
31,6
Średnia zmiana* w
6. tygodniu** (95% CI)
87,5 (52,1,
122,8)
34,9 (7,9,
61,9)
21,9 (-7,2,
51,1)
Średnia zmiana*w
12. tygodniu
45,2
55,8
12,9
Maksymalne ciśnienie mięśnia wypieracza podczas fazy magazynowania
(cmH2O)b
Średnia wartość na początku badania (SD)
56,7 (33,89)
56,5
(26.86)
58,2 (29,45)
0,0157
0,1737
Średnia zmiana* w
6. tygodniu** (95% CI)
-27,3 (-36,4, -
18,2)
-20,1 (-
27,3, -
12,8)
-12,9 (-20.4,
-5,3)
CI = Przedział ufności
*Zmiana średniej najmniejszych kwadratów (LS), CI =95% i poziom istotności p są oparte na modelu ANCOVA z wartością wyjściową jako współzmienną, a grupą leczoną w wieku (< 12 lat lub ≥ 12 lat), początkowymi epizodami nietrzymania moczu w ciągu dnia (≤ 6 lub > 6) oraz terapią antycholinergiczną (tak/nie) jako czynnikami początkowymi.
** Główny punkt czas aGłówny punkt końcowy bDrugi punkt końcowy
Średnia czasu trwania odpowiedzi w tym badaniu, w oparciu o żądania ponownej terapii wysuwane przez pacjenów wyniosła odpowiednio 214,0 (31 tygodni), 169,0 (24 tygodnie) i 207 dni (30 tygodni) dla BOTOX 50 jednostek, BOTOX 100 jednostek i BOTOX 200 jednostek.
Na 99 dzieci, które miały ujemny wynik początkowy dla przeciwciał wiążących lub przeciwciał neutralizujących i miały co najmniej jedną możliwą do oceny wartość początkową z jednego randomizowanego badania z podwójnie ślepą próbą i jednego badania z podwójnie ślepą próbą, u żadnego nie wytworzyły się przeciwciała neutralizujące po otrzymaniu od 50 do 200 jednostek BOTOX.
ZABURZENIA SKÓRY I JEJ PRZYDATKÓW
Pierwotna nadpotliwość pach
Przeprowadzono wieloośrodkowe badanie kliniczne metodą podwójnie ślepej próby z udziałem pacjentów z pierwotną, uporczywą, obustronną nadpotliwością pach, określoną na poziomie co najmniej 50 mg wydzielania potu z każdej pachy w ciągu 5 minut, w temperaturze pokojowej, w stanie spoczynku według pomiaru grawimetrycznego. 320 pacjentów otrzymało 50 jednostek
produktu BOTOX (n=242) lub placebo (n=78). Osoby u których wystąpiło zmniejszenie potliwości o 50 % w stosunku do stanu początkowego zostały określone jako reagujące na leczenie.
W momencie pierwszej oceny, w 4 tygodniu po podaniu produktu, 93,8% pacjentów w grupie otrzymujących BOTOX i 35,9% w grupie pacjentów otrzymujących placebo odpowiedziało na leczenie (p<0,001). Zależność ta utrzymywała się na wysokim poziomie w grupie pacjentów otrzymujących produkt BOTOX w porównaniu z grupą placebo przez cały okres po zabiegu, aż do 16 tygodnia (p<0,001).
Następnie przeprowadzono badanie otwarte z udziałem 207 wybranych pacjentów, którzy byli poddawani trzykrotnym zabiegom leczniczym produktem BOTOX. Ogólnie, 174 pacjentów ukończyło pełną terapię trwającą łącznie 16 miesięcy, gdyż uczestniczyli w 2 połączonych badaniach (4 miesięcznym badaniu metodą podwójnie ślepej próby i 12 miesięcznej kontynuacji metodą otwartej próby). W 16 tygodniu po pierwszym (n=287), drugim (n=123) i trzecim (n=30) podaniu produktu BOTOX, odpowiedź kliniczna utrzymywała się odpowiednio u 85%, 86,2% i 80% pacjentów. Średni czas utrzymywania się skutku terapeutycznego po podaniu pojedynczej dawki i kontynuacji leczenia w badaniu otwartym wynosił 7,5 miesiąca od pierwszego podania, a u 27,5% pacjentów skutek kliniczny utrzymywał się przez rok lub dłużej.
Zmarszczki typu „kurze łapki”
Do badania włączono 1362 pacjentów ze zmarszczkami typu „kurze łapki”, powstającymi podczas maksymalnego uśmiechu, o nasileniu umiarkowanym lub ciężkim, zarówno występującymi osobno (n=445, badanie 191622-098) bądź jednocześnie ze zmarszczkami gładzizny czołowej o nasileniu
umiarkowanym lub ciężkim, powstającymi przy maksymalnym zmarszczeniu brwi (n=917, badanie 191622-099).
Wstrzyknięcia produktu leczniczego BOTOX znacząco zmniejszyły nasilenie zmarszczek typu „kurze łapki” powstających podczas pełnego uśmiechu w porównaniu z placebo, we wszystkich punktach czasowych (p <0,001) przez okres do 5 miesięcy. Parametr ten mierzono za pomocą odsetka pacjentów, u których w obu badaniach głównych stopień nasilenia zmarszczek typu „kurze łapki” widocznych przy pełnym uśmiechu oceniano jako zerowy bądź łagodny (do 150. dnia (zakończenie badania) w badaniu 191622-098 oraz do 120. dnia (zakończenie pierwszego cyklu terapeutycznego) w badaniu 191622-099). W ocenie badacza oraz pacjenta, proporcja pacjentów uzyskujących zerowy bądź umiarkowany stopień nasilenia zmarszczek typu „kurze łapki” widocznych przy pełnym uśmiechu była większa wśród pacjentów z umiarkowanym nasileniem zmarszczek przed terapią niż pacjentów ze zmarszczkami o nasileniu ciężkim.
W Tabeli 1. przedstawiono podsumowanie wyników osiągniętych w 30. dniu, stanowiącym punkt czasowy pierwszorzędowego punktu końcowego oceny skuteczności.
W badaniu 191622-104 (stanowiącym rozszerzenie badania 191622-099), 101 pacjentów uprzednio poddanych randomizacji do grupy otrzymującej placebo włączono do grupy, która miała otrzymać pierwsze leczenie w dawce 44 jednostek. Pacjenci leczeni za pomocą BOTOX uzyskiwali statystycznie istotną poprawę w pierwszorzędowym punkcie końcowym oceny skuteczności
w porównaniu z placebo w 30. dniu podczas pierwszego badania. Odsetek odpowiedzi w 30. dniu po pierwszym podaniu produktu leczniczego był podobny do grupy otrzymującej 44 jednostki w ramach badania 191622-099. Łącznie 123 pacjentów było poddanych 4 cyklom leczenia w dawce 44 jednostek BOTOX stosowanego w jednoczesnym leczeniu zmarszczek typu „kurze łapki” oraz zmarszczek gładzizny czołowej.
Tabela 1. Dzień 30: Ocena badacza oraz pacjenta w zakresie poprawy wyglądu zmarszczek typu
„kurze łapki” widocznych przy pełnym uśmiechu - odsetek odpowiedzi (% pacjentów uzyskujących zerowy bądź łagodny stopień nasilenia zmarszczek typu „kurze łapki”)
Badanie kliniczne
Dawka
BOTOX
Placebo
BOTOX
Placebo
ocena badacza
ocena pacjenta
191622-098
24 jednostki
(zmarszczki typu “kurze łapki”)
66,7%*
(148/222)
6,7%
(15/223)
58,1%*
(129/222)
5,4%
(12/223)
191622-099
24 jednostki
(zmarszczki typu “kurze łapki”)
54,9%*
(168/306)
3,3%
(10/306)
45,8%*
(140/306)
3,3%
(10/306)
44 jednostki
(24 jednostki zmarszczki typu “kurze łapki”; 20 jednostek zmarszczki gładzizny
czołowej)
59,0%* (180/305)
3,3% (10/306)
48,5%* (148/305)
3,3% (10/306)
*p < 0,001 (BOTOX wobec placebo)
Oceniana przez pacjenta poprawa w zakresie wyglądu zmarszczek typu „kurze łapki” przy pełnym uśmiechu w stosunku do punktu początkowego była statystycznie istotna (p < 0,001) dla produktu leczniczego BOTOX (24 jednostki i 44 jednostki) w porównaniu z placebo, zarówno 30. w dniu jak i we wszystkich punktach czasowych po każdym cyklu leczenia w obu badaniach głównych.
Zaobserwowano poprawę w samoocenie wieku i atrakcyjności pacjentów po podaniu BOTOX (24 jednostki i 44 jednostki) w porównaniu z placebo stosując kwestionariusz wyników leczenia zmarszczek twarzy (ang. Facial Line Outcomes - FLO-11) w 30. dniu pierwszorzędowego punktu
czasowego oraz we wszystkich kolejnych punktach czasowych w obu badaniach głównych (p < 0,001).
W badaniach głównych, 3,9% pacjentów (53/1362) było w wieku powyżej 65 lat. U pacjentów w tej grupie wiekowej odsetek odpowiedzi, w ocenie badacza, wynosił 36% (30. w dniu) dla produktu leczniczego BOTOX (24 jednostki i 44 jednostki). Analiza grup wiekowych pacjentów ≤ 50 lat
i pacjentów > 50 lat wykazała statystycznie istotną poprawę w porównaniu z placebo. W ocenie badaczy, odpowiedź na leczenie po podaniu produktu BOTOX w dawce 24 jednostki, w grupie pacjentów w wieku > 50 lat była mniejsza niż w grupie pacjentów w wieku ≤ 50 lat (wynosząc odpowiednio 42% i 71,2%).
Ogólna odpowiedź po podaniu produktu BOTOX w leczeniu zmarszczek typu „kurze łapki” widocznych przy pełnym uśmiechu jest mniejsza (60%), niż odpowiedź na leczenie obserwowana w leczeniu zmarszczek gładzizny czoła widocznych przy maksymalnym zmarszczeniu brwi (80%).
U 916 pacjentów (517 pacjentów otrzymujących dawkę 24 jednostki oraz 399 pacjentów otrzymujących 44 jednostki) leczonych za pomocą produktu leczniczego BOTOX oznaczono przeciwciała. U żadnego z nich nie stwierdzono przeciwciał neutralizujących.
Zmarszczki poziome czoła
Do badania włączono 822 pacjentów ze zmarszczkami poziomymi czoła i zmarszczkami gładzizny czoła o nasileniu umiarkowanym lub ciężkim widocznymi przy maksymalnym napięciu, zarówno występującymi osobno (n=254, badanie 191622-142) lub jednocześnie z umiarkowanymi lub ciężkimi zmarszczkami typu „kurze łapki” widocznymi podczas maksymalnego uśmiechu (n=568, badanie 191622-143). Pacjentów tych włączono do głównych populacji badania w celu oceny wszystkich pierwszorzędowych i drugorzędowych punktów końcowych skuteczności. W badaniach klinicznych zmarszczki poziome czoła były leczone jednocześnie ze zmarszczkami gładzizny czoła.
Zarówno w ocenie badaczy, jak i pacjentów, odsetek pacjentów, u których po wstrzyknięciu produktu leczniczego BOTOX nie stwierdzono poziomych zmarszczek czoła lub łagodne ich nasilenie podczas maksymalnego uniesienia brwi, był większy niż odsetek pacjentów leczonych placebo w 30. dniu, stanowiącym punkt czasowy pierwszorzędowego punktu końcowego oceny skuteczności (patrz tabela poniżej). Podano również odsetek pacjentów osiągających co najmniej 1. stopniowe złagodzenie nasilenia zmarszczek poziomych czoła w porównaniu ze stanem wyjściowym w spoczynku oraz pacjentów, u których nie stwierdzono zmarszczek poziomych czoła lub o łagodnym nasileniu podczas maksymalnego napięcia.
Dzień 30: Ocena zmarszczek poziomych czoła oraz zmarszczek w górnej części twarzy przy maksymalnym napięciu oraz przy rozluźnionych mięśniach, dokonana przez badacza i pacjenta.
Badanie kliniczne
Punkt końcowy
BOTOX
Placebo
BOTOX
Placebo
Ocena badacza
Ocena pacjenta
Badanie 191622-142
40 jednostek
(20 jednostek na zmarszczki poziome czoła + 20 jednostek na zmarszczki gładzizny
czoła)
Zmarszczki poziome czoła przy maksymalnym
napięciua
94,8%
(184/194)
1,7%
(1/60)
87,6%
(170/194)
0,0%
(0/60)
p < 0,0005
p < 0,0005
Zmarszczki poziome czoła przy rozluźnionych mięśniachb
86,2% (162/188)
22,4% (13/58)
89,7% (174/194)
10,2% (6/59)
p < 0,0001
p < 0,0001
Zmarszczki poziome
czoła przy
90,5%
(201/222)
2,7%
(3/111)
81,5%
(181/222)
3,6%
(4/111)
Badanie 191622-143
40 jednostek
(20 jednostek na zmarszczki poziome czoła + 20 jednostek na zmarszczki
gładzizny czoła)
maksymalnym napięciua
p < 0,0005
p < 0,0005
Zmarszczki poziome czoła przy rozluźnionych mięśniachb
84,1% (185/220)
15,9% (17/107)
83,6% (184/220)
17,4% (19/109)
p < 0,0001
p < 0,0001
Badanie 191622-143
64 jednostki
(20 jednostek na zmarszczki poziome czoła + 20 jednostek na zmarszczki gładzizny czoła + 24 jednostki na zmarszczki typu „kurze
łapki”
Zmarszczki poziome czoła przy maksymalnym
napięciua
93,6%
(220/235)
2,7%
(3/111)
88,9%
(209/235)
3,6%
(4/111)
p < 0,0005
p < 0,0005
Zmarszczki górnej części twarzy przy maksymalnym napięciuc
56,6% (133/235)
0,9% (1/111)
n/d
p < 0,0001
a Odsetek pacjentów bez zmarszczek lub o łagodnym nasileniu zmarszczek poziomych czoła widocznych przy maksymalnym uniesieniu brwi.
b Odsetek pacjentów z co najmniej 1. stopniowym zmniejszeniem nasilenia zmarszczek poziomych czoła w spoczynku w stosunku do stanu wyjściowego.
c Odsetek pacjentów definiowanych jako ten sam pacjent bez zmarszczek lub o łagodnym nasileniu zmarszczek poziomych czoła, zmarszczek gładzizny czoła oraz zmarszczek typu „kurze łapki” w każdej części twarzy przy maksymalnym napięciu.
W porównaniu z placebo, wstrzyknięcia produktu leczniczego BOTOX znacząco zmniejszyły nasilenie zmarszczek poziomych czoła widocznych przy maksymalnym uniesieniu brwi w okresie do 6 miesięcy (p < 0,05). Określono to za pomocą odsetka pacjentów, u których w obu badaniach głównych dotyczących stopnia nasilenia zmarszczek poziomych czoła przy maksymalnym uniesieniu brwi nie stwierdzono zmarszczek lub oceniano je jako łagodne do 150. dnia w badaniu 191622-142 (21,6% pacjentów leczonych produktem leczniczym BOTOX w porównaniu z 0% pacjentów leczonych placebo) oraz do 180. dnia w badaniu 191622-143 (6,8% pacjentów leczonych produktem leczniczym BOTOX w porównaniu z 0% pacjentów leczonych placebo).
W przypadku jednoczesnego leczenia zmarszczek we wszystkich trzech częściach twarzy (badanie 191622-143) 64 jednostki produktu leczniczego BOTOX znacznie zmniejszyły nasilenie zmarszczek gładzizny czoła w okresie do 6 miesięcy (5,5% pacjentów leczonych produktem leczniczym BOTOX w porównaniu z 0% pacjentów leczonych placebo), zmarszczek typu „kurze łapki” w okresie do 6 miesięcy (3,4% pacjentów leczonych produktem leczniczym BOTOX w porównaniu z 0% pacjentów leczonych placebo) oraz zmarszczek poziomych czoła w okresie do 6 miesięcy (9,4% pacjentów leczonych produktem leczniczym BOTOX w porównaniu z 0% pacjentów leczonych placebo).
W ciągu roku, łącznie 116 i 150 pacjentów otrzymało 3 cykle leczenia produktem leczniczym BOTOX składające się z odpowiednio 40 jednostek (20 jednostek na zmarszczki poziome czoła i 20 jednostek na zmarszczki gładzizny czoła) oraz 64 jednostek (20 jednostek na zmarszczki poziome
czoła, 20 jednostek na zmarszczki gładzizny czoła i 24 jednostki na zmarszczki typu „kurze łapki”). Odsetek pacjentów uzyskujących mniejsze nasilenie zmarszczek poziomych czoła był podobny we wszystkich cyklach leczenia.
W badaniach 191622-142 i 191622-143, w pierwszorzędowym punkcie czasowym tj. 30. dnia, wykazano na podstawie kwestionariusza FLO-11, mniejsze nasilenia postrzeganych zmarszczek poziomych czoła, lepszą samoocenę wieku pacjentów i stopnia atrakcyjności u znacznie (p < 0,001) większego niż w przypadku placebo odsetka pacjentów otrzymujących 40 jednostek produktu leczniczego BOTOX (20 jednostek na zmarszczki poziome czoła i 20 jednostek na zmarszczki gładzizny czoła) oraz 64 jednostki (20 jednostek na zmarszczki poziome czoła i 20 jednostek na zmarszczki gładzizny czoła oraz 24 jednostki na zmarszczki typu „kurze łapki”).
Na podstawie kwestionariusza (ang. Facial Lines Satisfaction Questionnaire - FLSQ) ustalono, że 78,1% (150/192) pacjentów w badaniu 191622-142 oraz 62,7% (138/220) pacjentów w badaniu 191622-143 otrzymujących produkt leczniczy BOTOX w dawce 40 jednostek (20 jednostek na zmarszczki poziome czoła i 20 jednostek na zmarszczki gładzizny czoła) zgłosiło poprawę stanu emocjonalnego (definiowanych jako poczucia bycia starszym, niż się jest w rzeczywistości, negatywnej samooceny wyrazu twarzy świadczącej o zmęczeniu, znużeniu, zmartwieniu i złości)
w porównaniu z pacjentami leczonymi placebo – 19,0% (11/58) w badaniu 191622-142 oraz 18,9% (21/111) w badaniu 191622-143 w 30. dniu (p < 0,0001 w obydwu badaniach).
W tym samym kwestionariuszu FLSQ, w pierwszorzędowym punkcie czasowym tj. 60 dni, 90,2% (174/193) pacjentów w badaniu 191622-142 oraz 79,2% (175/221, 40 jednostek) pacjentów lub 86,4% (203/235, 64 jednostki) w badaniu 191622-143 zgłosiło, że byli „bardzo zadowoleni”/„przeważnie zadowoleni” ze stosowania 40 jednostek lub 64 jednostek produktu leczniczego BOTOX
w porównaniu z pacjentami leczonymi placebo (1,7% [1/58] i 3,6% [4/110] odpowiednio w badaniu 191622-142 oraz 191622-143) (p < 0,0001).
W badaniach głównych, 3,7% (22/587) pacjentów stanowiły osoby powyżej 65. roku życia. Według oceny badacza pacjenci w tej grupie wiekowej osiągali odpowiedź na leczenie produktem leczniczym BOTOX na poziomie 86,7% (13/15) (w 30. dniu) w porównaniu z odpowiedzią na leczenie placebo na poziomie 28,6% (2/7). Odsetek pacjentów w tej podgrupie leczonej produktem leczniczym BOTOX był podobny do odsetka w populacji całkowitej, znaczenie statystyczne nie zostało jednak osiągnięte
z uwagi na niewielką liczbę pacjentów.
Właściwości farmakokinetyczne
Ogólna charakterystyka substancji czynnej:
Badania dystrybucji prowadzone na szczurach wskazują na słabe rozproszenie mięśniowe znakowanego radioaktywnie J125 kompleksu neurotoksyny botulinowej typu A - w mięśniu brzuchatym łydki, a następnie szybkie metabolizowanie układowe i wydalanie z moczem. Okres półtrwania znakowanego radiologicznie materiału w mięśniu wynosił około 10 godzin. W miejscu wstrzyknięcia radioaktywność była związana z dużymi cząsteczkami białka, podczas gdy w osoczu była związana z małymi cząsteczkami, co wskazuje na szybki metabolizm układowy substratu.
W ciągu 24 godzin od podania dawki, substancja radioaktywna była usunięta wraz z moczem u 60% zwierząt. Produkt jest prawdopodobnie metabolizowany przez proteazy, a jego molekularne składniki włączane są w normalne ścieżki przemian metabolicznych.
Ze względu na rodzaj substancji czynnej, nie przeprowadzono typowych badań farmakokinetycznych: absorpcji, dystrybucji, biotransformacji i eliminacji.
Dystrybucja w organizmie pacjenta:
Przedkliniczne dane o bezpieczeństwie
jednostek/kg) została podana do sterczowej części cewki moczowej i proksymalnej części odbytnicy, pęcherzyka nasiennego i ściany pęcherza moczowego lub do macicy u małp (ok. 3 jednostki/kg) bez niepożądanych skutków klinicznych. W trwającym 9 miesięcy badaniu
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Fiolkę produktu leczniczego BOTOX zawierającą 100 jednostek poddać rekonstytucji, stosując 10 ml 9 mg/ml (0,9%) jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
Zakończyć rekonstytucję, pobierając 10 ml roztworu z fiolki do strzykawki o pojemności 10 ml.
W ten sposób uzyskuje się ogółem 100 jednostek produktu leczniczego BOTOX w strzykawce 10 ml. Wykorzystać natychmiast po rekonstytucji. Resztki niewykorzystanego roztworu soli fizjologicznej należy usunąć.
Instrukcja rozcieńczania w przypadku fiolek zawierających 200 jednostek
Fiolkę produktu leczniczego BOTOX zawierającą 200 jednostek poddać rekonstytucji, stosując 8ml 9mg/ml (0,9%) jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
Do strzykawki o pojemności 10 ml pobrać 4 ml roztworu z fiolki.
Zakończyć rekonstytucję, pobierając do strzykawki 6 ml 0,9% jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
W ten sposób uzyskuje się ogółem 100 jednostek produktu leczniczego BOTOX w strzykawce 10 ml. Wykorzystać natychmiast po rekonstytucji. Resztki niewykorzystanego roztworu soli fizjologicznej należy usunąć.
Nietrzymanie moczu wskutek neurogennej nadczynności mięśnia wypieracza
W przypadku tego wskazania zaleca się stosowanie fiolek zawierających 100 lub 200 jednostek produktu leczniczego BOTOX ze względu na łatwiejszą rekonstytucję.
Instrukcja rozcieńczania w przypadku fiolek zawierających 100 jednostek we wskazaniu nietrzymanie moczu wskutek neurogennej nadczynności mięśnia wypieracza:
Każdą z 2 fiolek produktu leczniczego BOTOX zawierających po 100 jednostek poddać rekonstytucji, stosując 6 ml 9 mg/ml (0,9%) jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) na każdą fiolkę i delikatnie wymieszać fiolki.
Do każdej z dwóch strzykawek o pojemności 10 ml pobrać 4 ml z każdej fiolki.
Do trzeciej strzykawki o pojemności 10 ml pobrać pozostałe 2 ml z obu fiolek.
Zakończyć rekonstytucję, pobierając do każdej strzykawki po 6 ml 0,9% jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
W ten sposób uzyskuje się ogółem 200 jednostek produktu leczniczego BOTOX po rekonstytucji
w 3 strzykawkach po 10 ml. Wykorzystać natychmiast po rekonstytucji. Resztki niewykorzystanego roztworu soli fizjologicznej należy usunąć.
Instrukcja rozcieńczania w przypadku fiolek zawierających 200 jednostek
Fiolkę leku BOTOX zawierającą 200 jednostek poddać rekonstytucji, stosując 6 ml 9 mg/ml (0,9%) jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
Do każdej z trzech strzykawek o pojemności 10 ml pobrać po 2 ml z fiolki.
Zakończyć rekonstytucję, pobierając do każdej z trzech strzykawek po 8 ml 0,9% jałowego roztworu soli fizjologicznej niezawierającego środków konserwujących (0,9% chlorek sodu do wstrzykiwań) i delikatnie wymieszać.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
BOTOX
100 jednostek Allergan
Proszek do sporządzania roztworu do wstrzykiwań Toxinum botulinicum typum A ad iniectabile Toksyna botulinowa typu A do wstrzykiwań
1 fiolka zawiera:
kompleks neurotoksyny Clostridium botulinum typu A (900 kD), 100 jednostek Allergan
Jedna jednostka odpowiada średniej dawce śmiertelnej (LD50), po podaniu dootrzewnowo rozpuszczonej toksyny myszom w określonych warunkach.
Jednostki są specyficzne dla produktu leczniczego BOTOX i nie są porównywalne z jednostkami innych produktów leczniczych toksyny botulinowej.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek do sporządzania roztworu do wstrzykiwań Biały proszek
Produkt leczniczy BOTOX ma wygląd białego osadu, który może być trudny do zaobserwowania na dnie fiolki
Wskazaniami do stosowania produktu BOTOX są:
Zaburzenia neurologiczne:
BOTOX jest wskazany do objawowego leczenia:
Dawkowanie
Zalecane jednostki Allergan odnoszą się do produktu BOTOX i nie są porównywalne z jednostkami innych produktów toksyny botulinowej.
Pacjenci w podeszłym wieku
Dawkowanie u pacjentów w podeszłym wieku jest takie samo, jak u młodszych, dorosłych pacjentów. Należy stosować najmniejszą zalecaną dawkę w danym wskazaniu. W przypadku wstrzykiwania kolejnych dawek zaleca się stosowanie najmniejszej skutecznej dawki produktu leczniczego, zachowując w oparciu o wskazania kliniczne możliwie najdłuższe odstępy między kolejnymi wstrzyknięciami. Ostrożność należy zachować w przypadku pacjentów o nietypowej historii
w wywiadzie oraz przyjmujących jednocześnie inne leki. Istnieją ograniczone dane kliniczne dotyczące pacjentów w wieku powyżej 65 lat po udarze przyjmujących produkt leczniczy BOTOX
z powodu leczenia spastyczności kończyny górnej i kończyny dolnej. Więcej informacji, patrz punkty 4.4, 4.8 i 5.1.
Dzieci i młodzież
Nie ustalono bezpieczeństwa i skuteczności produktu leczniczego BOTOX w innych wskazaniach niż opisane dla populacji pediatrycznej w punkcie 4.1. Nie można podać zaleceń dotyczących dawkowania w przypadku wskazań innych niż spastyczność ogniskowa u dzieci związana
z porażeniem mózgowym. Obecnie dostępne dane odnośnie wskazań opisano w punktach 4.2, 4.4, 4.8 i 5.1, jak pokazano w poniższej tabeli.
BOTOX powinien być podawany wyłącznie przez lekarzy z doświadczeniem w ocenie i leczeniu ogniskowej spastyczności u dzieci oraz w ramach zorganizowanego programu rehabilitacji.
Blefarospazm, połowiczy kurcz twarzy | 12 lat (patrz punkt 4.4 i 4.8) |
Dystonia szyjna | 12 lat (patrz punkt 4.4 i 4.8) |
Ogniskowa spastyczność u dzieci | 2 lata (patrz punkt 4.2, 4.4 i 4.8) |
Nadpotliwość pach | 12 lat (ograniczone dane u młodzieży w wieku między 12 i 17 lat, patrz punkt 4.4, 4.8 i 5.1) |
Poniższe informacje są istotne podczas rekonstytucji:
Jeśli podczas jednego zabiegu używane są różne wielkości fiolek produktu leczniczego BOTOX należy zwrócić uwagę, aby użyć właściwej ilości rozcieńczalnika, w celu uzyskania odpowiedniej liczby jednostek w 0,1 ml. Ilość rozcieńczalnika niezbędna do rekonstytucji produktu jest różna dla poszczególnych mocy. Każda strzykawka powinna być odpowiednio oznakowana.
Produkt leczniczy BOTOX poddać rekonstytucji, stosując jałowy roztwór soli fizjologicznej niezawierający środków konserwujących (0,9% chlorek sodu do wstrzykiwań).
Instrukcje dotyczące przygotowania produktu leczniczego do stosowania oraz stosowania, przenoszenia i usuwania fiolek podano w punkcie 6.6.
Sposób podawania
Szczegółowe zalecenia do poszczególnych wskazań zostały opisane poniżej.
BOTOX może być podawany jedynie przez lekarzy posiadających odpowiednie kwalifikacje i udokumentowane doświadczenie w prowadzeniu terapii i stosowaniu wymaganego sprzętu.
Ogólnie obowiązujący poziom optymalnej dawki oraz liczba miejsc wstrzyknięć do jednego mięśnia nie zostały ustalone do wszystkich wskazań. W takich przypadkach lekarz powinien opracować indywidualne schematy podawania produktu. Optymalny poziom dawki należy ustalić w wyniku prób z użyciem różnych stężeń produktu.
ZABURZENIA NEUROLOGICZNE:
Ogniskowa spastyczność kończyny dolnej u dzieci
Zalecana igła: Jałowa igła w odpowiednim rozmiarze. Długość igły należy określić na podstawie położenia i głębokości mięśni.
Wskazówki dotyczące podania: Zaleca się zlokalizowanie chorych mięśni za pomocą technik,
takich jak elektromiograficzne prowadzenie igły, stymulacja nerwów lub ultrasonografia. Przed wstrzyknięciem można zastosować znieczulenie miejscowe lub znieczulenie miejscowe w połączeniu z minimalną lub umiarkowaną sedacją, zgodnie z lokalną praktyką. Nie oceniano bezpieczeństwa i skuteczności produktu leczniczego BOTOX w leczeniu spastyczności u dzieci, w znieczuleniu ogólnym lub głębokiej sedacji / analgezji. Poniższy diagram wskazuje miejsca iniekcji w spastyczności kończyny dolnej u dzieci.
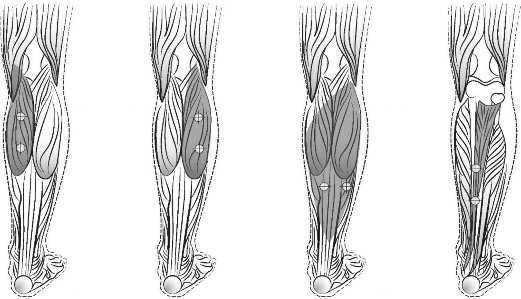
Głowa przyśrodkowa | Głowa boczna | Mięsień | Mięsień piszczelowy |
mięśnia brzuchatego łydki | mięśnia brzuchatego | płaszczkowaty | tylny |
łydki |
Zalecana dawka: Zalecana dawka w leczeniu spastyczności kończyny dolnej u dzieci wynosi 4 jednostki/kg do 8 jednostek/kg masy ciała i jest dzielona na mięśnie zajęte procesem chorobowym.
Dawkowanie produktu BOTOX w zależności od mięśnia w leczeniu spastyczności kończyny dolnej u dzieci:
Mięsień | BOTOX 4 j./kg* (maksymalna ilość jednostek na mięsień) | BOTOX 8 j./kg** (maksymalna ilość jednostek na mięsień) | Liczba ostrzyki wanych miejsc |
Obowiązkowe mięśnie stawu skokowego Głowa przyśrodkowa m. brzuchatego łydki | 1 j./kg (37.5 j.) | 2 j./kg (75 j.) | 2 |
Głowa boczna m. brzuchatego łydki | 1 j./kg (37.5 j.) | 2 j./kg (75 j.) | 2 |
Płaszczkowaty | 1 j./kg (37.5 j.) | 2 j./kg (75 j.) | 2 |
Piszczelowy tylny | 1 j./kg (37.5 j.) | 2 j./kg (75 j.) | 2 |
* nie przekraczać całkowitej dawki 150 jednostek
** nie przekraczać całkowitej dawki 300 jednostek
Maksymalna dawka całkowita: Całkowita dawka produktu leczniczego BOTOX,
podawana podczas jednej sesji terapeutycznej dla kończyny dolnej nie powinna przekraczać 8 jednostek/kg masy ciała lub 300 jednostek – w zależności, która dawka jest niższa. Jeżeli lekarz prowadzący uważa to za właściwe, u pacjenta należy rozważyć ponowne wstrzyknięcie, gdy efekt kliniczny poprzedniego wstrzyknięcia zmniejszył się, ale nie wcześniej niż 12 tygodni po poprzedniej iniekcji. W przypadku leczenia
obu kończyn dolnych, dawka całkowita nie powinna przekraczać niższej z następujących wartości: 10 jednostek/kg masy ciała lub 340 jednostek, w odstępie 12 tygodni.
Informacje dodatkowe: Leczenie produktem leczniczym BOTOX nie ma na celu
zastąpienia standardowych schematów leczenia rehabilitacyjnego. Kliniczna poprawa następuje zazwyczaj w ciągu pierwszych dwóch tygodni po wstrzyknięciu produktu. Kolejne dawki powinny być podane po ustąpieniu poprawy klinicznej od ostatniej dawki, jednak nie wcześniej niż po trzech miesiącach.
Ogniskowa spastyczność kończyny górnej u pacjentów dorosłych po udarze
Zalecana igła: Jałowa igła o rozmiarze 25, 27 lub 30 Ga. Długość igły należy dobrać w oparciu o głębokość i umiejscowienie mięśni.
Wskazówki dotyczące podania: W lokalizacji zajętych mięśni może być przydatna kontrola
EMG, techniki stymulacji nerwów lub techniki ultrasonograficzne. Dzięki licznym miejscom wstrzyknięcia, uzyskuje się równomierny kontakt produktu BOTOX
z unerwionymi obszarami mięśnia, co jest szczególnie ważne w przypadku większych mięśni.
Na poniższej rycinie przedstawiono miejsca wstrzyknięć w przypadku spastyczności kończyny górnej u pacjentów dorosłych:
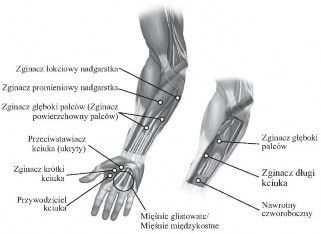
Zalecana dawka: Zalecana dawka w leczeniu spastyczności kończyny górnej
u pacjentów dorosłych wynosi do 240 jednostek podzielonych pomiędzy leczone mięśnie zgodnie z poniższą tabelą.
Maksymalna dawka podczas jednego zabiegu wynosi 240 jednostek.
Dokładna dawka i liczba miejsc wstrzyknięć powinna być ustalona indywidualnie w zależności od wielkości, liczby
i lokalizacji zaatakowanych mięśni, nasilenia spastyczności, miejscowego osłabienia siły mięśniowej oraz odpowiedzi pacjenta na wcześniejsze leczenie.
Mięsień | Zalecana dawka; liczba miejsc |
Przedramię Nawrotny czworoboczny | 10–50 jednostek; 1 miejsce |
Nadgarstek Zginacz promieniowy nadgarstka Zginacz łokciowy nadgarstka | 15–60 jednostek; 1–2 miejsca 10–50 jednostek; 1–2 miejsca |
Palce/dłoń Zginacz głęboki palców Zginacz głęboki/powierzchowny palców Mięśnie glistowate* Mięśnie międzykostne* | 15–50 jednostek; 1–2 miejsca 15–50 jednostek; 1–2 miejsca 5–10 jednostek; 1 miejsce 5–10 jednostek; 1 miejsce |
Kciuk Przywodziciel kciuka Zginacz długi kciuka Zginacz krótki kciuka Przeciwstawiacz kciuka | 20 jednostek; 1–2 miejsca 20 jednostek; 1–2 miejsca 5–25 jednostek; 1 miejsce 5–25 jednostek; 1 miejsce |
*W przypadku wstrzyknięć do mięśni glistowatych i (lub) międzykostnych zalecana dawka maksymalna wynosi 50 jednostek na dłoń.
Informacje dodatkowe: W kontrolowanych badaniach klinicznych pacjenci byli
obserwowani przez 12 tygodni po pojedynczym zabiegu. Poprawa kliniczna w zakresie napięcia mięśniowego występuje w okresie 2 tygodni, a maksymalny skutek uzyskuje się po 4 - 6 tygodniach. W otwartym badaniu, będącym kontynuacją poprzedniego badania, większości pacjentom ponownie wstrzyknięto produkt po przerwie 12 do 16 tygodni, gdy napięcie mięśniowe uległo zmniejszeniu.
Pacjenci ci otrzymali do 4 wstrzyknięć o maksymalnej skumulowanej dawce 960 jednostek w ciągu 54 tygodni. Leczenie może być powtórzone po zaniknięciu efektu klinicznego działania toksyny, jeżeli w opinii lekarza prowadzącego jest to zasadne. Nie należy podawać kolejnych wstrzyknięć przed upływem 12 tygodni od ostatniego wstrzyknięcia. Stopień i rodzaj spastyczności mięśni może wymagać dopasowania dawek produktu BOTOX w czasie ponownych wstrzykiwań. Należy stosować najmniejsze skuteczne dawki.
Ogniskowa spastyczność kończyn dolnych u pacjentów dorosłych po udarze
Zalecana igła: Jałowa igła o rozmiarze 25, 27 lub 30 Ga. Długość igły należy dobrać w oparciu o głębokość i umiejscowienie mięśni.
Wskazówki dotyczące podania: W lokalizacji zajętych mięśni może być przydatna kontrola
EMG lub techniki stymulacji nerwów. Dzięki licznym miejscom wstrzyknięcia, uzyskuje się równomierny kontakt
produktu BOTOX z unerwionymi obszarami mięśnia, co jest szczególnie ważne w przypadku większych mięśni.
Poniższe ryciny przedstawiają miejsca wstrzyknięć.
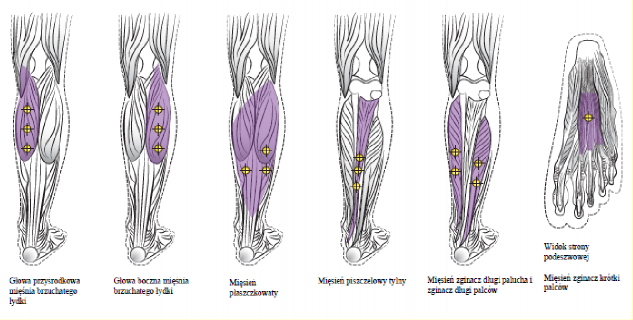
Zalecana dawka: Dawkowanie produktu leczniczego BOTOX w leczeniu kończyny dolnej u dorosłych z wyszczególnieniem mięśni:
Mięsień | Całkowita dawka; liczba miejsc |
Brzuchaty łydki | |
Głowa przyśrodkowa | 75 jednostek; 3 miejsca |
Głowa boczna | 75 jednostek; 3 miejsca |
Płaszczkowaty Piszczelowy tylny Zginacz długi palucha Zginacz długi palców Zginacz krótki palców | 75 jednostek; 3 miejsca 75 jednostek; 3 miejsca 50 jednostek; 2 miejsca 50 jednostek; 2 miejsca 25 jednostek;1 miejsce |
Zalecana dawka w leczeniu spastyczności kończyny dolnej u dorosłych, obejmującej staw skokowy i stopę, wynosi od
300 jednostek do 400 jednostek podzielonych na maksymalnie do 6 mięśni, zgodnie z powyższą tabelą. Maksymalna zalecana dawka podczas jednego zabiegu wynosi 400 jednostek.
Informacje dodatkowe: Leczenie może być powtórzone po zaniknięciu skutku
działania toksyny, jeżeli w opinii lekarza prowadzącego jest to zasadne. Nie należy wstrzykiwać kolejnych dawek przed upływem 12 tygodni.
Kurcz powiek (blefarospazm)/połowiczy kurcz twarzy
Zalecana igła: Jałowa igła o rozmiarze 27 - 30 Ga (0,40 – 0,30 mm)
Wskazówki dotyczące podania: Nie jest konieczne podawanie toksyny pod kontrolą
elektromiograficzną.
Zalecana dawka: Zalecaną wstępną dawką jest 1,25 - 2,5 jednostek podawanych do przyśrodkowej i bocznej części mięśnia okrężnego oczu powieki górnej i bocznej części mięśnia okrężnego oczu powieki dolnej. Niekiedy dodatkowo podaje się toksynę
w okolice brwi i mięśni górnej części twarzy, jeśli ich skurcze utrudniają patrzenie.
Dawka całkowita: Początkowa dawka nie powinna przekraczać 25 jednostek na jedno oko. W leczeniu kurczu powiek (blefarospazm) całkowita dawka podana w ciągu 12 tygodni nie powinna przekraczać 100 jednostek.
Informacje dodatkowe: W celu zmniejszenia ryzyka opadania powieki należy unikać
wstrzyknięć w pobliżu mięśnia dźwigacza powieki górnej. Aby zredukować występowanie komplikacji podwójnego widzenia należy unikać wstrzyknięć w przyśrodkową część dolnej powieki, w skutek czego zmniejsza się zewnętrzne skośne rozprowadzenie toksyny. Poniższe ryciny wskazują możliwe miejsca wstrzyknięcia produktu:
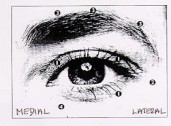
Przyśrodkowy boczny Przyśrodkowy boczny
Zazwyczaj pierwszy wynik działania toksyny jest zauważalny w ciągu trzech dni, zaś maksymalny skutek działania uzyskuje się w ciągu tygodnia lub dwóch po wstrzyknięciu. Uzyskany skutek utrzymuje się przez około 3 miesiące, po których można powtarzać leczenie. Przy kolejnych wstrzyknięciach dawkę można zwiększyć nawet dwukrotnie, jeżeli skuteczność pierwszego wstrzyknięcia została uznana za niewystarczającą. Wydaje się jednak, że nie uzyskuje się większego skutku po podaniu dawek większych niż 5 jednostek w jedno miejsce. Zazwyczaj nie uzyskuje się większej skuteczności przez podawanie produktu leczniczego częściej, niż co trzy miesiące.
Pacjenci z połowiczym kurczem twarzy lub zaburzeniami nerwu VII powinni być leczeni tak jak z jednostronnym kurczem powiek (blefarospazm), z dodatkowymi wstrzyknięciami w inne zaatakowane mięśnie twarzy zgodnie z wymaganiami.
Dystonia szyjna
Zalecana igła: Jałowa igła o rozmiarze 25 - 30 Ga (0,5 – 0,3 mm)
Wskazówki dotyczące podania: W badaniach klinicznych leczenie dystonii szyjnej zazwyczaj
obejmowało wstrzykiwanie produktu leczniczego BOTOX w mięśnie: sternocleidomastoideus, levator scapulae,
scalenus, splenius capitis, semispinalis, longissimus i (lub) trapezius. W razie konieczności leczeniu mogą być poddane również inne mięśnie odpowiedzialne za kontrolę pozycji głowy.
Masa mięśnia i stopień hipertrofii lub atrofii są czynnikami, jakie należy wziąć pod uwagę przy wyborze odpowiedniej dawki. W dystonii szyjnej rodzaj aktywności mięśnia może ulec spontanicznej zmianie bez zmian w obrazie klinicznym dystonii.
W przypadku trudności identyfikacji poszczególnych mięśni, należy dokonywać wstrzyknięć pod kontrolą EMG.
Zalecana dawka: Nie można podawać więcej niż 200 jednostek na początku leczenia. Ewentualne zmiany dawki są możliwe w kolejnych wstrzyknięciach w zależności od reakcji na leczenie.
We wstępnych kontrolowanych badaniach klinicznych dotyczących ustalenia bezpieczeństwa i skuteczności w przypadku dystonii szyjnej, dawki po rekonstytucji produktu BOTOX wahały się od 140 do 280 jednostek.
W najnowszych badaniach dawki wahały się od 95 do 360 jednostek (ze średnią 240 jednostek). Tak jak w wypadku każdego podawania produktu leczniczego, dawki początkowe u pacjenta pierwszy raz przyjmującego dany produkt powinny stanowić najmniejsze skuteczne dawki. Nie należy podawać więcej niż 50 jednostek w jedno miejsce. Nie można podawać więcej niż 100 jednostek do mięśnia sternocleidomastoideus. W celu ograniczenia przypadków zaburzeń połykania nie należy podawać toksyny do obydwu mięśni mostkowo- obojczykowo-sutkowych jednocześnie.
Dawka całkowita: Nie należy przekraczać całkowitej dawki 300 jednostek podanej podczas jednego zabiegu. Optymalna liczba miejsc wstrzyknięć zależy od wielkości mięśnia. Nie zaleca się krótszych odstępów pomiędzy wstrzyknięciami niż 10 tygodni.
Informacje dodatkowe: Kliniczna poprawa zauważalna jest zazwyczaj w ciągu
pierwszych dwóch tygodni po wstrzyknięciu produktu. Maksymalny skutek kliniczny uzyskuje się zwykle po około 6 tygodniach od wstrzyknięć. Nie zaleca się krótszych odstępów pomiędzy wstrzyknięciami niż 10 tygodni.
Skuteczność terapeutyczna, jak wykazały badania kliniczne, może utrzymywać się przez różny okres (od 2 do 33 tygodni), zazwyczaj jednak około 12 tygodni.
Przewlekła migrena
Zalecana igła: Jałowa igła o rozmiarze 30 G, 0,5 cala
Wskazówki dotyczące podania: Rozpoznanie chronicznej migreny, a także podanie produktu
leczniczego BOTOX powinno odbywać się wyłącznie pod nadzorem lekarza neurologa, specjalisty z zakresu leczenia migreny.
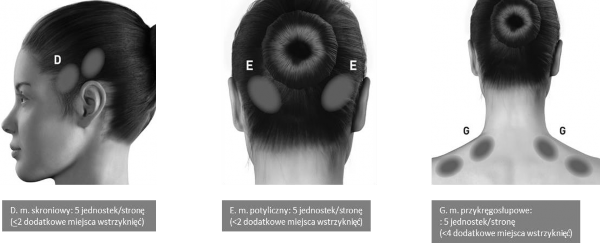
Wstrzyknięć dokonuje się w mięśnie 7 okolic głowy/szyi, zgodnie z rycinami i tabelą zamieszczoną poniżej.
U pacjentów o bardzo grubych mięśniach szyi może zachodzić potrzeba użycia igły 1-calowej. Z wyjątkiem mięśnia podłużnego, do którego produkt leczniczy należy wstrzyknąć w jedno miejsce (w linii środkowej mięśnia), wszystkie mięśnie należy ostrzyknąć obustronnie. Połowę zastrzyków należy podać w mięśnie po lewej, a drugą połowę w mięśnie po prawej stronie głowy i szyi. Jeśli ból dominuje w określonej okolicy/ach, można wykonać dodatkowe wstrzyknięcia po jednej lub obu stronach do 3 określonych grup mięśni (potylicznego, skroniowego i czworobocznego); do każdego mięśnia można wstrzyknąć dawkę maksymalną, podaną w poniższej tabeli.
Poniższe ryciny przedstawiają miejsca wstrzyknięć:
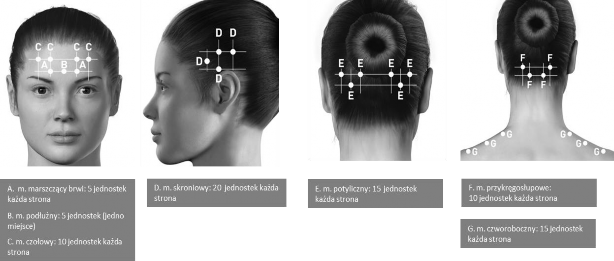
Poniższe ryciny przedstawiają zalecane grupy mięśni do dodatkowych, alternatywnych miejsc wstrzyknięć:
Zalecana dawka: W leczeniu przewlekłej migreny zaleca się od 155 do 195 jednostek (j.) po rekonstytucji produktu leczniczego BOTOX wstrzykiwanego domięśniowo (im.) po 0,1 ml (5 j.) do 31 maksymalnie 39 miejsc.
Dawkowanie produktu leczniczego BOTOX u pacjentów z przewlekłą migreną z wyszczególnieniem mięśni:
Zalecana dawka | |
Mięśnie głowy/ szyi | Dawka całkowita (liczba miejsc a) |
Mięsień marszczący brwib | 10 jednostek(2 miejsca) |
Podłużny | 5 jednostek(1 miejsce) |
Czołowyb | 20 jednostek(4 miejsca) |
Skroniowyb | 40 jednostek(8 miejsc) do 50 jednostek(do 10 miejsc) |
Potylicznyb | 30 jednostek(6 miejsc) do 40 jednostek(do 8 miejsc) |
Grupa mięśni przykręgosłupowychb | 20 jednostek(4 miejsca) |
Czworobocznyb | 30 jednostek(6 miejsc) do 50 jednostek(do 10 miejsc) |
Całkowity zakres dawkowania: | 155 jednostek do 195 jednostek 31 do 39 miejsc |
a 1 wstrzyknięcie im. = 0,1 ml = 5 jednostek produktu leczniczego BOTOX
b Dawka wstrzykiwana obustronnie
Informacje dodatkowe: Zaleca się powtarzanie leczenia co 12 tygodni.
ZABURZENIA CZYNNOŚCI PĘCHERZA MOCZOWEGO
Pacjenci nie mogą być leczeni, jeśli mają zakażenie dróg moczowych.
1-3 dni przed procedurą wstrzyknięcia, w dniu jej przeprowadzania oraz 1-3 dni po podaniu produktu leczniczego, należy stosować profilaktycznie antybiotykoterapię.
Należy zalecić pacjentom odstawienie leków przeciwpłytkowych co najmniej 3 dni przed wstrzyknięciem produktu leczniczego. Należy odpowiednio postępować w przypadku pacjentów stosujących terapię przeciwzakrzepową, aby zmniejszyć ryzyko krwawienia.
W leczeniu zaburzeń czynności pęcherza moczowego, BOTOX powinien być stosowany wyłącznie przez lekarzy, którzy posiadają odpowiednie doświadczenie w zakresie diagnozowania oraz leczenia zaburzeń czynności pęcherza moczowego (np. lekarze specjaliści w zakresie urologii
i uroginekologii).
Idiopatyczna nadreaktywność pęcherza moczowego
Zalecana igła: Może zostać użyty cystoskop elastyczny lub sztywny. Przed rozpoczęciem wstrzykiwania igłę iniekcyjną należy wypełnić ok. 1 ml produktu leczniczego (w zależności od długości igły), aby usunąć jakiekolwiek powietrze.
Wskazówki dotyczące podania: W zależności od lokalnej praktyki przed procedurą
wstrzyknięcia można zastosować dopęcherzowe wkroplenie rozcieńczonych środków znieczulających (z sedacją lub bez). W przypadku miejscowego wkraplania środków znieczulających, przed kolejnymi etapami procedury pęcherz należy odsączyć i wypłukać jałowym roztworem soli fizjologicznej.
Produkt leczniczy BOTOX po rekonstytucji (100 jednostek
w 10 ml) wstrzykiwać do mięśnia wypieracza przez cystoskop elastyczny lub sztywny, omijając trójkąt i dno pęcherza. Do pęcherza należy wkroplić wystarczająco dużo roztworu soli fizjologicznej, by osiągnąć odpowiednią wizualizację wstrzykiwania, ale należy unikać nadmiernego rozdęcia.
Igłę należy wprowadzać do mięśnia wypieracza na głębokość ok. 2 mm. Wstrzykiwać po 0,5 ml w 20 miejsc (całkowita objętość: 10 ml), zachowując odstępy ok. 1 cm (patrz schemat powyżej). Aby pełna dawka została podana, w ostatnim wstrzyknięciu należy podać ok. 1 ml sterylnego roztworu soli fizjologicznej. Po wykonaniu wstrzyknięć nie należy odsączać roztworu soli użytego w celu zwizualizowania ściany pęcherza, aby pacjent mógł zademonstrować zdolność do oddawania moczu przed opuszczeniem kliniki. Pacjenta należy obserwować przez co najmniej 30 minut po zakończeniu wstrzykiwań i do wystąpienia spontanicznego oddania moczu.
Miejsca wstrzyknięć
Ujście lewego moczowodu
Trójkąt pęcherza
moczowego
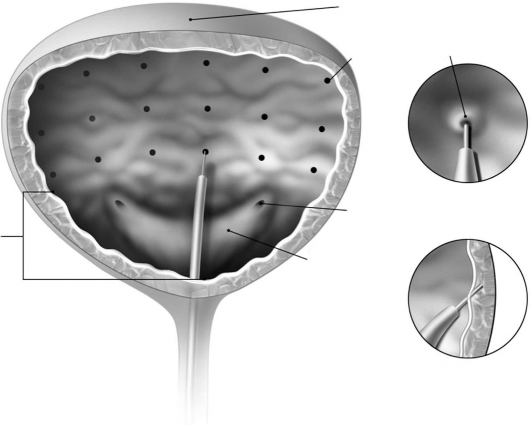
Szczyt pęcherza moczowego
Dno pęcherza moczowego
Zalecana dawka: Zalecana dawka to 100 jednostek produktu leczniczego BOTOX wstrzykiwanego po 0,5 ml (5 jednostek) w 20 miejsc w obrębie mięśnia wypieracza.
Informacje dodatkowe: Poprawa kliniczna na ogół występuje w ciągu 2 tygodni.
Należy rozważyć ponowne podanie produktu leczniczego, gdy skutek kliniczny wcześniejszego wstrzyknięcia uległ zmniejszeniu (w badaniach klinicznych III fazy mediana
czasu trwania w oparciu o ilość pacjentów zgłaszających się do ponownego leczenia, wyniosła 166 dni (ok. 24 tygodnie)), ale nie wcześniej niż po upływie 3 miesięcy od poprzedniego podania produktu leczniczego do pęcherza.
Nietrzymanie moczu u pacjentów z nadreaktywnością mięśnia wypieracza pęcherza moczowego
Zalecana igła: Może zostać użyty cystoskop elastyczny lub sztywny. Przed rozpoczęciem wstrzykiwania igłę iniekcyjną należy wypełnić ok. 1 ml produktu leczniczego (w zależności od długości igły), aby usunąć jakiekolwiek powietrze.
Wskazówki dotyczące podania: W zależności od lokalnej praktyki przed procedurą
wstrzyknięcia można zastosować dopęcherzowe wkroplenie rozcieńczonych środków znieczulających (z sedacją lub bez) lub znieczulenie ogólne. W przypadku miejscowego wkraplania środków znieczulających, przed kolejnymi etapami procedury pęcherz należy odsączyć i wypłukać jałowym roztworem soli fizjologicznej.
BOTOX po rekonstytucji (200 jednostek w 30 ml) wstrzykiwać do mięśnia wypieracza przez cystoskop elastyczny lub sztywny, omijając trójkąt i dno pęcherza. Do pęcherza należy wkroplić wystarczająco dużo roztworu soli fizjologicznej, by osiągnąć odpowiednią wizualizację wstrzykiwania, ale należy unikać nadmiernego rozdęcia.
Igłę należy wprowadzać do mięśnia wypieracza na głębokość ok. 2 mm. Wstrzykiwać po 1 ml w 30 miejsc (całkowita objętość: 30 ml), zachowując odstępy ok. 1 cm (patrz schemat poniżej). Aby pełna dawka została podana, w ostatnim wstrzyknięciu należy podać ok. 1 ml sterylnego roztworu soli fizjologicznej. Po wykonaniu wstrzyknięć należy odsączyć roztwór soli użyty w celu zwizualizowania ściany pęcherza.
Pacjenta należy obserwować przez co najmniej 30 minut po zakończeniu wstrzykiwań.
Zalecana dawka: Zalecana dawka to 200 jednostek produktu leczniczego BOTOX wstrzykiwanego po 1 ml (ok. 6,7 jednostki)
w 30 miejsc w obrębie mięśnia wypieracza.
Informacje dodatkowe: Poprawa kliniczna na ogół występuje w ciągu 2 tygodni.
Należy rozważyć ponowne podanie produktu leczniczego, gdy efekt kliniczny wcześniejszego wstrzyknięcia uległ zmniejszeniu (w badaniach klinicznych III fazy mediana czasu trwania w oparciu o ilość pacjentów zgłaszających się do ponownego leczenia, wyniosła 256–295 dni (~ 36-42 tygodni) w przypadku podania 200 jednostek produktu leczniczego BOTOX), ale nie wcześniej niż po upływie
3 miesięcy od poprzedniego podania produktu leczniczego do pęcherza.
ZABURZENIA SKÓRY I JEJ PRZYDATKÓW
Pierwotna nadpotliwość pach
Zalecana igła: Jałowa igła o rozmiarze 30 Ga
Wskazówki dotyczące podania: Obszar nadpotliwości określa się używając technik
barwieniowych np. testu jodynowo-skrobiowego Minora.
Zalecana dawka: 50 jednostek produktu BOTOX wstrzykiwane jest śródskórnie w wielu, równo rozmieszczonych punktach usytuowanych
w odległości około 1-2 cm od siebie, w obrębie obszaru nadpotliwości każdej pachy.
Dawka całkowita: Stosowanie dawek innych niż 50 jednostek podawanych do jednej pachy nie jest zalecane. Wstrzyknięcia nie powinny być stosowane częściej niż co 16 tygodni (patrz punkt 5.1).
Informacje dodatkowe: Zazwyczaj kliniczną poprawę obserwuje się w ciągu
pierwszego tygodnia po podaniu produktu. Kolejne wstrzyknięcia produktu BOTOX mogą być wykonane, kiedy obserwuje się zmniejszanie skutku klinicznego po wcześniejszym wstrzyknięciu oraz gdy lekarz prowadzący uzna je za konieczne.
Linie gładzizny czoła
Zalecana igła: Jałowa igła o rozmiarze 30 Ga
Wskazówki dotyczące podania: Aby zapobiec powikłaniu w postaci opadania powieki, należy
unikać wykonywania wstrzyknięć w okolicę mięśnia dźwigacza powieki górnej. Dotyczy to w sposób szczególny pacjentów z nasilonym zespołem opadania brwi.
Wstrzyknięcia w przyśrodkowe części mięśni marszczących brwi powinny być wykonywane co najmniej 1 cm powyżej części kostnych brzegu nadoczodołowego.
Podczas wstrzykiwania produktu leczniczego BOTOX w pionowe zmarszczki między brwiami widoczne przy
maksymalnym zmarszczeniu brwi (tzw. zmarszczki gładzizny czoła) należy zachować szczególną ostrożność, aby uniknąć wstrzyknięć w naczynia krwionośne, patrz punkt 4.4.
Ryc. 1.
Zalecana dawka: W pięć miejsc podaje się po 0,1 ml (4 jednostki) produktu.
Wykonuje się po dwa wstrzyknięcia w każdy z mięśni marszczących brwi i jedno w mięsień podłużny nosa, podając łącznie 20 jednostek produktu.
Informacje dodatkowe: Poprawa w zakresie wyglądu linii gładzizny czoła zwykle jest
zauważalna w ciągu tygodnia po leczeniu. Skutek terapii utrzymuje się przez okres do 4 miesięcy. Odstępy pomiędzy sesjami terapeutycznymi nie powinny być krótsze niż
3 miesiące.
Zmarszczki typu „kurze łapki”
Zalecana igła: Jałowa igła o rozmiarze 30 Ga
Wskazówki dotyczące podania: Aby ograniczyć ryzyko opadania powieki, nie należy
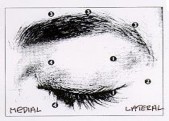
przekraczać maksymalnej, zalecanej dawki 4 jednostek produktu w każde miejsce wstrzyknięcia, ani zalecanej liczby miejsc wstrzyknięć. Ponadto wstrzyknięć należy dokonywać skroniowo w stosunku do krawędzi oczodołu, zachowując tym samym bezpieczną odległość do mięśni odpowiedzialnych za unoszenie powieki.
Podczas wstrzyknięcia igła powinna być ustawiona ukośnie w górę i skierowana w stronę przeciwną do oka. Pierwsza iniekcja (A) powinna być podana około 1,5 do 2,0 cm skroniowo w stosunku do bocznego kąta oka i bezpośrednio
skroniowo w stosunku do krawędzi oczodołu. Kolejne miejsca iniekcji zależeć będą od tego, czy zmarszczki znajdują się powyżej czy poniżej bocznego kąta oka (patrz Ryc. 2.), bądź też głównie poniżej bocznego kąta oka (patrz Ryc. 3.).
Podczas wstrzykiwania produktu leczniczego BOTOX w zmarszczki w okolicy bocznego kąta oka, widoczne podczas pełnego uśmiechu, należy zachować szczególną
ostrożność, aby uniknąć wstrzyknięć w naczynia krwionośne, patrz punkt 4.4.
Ryc. 2. Ryc. 3.
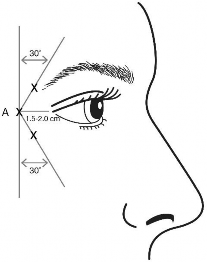
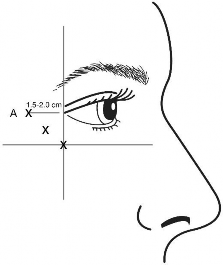
Zalecana dawka: W każde z 3 miejsc wstrzyknięć po każdej ze stron (łącznie 6 miejsc iniekcji) należy podać 0,1 ml (4 j.) produktu
w obrębie bocznej części mięśnia okrężnego oka, podając łącznie 24 jednostki w całkowitej objętości 0,6 ml
(12 jednostek po każdej ze stron).
W przypadku jednoczesnego leczenia zmarszczek gładzizny czoła widocznych podczas maksymalnego zmarszczenia brwi, dawka dla zmarszczek w okolicy bocznych kątów oczu, widocznych przy pełnym uśmiechu wynosi 24 jednostki oraz 20 jednostek dla zmarszczek gładzizny czołowej (patrz zalecenia dotyczące podawania w przypadku zmarszczek gładzizny czołowej), dając łączną dawkę 44 jednostek
w całkowitej objętości 1,1 ml.
Informacje dodatkowe: Zmniejszenie nasilenia zmarszczek typu „kurze łapki”
widocznych podczas pełnego uśmiechu, według oceny badacza, następowało w ciągu pierwszego tygodnia leczenia. Mediana czasu utrzymywania się działania terapeutycznego wynosiła 4 miesiące.
Odstępy pomiędzy sesjami terapeutycznymi nie powinny być krótsze niż 3 miesiące.
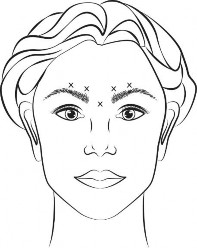
Zmarszczki poziome czoła widoczne przy maksymalnym uniesieniu brwi
Zalecana igła: Jałowa igła o rozmiarze 30 Ga
Wskazówki dotyczące podania: Aby określić odpowiednie miejsca wstrzyknięcia do mięśnia
czołowego, należy ocenić ogólną zależność pomiędzy rozmiarem czoła pacjenta a rozkładem aktywności mięśnia czołowego.
Za pomocą delikatnego badania palpacyjnego czoła przy rozluźnionych mięśniach oraz podczas maksymalnego uniesienia brwi należy zlokalizować następujące poziome linie leczenia:
Ryc. 4.
BOTOX jest przeciwwskazany:
Nie należy przekraczać zalecanych dawek ani częstości podawania produktu leczniczego BOTOX z uwagi na ryzyko przedawkowania, nadmiernego osłabienia mięśni, rozprzestrzeniania się toksyny w miejsca odległe od miejsca podania oraz powstania przeciwciał neutralizujących.
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na fiolkę, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
Dawkowanie początkowe u pacjentów dotychczas nieleczonych należy rozpocząć od najmniejszej dawki zalecanej w poszczególnych wskazaniach.
Zarówno lekarze przepisujący produkt, jak i pacjenci powinni być świadomi, że działania niepożądane mogą wystąpić mimo dobrej tolerancji wcześniejszych wstrzyknięć. Należy zatem zachować ostrożność za każdym razem kiedy produkt jest podawany.
Donoszono o występowaniu działań niepożądanych związanych z rozprzestrzenianiem się toksyny w miejsca odległe od miejsca podania (patrz: punkt 4.8). Niekiedy powodowały one śmierć pacjenta, w niektórych przypadkach związaną z utrudnieniem połykania (dysfagią), zapaleniem płuc i (lub) znacznym osłabieniem. Objawy te są zgodne z mechanizmem działania toksyny botulinowej, a ich występowanie stwierdzano w kilka godzin do kilku tygodni po podaniu. Ryzyko wystąpienia w/w objawów jest prawdopodobnie większe w przypadku pacjentów z chorobami współistniejącymi oraz zaburzeniami, które mogą predysponować do ich wystąpienia, w tym również u dzieci i dorosłych
leczonych z powodu spastyczności oraz w przypadku pacjentów otrzymujących duże dawki produktu leczniczego.
U pacjentów leczonych terapeutycznymi dawkami także może wystąpić nadmierne osłabienie mięśni.
Należy zachować ostrożność stosując produkt u pacjentów w podeszłym wieku oraz u pacjentów znacznie osłabionych. Zasadniczo, badania kliniczne z udziałem produktu leczniczego BOTOX nie wykazały różnic w odpowiedzi pomiędzy pacjentami w podeszłym wieku w porównaniu z pacjentami młodszymi. Należy ostrożnie ustalać dawkowanie u pacjentów w podeszłym wieku, zaczynając od dolnej granicy zakresu dawkowania.
Lekarz powinien decydować o podjęciu leczenia, w oparciu o stosunek korzyści do ryzyka, indywidualnie dla każdego pacjenta.
Donoszono o występowaniu zaburzeń połykania również wówczas, gdy produkt podawany był w inne miejsca, niż mięśnie szyi (patrz punkt 4.4. „Dystonia szyjna”).
U pacjentów z subklinicznymi lub klinicznymi objawami nieprawidłowości przewodzenia nerwowo- mięśniowego, jak myasthenia gravis czy zespół Lamberta-Eatona u pacjentów z obwodowymi neuropatiami ruchowymi (np. stwardnienie zanikowe boczne albo neuropatia ruchowa) produkt BOTOX powinien być stosowany z dużą ostrożnością i pod ścisłą kontrolą. Może u nich występować nadmierna wrażliwość na takie czynniki, jak toksyna botulinowa, co może prowadzić do nadmiernego osłabienia mięśni, zaś ryzyko wystąpienia klinicznie istotnych układowych działań niepożądanych
(w tym ciężkiej dysfagii i zaburzeń układu oddechowego) może być zwiększone nawet w wyniku zastosowania typowych dawek tego produktu. U takich pacjentów produkt powinien być stosowany pod ścisłą kontrolą lekarza specjalisty i tylko wówczas, gdy uważa się, że korzyści z leczenia przeważają nad ryzykiem.
Pacjenci z dysfagią i zachłyśnięciem w wywiadzie powinni być leczeni z zachowaniem największej ostrożności.
Pacjenci lub udzielający pomocy powinni wezwać natychmiast pomoc medyczną, jeżeli nastąpią trudności w połykaniu, mowie lub oddychaniu.
Tak jak w przypadku każdego leczenia, które umożliwia pacjentowi o siedzącym trybie życia powrót do aktywności, należy zwrócić uwagę aby aktywność zwiększać stopniowo.
Należy zapoznać się z anatomią i ewentualnymi zmianami wynikającymi z przebytych zabiegów chirurgicznych miejsca planowanego wstrzyknięcia przed podaniem produktu BOTOX.
Obserwowano przypadki odmy związane ze wstrzyknięciem produktu BOTOX w pobliżu klatki piersiowej. Należy zachować ostrożność podczas wstrzykiwania w bezpośredniej bliskości płuc, zwłaszcza w okolice wierzchołków płuc lub innych wrażliwych struktur anatomicznych.
Ciężkie przypadki działań niepożądanych, w tym przypadki zgonów obserwowano u pacjentów, którym BOTOX wstrzyknięto poza zarejestrowanymi wskazaniami, bezpośrednio w gruczoły ślinowe, okolice ustno-językowo-gardłową, w przełyk lub żołądek. Niektórzy z opisywanych pacjentów mieli wcześniej zaburzenia połykania lub znacznego stopnia osłabienie.
W rzadkich przypadkach donoszono o występowaniu ciężkich i (lub) natychmiastowych reakcji nadwrażliwości, w tym anafilaksji, choroby posurowiczej, pokrzywki, obrzęku tkanek miękkich
i duszności. Niektóre z nich występowały po zastosowaniu produktu BOTOX w monoterapii bądź w leczeniu skojarzonym z innymi lekami, których stosowanie związane jest z występowaniem podobnych reakcji. W przypadku wystąpienia takiej reakcji należy przerwać stosowanie produktu
BOTOX i bezzwłocznie rozpocząć odpowiednie leczenie farmakologiczne, np. podawanie epinefryny. Odnotowano jeden przypadek reakcji anafilaktycznej u pacjenta, który zmarł w skutek podania nieprawidłowo rozcieńczonego produktu razem z 5 ml 1% roztworu lidokainy.
Podobnie jak w przypadku innych wstrzyknięć, może dojść do wystąpienia zaburzeń w miejscu wstrzyknięcia związanego z podaniem. Wstrzyknięcie może spowodować miejscowe zakażenie, ból, stan zapalny, parestezje, niedoczulicę, tkliwość uciskową, obrzęk, rumień i (lub) krwawienie/siniak. Ból i (lub) lęk związany z podaniem może spowodować wystąpienie reakcji wazowagalnych, np. omdleń, niedociśnienia itp.
Należy zachować ostrożność podczas podawania produktu BOTOX, gdy w miejscu proponowanego wstrzyknięcia występuje stan zapalny, a także w tych przypadkach, w których występuje znaczne osłabienie lub zanik mięśni wybranych do iniekcji. Ponadto, należy zachować ostrożność stosując BOTOX u pacjentów z obwodowymi neuropatiami ruchowymi (np. stwardnieniem zanikowym bocznym lub neuropatią ruchową).
Odnotowano przypadki działań niepożądanych dotyczących układu sercowo-naczyniowego, łącznie z arytmią i zawałem mięśnia sercowego, niektóre ze skutkiem śmiertelnym. U niektórych pacjentów stwierdzono istniejące wcześniej czynniki ryzyka, w tym istniejącą chorobę układu sercowo- naczyniowego.
Donoszono o wystąpieniu nowych lub nawrocie drgawek, zazwyczaj u pacjentów, u których występuje predyspozycja do ich występowania. Nie ustalono dokładnego związku między nimi a podawaniem produktu BOTOX. Wśród dzieci dominowały zgłoszenia u pacjentów ze spastycznością (przykurczami) w mózgowym porażeniu dziecięcym.
Wytworzone przeciwciała przeciwko toksynie botulinowej typu A mogą zmniejszać skuteczność leczenia poprzez inaktywację tej toksyny. Wyniki niektórych badań sugerują, że do powstawania przeciwciał dochodzi częściej wówczas, gdy czas pomiędzy kolejnymi dawkami produktu BOTOX jest krótki oraz gdy stosowane są duże dawki. W stosownych przypadkach, możliwość powstawania przeciwciał może być zminimalizowana poprzez podawanie najmniejszej skutecznej dawki, zachowując w oparciu o ocenę kliniczną możliwie najdłuższe odstępy między kolejnymi wstrzyknięciami.
Kliniczne nieprawidłowości powstałe podczas powtórnego stosowania produktu BOTOX (dotyczy to wszystkich toksyn botulinowych) mogą być wynikiem różnych procedur rozpuszczania zawartości fiolki, przerw między wstrzyknięciami, różnych mięśni do których produkt jest wstrzykiwany
i nieznacznej różnicy mocy podanej dawki, która została określana metodą biologiczną.
Identyfikowalność
W celu poprawy identyfikowalności biologicznych produktów leczniczych, należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania ani skuteczności produktu BOTOX we wskazaniach innych, niż te, które wymieniono w punkcie 4.1, dotyczących dzieci i młodzieży. W okresie po wprowadzeniu produktu leczniczego do obrotu rzadko zgłaszano przypadki rozprzestrzeniania się toksyny w miejsca odległe od miejsca podania u dzieci i młodzieży, u których występowały choroby współistniejące, zazwyczaj dziecięce porażenie mózgowe. W zgłaszanych przypadkach zastosowana dawka zazwyczaj przekraczała dawkę zalecaną (patrz punkt 4.8).
Odnotowano rzadkie przypadki zgonów związane z zachłystowym zapaleniem płuc u dzieci z ciężkim dziecięcym porażeniem mózgowym, po zastosowaniu toksyny botulinowej, poza zarejestrowanymi
wskazaniami (off - label) (np. podanie w obszarze szyi). Należy zachować szczególną ostrożność podczas leczenia dzieci i młodzieży ze znacznym osłabieniem neurologicznym, utrudnieniem połykania (dysfagia) lub niedawno przebytym zachłystowym zapaleniem płuc lub inną chorobą płuc.
Leczenie pacjentów w złym stanie ogólnym możliwe jest tylko wówczas, jeżeli oceniono, że w przypadku danego pacjenta, potencjalne korzyści przeważają nad ryzykiem.
ZABURZENIA NEUROLOGICZNE:
Ogniskowa spastyczność stawu skokowego i stopy związana z dynamiczną deformacją stopy końsko- szpotawej u dzieci z mózgowym porażeniem dziecięcym oraz ogniskowa spastyczność stawu skokowego, stopy, nadgarstka i dłoni u pacjentów dorosłych po udarze.
Podawanie produktu BOTOX w leczeniu ogniskowej spastyczności zostało zbadane jedynie
w połączeniu ze standardowymi schematami leczenia i nie jest przeznaczone do zastąpienia tych procedur. BOTOX prawdopodobnie nie jest skuteczny w poprawie zakresu ruchu stawów zmienionych w wyniku stałego przykurczu.
Produkt leczniczy BOTOX nie powinien być stosowany w leczeniu ogniskowej spastyczności kończyny górnej (dłoni i nadgarstka) oraz kończyny dolnej (stawu skokowego i stopy) u dorosłych pacjentów po udarze, jeżeli nie oczekuje się aby zmniejszenie napięcia mięśni mogło spowodować poprawę czynności (np. poprawę chodu), lub poprawę objawów podmiotowych (np. zmniejszenie bólu) lub ułatwienie opieki nad pacjentem. W przypadku spastyczności kończyny dolnej poprawa w zakresie zaburzeń czynności stawu skokowego może być ograniczona jeżeli leczenie produktem leczniczym BOTOX rozpoczęto po okresie dłuższym niż 2 latach od udaru, lub u pacjentów ze spastycznością stawu skokowego o mniejszej ciężkości (Zmodyfikowana Skala Ashwortha (MAS)
<3).
Należy zachować ostrożność podczas leczenia dorosłych pacjentów ze spastycznością poudarową, z uwagi na większe ryzyko przewrócenia się pacjenta.
Po wprowadzeniu produktu do obrotu donoszono o przypadkach śmierci po podaniu toksyny botulinowej (w niektórych przypadkach związanych z zachłystowym zapaleniem płuc) związanych z rozprzestrzenianiem się toksyny w miejsca odległe od miejsca podania u dzieci z chorobami współistniejącymi, głównie porażeniem mózgowym.
Kurcz powiek (Blefarospazm)
Zmniejszenie częstości mrugania występujące po wstrzyknięciu toksyny botulinowej do mięśnia okrężnego oka, może prowadzić do patologicznych zmian rogówki. Należy dokładnie sprawdzać wrażliwość rogówki oka po podaniu produktu oraz unikać wstrzyknięć w dolną powiekę, aby nie doprowadzić do wywinięcia powieki, a w przypadku pojawienia się uszkodzeń nabłonka rogówki, należy je intensywnie leczyć. Leczenie może wymagać podania kropli ochronnych, maści, miękkich soczewek terapeutycznych, przymknięcia oka opaską lub innych zabiegów.
W miękkich tkankach powieki łatwo występują wybroczyny. Można temu zapobiegać przez łagodny ucisk w miejscu wstrzyknięcia.
Ze względu na antycholinergiczne działanie toksyny botulinowej, należy zachować szczególną ostrożność u pacjentów z ryzykiem jaskry z zamkniętym kątem przesączania.
Dystonia szyjna
Pacjentów z dystonią szyjną należy poinformować o możliwości wystąpienia zaburzenia połykania (dysfagii), które może być łagodne lub bardzo silne. Trudności w połykaniu mogą utrzymywać się przez dwa do trzech tygodni po wstrzyknięciu toksyny, ale były zgłaszane przypadki dysfagii utrzymujące się do pięciu miesięcy po wstrzyknięciu. W konsekwencji zaburzenia połykania może wystąpić duszność, aspiracja i czasami potrzeba karmienia przez zgłębnik. W rzadkich przypadkach donoszono o występowaniu zapalenia płuc a nawet przypadków śmiertelnych po aspiracji w przebiegu dysfagii.
Ograniczenie dawki toksyny botulinowej wstrzykiwanej do mięśnia sternocleidomastoideus do wartości poniżej 100 jednostek może zmniejszyć występowanie dysfagii. Wyższe ryzyko wystąpienia dysfagii obserwowano u pacjentów o mniejszej masie mięśni szyjnych lub u pacjentów, którym obustronnie wstrzyknięto toksynę w mięsień sternocleidomastoideus. Wystąpienie dysfagii jest przypisywane rozprzestrzenianiu toksyny w mięśniach przełyku. Podanie produktu do mięśnia dźwigacza łopatki może być związane ze zwiększonym ryzykiem wystąpienia zakażenia górnych dróg oddechowych i zaburzeń połykania.
Zaburzenia połykania mogą przyczyniać się do zmniejszenia ilości przyjmowanych płynów i pokarmów, co może prowadzić do odwodnienia i zmniejszenia masy ciała. U pacjentów
z subkliniczną dysfagią ryzyko wystąpienia cięższych zaburzeń połykania po zastosowaniu produktu BOTOX może być zwiększone.
Przewlekła migrena
Nie ustalono skuteczności w profilaktyce bólu głowy u pacjentów z migreną epizodyczną (bóle głowy
< 15 dni w miesiącu).
ZABURZENIA CZYNNOŚCI PĘCHERZA MOCZOWEGO
Podczas wykonywania cystoskopii należy stosować odpowiednie medyczne środki ostrożności.
U pacjentów niecewnikowanych należy oceniać objętość moczu zalegającego po mikcji w ciągu 2 tygodni po leczeniu oraz okresowo zgodnie z oceną lekarza przez okres do 12 tygodni. Należy poinstruować pacjentów, by skontaktowali się z lekarzem, jeśli odczują trudności w oddawaniu moczu, ponieważ może być konieczne cewnikowanie.
Idiopatyczna nadreaktywność pęcherza moczowego
Mężczyźni z idiopatyczną nadreaktywnością pęcherza moczowego i przedmiotowymi lub podmiotowymi objawami niedrożności dróg moczowych nie powinni być leczeni produktem leczniczym BOTOX.
Nietrzymanie moczu wskutek neurogennej nadczynności mięśnia wypieracza
Może wystąpić autonomiczna dysrefleksja związana z procedurą. Konieczne może być bezzwłoczne zapewnienie opieki medycznej.
ZABURZENIA SKÓRY I JEJ PRZYDATKÓW
Pierwotna nadpotliwość pach
Zaleca się przeprowadzenie wywiadu i badania pacjenta, jak również dodatkowych testów swoistych, w celu wykluczenia potencjalnych przyczyn wtórnej nadpotliwości (np. nadczynność tarczycy, guz chromochłonny). Pomoże to uniknąć leczenia objawowego nadpotliwości bez rozpoznania i (lub) leczenia choroby zasadniczej.
Zmarszczki gładzizny czoła widoczne przy maksymalnym zmarszczeniu brwi oraz zmarszczki typu
„kurze łapki” widoczne przy pełnym uśmiechu i (lub) zmarszczki poziome czoła widoczne przy maksymalnym uniesieniu brwi
Nie zaleca się stosowania produktu leczniczego BOTOX u osób w wieku poniżej 18 lat. Dane z badań klinicznych 3 fazy dotyczących stosowania BOTOX u pacjentów powyżej 65. roku życia są ograniczone.
Należy zachować ostrożność i upewnić się, że BOTOX nie zostanie podany do naczynia krwionośnego podczas iniekcji w obrębie zmarszczek gładzizny czoła widocznych podczas maksymalnego zmarszczenia brwi, zmarszczek w okolicy bocznego kąta oka widocznych przy pełnym uśmiechu oraz zmarszczek poziomych czoła widocznych przy maksymalnym uniesieniu brwi (patrz punkt 4.2).
W następstwie leczenia istnieje ryzyko wystąpienia opadania powieki. Informacje o sposobie ograniczenia ryzyka opadania powieki, patrz punkt 4.2. instrukcje dotyczące stosowania.
Teoretycznie, działanie toksyny botulinowej może być nasilone przez antybiotyki z grupy aminoglikozydów lub spektynomycyny oraz inne produkty lecznicze wpływające na przekaźnictwo nerwowo-mięśniowe (np. leki blokujące przekaźnictwo nerwowo-mięśniowe).
Skutek podawania różnych serotypów toksyny botulinowej w tym samym czasie oraz w odstępie kilku miesięcy jest nieznany. Nadmierne osłabienie mięśni może ulec nasileniu w wyniku podania innej toksyny botulinowej przed ustąpieniem działania wcześniej wstrzykniętej toksyny.
Nie przeprowadzono badań dotyczących interakcji. Nie zostały zgłoszone interakcje o istotnym znaczeniu klinicznym.
Dzieci i młodzież
Nie przeprowadzono badań dotyczących interakcji u dzieci.
Ciąża
Nie ma wystarczających danych na temat bezpieczeństwa stosowania toksyny botulinowej typu A u kobiet w ciąży. Badania na zwierzętach wykazały ryzyko uszkodzenia płodu (patrz: punkt 5.3). Potencjalne ryzyko stosowania u kobiet w ciąży nie zostało zbadane.
Produkt leczniczy BOTOX nie powinien być stosowany u kobiet w okresie ciąży oraz u kobiet w wieku rozrodczym, niestosujących skutecznych metod antykoncepcji, chyba że jest zdecydowanie konieczne.
Karmienie piersią
Nie ma informacji, czy produkt BOTOX przenika do mleka kobiecego. Nie zaleca się stosowania produktu BOTOX u kobiet karmiących piersią.
Płodność
Brak jest wystarczających danych dotyczących wpływu toksyny botulinowej typu A na płodność u kobiet w wieku rozrodczym. Badania na samcach i samicach szczurów wykazały zmniejszenie płodności (patrz punkt 5.3).
Ze względu na charakter schorzeń u leczonych pacjentów trudno jest ocenić wpływ produktu BOTOX na zdolność prowadzenia pojazdów i obsługiwania maszyn. Może on być oceniony jedynie po leczeniu.
Jednakże produkt leczniczy BOTOX może powodować astenię, osłabienie mięśni, zawroty głowy oraz zaburzenia widzenia, które mogą zaburzać zdolność prowadzenia pojazdów i obsługi maszyn.
Działania niepożądane przedstawiono według następujących kategorii, w zależności od częstości ich występowania:
Bardzo Często | (1/10) |
Często | (1/100 do <1/10) |
Niezbyt często | (1/1 000 do <1/100) |
Rzadko | (≥1/10 000 do <1/1000) |
Bardzo rzadko | (<1/10 000) |
Nieznana | częstość nie może być określona na podstawie dostępnych danych |
Poniżej przedstawiono działania niepożądane w zależności od miejsca podania produktu leczniczego BOTOX.
ZABURZENIA NEUROLOGICZNE:
Ogniskowa spastyczność kończyny dolnej u dzieci
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia skóry i tkanki podskórnej | wysypka | często |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | osłabienie mięśni | niezbyt często |
Urazy, zatrucia i powikłania po zabiegach | skręcenie więzadła, otarcie skóry | często |
Zaburzenia ogólne i stany w miejscu podania | zaburzenia chodu, ból w miejscu wstrzyknięcia | często |
Ogniskowa spastyczność kończyny górnej u pacjentów dorosłych po udarze.
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia żołądka i jelit | nudności | często |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | ból kończyny, osłabienie mięśni | często |
Zaburzenia ogólne i stany w miejscu podania | zmęczenie, obrzęk obwodowy | często |
Nie obserwowano zmiany ogólnego profilu bezpieczeństwa w przypadku kolejnych podań.
Ogniskowa spastyczność kończyn dolnych u pacjentów dorosłych po udarze.
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia skóry i tkanki podskórnej | wysypka | często |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | ból stawów, sztywność mięśniowo-szkieletowa, osłabienie mięśni | często |
Zaburzenia ogólne i stany w miejscu podania | obrzęki obwodowe | często |
Urazy, zatrucia i powikłania po zabiegach | przewracanie się | często |
Nie obserwowano zmian ogólnego profilu bezpieczeństwa w przypadku kolejnych podań.
Kurcz powiek (blefarospazm), połowiczy kurcz twarzy i związane z nim ogniskowe dystonie.
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia układu nerwowego | zawroty głowy, niedowład twarzy, porażenie nerwu twarzowego | niezbyt często |
Zaburzenia oka | opadanie powieki | bardzo często |
punkcikowe zapalenie rogówki, niedomykalność powieki, suchość oka, światłowstręt, nasilenie łzawienia, podrażnienie oka | często | |
zapalenie rogówki, wywinięcie powieki, podwójne widzenie, podwinięcie powieki, zaburzenia widzenia i niewyraźne widzenie | niezbyt często | |
obrzęk powiek | rzadko | |
wrzodziejące zapalenie rogówki, ubytek nabłonka i perforacja rogówki | bardzo rzadko | |
Zaburzenia skóry i tkanki podskórnej | wybroczyny | często |
wysypka/zapalenie skóry, | niezbyt często | |
Zaburzenia ogólne i stany w miejscu podania | podrażnienie i obrzęk twarzy | często |
zmęczenie | niezbyt często |
Dystonia szyjna
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | zapalenie śluzówki nosa, zakażenie górnych dróg oddechowych | często |
Zaburzenia układu nerwowego | zawroty głowy, wzmożone napięcie mięśniowe, niedoczulica, senność i ból głowy | często |
Zaburzenia oka | podwójne widzenie, opadanie powieki | niezbyt często |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | duszność i dysfonia | niezbyt często |
Zaburzenia żołądka i jelit | dysfagia (patrz podpunkt „Informacje dodatkowe”) | bardzo często |
suchość w jamie ustnej i nudności | często | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | osłabienie mięśni | bardzo często |
sztywność i bolesność układu mięśniowo-szkieletowego | często | |
Zaburzenia ogólne i stany w miejscu podania | ból | bardzo często |
astenia, choroba grypopodobna, złe samopoczucie | często | |
gorączka | niezbyt często |
Przewlekła migrena
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia układu nerwowego | ból głowy, migrena w tym nasilenie migreny, niedowład twarzy | często |
Zaburzenia oka | opadanie powieki | często |
Zaburzenia skóry i tkanki podskórnej | świąd, wysypka | często |
ból skóry | niezbyt często | |
Zaburzenia mięśniowo- szkieletowe, tkanki łącznej i kości | ból szyi, ból mięśni, ból mięśniowo-szkieletowy, sztywność mięśniowo- szkieletowa, skurcze mięśni, napięcie mięśni i osłabienie mięśni | często |
ból szczęki | niezbyt często | |
objaw Mefisto (nadmierne uniesienie zewnętrznej części brwi) | nieznana | |
Zaburzenia ogólne i stany w miejscu podania | ból w miejscu podania | często |
Zaburzenia żołądka i jelit | trudności z połykaniem | niezbyt często |
W badaniach fazy 3, wskaźnik przerwania terapii z powodu wystąpienia działań niepożądanych wynosił 3,8% w przypadku produktu BOTOX i 1,2% w przypadku placebo.
ZABURZENIA CZYNNOŚCI PĘCHERZA MOCZOWEGO:
Idiopatyczna nadreaktywność pęcherza moczowego
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | Zakażenie dróg moczowych | bardzo często |
Bakteriomocz | często | |
Zaburzenia nerek i dróg moczowych | Dysuria | bardzo często |
Zatrzymanie moczu, częstomocz, obecność leukocytów w moczu | często | |
Badania diagnostyczne | zaleganie moczu* | często |
* zwiększona objętość moczu zalegającego (ang. PVR) niewymagająca cewnikowania
Działania niepożądane związane z procedurą wstrzyknięcia występowały często i obejmowały dysurię i krwiomocz.
Czyste cewnikowanie przerywane wprowadzono u 6,5% pacjentów leczonych za pomocą 100 jednostek produktu leczniczego BOTOX oraz u 0,4% pacjentów otrzymujących placebo.
W badaniach klinicznych kontrolowanych placebo, spośród 1 242 pacjentów 41,4% pacjentów
(n = 514) było w wieku ≥ 65 lat, a 14,7% (n = 182) w wieku ≥ 75 lat. Nie zaobserwowano ogólnej
różnicy w profilu bezpieczeństwa po podaniu produktu leczniczego BOTOX pomiędzy pacjentami w wieku ≥ 65 lat a pacjentami w wieku <65 lat, z wyjątkiem zakażenia dróg moczowych, którego częstość występowania była większa u pacjentów w podeszłym wieku w obu grupach otrzymujących zarówno placebo jak i BOTOX, w porównaniu z młodszymi pacjentami.
Nie zaobserwowano zmiany w ogólnym profilu bezpieczeństwa, w przypadku ponownego podawania produktu leczniczego BOTOX.
Nietrzymanie moczu wskutek neurogennej nadczynności mięśnia wypieracza u dorosłych
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | zakażenie dróg moczowych a, b bakteriomocz b | bardzo często |
Badania diagnostyczne | zaleganie moczub** | bardzo często |
Zaburzenia psychiczne | bezsennośća | często |
Zaburzenia żołądka i jelit | zaparciea | często |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | osłabienie mięśnia, skurcz mięśnia | często |
Zaburzenia nerek i dróg moczowych | zatrzymanie moczua, b | bardzo często |
krwiomocz*a, b, dysuria*a, b, uchyłek pęcherzaa | często | |
Zaburzenia ogólne i stany w miejscu podania | zmęczeniea, zaburzenia chodua | często |
Urazy, zatrucia i powikłania po zabiegach | dysrefleksja autonomiczna*a, przewracanie sięa | często |
* działania niepożądane związane z procedurą wstrzyknięcia
** zwiększona objętość moczu zalegającego (ang. PVR) nie wymagająca cewnikowania
a działania niepożądane zgłaszane w ramach badań zasadniczych II i III fazy
b działania niepożądane zgłaszane w badaniach po wprowadzeniu produktu leczniczego BOTOX 100 jednostek do obrotu u pacjentów chorych na MS, którzy na początku badania nie byli cewnikowani
W badaniach klinicznych zakażenie dróg moczowych zgłosiło 49,2% pacjentów leczonych za pomocą 200 jednostek produktu leczniczego BOTOX oraz u 35,7% pacjentów leczonych placebo (53,0% pacjentów chorych na stwardnienie rozsiane leczonych za pomocą 200 jednostek produktu leczniczego BOTOX w porównaniu z 29,3% osób z grupy placebo; 45,4% pacjentów z urazem rdzenia kręgowego leczonych za pomocą 200 jednostek produktu leczniczego BOTOX w porównaniu z 41,7% w grupie placebo). Zatrzymanie moczu zgłosiło 17,2% pacjentów leczonych za pomocą 200 jednostek produktu leczniczego BOTOX oraz 2,9% pacjentów leczonych placebo (28,8% pacjentów chorych na stwardnienie rozsiane leczonych za pomocą 200 jednostek produktu leczniczego BOTOX w porównaniu z 4,5% osób z grupy placebo; 5,4% pacjentów z urazem rdzenia kręgowego leczonych za pomocą produktu leczniczego BOTOX w porównaniu z 1,4% w grupie placebo).
Nie zaobserwowano żadnej zmiany rodzaju działań niepożądanych po powtórzeniu dawkowania.
U pacjentów chorych na MS zakwalifikowanych do badań o zasadniczym znaczeniu nie zaobserwowano żadnych różnic w zakresie częstości nasilenia stwardnienia rozsianego (MS) w ujęciu rocznym (tj. liczby przypadków nasilenia MS na pacjentorok) (BOTOX = 0,23, placebo = 0,20), podobnie nie zaobserwowano żadnych różnic w badaniach po wprowadzeniu produktu leczniczego BOTOX 100 jednostek do obrotu u pacjentów chorych na MS, którzy na początku badania nie byli cewnikowani (BOTOX=0, placebo=0,07).
W badaniach zasadniczych u pacjentów, którzy na początku badania przed terapią nie byli cewnikowani, cewnikowanie rozpoczęto u 38,9% osób po terapii za pomocą 200 jednostek produktu leczniczego BOTOX w porównaniu z 17,3% osób z grupy placebo.
W badaniach po wprowadzeniu produktu leczniczego BOTOX 100 jednostek do obrotu u pacjentów chorych na MS, którzy na początku badania nie byli cewnikowani, cewnikowanie rozpoczęto u 15,2% osób po terapii za pomocą 100 jednostek produktu leczniczego BOTOX w porównaniu z 2,6% osób
z grupy placebo (patrz punkt 5.1).
Neurogenna nadczynność mięśnia wypieracza u dzieci i młodzieży
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | Bakteriomocz | Bardzo często |
Zakażenie dróg moczowych, obecność leukocytów w moczu | Często | |
Zaburzenia nerek i dróg moczowych | Krwiomocz, ból pęcherza* | Często |
* działania niepożądane związane z procedurą wstrzyknięcia
Nie zaobserwowano zmiany rodzaju działań niepożądanych po powtórzeniu dawkowania.
ZABURZENIA SKÓRY I JEJ PRZYDATKÓW:
Pierwotna nadpotliwość pach
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zaburzenia układu nerwowego | ból głowy, parestezje | często |
Zaburzenia naczyniowe | rozszerzenie naczyń krwionośnych (nagłe zaczerwienienie twarzy) | często |
Zaburzenia żołądka i jelit | nudności | niezbyt często |
Zaburzenia skóry i tkanki podskórnej | pocenie się poza okolicą pach, nieprawidłowy zapach skóry, świąd, guzki podskórne, łysienie | często |
świąd | niezbyt często | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | ból kończyn | często |
osłabienie mięśni, ból mięśni, dysfunkcje stawów, ból i osłabienie ramion | niezbyt często | |
Zaburzenia ogólne i stany w miejscu podania | reakcje i ból w miejscu wstrzyknięcia | często |
astenia, obrzęk i ból w miejscu wstrzyknięcia, krwiak w miejscu wstrzyknięcia, nadwrażliwość w miejscu wstrzyknięcia, podrażnienie w miejscu wstrzyknięcia, astenia, reakcje w miejscu wstrzyknięcia | niezbyt często |
W leczeniu pierwotnej nadpotliwości pach nasilenie pocenia się poza okolicą pach stwierdzono
u 4,5% pacjentów w okresie miesiąca od podania produktu. Nie zaobserwowano żadnej prawidłowości
w zakresie miejsc anatomicznych, w których zaburzenie występowało. U około 30% pacjentów zaburzenie ustąpiło w ciągu 4 miesięcy.
Niezbyt często (0,7%) donoszono również o osłabieniu ramion. Było ono łagodne, miało charakter przemijający, nie wymagało leczenia i ustępowało bez pozostawiania żadnych następstw. Objaw ten mógł być związany z samą chorobą, techniką podania lub obydwoma tymi czynnikami.
W występującym niezbyt często przypadku osłabienia mięśni można rozważyć przeprowadzenie badania neurologicznego. Ponadto zaleca się przeprowadzenie oceny techniki podawania produktu przed ponownym jego wstrzyknięciem w celu upewnienia się, że jest on podawany śródskórnie.
W niekontrolowanym badaniu dotyczącym bezpieczeństwa produktu leczniczego BOTOX (podanie 50 jednostek na każdą pachę) w grupie dzieci i młodzieży w wieku od 12 do 17 lat (n=144), działaniami niepożądanymi występującymi u więcej niż 1 pacjenta (po 2 pacjentów) były: ból
w miejscu wstrzyknięcia oraz pocenie się poza okolicą pach.
Zmarszczki gładzizny czoła
Poniższe niepożądane działania produktu obserwowano w badaniach klinicznych przeprowadzonych metodą podwójnie ślepej próby, z kontrolowanym placebo, po wstrzyknięciu 20 jednostek produktu leczniczego BOTOX wyłącznie w leczeniu zmarszczek gładzizny czoła:
Klasyfikacja układów i narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | zakażenie | niezbyt często |
Zaburzenia psychiczne | lęk | niezbyt często |
Zaburzenia układu nerwowego | ból głowy | często |
parestezje, zawroty głowy | niezbyt często | |
Zaburzenia oka | opadanie powiek | często |
zapalenie powiek, ból oka, zaburzenia widzenia | niezbyt często | |
Zaburzenia żołądka i jelit | nudności, suchość w jamie ustnej | niezbyt często |
Zaburzenia skóry i tkanki podskórnej | rumień | często |
napięcie skóry, obrzęk (twarzy, powiek, wokół oczodołu), reakcja nadwrażliwości na światło, świąd, suchość skóry | niezbyt często | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | miejscowe osłabienie mięśni | często |
drżenie mięśni, objaw Mefisto (nadmierne uniesienie zewnętrznej części brwi) | niezbyt często | |
Zaburzenia ogólne i stany w miejscu podania | ból twarzy | często |
zespół grypowy, astenia, gorączka | niezbyt często |
Zmarszczki typu „kurze łapki” wraz ze zmarszczkami gładzizny czoła lub bez nich
Poniższe niepożądane działania produktu obserwowano w badaniach klinicznych z podwójnie ślepą próbą, z kontrolowanym placebo, po wstrzyknięciu produktu leczniczego BOTOX w leczeniu kurzych łapek wraz z zmarszczkami gładzizny czoła lub bez nich:
Klasyfikacja układów i narządów | Działania niepożądane | Częstość |
Zaburzenia oka | obrzęk powiek | rzadko |
Zaburzenia ogólne i stany w miejscu podania | krwiak w miejscu wstrzyknięcia* | często |
krwawienie w miejscu wstrzyknięcia* | rzadko | |
ból w miejscu wstrzyknięcia* | rzadko |
parestezje w miejscu wstrzyknięcia | rzadko |
*działania niepożądane związane z procedurą wstrzyknięcia
Zmarszczki poziome czoła i zmarszczki gładzizny czoła ze zmarszczkami typu „kurze łapki” lub bez nich
Poniższe niepożądane działania produktu obserwowano w badaniach klinicznych z podwójnie ślepą próbą, z kontrolowanym placebo, po wstrzyknięciu produktu leczniczego BOTOX w jednoczesnym leczeniu zmarszczek na czole i zmarszczek gładzizny czoła wraz z kurzymi łapkami lub bez nich:
Klasyfikacja układów i narządów | Działania niepożądane | Częstość |
Zaburzenia układu nerwowego | ból głowy | często |
Zaburzenia oka | opadanie powiek1 | często |
Zaburzenia skóry i tkanki podskórnej | napięcie skóry | często |
opadanie brwi2 | często | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | objaw Mefisto (nadmierne uniesienie zewnętrznej części brwi) | często |
Zaburzenia ogólne i stany w miejscu podania | zasinienie w miejscu wstrzyknięcia* | często |
krwiak w miejscu wstrzyknięcia* | często | |
ból w miejscu wstrzyknięcia* | rzadko |
1Średni czas do wystąpienia opadania powiek wynosił 9 dni od daty zabiegu.
2Średni czas do wystąpienia opadania brwi wynosił 5 dni od daty zabiegu.
Nie zaobserwowano zmian w zakresie ogólnego profilu bezpieczeństwa w następstwie powtarzanych iniekcji.
Informacje dodatkowe
Poniższa lista obejmuje działania niepożądane lub inne medycznie istotne zdarzenia niepożądane, które zostały zgłoszone, po wprowadzeniu produktu do obrotu, niezależnie od wskazania terapeutycznego a nie zostały wymienione w punkcie 4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania oraz punkcie 4.8 Działania niepożądane.
Klasyfikacja układów i narządów | Działanie niepożądane |
Zaburzenia układu immunologicznego | wstrząs anafilaktyczny, obrzęk naczynioruchowy, choroba posurowicza i pokrzywka |
Zaburzenia metabolizmu i odżywiania | anoreksja |
Zaburzenia układu nerwowego | neuropatia splotu ramiennego, dysfonia, zaburzenia mowy, niedowład twarzy, zaburzenia czucia, osłabienie mięśni, miastenia, neuropatia obwodowa, parestezje, neuropatia korzeni rdzeniowych, drgawki, omdlenie oraz porażenie nerwu twarzowego |
Zaburzenia oka | jaskra z zamkniętym kątem (w przypadku leczenia kurczu powiek), opadanie powiek, niedomykalność powieki, zez, niewyraźne widzenie i zaburzenia widzenia, suchość oka, obrzęk powiek |
Zaburzenia ucha i błędnika | niedosłuch, szum w uszach i zawroty głowy |
Zaburzenia serca | zaburzenia rytmu serca, zawał mięśnia sercowego |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | zachłystowe zapalenie płuc (niektóre śmiertelne), duszność, depresja oddechowa i niewydolność oddechowa |
Zaburzenia żołądka i jelit | ból brzucha, biegunka, zaparcia, suchość w ustach, zaburzenia połykania, nudności i wymioty |
Zaburzenia skóry i tkanki podskórnej | łysienie, opadanie brwi, łuszczycowe zapalenie skóry, rumień wielopostaciowy, nadmierne pocenie się, wypadanie rzęs, świąd i wysypka |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | zanik mięśni i bóle mięśni, miejscowe drżenia mięśni/ mimowolne skurcze mięśni |
Zaburzenia ogólne i stany w miejscu podania | atrofia wskutek odnerwienia, złe samopoczucie i gorączka |
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C PL-02 222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Przedawkowanie produktu leczniczego BOTOX zależy od czynników takich jak wielkość podanej dawki, miejsca wstrzyknięcia oraz właściwości tkanek otaczających. Nie stwierdzono żadnego przypadku, w którym po przypadkowym podaniu produktu BOTOX wystąpiłyby objawy toksyczności ogólnoustrojowej. Zbyt duże dawki produktu mogą wywoływać miejscowy lub odległy od miejsca wstrzyknięcia, uogólniony paraliż nerwowo-mięśniowy. Nie stwierdzono żadnego przypadku doustnego przyjęcia produktu BOTOX.
Oznaki przedawkowania nie są widoczne bezpośrednio po wstrzyknięciu. Jeżeli przypadkowo nastąpi wstrzyknięcie lub połknięcie, lub podejrzewane jest przedawkowanie produktu, pacjent powinien być poddany obserwacji medycznej przez kilka dni w kierunku objawów uogólnionego osłabienia lub paraliżu mięśni, które mogą mieć charakter miejscowy lub odległy od miejsca wstrzyknięcia i mogą obejmować: opadanie powiek, podwójne widzenie, zaburzenia połykania, zaburzenia mowy, uogólnione osłabienie lub zaburzenia oddechowe. U takich pacjentów należy rozważyć konieczność przeprowadzenia dodatkowych badań, podjęcia natychmiastowego leczenia lub hospitalizacji.
Gdy mięśnie części ustnej gardła i przełyku są objęte może dojść do zachłyśnięcia się, co może prowadzić do wystąpienia zachłystowego zapalenia płuc. W przypadku porażenia mięśni oddechowych lub ich znacznego osłabienia, może być konieczna intubacja i wspomaganie oddychania do momentu wyzdrowienia. W uzupełnieniu leczenia podstawowego konieczna może okazać się tracheotomia i przedłużenie mechanicznego wspomagania oddychania.
Grupa farmakoterapeutyczna: leki zwiotczające mięśnie działające obwodowo, toksyna botulinowa
kod ATC: M03AX01.
Mechanizm działania
Toksyna botulinowa typu A hamuje uwalnianie acetylocholiny w presynaptycznych zakończeniach cholinergicznych nerwów poprzez rozszczepianie struktury SNAP-25, białka koniecznego dla skutecznego łączenia i uwalniania form acetylocholiny z pęcherzyków znajdujących się
w zakończeniach nerwów.
Działanie farmakodynamiczne
Po wstrzyknięciu, następuje szybkie wiązanie ze specyficznymi powierzchniowymi receptorami komórkowymi o wysokim powinowactwie do toksyny. Następnie toksyna jest przenoszona poprzez błonę komórkową z udziałem receptorów pośredniczących. Ostatecznie, toksyna jest uwalniana do cytozolu. Procesowi temu towarzyszy postępujące hamowanie uwalniania acetylocholiny, kliniczne objawy pojawiają się w ciągu 2-3 dni. Maksymalny efekt terapeutyczny uzyskuje się w 5 – 6 tygodniu po wstrzyknięciu. Dowody kliniczne z modelu syntetyzacji nerwu trójdzielnego, wskazują na zdolność do zmniejszenia indukowanego kapsaicyną bólu i neurogennego stanu zapalnego oraz zwiększania progu bólowego dla skórnych receptorów ciepła przez produkt leczniczy BOTOX.
Działanie produktu zwykle ustępuje w ciągu 12 tygodni po wstrzyknięciu, gdy ponownie będzie utworzone połączenie pomiędzy zakończeniami nerwowymi a płytką mięśniową. Po wstrzyknięciu śródskórnym, gdzie miejscem docelowym były potowe gruczoły ekrynowe, skutek działania utrzymywał się średnio 7,5 miesięcy po pierwszym wstrzyknięciu u pacjentów, którym podawano 50 jednostek do jednej pachy. Jednakże u 27,5% pacjentów efekt utrzymywał się przez okres 1 roku lub dłużej. Nie badano procesu odtwarzania się końcówek nerwów układu współczulnego, unerwiających gruczoły potowe po śródskórnym wstrzyknięciu produktu BOTOX.
Po wstrzyknięciu do mięśnia wypieracza, produkt leczniczy BOTOX wpływa na neurony dróg eferentnych odpowiedzialnych za czynność mięśnia wypieracza poprzez zahamowanie uwalniania acetylocholiny. Ponadto produkt leczniczy BOTOX może hamować czynność neurotransmiterów aferentnych i dróg czuciowych.
Skuteczność kliniczna i bezpieczeństwo stosowania
ZABURZENIA NEUROLOGICZNE
Ogniskowa spastyczność kończyny górnej u dzieci
Skuteczność i bezpieczeństwo produktu leczniczego BOTOX w leczeniu spastyczności kończyn górnych u dzieci w wieku 2 lat i starszych oceniano w randomizowanym, wieloośrodkowym, podwójnie zaślepionym, badaniu, kontrolowanym placebo. Badanie obejmowało 234 pacjentów pediatrycznych (77 BOTOX 6 jednostek / kg, 78 BOTOX 3 jednostki / kg i79 placebo) ze spastycznością kończyn górnych z powodu porażenia mózgowego (87%) lub udarem (13%)
i wyjściową oceną MAS łokcia lub nadgarstka co najmniej 2. Całkowita dawka 3 jednostek / kg (maksymalnie 100 jednostek) lub 6 jednostek / kg (maksymalnie 200 jednostek) lub placebo została wstrzyknięta domięśniowo i podzielona pomiędzy mięśnie łokcia lub nadgarstka i palców. Wszyscy pacjenci otrzymali standaryzowaną terapię zajęciową. Aby pomóc we właściwej lokalizacji mięśni do wstrzyknięć, konieczne było zastosowanie techniki elektromiografii, stymulacji nerwów lub technik ultradźwiękowych. Pierwszorzędowym punktem końcowym była średnia zmiany wyniku MAS
w stosunku do wartości wyjściowej w głównej grupie mięśni (łokieć lub nadgarstek) w 4. i 6. tygodniu, a kluczowym drugorzędowym punktem końcowym była średnia z klinicznego ogólnego wrażenia ogólnej zmiany według lekarza (CGI) w tygodniach 4 i 6. Skala Osiągania Celów (GAS) przez lekarza dla celów aktywnych i pasywnych została oceniona jako drugorzędowy punkt końcowy w tygodniach 8 i 12. Pacjenci byli obserwowani przez 12 tygodni.
Pacjenci spełniający wymagania mogli zostać włączeni do otwartego badania rozszerzonego,
w którym otrzymywali do 5 sesji terapeutycznych w dawkach do 10 jednostek/kg (maksymalnie 340 jednostek), w przypadku leczenia kończyny dolnej w połączeniu z kończyną górną.
Statystycznie istotną poprawę w porównaniu z placebo wykazano u pacjentów leczonych produktem BOTOX 3 i 6 jednostek / kg dla pierwszorzędowego punktu końcowego i we wszystkich punktach czasowych do tygodnia 12. Poprawa wyniku MAS była podobna w obu grupach leczenia produktem BOTOX. Jednak w żadnym momencie różnica w porównaniu z placebo wynosiła ≥ 1 punkt w skali MAS. Zobacz tabelę poniżej. Efekt leczenia w analizie odpowiedzi na leczenie wahał się od około 10- 20%.
Pierwszorzędowe i drugorzędowe punkty końcowe w ocenie skuteczności
BOTOX 3 jednostki/kg (N=78) | BOTOX 6 jednostek/kg (N=77) | Placebo (N=79) | |
Średnia zmiana w stosunku do wartości wyjściowej w grupie mięśni głównych (łokieć lub nadgarstek) na MASa | |||
Średnio 4 i 6 tydzień | -1,92* | -1,87* | -1,21 |
Średni wynik CGIb | |||
Średnio 4 i 6 tydzień | 1,88 | 1,87 | 1,66 |
Średni wynik GASc | |||
Cele pasywne w tygodniu 8 | 0,23 | 0,30 | 0,06 |
Cele pasywne w tygodniu 12 | 0,31 | 0,71* | 0,11 |
Cele aktywne w tygodniu 8 | 0,12 | 0,11 | 0,21 |
Cele aktywne w tygodniu 12 | 0,26 | 0,49 | 0,52 |
Średnia zmiana wyniku FPS w odniesieniu do wartości wyjściowej d | N=11 | N=11 | N=18 |
Tydzień 4 | -4.91 | -3,17 | -3,55 |
Tydzień 6 | -3,12 | -2,53 | -3,27 |
a Punktacja MAS jest 6-punktową skalą (0 [brak wzrostu napięcia mięśniowego], 1, 1+, 2, 3, i 4 [kończyna sztywna w zgięciu lub wyproście]), która mierzy siłę potrzebną do poruszania kończyną w stawie, przy czym zmniejszenie wyniku oznacza poprawę spastyczności.
b CGI oceniło odpowiedź na leczenie pod kątem tego, jak pacjent radził sobie w życiu, używając 9-stopniowej skali (-4 = bardzo wyraźne pogorszenie do +4 = bardzo wyraźna poprawa).
c GAS to 6-punktowa skala (-3 [gorzej niż na początku], -2 [tak jak na początku], -1 [mniej niż oczekiwano], 0 [oczekiwany cel], +1 [nieco więcej niż oczekiwano], +2 [znacznie więcej niż oczekiwano]).
d EVG to 11-pozycyjna skala oceniająca chód na podstawie postawy stopy (5 pozycji), postawy kolana (2 pozycje), wymachu stopy (2 pozycje) i wymachu kolana (2 pozycje) przy użyciu 3-punktowej skali porządkowej (0 [normalny], 1 [zgięcie 1 lub wyprost 1] i 2 [zgięcie 2 lub wyprost 2] odpowiednio dla każdej pozycji).
U dzieci i młodzieży ze spastycznością kończyn dolnych z analizowanymi próbkami z jednego badania fazy 3 i otwartego badania rozszerzonego, przeciwciała neutralizujące wytworzyły się
u 2 z 264 pacjentów (0,8%) leczonych produktem BOTOX przez maksymalnie 5 cykli leczenia. Obaj pacjenci nadal odczuwali korzyści kliniczne po kolejnych terapiach produktem leczniczym BOTOX.
Ogniskowa spastyczność kończyny górnej u pacjentów dorosłych po udarze
Skuteczność i bezpieczeństwo stosowania produktu BOTOX w leczeniu spastyczności kończyny górnej u pacjentów dorosłych oceniano w randomizowanym, wieloośrodkowym, podwójnie zaślepionym, kontrolowanym placebo badaniu.
Badanie to obejmowało 126 dorosłych pacjentów (64 pacjentów otrzymujących produkt BOTOX
i 62 pacjentów otrzymujących placebo) ze spastycznością kończyny górnej (wynik w skali Ashwortha co najmniej 3 dla napięcia zginaczy nadgarstka i co najmniej 2 dla napięcia zginaczy palców), którzy byli co najmniej 6 miesięcy po udarze. Produkt BOTOX (całkowita dawka od 200 do 240 jednostek) lub placebo wstrzykiwano domięśniowo do zginacza głębokiego palców, zginacza powierzchownego palców, zginacza promieniowego nadgarstka, zginacza łokciowego nadgarstka, a jeśli to konieczne do
przywodziciela kciuka i zginacza długiego kciuka. Zalecono użycie stymulatora EMG/nerwów, aby pomóc w prawidłowej lokalizacji mięśni do wstrzyknięć. Pacjenci byli obserwowani przez 12 tygodni.
Pierwszorzędowym punktem końcowym skuteczności było napięcie mięśni zginaczy nadgarstka w 6. tygodniu, mierzone za pomocą skali Ashwortha. Kluczowe drugorzędowe punkty końcowe
obejmowały ogólną ocenę przez lekarza, napięcie mięśni zginaczy palców i napięcie mięśni zginaczy kciuka w 6. tygodniu. Wyniki badania dotyczące pierwszorzędowego punktu końcowego i kluczowych drugorzędowych punktów końcowych przedstawiono w poniższej tabeli.
Pierwszorzędowe i drugorzędowe punkty końcowe w ocenie skuteczności w 6. tygodniu
BOTOX 200 do 240 jednostek (N=64) | Placebo (N=62) | |
Średnia zmiana napięcia mięśnia zginacza nadgarstka w skali Ashwortha w odniesieniu do wartości wyjścioweja | -1,7* | -0,5 |
Średnia odpowiedź na leczenie w skali PGAb | 1,8* | 0,6 |
Średnia zmiana napięcia mięśnia zginacza palców w skali Ashwortha w odniesieniu do wartości wyjścioweja | -1,3* | -0,5 |
Średnia zmiana napięcia mięśnia zginacza kciuka w skali Ashwortha w odniesieniu do wartości wyjścioweja | -1,66* | -0,48 |
* Istotna różnica względem placebo (p < 0,05)
a Skala Ashwortha jest 5-punktową skalą (0 [brak wzrostu napięcia mięśniowego], 1, 2, 3 i 4 [kończyna sztywna w zgięciu lub wyproście]), która mierzy siłę potrzebną do poruszania kończyną w stawie, przy czym zmniejszenie wyniku oznacza poprawę spastyczności.
b Ocena ogólna lekarza obejmowała odpowiedź na leczenie pod kątem tego, jak pacjent radził sobie w życiu, używając punktacji od -4 = bardzo wyraźne pogorszenie do +4 = bardzo wyraźna poprawa.
Ogniskowa spastyczność kończyny dolnej u pacjentów dorosłych związana z udarem
Skuteczność i bezpieczeństwo stosowania produktu leczniczego BOTOX w leczeniu spastyczności kończyny dolnej przeprowadzono w randomizowanym, wieloośrodkowym kontrolowanym placebo, metodą podwójnie ślepej próby badaniu, obejmującym 468 pacjentów po udarze (233 BOTOX i 235 placebo) ze spastycznością stawu skokowego (co najmniej 3 w zmodyfikowanej skali Ashwortha [MAS] ), którzy byli co najmniej 3 miesiące po udarze. W badaniu produkt leczniczy BOTOX albo placebo wstrzykiwano domięśniowo w dawce od 300 do 400 jednostek do wymaganych mięśni: brzuchatego łydki, mięśnia płaszczkowatego i mięśnia piszczelowego tylnego oraz do mięśni opcjonalnych w tym mięśnia prostownika palucha długiego, mięśnia zginacza długiego palców, mięśnia zginacza krótkiego palców, mięśnia prostownika palucha i mięśnia prostego uda.
Pierwszorzędowym punktem końcowym była średnia zmiana stawu skokowego w punktacji MAS w 4 i 6 tygodniu w stosunku do wartości wyjściowej, a kluczowym drugorzędowym punktem końcowym była średnia wartość CGI (ang. Physician Global Assessment of Response) w 4 i 6 tygodniu. Statystycznie i klinicznie istotne różnice między grupą otrzymującą produkt leczniczy BOTOX a placebo i wartości pierwszorzędowych punktów skuteczności MAS i wartości CGI kluczowego drugorzędowego punktu przedstawiono w poniższej tabeli. Na podstawie średniej wartości w punktacji MAS, pierwszorzędowego punktu końcowego, w 4 i 6 tygodniu leczenia stawu
skokowego, w porównaniu z wartością wyjściową nie zaobserwowano poprawy u pacjentów w wieku 65 lat i starszych, między grupą otrzymującą produkt BOTOX w porównaniu z placebo, prawdopodobnie z powodu małej liczby pacjentów.
Pierwszorzędowe i drugorzędowe punkty końcowe w ocenie skuteczności
BOTOX od 300 do 400 jednostek | Placebo (N=235) |
(ITT) (N=233) | ||
Średnia zmiana mięśnia zginacza podeszwowego kostki w punktacji MAS w odniesieniu do wartości wyjściowej | ||
Średnio 4 i 6 tydzień | -0,8* | -0,6 |
Średnia wartość Clinical Global Impression Score w ocenie badacza | ||
Średnio 4 i 6 tydzień | 0,9* | 0,7 |
Średnia zmiana w zginaczach palców w skali MAS | ||
FHaL średnio 4 i 6 tydzień | -1,02* | -0,6 |
FDL średnio 4 i 6 tydzień | -0,88 | - 0,77 |
Średnia zmiana mięśnia zginacza podeszwowego kostki w punktacji MAS u pacjentów w odniesieniu do wartości wyjściowej | ≥65 lat N=60 | ≥65 lat N=64 |
Średnio 4 i 6 tydzień | -0,7 | -0,7 |
*znaczące różnice od placebo (p<0,05)
Inne randomizowane, wieloośrodkowe, kontrolowane placebo badanie kliniczne fazy 3, prowadzone metodą podwójnie ślepej próby u dorosłych pacjentów po udarze, ze spastycznością kończyny dolnej, dotyczącą stawu skokowego. Ogółem, 120 pacjentów losowo przydzielono do grup otrzymujących albo BOTOX (n=58) (całkowita dawka 300 j.), albo placebo (n=62). Badanie to przeprowadzono wyłącznie na pacjentach z Japonii z punktacją w Zmodyfikowanej Skali Ashwortha (MAS) ≥3, będących średnio 6,5 roku po udarze.
Znaczną poprawę w porównaniu z placebo obserwowano w zakresie zasadniczego punktu końcowego dla całkowitej zmiany w punktacji dla kostek w skali MAS, od wartości wyjściowej do tygodnia 12, którą obliczono mierząc pole pod krzywą (AUC). Znaczną poprawę w porównaniu z placebo obserwowano także średniej zmiany kostki od wartości wyjściowej w punktacji MAS w czasie poszczególnych wizyt po leczeniu, po 4, 6 i 8 tygodniach. Odsetek osób reagujących (pacjentów
z poprawą o co najmniej 1 stopień) na tych wizytach także był znacznie wyższy niż wśród pacjentów leczonych placebo.
Leczenie produktem leczniczym BOTOX wiązało się także ze znaczną poprawą globalnego klinicznego wrażenia badacza (CGI) dotyczącego niesprawności czynnościowej (drugorzędowy punkt końcowy, bez korekty uwzględniającej liczebność) w porównaniu z placebo. Nie stwierdzono klinicznie znaczącej poprawy czynności, mierzonej Skalą Oceny przez Lekarza (ang. Physician’s Rating Scale, PRS) i szybkością chodu.
Wyniki badania fazy 3 przedstawiono poniżej
Pierwszorzędowe i drugorzędowe punkty końcowe w ocenie skuteczności
BOTOX (N=58) | Placebo (N=62) | Wartość P | |
Średnia wartość AUC w punktacji MAS | |||
AUC (od dnia 0 do tygodnia 12) | -8,5 | -5,1 | 0,006 |
Średnia zmiana od wartości wyjściowej w punktacji MAS | |||
Wyjściowo | 3,28 | 3,24 | |
Tydzień 1 | -0,61 | -0,52 | 0,222 |
Tydzień 4 | -0,88 | -0,43 | < 0,001 |
Tydzień 6 | -0,91 | -0,47 | < 0,001 |
Tydzień 8 | -0,82 | -0,43 | < 0,001 |
Tydzień 12 | -0,56 | -0,40 | 0,240 |
Odsetek osób reagujących* | |||
Tydzień 1 | 52,6% | 38,7% | 0,128 |
Tydzień 4 | 67,9% | 30,6% | < 0,001 |
Tydzień 6 | 68,4% | 36,1% | < 0,001 |
Tydzień 8 | 66,7% | 32,8% | < 0,001 |
Tydzień 12 | 44,4% | 34,4% | 0,272 |
*Pacjenci z poprawą o co najmniej 1 stopień w punktacji MAS w stosunku do wartości wyjściowej
Zbliżoną odpowiedź obserwowano po ponownym leczeniu.
Przewlekła migrena
Produkt BOTOX blokuje uwalnianie neuroprzekaźników związanych z powstawaniem bólu. Przedkliniczne i kliniczne badania farmakodynamiczne wskazują, że przypuszczalny mechanizm profilaktyki bólu głowy polega na blokowaniu sygnałów obwodowych do ośrodkowego układu nerwowego, co hamuje uwrażliwienie ośrodkowe.
W dwóch badaniach klinicznych fazy 3 uczestniczyli pacjenci z przewlekłą migreną niestosujący jednocześnie innej metody zapobiegania bólom głowy, u których w ciągu 28 dni przed badaniem wystąpiły przynajmniej 4 epizody i 15 dni z bólem głowy (trwającym co najmniej 4 godziny)
a w 50% przypadków określono je jako migrenę/prawdopodobną migrenę. Pacjenci mogli stosować leki doraźne do leczenia ostrego bólu. 66% pacjentów nadużywało leków doraźnych podczas okresu wstępnego.
W wyniku stosowania produktu leczniczego BOTOX co 12 tygodni w 2. cyklu badania z podwójnie ślepą próbą obserwowano statystycznie znamienne korzyści w stosunku do stanu wyjściowego
w porównaniu z placebo w następujących parametrach: średnia liczba dni z umiarkowanym/silnym bólem głowy, średnia liczba dni z migreną/prawdopodobną migreną, całkowita liczba godzin bólu głowy w dniach wystąpienia bólu oraz średnia liczba epizodów bólu głowy. Odsetek pacjentów
z 50% redukcją liczby dni, w których wystąpił ból głowy, wynosił 47% w przypadku produktu BOTOX i 35% w przypadku placebo (p<0,001). Funkcjonowanie pacjentów i ogólna jakość życia uległy znaczącej poprawie (p<0,001) w porównaniu z placebo, co wykazano za pomocą testu Headache Impact Test (HIT-6).
ZABURZENIA CZYNNOŚCI PĘCHERZA MOCZOWEGO
Idiopatyczna nadreaktywność pęcherza moczowego
Dwa randomizowane, wieloośrodkowe, dwudziestoczterotygodniowe badania kliniczne fazy trzeciej, prowadzone metodą podwójnie ślepej próby z kontrolą placebo, przeprowadzone zostały w grupie pacjentów z nadreaktywnym pęcherzem moczowym z objawami nietrzymania moczu, parcia naglącego oraz częstomoczu. Łącznie 1105 pacjentów, których objawy nie były dobrze kontrolowane przez przynajmniej jeden z preparatów antycholinergicznych (nieadekwatna odpowiedź lub źle tolerowane objawy uboczne), zostało dobranych losowo do jednej z grup: w jednej grupie stosowano 100 jednostek produktu leczniczego BOTOX (n=557), w drugiej grupie podawano placebo (n=548).
W obu badaniach obserwowano znamienną poprawę w zakresie dobowej częstości występowania epizodów nietrzymania moczu, w porównaniu ze stanem początkowym, w grupie otrzymującej BOTOX (100 jednostek) w stosunku do grupy placebo w początkowej ocenie po 12 tygodniach (5,49 dla BOTOX i 5,39 dla placebo), zawierającej proporcjonalnie się pacjentów niezgłaszających
problemów z trzymaniem moczu. Stosując Skalę Korzyści Leczenia, proporcja pacjentów pozytywnie reagujących na leczenie (ich kondycja „bardzo poprawiła się” lub „poprawiła się”) była istotnie większa w grupie BOTOX w porównaniu z grupą placebo w obu badaniach. Obserwowano znaczącą poprawę dotyczącą wszystkich objawów OAB (pęcherza nadreaktywnego) po dwóch tygodniach od zastosowania leczenia.
Leczenie produktem leczniczym BOTOX powodowało znaczącą poprawę jakości życia w porównaniu z grupą placebo, którą oceniano kwestionariuszem „Jakości Życia Związanej z Nietrzymaniem Moczu (I-QOL, zawierającym unikanie i ograniczanie różnych zachowań, wpływ psychologiczny i problemy
społeczne) oraz Królewskim Kwestionariuszem Zdrowia (ang. KHQ) badającym wpływ nietrzymania moczu na ograniczenie wykonywania różnych czynności, ograniczenia społeczne, ograniczenia fizyczne, relacje osobiste, emocje. Ogólnie nie stwierdzono różnicy w skuteczności leczenia produktem leczniczym BOTOX w zależności od wieku - porównano pacjentów w wieku powyżej
i poniżej 65 lat. Najważniejsze wyniki obu badań są zebrane i przedstawione poniżej:
Pierwszorzędowe i drugorzędowe punkty końcowe na początku badania oraz zmiana
w stosunku do wartości początkowych w zebranych danych z badań o zasadniczym znaczeniu:
BOTOX 100 jednostek (N=557) | Placebo (N=548) | Poziom istotności p | |
Dobowa częstość występowania epizodów nietrzymania moczu * | |||
Średnia wartość na początku badania | 5,49 | 5,39 | |
Średnia zmiana w 2. tygodniu | -2,85 | -1,21 | < 0,001 |
Średnia zmiana w 6. tygodniua | -3,11 | -1,22 | < 0,001 |
Średnia zmiana w 12. tygodniu | -2,80 | -0,95 | < 0,001 |
Pozytywna odpowiedź na zastosowane leczenie oceniana w Skali Skuteczności Leczenia (%) | |||
w 2. tygodniu | 64,4 | 34,7 | < 0,001 |
w 6. tygodniu | 68,1 | 32,8 | < 0,001 |
w 12. tygodniua | 61,8 | 28,0 | < 0,001 |
Dobowa częstość występowania epizodów moczenia | |||
Średnia wartość na początku badania | 11,99 | 11,48 | |
Średnia zmiana w 2. tygodniu | -1,53 | -0,78 | < 0,001 |
Średnia zmiana w 6. tygodniu | -2,18 | -0,97 | < 0,001 |
Średnia zmiana w 12. tygodniub | -2,35 | -0,87 | < 0,001 |
Dobowa częstość występowania epizodów parcia naglącego | |||
Średnia wartość na początku badania | 8,82 | 8,31 | |
Średnia zmiana w 2. tygodniu | -2,89 | -1,35 | < 0,001 |
Średnia zmiana w 6. tygodniu | -3,56 | -1,40 | < 0,001 |
Średnia zmiana w 12. tygodniub | -3,30 | -1,23 | < 0,001 |
Całkowity wynik w kwestionariuszu „Jakości Życia w Nietrzymaniu Moczu” (I- QOL) | < 0,001 | ||
Średnia wartość na początku badania | 34,1 | 34,7 | |
Średnia zmiana w ciągu 12 tygodnibc | +22,5 | +6,6 | |
Królewski Kwestionariusz Zdrowia (KHQ): ograniczenie czynności życiowych | < 0,001 | ||
Średnia podstawowa | 65,4 | 61,2 | |
Średnia zmiana w ciągu 12 tygodnibc | -25,4 | -3,7 | |
Królewski Kwestionariusz Zdrowia (KHQ): ograniczenie zachowań społecznych | < 0,001 | ||
Średnia wartość na początku badania | 44,8 | 42,4 | |
Średnia zmiana w ciągu 12 tygodnibc | -16,8 | -2,5 |
* Odsetek pacjentów, którzy nie zgłaszali nietrzymania moczu (nie moczyli się) w ciągu 12 tygodni, wynosił 27,1% dla grupy BOTOX i 8,4% dla placebo. Ostatecznie osiągnięto 75% i 50% redukcji epizodów nietrzymania moczu w stosunku do
stanu początkowego i wynosiły odpowiednio 46,0% i 60,5% w grupie leczonej produktem BOTOX w porównaniu z grupą placebo 17,7 % i 31,0%.
a Współistniejące pierwszorzędowe punkty oceny końcowej
b Drugorzędowe punkty oceny końcowej
c Początkowo zdefiniowana minimalna zmiana w stosunku do stanu początkowego wynosiła +10 punktów w kwestionariuszu I-QOL i -5 punktów w kwestionariuszu KHQ
Średni czas utrzymywania się wyników leczenia produktem leczniczym BOTOX, oceniany na podstawie zgłaszania się pacjentów na powtórne leczenie, wynosił 166 dni (ok. 24 tygodni).
W oparciu o wyniki badań pacjentów, którzy otrzymali produkt leczniczy BOTOX 100 jednostek w ramach otwartego badania rozszerzonego (n = 438), średni czas trwania odpowiedzi na leczenie wynosił 212 dni (~ 30 tygodni).
Również tylko ograniczona liczba pacjentów w wieku poniżej 40 lat (n=88, 8,0%), rasy niekaukaskiej (n=101, 9,1%) i płci męskiej (n=135, 12,2%) była badana w dwóch badaniach klinicznych 3 fazy, dane w tych podgrupach wykazywały pozytywne wyniki leczenia. Wyższy odsetek objawów ubocznych takich jak: zatrzymanie moczu, zaleganie moczu po mikcji, częstomocz był obserwowany u mężczyzn niż u kobiet. Wyniki porównawcze współistniejących pierwszorzędowych punktów oceny końcowej u mężczyzn są przedstawione poniżej:
Porównanie pierwszorzędowych punktów końcowych u mężczyzn na początku badania oraz zmiana w stosunku do wartości początkowych w oparciu o pulę danych z badań osiowych:
BOTOX 100 jednostek (N=61) | Placebo (N=74) | Poziom istotności p | |
Dobowa częstość występowania epizodów nietrzymania moczu | 0,612 | ||
Średnia wartość na początku badania | 5,61 | 4,33 | |
Średnia zmiana w 12. tygodniu | -1,86 | -1,23 | |
Pozytywna odpowiedź na leczenie oceniana w Skali Korzyści Leczenia (%) | 40,7 | 25,4 | 0,060 |
w 12. tygodniu |
Łącznie 839 pacjentów było ocenionych w długoterminowym, otwartym badaniu (n=758 kobiet, n=81 mężczyzn). W przypadku wszystkich punktów oceny końcowej, u pacjentów ponownie leczonych uzyskiwano spójną odpowiedź na leczenie. W podgrupie 345 pacjentów (n=316 kobiet, n=29 mężczyzn), którzy odbyli trzy dwunastotygodniowe cykle, średnie zmniejszenie epizodów nietrzymania moczu wynosiły odpowiednio -3,07, -3,49 i -3,49 epizodów w dwunastym tygodniu odpowiednio po pierwszym, drugim i trzecim podaniu 100 jednostek produktu leczniczego BOTOX.
Odpowiadająca powyższemu proporcja pacjentów z pozytywną odpowiedzią na leczenie, oceniana w Skali Korzyści Leczenie, wynosiła odpowiednio 63,6%, 76,9% i 77,3%.
W kluczowych badaniach u żadnego z 615 pacjentów w badanych próbkach nie stwierdzono przeciwciał neutralizujących. Analiza próbek uzyskanych od 954 pacjentów w ramach badań zasadniczych III fazy oraz w ramach otwartego badania rozszerzonego nie wykazała obecności przeciwciał neutralizujących u pacjentów otrzymujących BOTOX 100 jednostek (0,0%). U 3 z 260 pacjentów otrzymujących BOTOX w dawce co najmniej 150 jednostek (1,2%) stwierdzono obecność przeciwciał neutralizujących, przy czym u jednego z pacjentów kontynuacja leczenia przyniosła skutek kliniczny. W odniesieniu do całkowitej liczby pacjentów, w grupie pacjentów, u których wykryto przeciwciała neutralizujące, czas utrzymywania się odpowiedzi na leczenie był krótszy,
a produkt leczniczy podawany był częściej (patrz punkt 4.4).
Nietrzymanie moczu wskutek neurogennej nadczynności mięśnia wypieracza u dorosłych
Badania zasadnicze III fazy
Dwa randomizowane, wieloośrodkowe badania kliniczne III fazy prowadzone metodą ślepej próby z grupą kontrolną stosującą placebo przeprowadzono u pacjentów z nietrzymaniem moczu wskutek
neurogennej nadczynności mięśnia wypieracza, którzy albo oddawali mocz spontanicznie, albo
u których zastosowano cewnikowanie. Do badań zakwalifikowano ogółem 691 pacjentów chorych na uraz rdzenia kręgowego lub stwardnienie rozsiane, u których choroby nie można było odpowiednio kontrolować za pomocą co najmniej jednego środka antycholinergicznego. Pacjentów tych losowo przydzielono do grup otrzymujących 200 jednostek produktu leczniczego BOTOX (n = 227), 300 jednostek produktu leczniczego BOTOX (n = 223) lub placebo (n = 241).
W obu badaniach III fazy w porównaniu z grupą przyjmującą placebo zaobserwowano znaczącą poprawę w zakresie pierwszorzędowego parametru oceny skuteczności dotyczącego zmiany częstości występowania epizodów nieotrzymania moczu w porównaniu ze stanem początkowym, przy czym najlepsze wyniki (obejmujące odsetek pacjentów, u których nie wystąpiły epizody nietrzymania moczu) uzyskano dla produktu leczniczego BOTOX (200 jednostek i 300 jednostek) w punkcie czasowym 6 tygodni. Zaobserwowano znaczącą poprawę parametrów urodynamiki w tym wzrost maksymalnej pojemności cystometrycznej i spadek szczytowego ciśnienia wypieracza podczas pierwszego mimowolnego skurczu tego mięśnia. Obserwowano też znaczącą (w porównaniu
z placebo) poprawę zgłaszanych przez pacjenta wyników jakości życia związanej z nietrzymaniem moczu mierzonej za pomocą kwestionariusza I-QOL (ang. Incontinence Quality of Life [jakość życia związana z nietrzymaniem moczu]) obejmującego zachowania ograniczające unikanie, oddziaływanie psychosocjologiczne oraz skrępowanie w sytuacjach wymagających kontaktów społecznych. Nie wykazano żadnych dodatkowych korzyści ze stosowania 300 jednostek produktu leczniczego BOTOX w porównaniu z zastosowaniem 200 jednostek, natomiast stwierdzono o wiele korzystniejszy profil bezpieczeństwa po zastosowaniu 200 jednostek produktu leczniczego BOTOX.
Poniżej przedstawiono zebrane wyniki z badań o zasadniczym znaczeniu:
Pierwszorzędowe i drugorzędowe punkty końcowe na początku badania oraz zmiana w stosunku do wartości początkowych w zebranych danych z badań o zasadniczym znaczeniu:
BOTOX 200 jednostek (n = 227) | Placebo (n = 241) | Poziom istotności p | |
Tygodniowa częstość przypadków nietrzymania moczu* Średnia wartość na początku badania Średnia zmiana w 2. tygodniu Średnia zmiana w 6. tygodniua Średnia zmiana w 12. tygodniu | 32,4 –17,7 –21,3 –20,6 | 31,5 –9,0 –10,5 –9,9 | p < 0,001 p < 0,001 p < 0,001 |
Maksymalna pojemność cystometryczna (ml) Średnia wartość na początku badania Średnia zmiana w 6. tygodniub | 250,2 +153,6 | 253,5 +11,9 | p < 0,001 |
Maksymalne ciśnienie wypieracza podczas pierwszego mimowolnego skurczu tego mięśnia (cm H2O) Średnia wartość na początku badania Średnia zmiana w 6. tygodniub | 51,5 –32,4 | 47,3 +1,1 | p < 0,001 |
Całkowita ocena jakości życia w aspekcie nietrzymania moczuc,d Średnia wartość na początku badania Średnia zmiana w 6. tygodniub Średnia zmiana w 12. tygodniu | 35,37 +25,89 +28,89 | 35,32 +11,15 +8,86 | p < 0,001 p < 0,001 |
* Odsetek pacjentów, u których nie doszło do incydentu nietrzymania moczu w całym 6. tygodniu
wyniósł 37% w grupie stosującej produkt leczniczy BOTOX 200 jednostek oraz 9% w grupie placebo. Odsetki osób, u których ograniczono epizody nietrzymania moczu o co najmniej 75% w porównaniu ze stanem początkowym, wynosiły odpowiednio 63% i 24%. Natomiast odsetki osób, u których
ograniczono takie epizody o co najmniej 50% w porównaniu ze stanem początkowym, wynosiły odpowiednio 76% i 39%.
a Pierwszorzędowy punkt końcowy
b Drugorzędowe punkty końcowe
c Całkowite wyniki w skali I-QOL mieszczą się w zakresie od 0 (maksymalne nasilenie problemu) do 100 (całkowity brak problemu).
d W badaniach o zasadniczym znaczeniu wstępnie określona najmniejsza istotna różnica (NIR) dla
całkowitego wyniku I-QOL wynosiła 8 punktów w oparciu o szacunkowe wartości NIR wynoszące od 4 do 11 punktów, jakie zgłaszano u pacjentów z neurogenną nadczynnością mięśnia wypieracza.
Mediana czasu trwania odpowiedzi w tych dwóch badaniach o zasadniczym znaczeniu obliczona w oparciu o żądania ponownej terapii wysuwane przez pacjentów wyniosła 256–295 dni (36-42 tygodnie) w grupie przyjmującej dawkę 200 jednostek w porównaniu z 92 dniami (13 tygodniami) w grupie placebo. W oparciu o wyniki badań pacjentów zgłaszających się do ponownego leczenia, którzy otrzymali produkt leczniczy BOTOX 100 jednostek w ramach otwartego badania
rozszerzonego (n = 174), średni czas trwania odpowiedzi na leczenie wynosił 253 dni (~ 36 tygodni). W przypadku wszystkich punktów końcowych skuteczności u pacjentów konsekwentnie występowała odpowiedź podczas ponownej terapii.
W badaniach o zasadniczym znaczeniu u żadnego z 475 pacjentów z neurogenną nadczynnością mięśnia wypieracza, od których zostały przeanalizowane próbki, nie doszło do rozwoju przeciwciał neutralizujących. Analiza próbek uzyskanych w ramach badań zasadniczych III fazy oraz w ramach otwartego badania rozszerzonego wykazała obecności przeciwciał neutralizujących u 3 z 300 pacjentów otrzymujących BOTOX w dawce 200 jednostek (1,0%) oraz u 5 z 258 pacjentów otrzymujących BOTOX w dawce 300 jednostek (1,9%). U czterech z pacjentów kontynuacja leczenia przyniosła skutek kliniczny. W odniesieniu do całkowitej liczby pacjentów, w grupie pacjentów,
u których wykryto przeciwciała neutralizujące, czas utrzymywania się odpowiedzi na leczenie był krótszy, a produkt leczniczy podawany był częściej (patrz punkt 4.4).
Badania po wprowadzeniu produktu leczniczego do obrotu
Przeprowadzono badanie kliniczne po wprowadzeniu produktu leczniczego do obrotu, prowadzone metodą ślepej próby z grupą kontrolną stosującą placebo przeprowadzono u pacjentów ze stwardnieniem rozsianym (MS), z nietrzymaniem moczu wskutek neurogennej nadczynności mięśnia wypieracza, u których choroby nie można było odpowiednio kontrolować za pomocą co najmniej jednego środka antycholinergicznego i którzy na początku badania nie byli cewnikowani. Pacjentów tych losowo przydzielono do grup otrzymujących 100 jednostek produktu leczniczego BOTOX
(n = 66) lub placebo (n = 78).
Obserwowano też znaczącą (w porównaniu z placebo) poprawę zgłaszanych przez pacjentów wyników w zakresie częstości przypadków nietrzymania moczu w pierwszorzędowym punkcie końcowym w 6 tygodniu u pacjentów otrzymujących BOTOX w dawce 100 jednostek, włączając odsetek pacjentów bez epizodów nietrzymania moczu. Zaobserwowano znaczącą poprawę parametrów urodynamiki oraz zgłaszanych przez pacjenta wyników jakości życia związanej
z nietrzymaniem moczu mierzonej za pomocą kwestionariusza I-QOL (ang. Incontinence Quality of Life [jakość życia związana z nietrzymaniem moczu]), obejmującego zachowania ograniczające unikanie, oddziaływanie psychosocjologiczne oraz skrępowanie w sytuacjach wymagających kontaktów społecznych.
Wyniki badań po wprowadzeniu produktu leczniczego do obrotu przedstawiono poniżej:
Pierwszorzędowe i drugorzędowe punkty końcowe w badaniach po wprowadzeniu produktu leczniczego BOTOX 100 jednostek do obrotu u pacjentów chorych na MS, którzy na początku badania nie byli cewnikowani:
BOTOX 100 jednostek (n = 66) | Placebo (n = 78) | Poziom istotności p |
Tygodniowa częstość przypadków nietrzymania moczu* Średnia wartość na początku badania Średnia zmiana w 2. tygodniu Średnia zmiana w 6. tygodniua Średnia zmiana w 12. tygodniu | 4,2 –2,9 –3,3 –2,8 | 4,3 –1,2 –1,1 –1,1 | p < 0,001 p < 0,001 p < 0,001 |
Maksymalna pojemność cystometryczna (ml) Średnia wartość na początku badania Średnia zmiana w 6. tygodniub | 246,4 +127,2 | 245,7 -1,8 | p < 0,001 |
Maksymalne ciśnienie wypieracza podczas pierwszego mimowolnego skurczu tego mięśnia (cm H2O) Średnia wartość na początku badania Średnia zmiana w 6. tygodniub | 35,9 –19,6 | 36,1 +3,7 | p < 0,007 |
Całkowita ocena jakości życia w aspekcie nietrzymania moczuc,d Średnia wartość na początku badania Średnia zmiana w 6. tygodniub Średnia zmiana w 12. tygodniu | 32,4 +40,4 +38,8 | 34,2 +9,9 +7,6 | p < 0,001 p < 0,001 |
* Odsetek pacjentów, u których nie doszło do incydentu nietrzymania moczu w całym 6. tygodniu wyniósł 53% w grupie stosującej produkt leczniczy BOTOX 100 jednostek oraz 10,3% w grupie placebo.
a Pierwszorzędowy punkt końcowy
b Drugorzędowe punkty końcowe
c Całkowite wyniki w skali I-QOL mieszczą się w zakresie od 0 (maksymalne nasilenie problemu) do 100 (całkowity brak problemu).
d W badaniach o zasadniczym znaczeniu wstępnie określona najmniejsza istotna różnica (NIR) dla całkowitego wyniku I-QOL wynosiła11 punktów w oparciu o szacunkowe wartości NIR wynoszące od 4 do 11 punktów, jakie zgłaszano u pacjentów z neurogenną nadczynnością mięśnia wypieracza.
Średni czas trwania odpowiedzi na leczenie w oparciu o żądania ponownej terapii wysuwane przez pacjentów wynosił 362 dni (~ 52 tygodnie) w grupie pacjentów otrzymujących BOTOX w dawce 100 jednostek, w porównaniu do 88 dni (~ 13 tygodni) w grupie pacjentów otrzymujących placebo.
Neurogenna nadczynność mięśnia wypieracza u dzieci i młodzieży
Przeprowadzono jedno randomizowane, wieloośrodkowe badanie kliniczne z podwójnie ślepą próbą w grupach równoległych (191622-120) u pacjentów w wieku od 5 do 17 lat z nietrzymaniem moczu
z powodu nadczynności mięśnia wypieracza związanej ze stanem neurologicznym i stosujących czyste okresowe cewnikowanie pęcherza moczowego. Zbadano łącznie 113 pacjentów (w tym 99
z dysrafizmem kręgosłupa, takim jak rozszczep kręgosłupa, 13 z urazem rdzenia kręgowego i 1 z poprzecznym zapaleniem rdzenia kręgowego), u których nie uzyskano wystarczającej
odpowiedzi na co najmniej jeden lek antycholinergiczny lub u których wystąpiła nietolerancja na ten lek. Pacjentów losowo przydzielono do grup otrzymujących 50 jednostek, 100 jednostek lub
200 jednostek produktu leczniczego, z równoczesnym ograniczeniem do 6 jednostek/kg masy ciała. Pacjenci otrzymujący dawkę mniejszą niż dawka randomizowana ze względu na ograniczenie do
Uważa się, że po zastosowaniu dawek leczniczych produktu BOTOX, następuje niewielka dystrybucja systemowa. Badania kliniczne z użyciem techniki elektromiografii, wykazały zwiększoną elektrofizjologiczną aktywność nerwowo-mięśniową w mięśniach odległych od miejsca wstrzyknięcia, któremu nie towarzyszyły żadne objawy kliniczne.
Badania nad reprodukcją
Po podaniu domięśniowo produktu BOTOX ciężarnym myszom, szczurom i królikom podczas okresu organogenezy, najwyższa dawka niewywołująca skutków szkodliwych (NOAEL), wynosiła odpowiednio 4, 1 i 0,125 jednostek/kg. Większe dawki były związane z obniżeniem masy ciała płodu i (lub) opóźnionym kostnieniem, a u królików obserwowano poronienia.
Inne badania
Oprócz badań nad toksycznością w okresie płodowym, zostały przeprowadzone następujące badania nad bezpieczeństwem stosowania produktu BOTOX: ostrej toksyczności, toksyczności po podaniu wielokrotnym, miejscowej tolerancji, mutagenności, antygenowości, zgodności z ludzką krwią.
Badania te nie wykazały istotnego ryzyka dla ludzi po podaniu odpowiednich dawek klinicznych.
W leczeniu kończyny dolnej pacjentów pediatrycznych maksymalna skumulowana dawka w okresie 3 miesięcy nie powinna zasadniczo przekraczać 6,0 jednostek / kg masy ciała lub 200 jednostek,
w zależności od tego, która wartość jest mniejsza.
Według opublikowanych danych, dawka LD50 podana domięśniowo młodocianym małpom, wynosi 39 jednostek/kg.
W badaniu, w którym młode szczury otrzymywały domięśniowe iniekcje produktu leczniczego BOTOX co dwa tygodnie od 21 dnia po urodzeniu przez 3 miesiące w dawkach 8, 16 lub 24 jednostek/kg, obserwowano zmiany wielkości/geometrii kości związane ze zmniejszoną gęstością kości i masą kostną wtórnymi do nieużywania kończyny, brak skurczów mięśni i zmniejszenie przyrostu masy ciała. Zmiany były mniej poważne przy najniższej badanej dawce, z oznakami odwracalności przy wszystkich poziomach dawek. Dawka, przy której nie obserwowano działań niepożądanych u młodych zwierząt (8 jednostek/kg), jest podobna do maksymalnej dawki dla dorosłych (400 jednostek) i niższa niż maksymalna dawka u dzieci (340 jednostek) przy przeliczeniu na kg masy ciała (kg).
Po pojedynczym wstrzyknięciu szczurom mniej niż 50 jednostek produktu leczniczego BOTOX na 1kg masy ciała nie zaobserwowano żadnej toksyczności układowej. W celu zasymulowania niezamierzonego wstrzyknięcia, pojedyncza dawka produktu leczniczego BOTOX (ok.
powtarzanych dawek podawanych do mięśnia wypieracza (4 wstrzyknięcia) opadanie powieki zaobserwowano po dawce 24 jednostki/kg a umieralność po dawkach ≥ 24 jednostki/kg.
Degenerację/regenerację włókien mięśniowych obserwowano w mięśniu szkieletowym u zwierząt, którym podawano dawki co najmniej 24 jednostki/kg. Te zmiany miopatyczne uznawano za wtórne skutki narażenia ogólnoustrojowego. Ponadto zaobserwowano degenerację włókien mięśniowych
u jednego zwierzęcia, któremu podano 12 jednostek/kg. W przypadku tego zwierzęcia zmiana chorobowa miała minimalne nasilenie i nie została uznana za powiązaną z jakimikolwiek objawami klinicznymi. Nie można było z całą pewnością określić, czy miało to związek z terapią produktem leczniczym BOTOX. Dawka 12 jednostek/kg odpowiadała trzykrotnie większemu narażeniu na BOTOX niż po zalecanej dawce klinicznej wynoszącej 200 jednostek w przypadku nietrzymania moczu wskutek neurogennej nadczynności mięśnia wypieracza (na podstawie masy ciała człowieka wynoszącej 50 kg).
Albumina ludzka Sodu chlorek
Ponieważ nie ma badań dotyczących niezgodności, niniejszy produkt leczniczy nie powinien być mieszany z innymi produktami leczniczymi.
3 lata
Wykazano, że po rekonstytucji roztwór jest stabilny przez 24 godziny w temperaturze 2oC - 8oC.
Fiolka z bezbarwnego szkła o pojemności 10 ml zamknięta korkiem z gumy chlorobutylowej i aluminiowym kapslem zabezpieczającym.
W opakowaniu znajduje się 1, 2, 3 lub 6 fiolek w tekturowym pudełku. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Produkt leczniczy BOTOX przygotowuje się przez dodanie jałowego, pozbawionego konserwantów roztworu soli fizjologicznej (0,9% roztwór chlorku sodu do wstrzykiwań). Zaleca się przygotowanie roztworu i napełnianie strzykawek na papierowych, laminowanych plastikiem serwetach w celu uniknięcia przypadkowych zanieczyszczeń otoczenia.
Jeśli podczas jednego zabiegu używane są różne wielkości fiolek produktu leczniczego BOTOX należy zwrócić uwagę, aby użyć właściwej ilości rozcieńczalnika, w celu uzyskania odpowiedniej liczby jednostek w 0,1 ml. Ilość rozcieńczalnika niezbędna do rekonstytucji produktu jest różna dla poszczególnych mocy. Każda strzykawka powinna być odpowiednio oznakowana.
Instrukcja rozcieńczania dla fiolek 100 i 200 jednostek we wszystkich wskazaniach, z wyjątkiem leczenia zaburzeń pęcherza moczowego;
Fiolka 100 jednostek | Fiolka 200 jednostek | |
Uzyskana dawka | Objętość dodanego | Objętość dodanego |
(w jednostkach na | rozpuszczalnika | rozpuszczalnika |
0,1 ml) | (jałowy 0,9% chlorek | (jałowy 0,9% chlorek |
sodu niezawierający | sodu niezawierający | |
środków | środków | |
konserwujących) do | konserwujących) do | |
fiolki 100 jednostek | fiolki 200 jednostek | |
20 jednostek | 0,5 ml | 1 ml |
10 jednostek | 1 ml | 2 ml |
5 jednostek | 2 ml | 4 ml |
4 jednostki | 2,5 ml | 5 ml |
2,5 jednostek | 4 ml | 8 ml |
1,25 jednostek | 8 ml | N/A |
Idiopatyczna nadreaktywność pęcherza moczowego
W przypadku tego wskazania zaleca się stosowanie fiolek zawierających 100 jednostek produktu leczniczego BOTOX ze względu na łatwiejszą rekonstytucję.
Instrukcja rozcieńczania w przypadku fiolek zawierających 100 jednostek
W ten sposób uzyskuje się ogółem 200 jednostek produktu leczniczego BOTOX po rekonstytucji
w 3 strzykawkach po 10 ml. Wykorzystać natychmiast po rekonstytucji. Resztki niewykorzystanego roztworu soli fizjologicznej należy usunąć.
Produkt leczniczy BOTOX jest przeznaczony wyłącznie do jednorazowego użycia, wszelkie pozostałości niewykorzystanego roztworu należy zniszczyć.
BOTOX może ulec denaturacji w wyniku tworzenia pęcherzyków lub gwałtownych ruchów podczas rozpuszczania proszku, dlatego sól fizjologiczną należy powoli wstrzykiwać do fiolki. Jeżeli po przekłuciu korka sól fizjologiczna nie jest zasysana przez podciśnienie fiolki, należy fiolkę zniszczyć. Odtworzony roztwór jest przejrzysty lub lekko żółtawy bez cząstek stałych. Przed zastosowaniem należy obejrzeć czy roztwór jest przezroczysty i czy nie zawiera cząstek stałych. Po rekonstytucji produkt może być przechowywany do 24 godzin w lodówce (2C - 8C). Jeżeli roztwór nie zostanie zużyty w ciągu 24 godzin należy go zniszczyć.
Do fiolek z niewykorzystaną toksyną należy dodać niewielką ilość wody, a następnie włożyć do autoklawu. Wszystkie zużyte fiolki, strzykawki itp. powinny być również autoklawowane.
Niewykorzystany roztwór można też inaktywować przez dodanie roztworu podchlorynu (0,5%) na 5 minut.
Wszelkie niewykorzystane resztki produktu lub jego odpady należy usunąć w sposób zgodny z lokalnymi przepisami.
AbbVie Sp. z o.o. ul. Postępu 21B 02-676 Warszawa
R/6748
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 9 lipca 1996 Data ostatniego przedłużenia pozwolenia: 21 maja 2014
02/2023







