







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTRYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
zakażenia dolnych dróg oddechowych, w tym zapalenie płuc, wywołane przez Streptococcus pneumoniae (o obniżonej wrażliwości na penicylinę), Escherichia coli, Klebsiella spp., Haemophilus influenzae (w tym szczepy oporne na ampicylinę), Proteus mirabilis, Serratia marcescens i Enterobacter spp.;
zakażenia układu moczowego wywołane m.in. przez Citrobacter spp., Enterobacter spp., Escherichia coli, Klebsiella spp., Proteus mirabilis, Proteus vulgaris, Morganella morganii, Providencia rettgeri i Serratia marcescens; również niepowikłane przypadki rzeżączki, w tym wywołane przez szczepy Neisseria gonorrhoeae wytwarzające penicylinazy;
zakażenia w obrębie miednicy mniejszej, w tym wywołane przez Escherichia coli, Proteus mirabilis, beztlenowe ziarenkowce - Peptostreptococcus spp. i Peptococcus spp. oraz niektóre szczepy Bacteroides spp. w tym B. fragilis, Clostridium spp.;
posocznica wywołana m.in. przez szczepy Escherichia coli, Klebsiella spp., Serratia marcescens;
zakażenia skóry i tkanek miękkich wywołane przez Escherichia coli, Enterobacter spp., Klebsiella spp., Proteus mirabilis, Proteus vulgaris, Morganella morganii, Providencia rettgeri, Pseudomonas spp., Serratia marcescens, Bacteroides spp. i beztlenowe ziarenkowce - Peptostreptococcus spp. i Peptococcus spp.;
zakażenia wewnątrz jamy brzusznej, w tym zapalenie otrzewnej, wywołane przez Escherichia coli, Klebsiella spp., Bacteroides spp. oraz Peptostreptococcus spp. i Peptococcus spp.;
zakażenia kości i stawów, w tym wywołane przez szczepy Haemophilus influenzae, Streptococcus spp., Proteus mirabilis;
zakażenia ośrodkowego układu nerwowego, w tym zapalenie opon mózgowo-rdzeniowych, wywołane przez Neisseria meningitidis, Haemophilus influenzae, Streptococcus pneumoniae, Klebsiella pneumoniae i Escherichia coli;
zapobieganiu zakażeniom okołooperacyjnym.
Cefotaksym z antybiotykami aminoglikozydowymi wykazuje in vitro synergizm działania wobec bakterii Gram-ujemnych, m.in. niektórych szczepów Pseudomonas aeruginosa.
Należy uwzględnić oficjalne, krajowe wytyczne dotyczące oporności bakterii oraz prawidłowego stosowania leków przeciwbakteryjnych.
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na cefotaksym lub inne cefalosporyny. U osób uczulonych na penicyliny może wystąpić reakcja alergiczna na cefalosporyny (nadwrażliwość krzyżowa) (patrz punkt 4.4).
Cefotaksymu rozpuszczonego w roztworze lidokainy nigdy nie należy podawać w następujących przypadkach:
nadwrażliwość na lidokainę lub inne amidowe leki do znieczulenia miejscowego;
blok serca (bez rozrusznika);
ciężka niewydolność mięśnia sercowego;
dożylna droga podania;
niemowlęta w wieku poniżej 30 miesięcy.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
hydroliza z udziałem β-laktamaz; cefotaksym może być hydrolizowany przez niektóre β-laktamazy o rozszerzonym spektrum substratowym, może być również hydrolizowany przez aktywację i (lub) utworzenie zakodowanego chromosomalnie enzymu (AmpC);
mechanizm oporności oparty na braku przepuszczalności ściany komórkowej;
aktywne usuwanie leku z komórki bakterii.
Drobnoustroje oporne na cefotaksym mogą być w różnym stopniu oporne również na inne antybiotyki β-laktamowe. Gram-ujemne drobnoustroje oporne na cefotaksym wykazują pełną oporność krzyżową na inne cefalosporyny trzeciej generacji o szerokim spektrum działania (np. ceftazydym, ceftriakson).
Zakres działania cefotaksymu
Drobnoustroje wrażliwe
Tlenowe bakterie Gram-dodatnie
Staphylococcus aureus* wrażliwe na metycylinę Staphylococcus epidermidis* wrażliwe na metycylinę Staphylococcus haemolyticus wrażliwe na metycylinę Staphylococcus spp. wrażliwe na metycylinę, koagulazo-ujemne Streptococcus spp. grupa A (w tym Streptococcus pyogenes)* Streptococcus spp. grupa B
Streptococcus pneumoniae*
Streptococcus viridans*
Tlenowe bakterie Gram-ujemne
Citrobacter freundii
Enterobacter spp. (bez Enterobacter cloacae)* Escherichia coli*
Haemophilus influenzae* Haemophilus parainfluenzae* Moraxella catarrhalis* Neisseria gonorrhoeae* Neisseria meningitidis* Klebsiella spp.*
Morganella morganii Proteus mirabilis*
Serratia spp.* Yersinia enterocolitica
Bakterie beztlenowe
Clostridium spp. (bez Clostridium difficile) Peptostreptococcus spp.
Propionibacterium spp.
Inne
Borrelia spp.
Drobnoustroje, wśród których może wystąpić problemem oporności nabytej
Tlenowe bakterie Gram-dodatnie
Enterobacter cloacae
Staphylococcus aureus# oporne na metycylinę
Staphylococcus spp.# oporne na metycylinę, koagulazo-ujemne
Tlenowe bakterie Gram-ujemne
Acinetobacter spp. Pseudomonas aeruginosa# Stenotrophomonas maltophilia# Bakterie beztlenowe Bacteroides fragilis Clostridium difficile#
Drobnoustroje oporne
Tlenowe bakterie Gram-dodatnie
Enterococcus spp.
Inne
Chlamydia spp.
Legionella pneumophila Listeria spp.
Mycoplasma spp.
* Skuteczność kliniczną wykazano w stosunku do wrażliwych szczepów wyizolowanych podczas stosowania w potwierdzonych wskazaniach.
# Należy traktować jako oporne.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
roztworami o pH większym od 7,5, np. wodorowęglanem sodu;
aminoglikozydami, podczas mieszania w roztworach do podawania parenteralnego; podczas równoczesnego stosowania leki te należy wstrzykiwać w różne miejsca ciała.
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTRYSTYKI PRODUKTU LECZNICZEGO
BIOTAKSYM, 1 g, proszek do sporządzania roztworu do wstrzykiwań lub infuzji BIOTAKSYM, 2 g, proszek do sporządzania roztworu do wstrzykiwań lub infuzji
Każda fiolka zawiera odpowiednio 1 g lub 2 g cefotaksymu (Cefotaximum) w postaci cefotaksymu sodowego.
Produkt leczniczy zawiera sód: 1 g produktu zawiera 48 mg sodu (2,09 mmol).
Proszek do sporządzania roztworu do wstrzykiwań lub infuzji Biały lub nieznacznie żółty, higroskopijny proszek.
Cefotaksym stosuje się w leczeniu ciężkich zakażeń wywołanych przez wrażliwe bakterie:
Cefotaksym jest przeznaczony do podawania pozajelitowego - domięśniowego lub dożylnego. Dawkowanie i sposób podawania ustala się w zależności od ciężkości i rodzaju zakażenia, wieku, masy ciała oraz wydolności nerek pacjenta.
Dawkowanie
Dorośli i dzieci w wieku powyżej 12 lat
W zakażeniach lekkich i umiarkowanych stosuje się 1 g co 12 godzin.
W ciężkich zakażeniach dawkę dobową można zwiększyć do 12 g w 3 lub 4 dawkach podzielonych. Dawki dobowe do 6 g należy podzielić na co najmniej dwie dawki, podawane co 12 godzin. Większe dawki dobowe należy podzielić na co najmniej 3 do 4 dawek podawanych co 8 lub 6 godzin.
Wytyczne dotyczące dawkowania podano w poniższej tabeli.
Rodzaj zakażenia | Pojedyncza dawka cefotaksymu | Odstęp pomiędzy dawkami | Dobowa dawka cefotaksymu |
Typowe zakażenie, gdy potwierdzono lub podejrzewa się wrażliwość drobnoustroju | 1 g | 12 godz. | 2 g |
Zakażenia, w których potwierdzono lub podejrzewa się kilka drobnoustrojów o dużej lub średniej wrażliwości | 2 g | 12 godz. | 4 g |
Niewyjaśnione zakażenia bakteryjne, w których nie udało się zlokalizować miejsca zakażenia i w których stan pacjenta jest krytyczny | 2 do 3 g | 8 godz. 6 godz. | od 6 g do 9 g od 8 g do 12 g |
Niemowlęta i dzieci w wieku do 12 lat
W zależności od ciężkości zakażenia stosuje się od 50 mg do 150 mg/kg mc. na dobę cefotaksymu w 2 lub 4 dawkach podzielonych.
W bardzo ciężkich zakażeniach, zwłaszcza zagrażających życiu, może być konieczne zwiększenie dawki dobowej cefotaksymu do 200 mg/kg mc. na dobę w dawkach podzielonych.
Wcześniaki i noworodki urodzone w terminie
Nie należy przekraczać dawki 50 mg/kg mc. na dobę cefotaksymu w 2 do 4 dawkach podzielonych.
W ciężkich zakażeniach zagrażających życiu może być konieczne zwiększenie dawki dobowej
i podawanie od 150 do 200 mg/kg mc. na dobę, w dawkach podzielonych. W takich sytuacjach zaleca się dawkowanie podane w tabeli poniżej.
Wiek dziecka | Dobowa dawka cefotaksymu |
Od 0 do 7. doby życia | 50 mg/kg mc. co 12 godzin dożylnie |
Od 8. doby do 1. miesiąca życia | 50 mg/kg mc. co 8 godzin dożylnie |
Rzeżączka
Niepowikłana rzeżączka: pojedyncze wstrzyknięcie domięśniowe 1 g cefotaksymu. Przed rozpoczęciem leczenia cefotaksymem należy wykonać odpowiednie badania w celu upewnienia się, czy nie występuje jednoczesne zakażenie kiłą.
Zapobieganie zakażeniom okołooperacyjnym
Należy podać pojedynczą dawkę 1 g cefotaksymu na 30 do 90 minut przed zabiegiem chirurgicznym. W zależności od ryzyka zakażenia można kontynuować podawanie produktu po zabiegu.
Dawkowanie u pacjentów z niewydolnością nerek
Zmniejszenie dawki produktu jest konieczne tylko u pacjentów z ciężką niewydolnością nerek (klirens kreatyniny mniejszy niż 5 ml/min, stężenie kreatyniny w surowicy 751 mikromoli/l). Po podaniu początkowej, nasycającej dawki 1 g, dawkę dobową należy zmniejszyć o połowę bez zmiany częstości podawania produktu, np. dawkę 1 g co 12 godzin zmniejszyć do 500 mg co 12 godzin, dawkę 1 g co
8 godzin zmniejszyć do 500 mg co 8 godzin, dawkę 2 g co 8 godzin zmniejszyć do 1 g co 8 godzin itd. U niektórych pacjentów może być konieczna dalsza modyfikacja dawki w zależności od przebiegu zakażenia i ogólnego stanu pacjenta.
Sposób podawania
Nie podawać dożylnie roztworów cefotaksymu z lidokainą.
Produkt, po odpowiednim rozcieńczeniu, podaje się dożylnie w 3-5 minutowym wstrzyknięciu lub domięśniowo - głęboko w górny, zewnętrzny kwadrant mięśnia pośladkowego większego lub boczną część uda.
Produkt można podawać w infuzji dożylnej, trwającej od 20 do 60 minut. Szczegółowa instrukcja, patrz punkt 6.6.
W okresie po wprowadzeniu produktu do obrotu odnotowano potencjalnie zagrażające życiu zaburzenia rytmu serca u bardzo niewielu pacjentów, którzy otrzymali cefotaksym w szybkim wstrzyknięciu przez cewnik do żyły centralnej.
Cefotaksymu nie należy podawać razem z aminoglikozydami w jednej strzykawce ani w płynie do infuzji.
Reakcje anafilaktyczne
Przed rozpoczęciem leczenia cefotaksymem należy ustalić, czy pacjent jest uczulony na cefotaksym, cefalosporyny lub penicyliny (i inne antybiotyki β-laktamowe). Nadwrażliwość krzyżowa występuje u 5-10% pacjentów.
U pacjentów otrzymujących cefotaksym obserwowano ciężkie, w tym zakończone zgonem, reakcje nadwrażliwości (patrz punkt 4.3 i 4.8).
Jeśli wystąpi reakcja alergiczna należy bezwzględnie przerwać stosowanie cefotaksymu i w razie konieczności zastosować odpowiednie leczenie. Szczególną ostrożność zachować podczas stosowania cefotaksymu u pacjentów ze skazą alergiczną lub astmą.
Stosowanie cefotaksymu jest bezwzględnie przeciwwskazane u pacjentów, u których wcześniej wystąpiła natychmiastowa reakcja nadwrażliwości na cefalosporyny.
Pacjenci uczuleni na penicyliny mogą być uczuleni również na cefalosporyny. Istnieje wówczas niebezpieczeństwo wystąpienia ciężkiej reakcji alergicznej. Należy zachować szczególną ostrożność podczas podawania cefotaksymu pacjentom, u których wystąpiły reakcje nadwrażliwości (szczególnie jeśli wystąpiła reakcja anafilaktyczna) na penicyliny lub inne antybiotyki β-laktamowe.
Wzrost niewrażliwych drobnoustrojów
Podobnie jak w przypadku innych antybiotyków, podczas stosowania cefotaksymu przez dłuższy czas, pierwotnie wrażliwe szczepy mogą stać się oporne oraz może wystąpić wzrost bakterii z rodziny Enterococcus spp. Niezbędne jest przeprowadzenie oceny stanu pacjenta, zaleca się wykonać antybiogram.
Ciężkie skórne reakcje pęcherzowe
Zgłaszano przypadki wystąpienia ciężkich skórnych reakcji pęcherzowych, takich jak zespół Stevensa-Johnsona lub toksyczna nekroliza naskórka, w związku ze stosowaniem cefotaksymu (patrz punkt 4.8). Należy poinstruować pacjenta, że w razie wystąpienia takich reakcji nie należy kontynuować leczenia bez wcześniejszego omówienia tego z lekarzem.
Powikłania związane z Clostridium difficile
W trakcie lub po zakończeniu leczenia może wystąpić rzekomobłoniaste zapalenie jelita grubego na skutek nadmiernego wzrostu niewrażliwych na produkt bakterii Clostridium difficile. W razie wystąpienia biegunki, należy brać pod uwagę możliwość tego powikłania. W lżejszych przypadkach wystarczy odstawić produkt, w cięższych, po odstawieniu produktu, należy właściwie nawodnić pacjenta, uzupełnić elektrolity i wykonać badanie w celu wykrycia bakterii.
Jeśli wykryje się C. difficile, należy zastosować doustnie metronidazol lub wankomycynę. Nie należy podawać leków hamujących perystaltykę ani innych działających zapierająco.
Należy zachować ostrożność podczas stosowania cefotaksymu u pacjentów, u których w przeszłości występowało zapalenie jelit, zwłaszcza zapalenie okrężnicy.
Pacjenci z niewydolnością nerek
Pacjenci z niewydolnością nerek powinni otrzymać dawkę odpowiednio zmniejszoną w zależności od klirensu kreatyniny (patrz pkt. 4.2).
Należy zachować ostrożność podczas podawania cefotaksymu razem z aminoglikozydami, probenecydem lub innymi lekami nefrotoksycznymi (patrz punkt 4.5).
U pacjentów tych, a także u pacjentów w podeszłym wieku oraz u pacjentów, u których wcześniej występowało zaburzenie czynności nerek, należy kontrolować czynność nerek.
Neurotoksyczność
Duże dawki antybiotyków beta-laktamowych, w tym cefotaksymu, zwłaszcza stosowane u pacjentów z niewydolnością nerek, mogą powodować encefalopatię, tj. zaburzenia świadomości, nieprawidłowe ruchy i drgawki (patrz punkt 4.8).
Należy poinstruować pacjenta, że w razie wystąpienia takich reakcji nie należy kontynuować leczenia bez wcześniejszego omówienia tego z lekarzem.
Reakcje hematologiczne
Podczas terapii cefotaksymem, szczególnie jeśli jest on podawany jest przez długi okres, może rozwinąć się leukopenia, neutropenia i, rzadziej, agranulocytoza. Jeśli cykl leczenia trwa dłużej niż 7-10 dni, należy kontrolować liczbę białych krwinek i wstrzymać podawanie produktu w przypadku pojawienia się neutropenii. Zgłoszono kilka przypadków eozynofilii i trombocytopenii, szybko ustępujących po zaprzestaniu leczenia. Zgłaszano również przypadki niedokrwistości hemolitycznej (patrz punkt 4.8).
Kontrolowanie czynności wątroby
Jeśli produkt stosowany jest długotrwale, należy kontrolować czynność wątroby.
Szybkość podawania
Cefotaksym podawany zbyt szybko dożylnie (krócej niż przez 1 minutę) może powodować zaburzenia rytmu serca.
Produkt leczniczy zawiera sód
Produkt leczniczy zawiera 48 mg sodu na 1 g produktu co odpowiada 2,4% zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych. Należy to wziąć pod uwagę u pacjentów ze zmniejszoną czynnością nerek i u pacjentów kontrolujących zawartość sodu w diecie.
Produkt podaje się wyłącznie po rekonstytucji - patrz punkt 6.6. Przy obliczaniu całkowitej zawartości sodu w przygotowanym rozcieńczeniu produktu należy brać pod uwagę ilość sodu pochodzącego
z rozcieńczalnika. W celu uzyskania dokładnej informacji dotyczącej zawartości sodu w roztworze wykorzystanym do rozcieńczenia produktu, należy zapoznać się z charakterystyką produktu leczniczego stosowanego rozcieńczalnika.
Nie należy stosować cefotaksymu w skojarzeniu z substancjami o działaniu bakteriostatycznym
(np. tetracyklina, erytromycyna, chloramfenikol, sulfonamidy), ponieważ in vitro zaobserwowano ich antagonistyczne działanie przeciwbakteryjne. Działanie synergiczne może występować podczas leczenia skojarzonego cefotaksymem i aminoglikozydami.
Antybiotyki aminoglikozydowe i leki moczopędne. Jak w przypadku innych cefalosporyn, stosowanie cefotaksymu jednocześnie z lekami działającymi nefrotoksycznie, takimi jak antybiotyki aminoglikozydowe lub silnie działające leki moczopędne (np. furosemid), może nasilać ich szkodliwy wpływ na czynność nerek. Należy wówczas kontrolować u pacjenta czynność nerek (patrz punkt 4.4).
Produkty zwiększające wydalanie kwasu moczowego. Probenecyd zakłóca wydzielanie cefotaksymu w kanalikach nerkowych, zwiększając tym samym ogólny wpływ cefotaksymu na organizm około 2-krotnie i hamując klirens nerkowy o około połowę, po podaniu w dawkach terapeutycznych. Ze względu na szeroki przedział terapeutyczny cefotaksymu, nie jest konieczna zmiana dawkowania
u pacjentów z prawidłową czynnością nerek. U pacjentów z zaburzeniami czynności nerek może być konieczna modyfikacja dawki (patrz punkt 4.4 i 4.2).
Wpływ na wyniki badań laboratoryjnych
Cefotaksym nie wpływa na wynik testów enzymatycznych wykrywających cukier w moczu. Cefotaksym, podobnie jak inne cefalosporyny, może powodować fałszywie dodatni wynik testu Coombsa.
Wyniki oznaczania stężenia glukozy w moczu z użyciem niespecyficznych czynników redukcyjnych mogą być fałszywie pozytywne. Zjawisko to nie jest obserwowane, jeśli używa się testu oksydazowego.
Ciąża
Bezpieczeństwo stosowania cefotaksymu u kobiet w ciąży nie zostało ustalone.
Badania na zwierzętach nie wykazały bezpośredniego ani pośredniego szkodliwego wpływu na rozmnażanie. Nie ma jednak odpowiednich i dobrze kontrolowanych badań u kobiet w ciąży. Cefotaksym przenika przez barierę łożyska. Dlatego cefotaksymu nie należy stosować podczas ciąży, chyba że przewidywane korzyści przewyższają ryzyko.
Karmienie piersią
Cefotaksym przenika do mleka kobiecego.
Nie można wykluczyć wpływu na fizjologiczną florę jelitową u noworodków karmionych piersią, co prowadzi do biegunki i kolonizacji grzybami drożdżakopodobnymi. U noworodka może też wystąpić reakcja uczuleniowa.
Dlatego też należy podjąć decyzję, czy zaprzestać karmienia piersią, czy przerwać leczenie, biorąc pod uwagę korzyść z karmienia piersią dla dziecka i korzyść wynikającą z leczenia dla matki.
Nie ma dowodów na to, że cefotaksym bezpośrednio zaburza zdolność prowadzenia pojazdów lub obsługiwania maszyn. Duże dawki cefotaksymu, szczególnie u pacjentów z niewydolnością nerek, mogą powodować encefalopatię (np. zaburzenia świadomości, zaburzenia ruchu i drgawki) (patrz punkt 4.8). Należy ostrzec pacjenta, że jeśli wystąpią takie objawy, nie powinien on prowadzić pojazdów ani obsługiwać maszyn.
Częstość występowania działań niepożądanych określono w następujący sposób: bardzo często 1/10;
często 1/100 do 1/10;
niezbyt często 1/1000 do 1/100; rzadko 1/10 000 do 1/1000; bardzo rzadko 1/10 000;
częstość nieznana (nie może być określona na podstawie dostępnych danych)*.
Układy i narządy | Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko | Częstość nieznana |
Zakażenia i zarażenia pasożytnicze | nadkażenia (patrz punkt 4.4) | |||||
Zaburzenia krwi i układu chłonnego | leukopenia, eozynofilia, trombocytopenia | neutropenia, agranulocytoza (patrz punkt 4.4), niedokrwistość hemolityczna | ||||
Zaburzenia układu immunologiczn ego | reakcja Herxheimera | reakcje anafilaktyczne, obrzęk naczyniorucho- wy, skurcz oskrzeli, wstrząs anafilaktyczny | ||||
Zaburzenia układu nerwowego | drgawki (patrz punkt 4.4) | bóle i zawroty głowy, encefalopatia (np. zaburzenia świadomości, nieprawidłowe ruchy) (patrz punkt 4.4) | ||||
Zaburzenia serca | arytmia spowodowana szybką infuzją przez cewnik do żyły centralnej |
Układy i narządy | Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko | Częstość nieznana |
Zaburzenia żołądka i jelit | biegunka | nudności, wymioty, bóle brzucha, rzekomo- błoniaste zapalenie jelita grubego (patrz punkt 4.4) | ||||
Zaburzenia wątroby i dróg żółciowych | zwiększenie aktywności enzymów wątrobowych (AlAT, AspAT, LDH, GGTP, fostatazy alkalicznej) i (lub) stężenia bilirubiny | zapalenie wątroby* (czasami z żółtaczką) | ||||
Zaburzenia skóry i tkanki podskórnej | wysypka, świąd skóry, pokrzywka | rumień wielo- postaciowy, zespół Stevensa- Johnsona, toksyczna nekroliza naskórka (patrz punkt 4.4) | ||||
Zaburzenia nerek i dróg moczowych | pogorszenie czynności nerek i (lub) zwiększenie stężenia kreatyniny (podczas stosowania z aminoglikozy- dami) | śródmiąższowe zapalenie nerek | ||||
Zaburzenia ogólne i stany w miejscu podania | Po podaniu domięśniowym ból w miejscu podania | gorączka, reakcje zapalne w miejscu podania w tym zapalenie żył lub zakrzepowe zapalenie żył | reakcje ogólnoustrojo- we na lidokainę (jeśli domięśniowo podano roztwór zawierający lidokainę) |
* na podstawie doświadczeń uzyskanych po wprowadzeniu produktu do obrotu.
Reakcja Herxheimera
W ciągu kilku pierwszych dni leczenia boreliozy może rozwinąć się reakcja Herxheimera.
Występowanie jednego lub kilku z poniżej wymienionych objawów zanotowano w ciągu kilku tygodni po rozpoczęciu leczenia boreliozy: wysypka na skórze, świąd, gorączka, leukopenia, zwiększenie aktywności enzymów wątrobowych, trudności w oddychaniu, dolegliwości stawowe.
Zaburzenia wątroby i dróg żółciowych
Obserwowano zwiększenie aktywności enzymów wątrobowych (AlAT, AspAT, LDH, GGTP, fosfatazy alkalicznej) i (lub) stężenia bilirubiny. Te nieprawidłowości w wynikach badań laboratoryjnych rzadko mogą przekroczyć dwukrotnie górną granicę prawidłowego stężenia
i wskazywać na uszkodzenie wątroby, zwykle polegające na zatrzymaniu żółci i najczęściej przebiegające bezobjawowo.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Objawy przedawkowania w dużym stopniu odpowiadają profilowi działań niepożądanych. Podanie dużych dawek antybiotyków β-laktamowych, w tym cefotaksymu, może spowodować odwracalną encefalopatię.
W razie przedawkowania należy przerwać podawanie cefotaksymu i wdrożyć postępowanie polegające na przyspieszeniu eliminacji produktu z organizmu i leczeniu objawowym objawów niepożądanych (np. drgawek).
Nie ma specyficznego antidotum. Stężenie cefotaksymu w surowicy można obniżyć stosując hemodializę lub dializę otrzewnową.
Grupa farmakoterapeutyczna: produkt przeciwbakteryjny do stosowania ogólnego; antybiotyki β-laktamowe; cefalosporyny III generacji, kod ATC: J01DD01
Cefotaksym jest półsyntetyczną cefalosporyną III generacji o szerokim zakresie działania bakteriobójczego, do stosowania pozajelitowego. Mechanizm działania polega na hamowaniu syntezy ściany komórkowej bakterii. Cefotaksym jest w organizmie rozkładany przez nieswoiste esterazy do aktywnego produktu - deacetylocefotaksymu. Właściwość ta znacznie przedłuża bakteriobójcze działanie i zwiększa aktywność przeciwbakteryjną produktu.
Mechanizmy powstawania oporności
Oporność bakterii na cefotaksym może być spowodowana przez:
Cefotaksym podaje się pozajelitowo. Wchłanianie
Po podaniu dożylnym 1 g cefotaksymu stężenie w surowicy wynosiło po 5 minutach 81-102 mg/l,
a po 15 minutach do 46 mg/l. Po 8 minutach od dożylnego wstrzyknięcia 2 g cefotaksymu, stężenie w surowicy wyniosło 167-214 mg/l. Po podaniu domięśniowym 1 g cefotaksymu, maksymalne stężenie w surowicy wynosiło około 20 mg/l i występowało w ciągu 30 minut.
Dystrybucja
Pozorna objętość dystrybucji wynosi 21-37 l. Cefotaksym w 25-40% wiąże się z białkami surowicy.
Stężenie cefotaksymu (zwykle oznaczane niespecyficznymi metodami) badano w próbkach wielu tkanek i płynów ustrojowych pochodzących od ludzi. Stężenie w płynie mózgowo-rdzeniowym jest niskie, kiedy nie ma stanu zapalnego, ale osiąga poziom pomiędzy 3 a 30 μg/ml u dzieci z zapaleniem opon mózgowych.
Cefotaksym zwykle przenika przez barierę krew-mózg. W stanie zapalnym opon mózgowych osiąga w płynie mózgowo-rdzeniowym stężenie powyżej wartości MIC dla powszechnie wrażliwych patogenów.
Stężenia (0,2-5,4 μg/ml) hamujące rozwój większości bakterii Gram-ujemnych są osiągane w ropnej plwocinie, wydzielinie oskrzelowej i płynie opłucnej po podaniu 1 g lub 2 g.
Stężenie, które może być skuteczne wobec najbardziej wrażliwych drobnoustrojów, jest podobnie osiągane w obrębie żeńskich narządów płciowych, w wysięku z ucha środkowego w stanie zapalnym, w gruczole krokowym, płynie śródmiąższowym, tkankach nerki, otrzewnej i w ścianie pęcherzyka żółciowego, po podaniu zwykle stosowanych dawek terapeutycznych. Duże stężenia cefotaksymu
i deacetylocefotaksymu są osiągane w żółci.
Cefotaksym przenika także przez barierę łożyska i do mleka matki. Osiąga duże stężenie w tkankach płodu, do 6 mg/kg mc.
Metabolizm
Cefotaksym w dużym stopniu ulega przemianom metabolicznym. Około 15-25% dawki podanej pozajelitowo zostaje wydalone w postaci O-deacetylocefotaksymu (wykazuje działanie przeciwbakteryjne). Powstałe dwa inne nieczynne laktony są prawdopodobnie wydalane z moczem.
Eliminacja
Wydalanie cefotaksymu i deacetylocefotaksymu następuje głównie przez nerki. Około 40-60% podanej dawki cefotaksymu jest wydalane z moczem w postaci niezmienionej w ciągu 24 godzin, a około 20% w postaci deacetylocefotaksymu.
Klirens osoczowy cefotaksymu wynosi 260-390 ml/min, a klirens nerkowy 145-217 ml/min.
Okres półtrwania cefotaksymu w surowicy wynosi 50-80 minut, a jego aktywnego metabolitu - 125 minut. U pacjentów w podeszłym wieku (>80 lat) stwierdzony okres półtrwania cefotaksymu wynosił 120-150 minut, a aktywnego metabolitu - 5 godzin.
W ciężkiej niewydolności nerek (klirens kreatyniny 3-10 ml/min) okres półtrwania cefotaksymu może wynosić od 2,5 do 10 godzin.
Cefotaksym nie ma właściwości mutagennych. Badania na szczurach i myszach nie wykazały wpływu teratogennego cefotaksymu. Nie odnotowano zmniejszenia płodności zwierząt.
Nie zawiera.
Cefotaksym wykazuje niezgodności z:
2 lata
Przechowywać w temperaturze do 25ºC. Chronić od światła.
Przygotowany roztwór można przechowywać 24 godziny w lodówce, tj. w temperaturze od 2C do 8C.
Zaleca się podanie roztworu produktu leczniczego Biotaksym bezpośrednio po sporządzeniu.
Szklana fiolka, zamknięta korkiem z gumy bromobutylowej i zabezpieczona aluminiowym kapslem lub aluminiowym kapslem z kapturkiem, umieszczona jest w tekturowym pudełku z ulotką informacyjną dla pacjenta.
Fiolka o pojemności 26 ml zawiera 1 g lub 2 g cefotaksymu. Opakowanie zawiera 1 lub 10 fiolek.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Przed zastosowaniem produktu sprawdzić termin ważności. Nie stosować po terminie podanym na opakowaniu.
Po dodaniu do fiolki rozpuszczalnika należy ją potrząsnąć aż do rozpuszczenia się proszku, po 1-2 minutach roztwór powinien być klarowny. Przed podaniem produktu należy sprawdzić, czy roztwór jest klarowny i nie zawiera cząstek nierozpuszczalnych. Jeśli jest mętny lub zawiera stałe cząstki, nie nadaje się do użycia. Roztwór produktu może być bezbarwny lub jasnożółty.
Dzieciom do 2 miesiąca życia produkt podawać wyłącznie dożylnie.
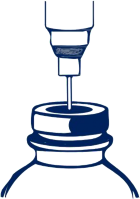
W celu nakłucia korka należy użyć igły o średnicy nie większej niż 0,8 mm (21 G w skali Gauge [G]). Igłę należy wbić w centralnie wyznaczonym polu pod kątem 90°.
Przygotowanie roztworów do wstrzykiwań i infuzji
Zawartość antybiotyku w fiolce | Objętość rozpuszczalnika | ||
Wstrzyknięcie domięśniowe | Wstrzyknięcie dożylne | Infuzja dożylna | |
1 g | 4 ml | 10 ml | 50-100 ml |
2 g | - | 10 ml | 50-100 ml |
Wstrzyknięcie domięśniowe
Produkt należy podawać głęboko domięśniowo po rozpuszczeniu w odpowiedniej ilości wody do wstrzykiwań lub 1% roztworu lidokainy. Nie podawać dożylnie roztworu produktu z lidokainą.
Wstrzyknięcie dożylne (od 3 do 5 minut)
Zawartość fiolki rozpuszcza się w wodzie do wstrzykiwań, w objętości zależnej od dawki, tak jak to przedstawiono w powyższej tabeli.
Infuzja dożylna (od 20 do 60 minut)
W celu sporządzenia roztworów do infuzji dożylnej proszek rozpuszcza się w wodzie do wstrzykiwań (jak do wstrzyknięć dożylnych). Otrzymany roztwór należy następnie rozcieńczyć jednym
z następujących roztworów:
0,9% roztwór chlorku sodu, 5% roztwór glukozy,
5% roztwór glukozy z 0,45% roztworem chlorku sodu, 5% roztwór glukozy z 0,2% roztworem chlorku sodu, mleczanowy roztwór Ringera,
mleczan sodu do wstrzykiwań (M/6).
Nie zaleca się stosowania płynów infuzyjnych zawierających wodorowęglan sodu, gdyż cefotaksym jest niestabilny w tych roztworach.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Zakłady Farmaceutyczne POLPHARMA S.A. ul. Pelplińska 19
83-200 Starogard Gdański
Biotaksym, 1 g: pozwolenie nr R/0593 Biotaksym, 2 g: pozwolenie nr 16527
Biotaksym, 1 g
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 29.11.1990 r. Data ostatniego przedłużenia pozwolenia: 22.07.2013 r.
Biotaksym, 2 g
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 18.02.2010 r. Data ostatniego przedłużenia pozwolenia: 30.07.2014 r.
24.03.2023 r.







