







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO Berinert 500
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMER (-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO Berinert 500
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Berinert 500 powinien być bezbarwny i przezroczysty
Berinert 1500 powinien być bezbarwny i przezroczysty do lekko opalizującego.
Roztwór powinien być podawany powoli we wstrzyknięciu dożylnym, Berinert 500 może być podany
we wlewie (4 ml/min).
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność do prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
zestaw do wkłucia,
waciki nasączone alkoholem,
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Roztwór produktu Berinert 500 powinien być bezbarwny lub przezroczysty.
Roztwór produktu Berinert 1500 powinien być bezbarwny lub przezroczysty do lekko
opalizującego.
Po filtracji/opróżnieniu fiolki (patrz poniżej) produkt leczniczy po rekonstytucji przed podaniem powinien być poddany wizualnej ocenie; należy sprawdzić czy nie pojawiły się cząstki stałe i czy nie nastąpiła zmiana zabarwienia.
Nie stosować mętnych roztworów i takich, które zawierają osad.
Podczas rekonstytucji i opróżniania fiolki muszą być zachowane aseptyczne warunki. Należy
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMER (-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
500 j.m.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań/ do infuzji.
Berinert 1500
1500 j.m.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Substancja czynna: ludzki inhibitor C1-esterazy (z ludzkiego osocza).
W jednej fiolce Berinert 500 znajduje się 500 j.m. inhibitora C1-esterazy.
W jednej fiolce Berinert 1500 znajduje się 1500 j.m. inhibitora C1-esterazy.
Aktywność ludzkiego inhibitora C1-esterazy jest wyrażona w jednostkach międzynarodowych (j.m.), które odnoszą się do aktualnych standardów WHO dotyczących produktów inhibitora C1-esterazy.
Po rekonstytucji w 10 ml wody do wstrzykiwań Berinert 500 zawiera 50 j.m./ml ludzkiego inhibitora C1-esterazy.
Po rekonstytucji w 3 ml wody do wstrzykiwań Berinert 1500 zawiera 500 j.m./ml ludzkiego inhibitora C1-esterazy.
Zawartość białka całkowitego w roztworze po rekonstytucji produktu 500 j.m. wynosi 6,5 mg/ml. Zawartość białka całkowitego w roztworze po rekonstytucji produktu 1500 j.m. wynosi 65 mg/ml.
Substancje pomocnicze o znanym działaniu:
Sód do 486 mg (około 21 mmol) na 100 ml roztworu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Berinert 500:
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań/ do infuzji. Berinert 1500:
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Biały proszek.
Przeźroczysty, bezbarwny rozpuszczalnik.
Wrodzony obrzęk naczynioruchowy typu I i II (hereditary angioedema – HAE).
Leczenie i przedzabiegowe zapobieganie stanom ostrym.
Leczenie powinno zostać rozpoczęte pod kontrolą lekarza doświadczonego w terapii niedoboru inhibitora C-1 esterazy.
Dawkowanie
Dorośli
Leczenie ostrego napadu obrzęku naczynioruchowego:
20 j.m. na kilogram masy ciała (20 j.m./kg m.c.)
Przedzabiegowe zapobieganie wystąpieniu obrzęku naczynioruchowego:
1000 j.m. na mniej niż 6 godzin przed zabiegiem medycznym, stomatologicznym lub chirurgicznym.
Dzieci i młodzież
Leczenie ostrego napadu obrzęku naczynioruchowego:
20 j.m. na kilogram masy ciała (20 j.m./kg m.c.)
Przedzabiegowe zapobieganie wystąpieniu obrzęku naczynioruchowego:
15 do 30 j.m. na kilogram masy ciała (15-30 j.m./kg m.c.) na mniej niż 6 godzin przed zabiegiem medycznym, stomatologicznym lub chirurgicznym. Dawka powinna być dobrana z uwzględnieniem okoliczności klinicznych (np. typ zabiegu i ciężkość schorzenia).
Sposób podawania
Berinert musi być rekonstytuowany zgodnie z informacją podaną w punkcie 6.6. Roztwór po rekonstytucji:
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
U pacjentów z potwierdzoną skłonnością do alergii należy profilaktycznie stosować leki
antyhistaminowe i kortykosteroidy.
Jeżeli wystąpią objawy alergiczne lub reakcja anafilaktyczna, należy natychmiast zaprzestać podawania leku Berinert (tzn. przerwać wstrzyknięcie/infuzję) i rozpocząć odpowiednie leczenie. Postępowanie lecznicze zależy od rodzaju i stopnia nasilenia działań niepożądanych. Powinno zostać włączone leczenie przeciwwstrząsowe zgodne z aktualnymi standardami.
Pacjenci z obrzękiem krtani wymagają szczególnej obserwacji z zabezpieczeniem możliwości
natychmiastowego intensywnego leczenia w szpitalu.
Użycie niezgodnie z zarejestrowanymi wskazaniami lub w leczeniu zespołu przeciekania włośniczkowego (Capillary Leak Syndrome, CLS) nie jest polecane (patrz także punkt 4.8 „Działania niepożądane”).
Produkt leczniczy Berinert 500 zawiera 49 mg sodu na fiolkę co odpowiada 2.5% zalecanej przez
WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych.
Produkt leczniczy Berinert 1500 zawiera mniej niż 1 mmol (23 mg) sodu na fiolkę, to znaczy lek
uznaje się za „wolny od sodu.
Leczenie domowe i samodzielne podawanie
Istnieją ograniczone dane dotyczące podawania tego produktu leczniczego w domu lub samodzielnego podawania.
Potencjalne ryzyko towarzyszące terapii domowej jest związane z samodzielnym podawaniem jak
i radzeniem sobie z działaniami niepożądanymi, w szczególności z nadwrażliwością.
Decyzja odnośnie zastosowania terapii domowej dla indywidualnego pacjenta powinna być podjęta przez lekarza prowadzącego, który powinien zapewnić odpowiednie przeszkolenie pacjenta oraz weryfikację terapii domowej w odstępach czasu.
Bezpieczeństwo wirusowe
Standardowe postępowanie zabezpieczające przed przenoszeniem zakażeń poprzez podawanie produktów leczniczych otrzymywanych z krwi lub osocza ludzkiego obejmuje: selekcję dawców, badanie pojedynczych donacji i pul osocza na obecność markerów wirusów oraz zastosowanie skutecznych metod inaktywacji/usuwania wirusów podczas procesu wytwarzania. Pomimo tego, nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych po podaniu produktu leczniczego pochodzącego z krwi lub osocza ludzkiego. To ryzyko dotyczy nieznanych lub nowo odkrytych wirusów i innych czynników zakaźnych.
Podjęte środki zabezpieczające są uważane za skuteczne w stosunku do otoczkowych wirusów takich
jak: HIV, HBV, HCV oraz wirusów bezotoczkowych HAV i parwowirusa B 19.
U pacjentów regularnie lub wielokrotnie otrzymujących produkty pochodzące z ludzkiego osocza należy rozważyć dodatkowo szczepienia przeciwko wirusowemu zapaleniu wątroby typu A i B.
Zdecydowanie zaleca się, aby przy każdorazowym podaniu pacjentowi produktu leczniczego Berinert
odnotować nazwę i numer serii produktu leczniczego, aby móc powiązać pacjenta z daną serią leku.
Nie przeprowadzono badań dotyczących interakcji.
Ciąża
Istnieje ograniczona ilość danych stwierdzających brak zwiększonego ryzyka związanego ze stosowaniem Berinert u kobiet w ciąży. Berinert jest fizjologicznym składnikiem ludzkiego osocza. Z tego powodu nie przeprowadzano badań w kierunku toksyczności Berinert w okresie ciąży i rozwoju płodowego u zwierząt oraz nie należy spodziewać się wystąpienia działań niepożądanych związanych z płodnością oraz rozwojem przed- i pourodzeniowym u ludzi. Dlatego też u kobiet w ciąży Berinert powinien być stosowany tylko w przypadku, jeśli istnieją ścisłe wskazania.
Karmienie piersią
Nie jest znany stopień przenikania Berinert do ludzkiego mleka, jednak zważywszy jego wysoką masę cząsteczkową prawdopodobieństwo wydzielania z mlekiem matki jest bardzo niskie. Jednakże karmienie piersią u kobiet cierpiących z powodu wrodzonego obrzęku naczynioruchowego jest niewskazane. Biorąc pod uwagę zalety karmienia piersią dla dziecka oraz korzyści wynikające z terapii Berinert dla matki należy rozważyć czy zalecić zaprzestanie karmienia piersią, czy zaprzestać terapii przy użyciu Berinert.
Płodność
Berinert jest fizjologicznym składnikiem ludzkiego osocza. W związku z tym nie przeprowadzono badań na zwierzętach dotyczących wpływu na reprodukcję i toksycznego wpływu na rozwój i nie oczekuje się niekorzystnego działania na płodność, rozwój płodu i noworodka u ludzi.
Berinert nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów
i obsługiwania maszyn.
W oparciu o doświadczenia po wprowadzeniu do obrotu i naukowe piśmiennictwo podział reakcji ze względu na częstość występowania przedstawia się w sposób następujący:
bardzo często: ≥ 1/10 często: ≥ 1/100 do< 1/10
niezbyt często: ≥ 1/1 000 do < 1/100 rzadko: ≥ 1/10 000 do < 1/1 000
bardzo rzadko: < 1/10 000 (w tym zawarte są pojedyncze przypadki) Działania niepożądane związane z zastosowaniem Berinert występują rzadko.
Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko | |
Zaburzenia naczyniowe: | Rozwój zakrzepicy* | ||||
Zaburzenia ogólne i stany w miejscu podania leku: | Wzrost temperatury i/lub reakcje w miejscu wkłucia. | ||||
Zaburzenia układu immunologicznego: | Reakcje alergiczne lub anafilaktyczne (np. tachykardia, hiper- lub hipotensja, zaczerwienienie, pokrzywka, duszność, bóle i zawroty głowy, nudności). | Wstrząs |
* w próbach leczenia wysokimi dawkami Berinert w profilaktyce i terapii zespołu przeciekania włośniczkowego (Capillary Leak Syndrome, CLS), przed, w czasie lub po operacji kardiochirurgicznej z użyciem krążenia pozaustrojowego (wskazanie i dawka niezarejestrowane), w pojedynczych przypadkach, zakończoną zgonem.
Możliwość przeniesienia czynników zakaźnych przedstawiono w punkcie 4.4.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie zgłoszono przypadków przedawkowania.
Grupa farmakoterapeutyczna: C1-inhibitor
ATC kod: B06AC01
Inhibitor C1-esterazy jest osoczową glikoproteiną o masie cząsteczkowej 105 kDa zawierającą 40% węglowodanów. Stężenie w ludzkim osoczu wynosi około 240 mg/l. Poza osoczem inhibitor C1- esterazy wykrywany jest także w łożysku, komórkach wątroby, monocytach i płytkach krwi.
Inhibitor C1-esterazy należy do ludzkiego osoczowego układu inhibitorów proteaz serynowych (serpin) i działa jak inne białka tej grupy, takie jak: antytrombina III, alpha-2-antyplazmina, alpha-1- antytrypsyna i inne.
W warunkach fizjologicznych inhibitor C1-esterazy blokuje klasyczną drogę układu dopełniacza poprzez inaktywację aktywnych enzymatycznie składników C1s i C1r. Aktywny enzym tworzy kompleks z inhibitorem w stechiometrycznym stosunku 1:1.
Poza tym inhibitor C1-esterazy stanowi najważniejszy inhibitor aktywatorów układu krzepnięcia poprzez hamowanie czynnika XIIa i jego fragmentów. Dodatkowo, oprócz alpha-2-makroglobuliny, jest głównym inhibitorem osoczowej kallikreiny.
Efekt leczniczy produktu Berinert we wrodzonym obrzęku naczynioruchowym polega na substytucji brakującej aktywności inhibitora C1-esterazy.
Produkt podawany jest drogą dożylną i natychmiast po podaniu osiąga w osoczu stężenie zależne od
dawki leku.
Właściwości farmakokinetyczne produktu leczniczego Berinert oceniano w dwóch badaniach.
Badanie I fazy zostało przeprowadzone u 15 zdrowych, dorosłych pacjentów i otrzymane dane farmakokinetyczne zostały użyte do oceny biodostępności produktu leczniczego Berinert 1500 i Berinert 500. Została wykazana porównywalna biodostępność obu dawek produktu leczniczego Berinert. Dla stężeń Cmax. i AUC 0-last. antygenu C1-INH stosunek średniej geometrycznej (90%Cis) wynosił odpowiednio 1,02 (0,99; 1,04) oraz 1,02 (0,99; 1,05). Okres półtrwania został określony na podgrupie pacjentów z zastosowaniem bezprzedziałowej analizy farmakokinetycznej. Średni okres półtrwania dla Berinert 1500 oraz Berinert 500 wynosił odpowiednio 87,7 i 91,4 godziny.
Właściwości farmakokinetyczne były badane u pacjentów z wrodzonym obrzękiem naczynioruchowym (34 pacjentów > 18 lat, 6 pacjentów < 18 lat). Dotyczyły one 15 pacjentów leczonych profilaktycznie (z częstymi/ciężkimi atakami), a także 25 pacjentów z mniej częstymi/łagodnymi atakami i leczonymi „na żądanie”. Dane zostały uzyskane w przerwach pomiędzy atakami.
Średnia poprawy in vivo (in vivo recovery, IVR) wynosiła 86,7% (zakres: 54,0-254,1%), przy czym była nieznacznie wyższa u dzieci 98,2% (zakres: 69,2- 106,8%), w porównaniu z dorosłymi 82,5 % (zakres: 54,0-254,1%). U pacjentów z ciężkimi atakami, średnia IVR była jeszcze wyższa i wynosiła 101,4 % w porównaniu z grupą pacjentów z łagodnymi atakami choroby 75,8 % (zakres: 57,2- 195,9%).
Średnia wzrostu aktywności wynosiła 2,3%/j.m./kg mc (zakres: 1,4- 6,9%/j.m./kg mc). Nie stwierdzono statystycznie znamiennych różnic pomiędzy dziećmi i dorosłymi. U pacjentów, u których występowały ciężkie ataki obserwowano nieznacznie wyższy wzrost aktywności (2,9%/j.m./kg mc, zakres: 1,4-6,9%/j.m./kg mc), w porównaniu z grupą pacjentów z łagodnymi atakami (2,1%/j.m./kg mc, zakres: 1,5- 5,1 %/j.m./kg mc).
Maksymalne stężenie ludzkiego inhibitora C1-esterazy w osoczu uzyskiwano w ciągu 0,8 godziny po
podaniu Berinert (bez istotnych różnic między poszczególnymi grupami pacjentów).
Średni okres półtrwania wynosił 36,1 godzin i był nieznacznie krótszy u dzieci w porównaniu z grupą dorosłych (32,9 vs. 36,1 godzin) oraz w grupie pacjentów z ciężkimi atakami w porównaniu z grupą pacjentów z łagodnymi atakami (30,9 vs. 37,0 godzin).
Berinert zawiera aktywny składnik ludzki inhibitor C1-esterazy. Otrzymywany jest on z ludzkiego osocza i działa jak endogenny składnik osocza. W badaniach na zwierzętach nie wykazano toksycznego działania ani po podaniu pojedynczej dawki Berinert u szczurów i myszy, ani po podaniu wielokrotnym dawki tego produktu szczurom.
Nie prowadzono na modelach zwierzęcych badań przedklinicznych dotyczących wpływu wielokrotnych podań leku na proces rozmnażania oraz działania kancerogennego, z uwagi na powstawanie przeciwciał do heterogennego ludzkiego białka.
Test in vitro Ouchterlony’ego i model in vivo PCA u świnek morskich nie wykazał dowodów na nowo powstające w Berinert determinanty antygenowe będące skutkiem pasteryzacji.
Badania zakrzepowości In-vivo przeprowadzono na królikach z zastosowaniem dawek produktu leczniczego Berinert do 800 j.m./kg. Zastosowaniu produktu leczniczego Berinert we wstrzyknięciach dożylnych, w dawkach do 800 j.m./kg nie towarzyszyło żadne ryzyko zakrzepicy.
Badania miejscowej tolerancji przeprowadzane na królikach wykazały dobrą tolerancję kliniczną, miejscową i histologiczną po podaniu dożylnym, podskórnym, dotętniczym oraz domięśniowym.
Proszek:
Glicyna Sodu chlorek
Sodu cytrynian
Rozpuszczalnik:
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
36 miesięcy
Po rekonstytucji stabilność fizyko-chemiczna Berinert 500 jest zachowana przez 48 godzin w temperaturze pokojowej (maks. 30ºC). Dla Berinert 1500, stabilność fizyko-chemiczna jest zachowana przez 48 godzin w temperaturze pokojowej (maks. 25 °C). Z mikrobiologicznego punktu widzenia oraz z uwagi na to, że Berinert nie zawiera środków konserwujących, produkt po rekonstytucji powinien być zużyty natychmiast. Jeżeli produkt nie został natychmiast podany, czas przechowywania w temperaturze pokojowej nie może przekroczyć 8 godzin. Produkt leczniczy po rekonstytucji powinien być przechowywany wyłącznie w fiolce.
Nie przechowywać w temperaturze powyżej 30 °C. Nie zamrażać.
Przechowywać fiolkę w opakowaniu zewnętrznym w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Opakowanie bezpośrednie Berinert 500:
Proszek (500j.m.) w fiolce ze szkła typu II, zamkniętej korkiem z gumy bromobutylowej z aluminiowym kapslem w kolorze starego złota i limonkowym plastikowym wieczkiem typu flip-off.
Rozpuszczalnik (10 ml) w fiolce ze szkła typu I, zamkniętej korkiem z gumy chlorobutylowej lub bromobutylowej z niebieskim aluminiowym kapslem i niebieskim plastikowym wieczkiem typu flip- off.
Berinert 1500:
Proszek (1500j.m.) w fiolce ze szkła typu I, zamkniętej korkiem z gumy bromobutylowej z niebieskim aluminiowym kapslem i pomarańczowym plastikowym wieczkiem typu flip-off.
Rozpuszczalnik (3 ml) w fiolce ze szkła typu I, zamkniętej korkiem z gumy chlorobutylowej lub bromobutylowej z niebieskim aluminiowym kapslem i pomarańczowym plastikowym wieczkiem typu flip-off.
Dostępne opakowania:
Opakowanie zawiera:
1 fiolka z proszkiem
1 fiolka z rozpuszczalnikiem (Berinert 500: 10 ml, Berinert 1500: 3 ml) 1 system do transferu 20/20 z filtrem
Zestaw do podawania (opakowanie wewnętrzne)
1 strzykawka jednorazowego użytku o pojemności (Berinert 500: 10 ml, Berinert 1500: 5ml),
1 niejałowy plaster.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Sposób podawania
Wskazówki ogólne
używać strzykawkę dołączoną do produktu.
Rekonstytucja
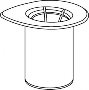
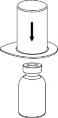
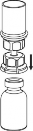

Doprowadzić rozpuszczalnik do temperatury pokojowej. Zdjąć z fiolek zawierających proszek i rozpuszczalnik plastikowe wieczka i przemyć korki w miejscach wkłucia aseptycznym roztworem. Po wyschnięciu otworzyć zestaw zawierający Mix2Vial.
1 | 1. Otworzyć opakowanie zawierające Mix2Vial poprzez usunięcie wieczka. Nie wyjmować Mix2Vial z blistra. |
2 | 2. Umieścić fiolkę z rozpuszczalnikiem na równej, czystej powierzchni i mocno przytrzymać. Nie wyjmując z blistra zestawu Mix2Vial nałożyć jego niebieską końcówkę z ostrzem na korek fiolki rozpuszczalnika i naciskając pionowo w dół przebić korek fiolki rozpuszczalnika. |
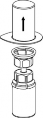
3 | 3. Przytrzymując krawędź zestawu Mix2Vial ostrożnie zdjąć blister pociągając go pionowo do góry. Należy zwrócić uwagę, aby zdjąć jedynie blister a nie cały zestaw Mix2Vial. |
4 | 4. Umieścić fiolkę z proszkiem na równej i twardej powierzchni. Odwrócić do góry dnem fiolkę z rozpuszczalnikiem i dołączonym do niej zestawem Mix2Vial i wbić w korek fiolki z proszkiem, ruchem pionowo w dół, ostrze przezroczystego końca. Rozpuszczalnik samoczynnie zostanie przeniesiony do fiolki z proszkiem. |
5 | 5. Jedną ręką chwycić część zestawu Mix2Vial w fiolce zawierającej obecnie roztwór, drugą zaś ręką przytrzymać część łącznika od strony fiolki po rozpuszczalniku i ostrożnie odkręcając oddzielić od siebie obie części łącznika. Usunąć fiolkę po rozpuszczalniku z przyczepionym do niej niebieskim końcem zestawu Mix2Vial. |
6 | 6. Fiolkę z doczepionym przezroczystym końcem zestawu Mix2Vial, zawierającą roztwór, łagodnie mieszać ruchem wirowym do całkowitego rozpuszczenia leku. Nie wstrząsać. |

7 | 7. Nabrać powietrza do pustej, jałowej strzykawki. Stosować strzykawkę dołączoną do produktu. Trzymając fiolkę z produktem leczniczym pionowo korkiem do góry, przyłączyć strzykawkę do połączenia Luer Lock zestawu Mix2Vial. Wstrzyknąć powietrze do fiolki z produktem. |
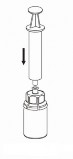

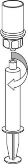
Pobieranie i sposób podawania
8 | 8. Przytrzymując tłok strzykawki odwrócić fiolkę wraz ze strzykawką do góry dnem i nabrać roztwór do strzykawki, powoli odciągając tłok. |
9 | 9. Po napełnieniu strzykawki roztworem, mocno uchwycić cylinder strzykawki (utrzymując strzykawkę tłokiem do dołu) odłączyć od niej przezroczysty koniec zestawu Mix2Vial. |
DOPUSZCZENIE DO OBROTU
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg Niemcy
500 j.m. - pozwolenie nr: 15705
1500 j.m. - pozwolenie nr: 22352
PRZEDŁUŻENIA POZWOLENIA
500 j.m. - Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 17.06.2009 r. 500 j.m. - Data ostatniego przedłużenia pozwolenia: 20.01.2014 r.
1500 j.m. - Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:18.03.2015 r.
1500 j.m. - Data ostatniego przedłużenia pozwolenia: 13.02.2020 r.
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Październik 2021







