







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Jeżeli w ostatnim czasie nie stosowano hormonalnych produktów antykoncepcyjnych (w ostatnim miesiącu)
Przyjmowanie tabletek należy rozpocząć w 1. dniu cyklu miesiączkowego (tzn. w pierwszym dniu wystąpienia krwawienia miesiączkowego).
Zmiana z innego złożonego produktu antykoncepcyjnego (doustnego złożonego produktu antykoncepcyjnego (COC), systemu terapeutycznego dopochwowego lub systemu transdermalnego)
Pacjentka powinna rozpocząć przyjmowanie produktu leczniczego Asubtela następnego dnia po przyjęciu ostatniej aktywnej tabletki (ostatniej tabletki zawierającej substancje czynne) dotychczas stosowanego COC, ale nie później niż następnego dnia po zakończeniu przerwy lub po przyjęciu tabletek placebo poprzedniego COC. W przypadku stosowania przez pacjentkę systemu terapeutycznego dopochwowego lub systemu transdermalnego pacjentka powinna rozpocząć stosowanie produktu leczniczego Asubtela najlepiej w dniu ich usunięcia, ale najpóźniej w chwili, gdy miały być one ponownie założone.
Zmiana z metody antykoncepcyjnej opartej jedynie na stosowaniu progestagenu (minitabletka, iniekcja, implant) lub systemu terapeutycznego domacicznego uwalniającego progestagen (IUS)
Kobiety mogą zmienić metodę antykoncepcji z minitabletki zawierającej tylko progestagen w dowolnym dniu cyklu (z implantu lub systemu terapeutycznego domacicznego – w dniu ich usunięcia, z produktu podawanego we wstrzyknięciu – w dniu, w którym miało być wykonane kolejne wstrzyknięcie), ale w każdym z tych przypadków należy zalecić stosowanie dodatkowo metody mechanicznej przez pierwsze 7 dni przyjmowania tabletek.
Po poronieniu w pierwszym trymestrze ciąży
Pacjentka może natychmiast rozpocząć przyjmowanie produktu leczniczego. W takim przypadku nie ma potrzeby stosowania innych metod antykoncepcyjnych.
Po porodzie lub po poronieniu w drugim trymestrze ciąży
Tydzień 1
Tydzień 2
Należy przyjąć ostatnią pominiętą tabletkę natychmiast po przypomnieniu sobie o pominięciu dawki, nawet jeśli oznacza to przyjęcie dwóch tabletek na raz. Następnie kontynuuje się przyjmowanie tabletek
o zwykłej porze. Jeżeli w ciągu 7 dni poprzedzających pominięcie tabletki, produkt leczniczy był stosowany prawidłowo, nie ma konieczności stosowania dodatkowych produktów antykoncepcyjnych. Jeżeli natomiast pominięto więcej niż 1 tabletkę, należy zalecić stosowanie dodatkowych produktów antykoncepcyjnych przez 7 dni.
Tydzień 3
Ryzyko zmniejszenia skuteczności produktu antykoncepcyjnego jest duże z powodu zbliżającej się 7- dniowej przerwy w przyjmowaniu tabletek. Można jednak nadal zapobiec zmniejszeniu ochrony antykoncepcyjnej poprzez odpowiednie dostosowanie schematu przyjmowania tabletek. Postępowanie według jednej z dwóch poniższych opcji wyklucza konieczność stosowania dodatkowej metody antykoncepcji, o ile wszystkie tabletki były przyjmowane prawidłowo w ciągu 7 dni poprzedzających pominięcie przyjęcia tabletki po raz pierwszy. W przeciwnym razie należy zastosować się do opcji pierwszej, a ponadto przez kolejne 7 dni stosować dodatkowe zabezpieczenia.
Zalecenia w przypadku zaburzeń żołądkowo-jelitowych
W przypadku ciężkich zaburzeń żołądkowo-jelitowych (np. wymioty lub ciężka biegunka), wchłanianie może być niepełne i należy stosować dodatkowe metody antykoncepcyjne. Jeśli wymioty wystąpiły
w ciągu 3-4 godzin po przyjęciu tabletki, należy jak najszybciej przyjąć nową tabletkę. Jeśli to możliwe nową tabletkę należy przyjąć w ciągu 12 godzin od zwykłej pory przyjmowania produktu leczniczego. Jeżeli upłynęło więcej niż 12 godzin należy postępować zgodnie z zaleceniami dotyczącymi pominięcia tabletek, podanymi w punkcie 4.2. „Postępowanie w przypadku pominięcia tabletek”. Jeżeli pacjentka nie
chce zmieniać ustalonego schematu dawkowania, powinna przyjąć dodatkową tabletkę (tabletki) z innego opakowania.
W jaki sposób opóźnić wystąpienie krwawienia z odstawienia
Aby opóźnić wystąpienie krwawienia z odstawienia, po dokończeniu aktualnego opakowania należy bez zachowania przerwy rozpocząć przyjmowanie tabletek z następnego blistra produktu leczniczego Asubtela. Tabletki mogą być przyjmowane tak długo jak chce tego pacjentka nawet do wyczerpania drugiego opakowania. W tym czasie może wystąpić krwawienie lub plamienie. Kolejne regularne przyjmowanie produktu leczniczego Asubtela należy rozpocząć po 7-dniowej przerwie.
Aby przesunąć termin wystąpienia krwawienia na inny dzień tygodnia, niż wynikający z obecnego schematu przyjmowania produktu leczniczego, należy zalecić skrócenie kolejnej przerwy w przyjmowaniu tabletek o tyle dni, o ile zamierza się przesunąć termin wystąpienia krwawienia. Im krótsza przerwa w przyjmowaniu tabletek tym większe prawdopodobieństwo, że krwawienie z odstawienia nie wystąpi, a podczas przyjmowania tabletek z następnego opakowania może wystąpić plamienie lub krwawienie śródcykliczne (tak jak w przypadku opóźnienia wystąpienia krwawienia).
Dzieci i młodzież
Produkt leczniczy Asubtela jest wskazany do stosowania wyłącznie u pacjentek po pierwszej miesiączce. Dane epidemiologiczne zebrane od ponad 2000 nastoletnich pacjentek w wieku poniżej 18 lat nie wskazuje na różnice dotyczące bezpieczeństwa i skuteczności w tej grupie wiekowej w porównaniu z kobietami w wieku powyżej 18 lat.
Droga padania Podanie doustne.
Przeciwwskazania
Nadwrażliwość na substancje czynne lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
Występowanie lub ryzyko żylnej choroby zakrzepowo-zatorowej (ang. venous thromboembolism, VTE)
Żylna choroba zakrzepowo-zatorowa - czynna (leczona przeciwzakrzepowymi produktami leczniczymi) lub przebyta żylna choroba zakrzepowo-zatorowa, np. zakrzepica żył głębokich (ang. deep venous thrombosis, DVT), zatorowość płucna (ang. pulmonary embolism, PE).
Znana dziedziczna lub nabyta predyspozycja do występowania żylnej choroby zakrzepowo- zatorowej np. oporność na aktywowane białko C (ang. activated protein C, APC) (w tym czynnik V Leiden), niedobór antytrombiny III, niedobór białka C, niedobór białka S.
Rozległy zabieg operacyjny związany z długotrwałym unieruchomieniem (patrz punkt 4.4).
Wysokie ryzyko żylnej choroby zakrzepowo-zatorowej wskutek występowania wielu czynników ryzyka (patrz punkt 4.4).
Występowanie lub ryzyko tętniczych zaburzeń zakrzepowo-zatorowych (ang. arterial thromboembolism, ATE)
Tętnicze zaburzenia zakrzepowo-zatorowe - czynne lub w wywiadzie (np. zawał mięśnia sercowego) lub objawy prodromalne (np. dławica piersiowa).
Choroby naczyń mózgowych - czynny udar, przebyty udar lub objawy prodromalne
w wywiadzie (np. przemijający napad niedokrwienny, ang. transient ischaemic attack, TIA).
Stwierdzona dziedziczna lub nabyta skłonność do występowania tętniczych zaburzeń zakrzepowo-zatorowych np. hiperhomocysteinemia i obecność przeciwciał. antyfosfolipidowych (przeciwciała antykardiolipinowe, antykoagulant toczniowy)
Migrena z ogniskowymi objawami neurologicznymi w wywiadzie.
Wysokie ryzyko zaburzeń zakrzepowo-zatorowych tętnic z powodu występowania wielu czynników ryzyka (patrz punkt 4.4) lub występowania jednego z poważnych czynników ryzyka, takich jak:
cukrzyca z powikłaniami naczyniowymi
ciężkie nadciśnienie tętnicze
ciężka dyslipoproteinemia
Występująca obecnie lub w przeszłości ciężka choroba wątroby, do czasu powrotu parametrów czynności wątroby do normy
Ciężka lub ostra niewydolność nerek
Stwierdzony obecnie lub w wywiadzie guz wątroby (łagodny lub złośliwy)
Rozpoznane lub podejrzewane nowotwory złośliwe (np. narządów rodnych lub piersi) zależne od wpływu hormonów płciowych
Krwawienie z dróg rodnych o nieustalonej etiologii
Stosowanie produktu leczniczego Asubtela jest przeciwwskazane w przypadku jednoczesnego stosowania produktów leczniczych zawierających ombitaswir/parytaprewir/rytonawir lub dasabuwir lub produktów leczniczych zawierających glekaprewir/pibrentaswir oraz sofosbuwir z welpataswirem i woksylaprewirem (patrz punkt 4.5).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Ostrzeżenia
Częstość została oszacowana na podstawie wszystkich dostępnych danych epidemiologicznych, z wykorzystaniem relatywnego ryzyka dla różnych produktów leczniczych w porównaniu do złożonych hormonalnych produktów antykoncepcyjnych zawierających lewonorgestrel.
Punkt środkowy z zakresu od 5 do 7 na 10 000 kobiet w okresie roku, w oparciu o relatywne ryzyko wynoszące około 2,3 do 3,6 d la złożonych hormonalnych produktów antykoncepcyjnych zawierających lewonorgestrel w porównaniu do sytuacji, gdy terapia nie jest stosowana.
obrzęk nogi i (lub) stopy lub obrzęk wzdłuż żyły w nodze;
ból lub tkliwość w nodze, które mogą być odczuwane wyłącznie w czasie stania lub chodzenia;
zwiększona temperatura w zmienionej chorobowo nodze; czerwona lub przebarwiona skóra nogi.
Objawy zatorowości płucnej (ang. pulmonary embolism, PE) mogą obejmować:
nagły napad niewyjaśnionego spłycenia oddechu lub przyspieszenia oddechu;
nagły napad kaszlu, który może być połączony z krwiopluciem;
ostry ból w klatce piersiowej;
ciężkie zamroczenie lub zawroty głowy;
przyspieszone lub nieregularne bicie serca.
nagłe zdrętwienie lub osłabienie twarzy, rąk lub nóg, szczególnie po jednej stronie ciała;
nagłe trudności z chodzeniem, zawroty głowy, utratę równowagi lub koordynacji;
nagłe splątanie, trudności z mówieniem lub rozumieniem;
nagłe zaburzenia widzenia w jednym oku lub obydwu oczach;
nagłe, ciężke lub długotrwałe ból głowy bez przyczyny;
utratę przytomności lub omdlenie z drgawkami lub bez drgawek.
Przejściowe objawy sugerujące, że zdarzenie jest przemijającym napadem niedokrwiennym (ang.
transient ischaemic attack, TIA).
Objawy zawału serca (ang. myocardial infarction, MI) mogą być następujące:
ból, uczucie dyskomfortu, ociężałość, uczucie ściskania lub pełności w klatce piersiowej, ramieniu lub poniżej mostka;
uczucie dyskomfortu promieniujące do pleców, szczęki, gardła, ramienia, żołądka;
uczucie pełności, niestrawności lub zadławienia;
pocenie się, nudności, wymioty lub zawroty głowy;
skrajne osłabienie, niepokój lub spłycenie oddechu;
przyspieszone lub nieregularne bicie serca.
Nowotwory
Niektóre badania epidemiologiczne wykazały, że w czasie długotrwałego stosowania złożonych doustnych produktów antykoncepcyjnych (> 5 lat) istnieje zwiększone ryzyko wystąpienia raka szyjki macicy. Jednak pozostaje nadal kwestią sporną, czy nie wynika to z określonych zachowań seksualnych oraz innych czynników takich jak zakażenie wirusem brodawczaka ludzkiego (ang. Human Papilloma Virus, HPV).
W metaanalizie 54 badań epidemiologicznych odnotowano stosunkowo niewielkie zwiększenie ryzyka względnego (ang. Relative Risk, RR = 1,24) zdiagnozowania raka piersi u kobiet obecnie stosujących złożone doustne produkty antykoncepcyjne. Ryzyko to zmniejsza się stopniowo w ciągu 10 lat po odstawieniu złożonych doustnych produktów antykoncepcyjnych. Ponieważ u kobiet w wieku poniżej 40 lat rak piersi występuje rzadko, wzrost liczby rozpoznań raka piersi u kobiet obecnie lub niedawno stosujących złożone doustne produkty antykoncepcyjne jest niewielki w stosunku do całkowitego ryzyka wystąpienia tego nowotworu. Badania te nie dostarczają dowodów na istnienie związku przyczynowo- skutkowego. Zaobserwowany zespół czynników zwiększonego ryzyka może wynikać z wcześniejszego wykrywania raka piersi u kobiet stosujących złożone doustne produkty antykoncepcyjne, z działania biologicznego tych produktów lub ze współdziałania obu tych czynników. U kobiet, które stosowały złożone doustne produkty antykoncepcyjne częściej rozpoznaje się raka piersi o mniejszym zaawansowaniu klinicznym, niż u kobiet, które nigdy nie stosowały tego typu produktów leczniczych.
Istnieją doniesienia o rzadkich przypadkach łagodnych nowotworów wątroby i jeszcze rzadszych przypadkach złośliwych nowotworów wątroby u kobiet stosujących złożone doustne produkty antykoncepcyjne (COC). Sporadycznie nowotwory te mogą prowadzić do zagrażających życiu krwotoków do jamy brzusznej. Nowotwór wątroby należy wziąć pod uwagę w rozpoznaniu różnicowym silnego bólu w nadbrzuszu, hepatomegalii lub objawów krwotoku do jamy brzusznej u kobiet stosujących złożone doustne produkty antykoncepcyjne.
Podczas stosowania złożonych doustnych produktów antykoncepcyjnych zawierających większe dawki hormonów (50 µg etynyloestradiolu) zmniejsza się ryzyko raka endometrium i raka jajnika. Nie potwierdzono, czy dotyczy to również COC zawierających mniejsze dawki hormonów.
Zwiększenie aktywności aminotransferazy alaninowej (AlAT)
W trakcie badań klinicznych u pacjentów leczonych z powodu zakażenia wirusem zapalenia wątroby typu C (HCV) produktami leczniczymi zawierającymi ombitaswir/ parytaprewir/ rytonawir lub dasabuwir
w monoterapii lub w połączeniu z rybawirynem, większe niż 5-krotne zwiększenie aktywności transaminazy (AlAT) powyżej górnej granicy normy (ang. upper limit of normal, ULN), występowało istotnie statystycznie częściej u kobiet stosujących produkty lecznicze zawierające etynyloestradiol takie, jak złożone hormonalne środki antykoncepcyjne. Dodatkowo, również u pacjentek leczonych produktem leczniczym zawierającym glekaprewir/pibrentaswir, obserwowano zwiększenie aktywności AlAT u kobiet stosujących produkty lecznicze zawierające etynyloestradiol takie, jak złożone hormonalne leki antykoncepcyjne (patrz punkt 4.3 i 4.5).
Inne zaburzenia
Progestagenowy składnik produktu leczniczego Asubtela jest antagonistą aldosteronu o właściwościach oszczędzających potas. W większości przypadków nie należy oczekiwać zwiększenia stężenia potasu. Jednak w badaniu klinicznym u niektórych pacjentek z lekkimi lub umiarkowanymi zaburzeniami czynności nerek, przyjmujących jednocześnie produkty lecznicze oszczędzające potas, stężenie potasu w surowicy zwiększało się nieznacznie, choć w nieistotnym stopniu, podczas stosowania drospirenonu. Dlatego zaleca się kontrolowanie stężenia potasu w surowicy w pierwszym cyklu leczenia u pacjentek z objawami niewydolności nerek, u których stężenie potasu przed leczeniem znajdowało się w górnym zakresie normy, w szczególności podczas jednoczesnego stosowania produktów leczniczych oszczędzających potas. Patrz też punkt 4.5.
U kobiet z hipertrójglicerydemią, lub dodatnim wywiadem rodzinnym w kierunku hipertrójglicerydemii, może występować zwiększone ryzyko zapalenia trzustki podczas stosowania złożonych doustnych produktów antykoncepcyjnych.
Mimo że, u wielu kobiet stosujących złożone doustne produkty antykoncepcyjne odnotowano niewielki wzrost ciśnienia krwi, jednak w nielicznych przypadkach stwierdzono wzrost ciśnienia tętniczego, który byłby klinicznie istotny. Tylko w tych nielicznych przypadkach odstawienie złożonych doustnych produktów antykoncepcyjnych jest uzasadnione. Jeśli, podczas stosowania złożonych doustnych produktów antykoncepcyjnych u kobiet z wcześniej stwierdzonym nadciśnieniem tętniczym, stwierdza się ciągły wzrost ciśnienia tętniczego lub znaczne zwiększenie ciśnienia tętniczego nie reagujące na leczenie przeciwnadciśnieniowe, konieczne jest odstawienie złożonych doustnych produktów antykoncepcyjnych. W uzasadnionych przypadkach, jeśli podczas leczenia przeciwnadciśnieniowego wartość ciśnienia tętniczego krwi została unormowana, można ponownie rozpocząć stosowanie złożonych doustnych produktów antykoncepcyjnych.
Odnotowano występowanie następujących stanów lub pogorszenie ich przebiegu zarówno podczas ciąży oraz podczas stosowania złożonych doustnych produktów antykoncepcyjnych, jednak nie potwierdzono związku ich występowania ze stosowaniem złożonych doustnych produktów antykoncepcyjnych: żółtaczka i (lub) świąd związany z cholestazą; kamica żółciowa; porfiria; toczeń rumieniowaty układowy; zespół hemolityczno-mocznicowy; pląsawica Sydenhama; opryszczka ciężarnych; utrata słuchu związana z otosklerozą.
Egzogenne estrogeny mogą wywoływać lub nasilać objawy wrodzonego lub nabytego obrzęku naczynioworuchowego.
Ostre lub przewlekłe zaburzenia czynności wątroby mogą spowodować konieczność odstawienia złożonych doustnych produktów antykoncepcyjnych do czasu, aż parametry czynności wątroby powrócą do normy. Nawrót żółtaczki cholestatycznej i (lub) świądu związanego z cholestazą, które występowały wcześniej podczas ciąży lub podczas wcześniejszego stosowania hormonów płciowych wymaga odstawienia złożonych doustnych produktów antykoncepcyjnych.
Mimo, iż złożone doustne produkty antykoncepcyjne mogą mieć wpływ na oporność na insulinę tkanek obwodowych lub na tolerancję glukozy, nie ma dowodów na konieczność zmiany schematu leczenia cukrzycy u osób stosujących złożone doustne produkty antykoncepcyjne zawierające małą dawkę estrogenów (zawierające < 0,05 mg etynyloestradiolu). Jednakże, kobiety cierpiące na cukrzycę powinny być pod stałą obserwacją, szczególnie w początkowym okresie stosowania złożonych doustnych produktów antykoncepcyjnych.
Podczas stosowania złożonych doustnych produktów antykoncepcyjnych odnotowano przypadki nasilenia przebiegu depresji endogennej, padaczki, choroby Leśniowskiego-Crohna oraz wrzodziejącego zapalenia jelita grubego.
Niekiedy może wystąpić ostuda (plamy barwnikowe na twarzy), zwłaszcza u kobiet, u których w przeszłości wystąpiła ostuda ciążowa. Kobiety z tendencją do ostudy powinny unikać ekspozycji na światło słoneczne lub promieniowanie ultrafioletowe podczas stosowania złożonych doustnych produktów antykoncepcyjnych.
Obniżony nastrój i depresja to dobrze znane działania niepożądane stosowania hormonalnych środków antykoncepcyjnych (patrz punkt 4.8). Depresja może mieć ciężki przebieg i jest dobrze znanym czynnikiem ryzyka zachowań samobójczych i samobójstw. Jeśli u pacjentki wystąpią zmiany nastroju lub objawy depresji, również krótko po rozpoczęciu leczenia, zaleca się, aby skonsultowała się z lekarzem.
Badania lekarskie, konsultacje
Przed rozpoczęciem stosowania produktu leczniczego Asubtela lub przed ponownym zastosowaniem produktu leczniczego Asubtela należy przeprowadzić pełny wywiad lekarski (w tym wywiad rodzinny) oraz wykluczyć ciążę. Należy również zmierzyć ciśnienie krwi oraz przeprowadzić badanie przedmiotowe, w celu wykluczenia przeciwwskazań (patrz punkt 4.3) oraz stanów wymagających szczególnej ostrożności (patrz punkt 4.4). Ważne jest zwrócenie uwagi kobiety na informacje dotyczące zakrzepicy żył i tętnic, w tym na ryzyko stosowania produktu leczniczego Asubtela w porównaniu z innymi złożonymi hormonalnymi produktami antykoncepcyjnymi, objawy żylnej choroby zakrzepowo- zatorowej oraz zaburzeń zakrzepowo-zatorowych tętnic, znane czynniki ryzyka oraz co należy robić w przypadku podejrzenia zakrzepicy.
Należy również polecić kobietom dokładne przeczytanie ulotki i stosowanie się do znajdujących się w niej zaleceń. Częstość i rodzaj badań powinny zostać dobrane na podstawie przyjętych zaleceń postępowania
i dostosowane dla każdej pacjentki.
Należy poinformować kobietę, że doustne produkty antykoncepcyjne nie chronią przed zakażeniem wirusem HIV (AIDS) oraz przed innymi chorobami przenoszonymi droga płciową.
Zmniejszenie skuteczności działania
Skuteczność działania złożonych doustnych produktów antykoncepcyjnych może być zmniejszona
w przypadku, np. pominięcia tabletki (patrz punkt 4.2), zaburzeń żołądkowo-jelitowych (patrz punkt 4.2) lub jednoczesnego stosowania innych produktów leczniczych (patrz punkt 4.5).
Zaburzenia cyklu miesiączkowego
Podczas stosowania wszystkich złożonych doustnych produktów antykoncepcyjnych, może wystąpić nieregularne krwawienie (plamienie lub krwawienie śródcykliczne), zwłaszcza w pierwszych miesiącach stosowania tabletek. Dlatego też prawidłowa ocena występowania nieregularnych krwawień możliwa jest dopiero po upływie okresu adaptacyjnego, trwającego około 3 cykle.
Jeśli nieregularne krwawienia utrzymują się lub występują po uprzednich regularnych cyklach, należy rozważyć przyczyny nie związane z działaniem hormonów i przeprowadzić odpowiednie badania diagnostyczne w celu wykluczenia nowotworów złośliwych lub ciąży. Badania te mogą obejmować łyżeczkowanie jamy macicy.
U niektórych kobiet krwawienie z odstawienia może nie wystąpić podczas przerwy w stosowaniu tabletek Asubtela. Jeśli złożone doustne produkty antykoncepcyjne były stosowane zgodnie z wskazówkami opisanymi w punkcie 4.2, prawdopodobieństwo, że kobieta zaszła w ciążę jest niewielkie. Jednakże, jeśli stosowanie złożonych doustnych produktów antykoncepcyjnych nie przebiegało zgodnie ze wskazówkami, a w przerwie między przyjmowaniem tabletek nie wystąpiło krwawienie z odstawienia lub jeśli nie wystąpiły dwa kolejne krwawienia z odstawienia, przed kontynuacją stosowania złożonego doustnego produktu antykoncepcyjnego należy wykluczyć ciążę.
Substancje pomocnicze
Laktoza
Ten produkt medyczny zawiera 62 mg laktozy w jednej tabletce. Lek nie powinien być stosowany u pacjentek z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ innych produktów leczniczych na działanie produktu leczniczego Asubtela
Interakcje mogą wystąpić z produktami leczniczymi indukującymi enzymy mikrosomalne w wątrobie, co może powodować zwiększenie klirensu hormonów płciowych i co może prowadzić do występowania krwawienia śródcyklicznego i (lub) zmniejszenia skuteczności antykoncepcyjnej produktu leczniczego.
Postępowanie
Indukcja enzymów może być widoczna po kilku dniach leczenia. Maksymalna indukcja enzymów występuje zwykle po kilku tygodniach. Po zaprzestaniu leczenia indukcja enzymów może utrzymywać się przez mniej więcej 4 tygodnie.
Leczenie krótkoterminowe
Kobiety leczone produktami leczniczymi indukującymi enzymy powinny tymczasowo stosować dodatkowo mechaniczną metodę antykoncepcji lub inną metodę antykoncepcji. Mechaniczna metoda antykoncepcji musi być stosowana podczas całego okresu leczenia skojarzonego oraz przez 28 dni po jego zakończeniu. Jeśli leczenie to trwa dłużej niż koniec przyjmowania tabletek z opakowania złożonego doustnego produktu antykoncepcyjnego, następne opakowanie należy rozpocząć zaraz po poprzednim, bez zazwyczaj stosowanej przerwy w przyjmowaniu tabletek.
Leczenie długoterminowe
U kobiet długotrwale przyjmujących substancje czynne indukujące enzymy wątrobowe, zaleca się stosowanie innej, skutecznej, niehormonalnej metody antykoncepcji.
W literaturze opisywano następujące interakcje.
Substancje zwiększające klirens złożonych doustnych produktów antykoncepcyjnych (zmniejszające skuteczność złożonych doustnych produktów antykoncepcyjnych poprzez indukcję enzymów), np. barbiturany, bozentan, karbamazepina, fenytoina, prymidon, ryfampicyną, produkty lecznicze stosowane w leczeniu zakażeń wirusem HIV (tj. rytonawir, newirapina, efawirenz) i prawdopodobnie także felbamat, gryzeofulwina, okskarbazepina, topiramat oraz produkty zawierające ziele dziurawca zwyczajnego (Hypericum perforatum).
Produkty lecznicze wywierające wpływ na Klirens doustnych produktów antykoncepcyjnych:
Jednoczesne stosowanie złożonych doustnych produktów antykoncepcyjnych z wieloma inhibitorami proteazy HIV i nienukleozydowymi inhibitorami odwrotnej transkryptazy, w tym jednoczesne stosowanie z inhibitorami HCV, może zwiększać lub zmniejszać stężenie estrogenów lub progestagenów w osoczu. W niektórych przypadkach zmiany te mogą mieć znacznie kliniczne.
Dlatego też, zalecając równoczesne stosowanie produktów leczniczych przeciwko HIV i (lub) HCV należy wziąć pod uwagę możliwe interakcje oraz odpowiednie zalecenia. W przypadku jakichkolwiek wątpliwości, u kobiet podczas leczenia inhibitorami proteazy lub nienukleozydowymi inhibitorami odwrotnej transkryptazy należy stosować dodatkową, mechaniczną metodę antykoncepcji.
Produkty lecznicze zmniejszające klirens złożonych doustnych produktów antykoncepcyjnych (inhibitory enzymów):
Znaczenie kliniczne potencjalnych interakcji z inhibitorami enzymów pozostaje nieznane. Jednoczesne stosowanie silnych inhibitorów CYP3A4 może zwiększyć stężenie estrogenów lub progestagenów lub obu substancji w osoczu.
W wielodawkowym badaniu z zastosowaniem połączenia drospirenonuu (3 mg na dobę) i etynyloestradiolu (0,02 mg na dobę) z silnym inhibitorem CYP2A4, ketokonazolu przez 10 dni spowodowało wzrost AUC (0-24h) drospirenonu i etynyloestradiolu odpowiednio 2,7- i 1,4-krotnie.
Dawka etorykoksybu wynosząca 60 do 120 mg na dobę, podawana jednocześnie ze złożoną antykoncepcją doustną zawierającą 0,035 mg etynyloestradiolu, spowodowała zwiększenie stężenia etynyloestradiolu w osoczu od 1,4- do 1,6-krotnie.
Główne metabolity drospirenonu w ludzkim osoczu są wytwarzane bez udziału układu enzymatycznego cytochromu P450. Istnieje zatem niewielkie prawdopodobieństwo, że inhibitory tego układu enzymatycznego wpływają na metabolizm drospirenonu.
Wpływ produktu leczniczego Asubtela na inne produkty lecznicze
Doustne produkty antykoncepcyjne mogą wpływać na metabolizm innych substancji czynnych. Zatem, stężenia osoczowe jak i tkankowe mogą zarówno zwiększać się (np. cyklosporyna) lub zmniejszać się (np. lamotrygina).
Na podstawie badań inhibicji, przeprowadzonych in vitro oraz badań interakcji in vivo w grupie ochotniczek przyjmujących omeprazol, symwastatynę oraz midazolam jako substraty markerowe stwierdzono, że ryzyko wpływu drospirenonu w dawce 3 mg na metabolizm innych substancji czynnych jest niewielkie.
Dane kliniczne sugerują, że etynyloestradiol spowalnia klirens substratów CYP1A2, co prowadzi do niewielkiego (np. teofilina) lub umiarkowanego (np. tizanidyna) zwiększenia ich stężenia w osoczu.
Inne rodzaje interakcji
U pacjentek bez zaburzeń czynności nerek, jednoczesne stosowanie drospirenonu oraz inhibitorów konwertazy angiotensyny lub niesterydowych leków przeciwzapalnych nie wykazało znaczącego wpływu na stężenie potasu w surowicy. Nie prowadzono jednak badań nad wpływem jednoczesnego stosowania produktu leczniczego Asubtela z antagonistami aldosteronu lub moczopędnymi produktami leczniczymi oszczędzającymi potas. W takim przypadku, podczas pierwszego cyklu leczenia należy oznaczyć stężenie potasu w surowicy. Patrz również punkt 4.4.
Wyniki badań laboratoryjnych
Wpływ na płodność, ciążę i laktację Ciąża
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Zaburzenia zakrzepowo-zatorowe żył.
Zaburzenia zakrzepowo-zatorowe tętnic.
Nadciśnienie tętnicze.
Nowotwory wątroby.
Występowanie lub nasilanie się stanów, których związek ze stosowaniem złożonych doustnych produktów antykoncepcyjnych nie jest przesądzony: choroba Leśniowskiego-Crohna, wrzodziejące zapalenie jelita grubego, padaczka, migrena, mięśniak macicy, porfiria, toczeń rumieniowaty
układowy, opryszczka ciężarnych, pląsawica Sydenhama, zespół hemolityczno-mocznicowy, żółtaczka cholestatyczna.
Ostuda.
Ostre lub przewlekłe zaburzenia czynności wątroby mogą wymagać odstawienia złożonych doustnych produktów antykoncepcyjnych aż do normalizacji parametrów czynności wątroby.
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne Drospirenon
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych Rdzeń tabletki:
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywaniu
Rodzaj i zawartość opakowania
x 21 tabletek powlekanych
x 21 tabletek powlekanych
x 21 tabletek powlekanych
Szczególne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Asubtela, 3 mg + 0,03 mg, tabletki powlekane
Każda tabletka powlekana zawiera 0,03 mg etynyloestradiolu i 3 mg drospirenonu. Substancja pomocnicza o znanym działaniu:
Laktoza jednowodna 62 mg
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana.
Żółte, okrągłe tabletki powlekane o średnicy około 5,7 mm.
Antykoncepcja doustna.
Decyzja o przepisaniu produktu leczniczego Asubtela powinna zostać podjęta na podstawie indywidualnej oceny czynników ryzyka u kobiety, zwłaszcza ryzyka żylnej choroby zakrzepowo-zatorowej oraz ryzyka żylnej choroby zakrzepowo-zatorowej związanego ze stosowaniem produktu leczniczego Asubtela, w odniesieniu do innych złożonych hormonalnych produktów antykoncepcyjnych (patrz punkty 4.3oraz 4.4).
Dawkowanie
Jak stosować produkt leczniczy Asubtela
Tabletki należy przyjmować codziennie, mniej więcej o tej samej porze, popijając w razie potrzeby niewielką ilością płynu, w kolejności wskazanej na blistrze. Należy przyjmować jedną tabletkę na dobę przez 21 kolejnych dni. Każde kolejne opakowanie rozpoczyna się po 7-dniowej przerwie, podczas której występuje zazwyczaj krwawienie z odstawienia. Krwawienie rozpoczyna się zazwyczaj 2. lub 3. dnia po przyjęciu ostatniej tabletki i może nie skończyć się przed rozpoczęciem kolejnego opakowania.
Jak rozpocząć stosowanie produktu leczniczego Asubtela
Należy zalecić rozpoczęcie przyjmowania produktu leczniczego między 21. a 28. dniem po porodzie lub po poronieniu w drugim trymestrze ciąży. W przypadku późniejszego rozpoczęcia stosowania produktu leczniczego należy zalecić dodatkowe stosowanie mechanicznej metody antykoncepcji przez pierwsze 7 dni przyjmowania tabletek. Jeśli jednak wcześniej doszło do stosunku płciowego, przed rozpoczęciem stosowania COC należy wykluczyć ciążę lub poczekać do pierwszej miesiączki.
Kobiety karmiące piersią, patrz punkt 4.6.
Postępowanie w przypadku pominięcia tabletek
Jeśli minęło mniej niż 12 godzin od pominięcia tabletki, ochrona antykoncepcyjna nie jest zmniejszona. Pacjentka powinna przyjąć tabletkę tak szybko, jak sobie przypomni o pominięciu dawki, a kolejne tabletki przyjmować o zwykłej porze.
Jeśli minęło więcej niż 12 godzin od pominięcia tabletki, ochrona antykoncepcyjna może być zmniejszona. Postępowanie w przypadku pominięcia tabletek może przebiegać według dwóch podstawowych zasad:
Należy przyjąć ostatnią pominiętą tabletkę natychmiast po przypomnieniu sobie o pominięciu dawki, nawet jeśli oznacza to przyjęcie dwóch tabletek na raz. Następnie kontynuuje się przyjmowanie tabletek
o zwykłej porze. Ponadto przez kolejne 7 dni należy stosować dodatkowo mechaniczną metodę antykoncepcji np. prezerwatywę. Jeżeli w ciągu minionych 7 dni doszło do stosunku płciowego, należy wziąć pod uwagę możliwość zajścia w ciążę. Ryzyko ciąży jest tym większe, im więcej tabletek pominięto i im mniej czasu pozostało do normalnej przerwy w stosowaniu tabletek.
Nie należy stosować złożonych doustnych produktów antykoncepcyjnych (COC), jeżeli występuje którykolwiek ze stanów wymienionych poniżej. Jeżeli którykolwiek z nich wystąpi po raz pierwszy podczas stosowania COC, należy je natychmiast odstawić.
Jeśli występuje którykolwiek z poniższych stanów lub czynników ryzyka, należy omówić z pacjentką zasadność stosowania produktu leczniczego Asubtela.
W razie pogorszenia lub wystąpienia po raz pierwszy któregokolwiek z wymienionych stanów lub czynników ryzyka kobieta powinna zgłosić się do lekarza prowadzącego, który zadecyduje, czy konieczne jest przerwanie stosowania produktu leczniczego Asubtela.
W przypadku podejrzenia lub potwierdzenia żylnych zaburzeń zakrzepowo-zatorowych lub tętniczych zaburzeń zakrzepowo-zatorowych, należy przerwać stosowanie złożonych hormonalnych produktów antykoncepcyjnych. Jeśli pacjentka rozpoczęła stosowania przeciwzakrzepowych produktów leczniczych, powinna zastosować inną metodę antykoncepcji ze względu na działanie teratogenne przeciwzakrzepowych produktów leczniczych (pochodnymi kumaryny).
Zaburzenia układu krążenia
Ryzyko żylnej choroby zakrzepowo-zatorowej
Stosowanie jakichkolwiek złożonych hormonalnych produktów antykoncepcyjnych wiąże się ze zwiększonym ryzykiem żylnej choroby zakrzepowo-zatorowej w porównaniu do sytuacji, gdy leczenie nie jest stosowane. Stosowanie produktów leczniczych zawierających lewonorgestrel, norgestimat lub noretisteron jest związane z najmniejszym ryzykiem żylnej choroby zakrzepowo-zatorowej.
Stosowanie innych produktów leczniczych, takich jak produkt leczniczy Asubtela może być związane z dwukrotnie większym ryzykiem. Decyzja o zastosowaniu produktu leczniczego spoza grupy najmniejszego ryzyka żylnej choroby zakrzepowo-zatorowej powinna zostać podjęta wyłącznie po rozmowie z pacjentką, w celu zapewnienia, że rozumie ona ryzyko żylnej choroby
zakrzepowo-zatorowej związane z produktem leczniczym Asubtela, jak obecne czynniki ryzyka wpływają na to ryzyko oraz, że ryzyko żylnej choroby zakrzepowo-zatorowej jest największe w pierwszym roku stosowania. Istnieją pewne dowody, świadczące, że ryzyko zwiększa się, gdy złożone hormonalne produkty antykoncepcyjne są przyjmowane ponownie po przerwie w stosowaniu wynoszącej 4 tygodnie lub więcej.
U około 2 na 10 000 kobiet, które nie stosują złożonych hormonalnych produktów antykoncepcyjnych i nie są w ciąży, w okresie roku rozwinie się żylna choroba zakrzepowo-zatorowa. Jakkolwiek, ryzyko to może być znacznie wyższe, w zależności od czynników ryzyka występujących u danej pacjentki (patrz poniżej).
Szacuje się1, że spośród 10 000 kobiet, które stosują złożone hormonalne produkty antykoncepcyjne zawierające drospirenon, u około 9 do 12 kobiet w okresie roku rozwinie się żylna choroba zakrzepowo- zatorowa, w porównaniu do około 62 kobiet stosujących złożone hormonalne produkty antykoncepcyjne zawierające lewonorgestrel.
W obydwu sytuacjach, liczba przypadków żylnej choroby zakrzepowo-zatorowej przypadających na okres roku jest mniejsza niż oczekiwana liczba przypadków u kobiet w ciąży lub w okresie poporodowym.
Żylna choroba zakrzepowo-zatorowa może być śmiertelna w 1-2% przypadków.
Liczba przypadków żylnej choroby zakrzepowo-zatorowej przypadających na 10 000 kobiet w okresie roku
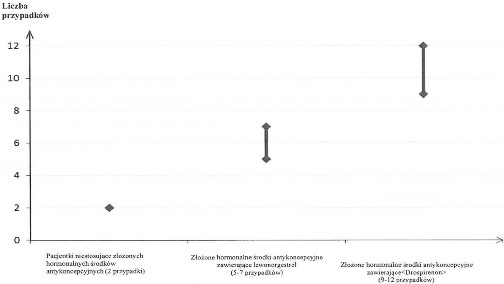
Badania epidemiologiczne wykazały, że ryzyko ŻChZZ dla doustnych produktów antykoncepcyjnych (ang. oral contraceptive, OC) zawierających drospirenon jest większe niż dla OC zawierających lewonorgestrel (tzw. produktów antykoncepcyjnych drugiej generacji) i może być podobne do ryzyka dla OC zawierających połączenie dezogestrel + gestoden (tzw. produktów antykoncepcyjnych trzeciej generacji).
Ponadto badania epidemiologiczne powiązały stosowanie złożonych doustnych produktów antykoncepcyjnych ze zwiększonym ryzykiem tętniczej choroby zakrzepowo-zatorowej (zawał mięśnia sercowego, przemijający napad niedokrwienny).
U pacjentek stosujących złożone hormonalne produkty antykoncepcyjne niezwykle rzadko zgłaszano przypadki zakrzepicy w innych naczyniach krwionośnych, np. wątrobowych, krezkowych, nerkowych lub w żyłach i tętnicach siatkówki.
Czynniki ryzyka żylnej choroby zakrzepowo-zatorowej
Ryzyko żylnych powikłań zakrzepowo-zatorowych u pacjentek stosujących złożone hormonalne produkty antykoncepcyjne może znacząco wzrosnąć w przypadku występowania dodatkowych czynników ryzyka, szczególnie, jeśli występuje kilka czynników ryzyka jednocześnie (patrz tabela).
Stosowanie produktu leczniczego Asubtela jest przeciwwskazane, jeśli u pacjentki występuje jednocześnie kilka czynników ryzyka, zwiększających ryzyko zakrzepicy żylnej (patrz punkt 4.3). Jeśli u kobiety wstępuje więcej niż jeden czynnik ryzyka, możliwe jest, że zwiększenie ryzyka jest większe niż suma pojedynczych czynników – w tym przypadku należy ocenić całkowite ryzyko żylnej choroby zakrzepowo- zatorowej. Jeśli ocena stosunku korzyści do ryzyka jest negatywna, nie należy przepisywać złożonych hormonalnych produktów antykoncepcyjnych (patrz punkt 4.3).
Tabela: Czynniki ryzyka żylnej choroby zakrzepowo-zatorowej
Czynnik ryzyka | Uwagi |
Otyłość (wskaźnik masy ciała (BMI) powyżej 30 kg/m2) | Ryzyko istotnie zwiększa się ze wzrostem BMI. Jest to szczególnie istotne do oceny, jeśli występują również inne czynniki ryzyka. |
Długotrwałe unieruchomienie, rozległy zabieg operacyjny, jakikolwiek zabieg operacyjny w obrębie kończyn dolnych lub miednicy, zabieg neurochirurgiczny lub poważny uraz Uwaga: tymczasowe unieruchomienie, w tym podróż samolotem >4 godzin może również stanowić czynnik ryzyka żylnej choroby zakrzepowo-zatorowej, szczególnie u kobiet ze współistniejącymi innymi czynnikami ryzyka. | W powyższych sytuacjach zaleca się przerwanie stosowania tabletek na co najmniej 4 tygodnie przed planowanym zabiegiem chirurgicznym i nie wznawianie stosowania produktu leczniczego przed upływem 2 tygodni od czasu powrotu do sprawności ruchowej. Należy stosować inną metodę antykoncepcji, aby uniknąć niezamierzonego zajścia w ciążę. Należy rozważyć leczenie przeciwzakrzepowe, jeśli stosowania produktu leczniczego Asubtela nie przerwano odpowiednio wcześnie. |
Dodatni wywiad rodzinny (występowanie żylnych zaburzeń zakrzepowo-zatorowych u rodzeństwa bądź rodziców, szczególnie w stosunkowo młodym wieku, np. przed 50 rokiem życia). | Jeśli podejrzewa się predyspozycję genetyczną, przed podjęciem decyzji o stosowaniu złożonego hormonalnego produktu antykoncepcyjnego kobieta powinna zostać skierowana na konsultację u specjalisty |
Inne schorzenia związane z żylną chorobą zakrzepowo-zatorową | Nowotwór, toczeń rumieniowaty układowy, zespół hemolityczno-mocznicowy, przewlekłe zapalne choroby jelit (np. choroba Leśniowskiego-Crohna lub wrzodziejące zapalenie jelita grubego) oraz niedokrwistość sierpowatokrwinkowa |
Wiek | Szczególnie w wieku powyżej 35 lat |
Nie osiągnięto konsensusu, co do możliwej roli żylaków oraz zakrzepowego zapalenia żył powierzchniowych na wystąpienie lub progresję żylnej choroby zakrzepowo-zatorowej.
Należy uwzględnić zwiększone ryzyko wystąpienia choroby zakrzepowo-zatorowej w ciąży oraz
w szczególności w 6-tygodniowym okresie poporodowym (więcej informacji w punkcie „Ciąża i laktacja” patrz punkt 4.6).
Objawy żylnej choroby zakrzepowo-zatorowej (zakrzepicy żył głębokich oraz zatorowości płucnej)
Należy poinformować pacjentkę, że w razie wystąpienia następujących objawów należy natychmiast zgłosić się do lekarza i powiedzieć personelowi medycznemu, że stosuje się złożone hormonalne produkty antykoncepcyjne.
Objawy zakrzepicy żył głębokich (ang. deep vein thrombosis, DVT) mogą obejmować:
Niektóre z tych objawów (np. „spłycenie oddechu”, „kaszel”) są niespecyficzne i mogą być niepoprawnie zinterpretowane jako występujące częściej lub mniej poważne stany (np. zakażenia układu oddechowego).
Inne objawy zamknięcia naczyń mogą obejmować: nagły ból, obrzęk oraz lekko niebieskie przebarwienie kończyn.
Jeżeli zamknięcie naczynia wystąpi w oku, objawy mogą obejmować bezbolesne zaburzenia widzenia, które mogą przekształcić się w utratę widzenia. W niektórych przypadkach utrata widzenia może nastąpić niemal natychmiast.
Ryzyko tętniczych zaburzeń zakrzepowo-zatorowych
Badania epidemiologiczne wykazały związek pomiędzy stosowaniem hormonalnych produktów antykoncepcyjnych, a zwiększonym ryzykiem tętniczych zaburzeń zakrzepowo-zatorowych (zawału mięśnia sercowego) lub incydentów naczyniowo-mózgowych (np. przemijającego napadu niedokrwiennego, udaru). Przypadki tętniczych zaburzeń zakrzepowo-zatorowych mogą być śmiertelne.
Czynniki ryzyka tętniczych zaburzeń zakrzepowo-zatorowych
Ryzyko wystąpienia tętniczych powikłań zakrzepowo-zatorowych lub napadów naczyniowo-mózgowych u pacjentek stosujących złożone hormonalne produkty antykoncepcyjne jest zwiększone u kobiet, u których występują czynniki ryzyka (patrz tabela). Stosowanie produktu leczniczego Asubtela jest przeciwwskazane, jeżeli u pacjentki występuje jeden poważny lub jednocześnie kilka czynników ryzyka tętniczych zaburzeń zakrzepowo-zatorowych, które stawiają pacjentkę w grupie wysokiego ryzyka zakrzepicy tętniczej (patrz punkt 4.3). Jeśli u kobiety wstępuje więcej niż jeden czynnik ryzyka, możliwe jest, że zwiększenie ryzyka jest większe niż suma pojedynczych czynników – w tym przypadku należy ocenić całkowite ryzyko. Jeśli ocena stosunku korzyści do ryzyka jest negatywna, nie należy przepisywać złożonych hormonalnych produktów antykoncepcyjnych (patrz punkt 4.3).
Tabela: Czynniki ryzyka tętniczych zaburzeń zakrzepowo-zatorowych
Czynnik ryzyka | Uwagi |
Wiek | Szczególnie w wieku powyżej 35 lat |
Palenie | Należy dokładnie pouczyć kobiety, aby nie paliły, jeśli zamierzają stosować złożone hormonalne produkty antykoncepcyjne. Kobiety w wieku powyżej 35 lat, które nie zaprzestały palenia, należy dokładnie pouczyć, aby stosowały inną metodę antykoncepcji. |
Nadciśnienie tętnicze | |
Otyłość (wskaźnik masy ciała (BMI) powyżej 30 kg/m2); | Ryzyko istotnie wzrasta wraz ze wzrostem BMI. Jest to szczególnie ważne dla kobiet, u których występują również inne czynniki ryzyka |
Dodatni wywiad rodzinny (występowanie tętniczych zaburzeń zakrzepowo-zatorowych u rodzeństwa bądź rodziców, szczególnie w stosunkowo młodym wieku, np. przed 50 rokiem życia). | Jeśli podejrzewa się predyspozycję genetyczną, przed podjęciem decyzji o stosowaniu złożonego hormonalnego produktu antykoncepcyjnego kobieta powinna zostać skierowana na konsultację u specjalisty |
Migrena | Zwiększenie częstości występowania lub nasilenia migreny w trakcie stosowania złożonych hormonalnych produktów antykoncepcyjnych (która może zapowiadać wystąpienie incydentu naczyniowo-mózgowego) może być powodem do natychmiastowego przerwania stosowania |
Inne schorzenia związane ze działaniami niepożądanymi w obrębie naczyń | Cukrzyca, hiperhomocysteinemia, wady zastawkowe serca, migotanie przedsionków, dyslipoproteinemia oraz toczeń rumieniowaty układowy |
Objawy tętniczych zaburzeń zakrzepowo-zatorowych
Należy poinformować pacjentkę, że w razie wystąpienia następujących objawów należy natychmiast zgłosić się do lekarza i powiedzieć personelowi medycznemu, że stosuje się złożone hormonalne produkty antykoncepcyjne.
Objawy napadu naczyniowo-mózgowego mogą obejmować:
Uwaga: Należy zapoznać się z informacją o jednocześnie stosowanych produktach leczniczych w celu sprawdzenia możliwości wystąpienia interakcji.
Interakcje farmakodynamiczne
Podczas badań klinicznych u pacjentów leczonych z powodu zakażenia wirusem zapalenia wątroby typu C (HCV) produktami leczniczymi zawierającymi ombitaswir z parytaprewirem i rytonawirem oraz dazabuwir z rybawiryna lub bez, zwiększenie aktywności aminotransferaz (AlAT) do wartości ponad pięciokrotnie większych niż górna granica normy występowało znacząco częściej u kobiet stosujących produkty lecznicze zawierające etynyloestradiol, takie jak złożone hormonalne produkty antykoncepcyjne. Dodatkowo, również u pacjentów leczonych glekaprewirem z pibrentaswirem lub sofosbuwirem z welpataswirem i woksylaprewirem, obserwowano zwiększenie aktywności AlAT u kobiet stosujących leki zawierające etynyloestradiol, takie jak złożone hormonalne środki antykoncepcyjne (patrz punkt 4.3).
Dlatego kobiety stosujące produkt leczniczy Asubtela muszą, przed rozpoczęciem leczenia za pomocą tych połączeń przeciwwirusowych, zmienić metodę stosowanej antykoncepcji na alternatywną (np. antykoncepcja zawierająca wyłącznie progestagen lub metody niehormonalne).
Ponowne stosowanie produktu leczniczego Asubtela można rozpocząć po 2 tygodniach od zakończenia leczenia za pomocą wcześniej opisanych połączeń przeciwwirusowych.
Stosowanie steroidowych produktów antykoncepcyjnych może wpływać na wyniki niektórych badań laboratoryjnych, w tym na biochemiczne parametry czynności wątroby, tarczycy, nadnerczy i nerek, stężenia osoczowe białek (nośnikowych), np. globuliny wiążącej kortykosteroidy oraz stężenia frakcji lipidów i lipoprotein, parametry metabolizmu węglowodanów oraz wskaźniki krzepnięcia i fibrynolizy. Zmienione wyniki badań laboratoryjnych zazwyczaj pozostają w granicach wartości prawidłowych.
Drospirenon powoduje zwiększenie aktywności reninowej osocza oraz stężenie aldosteronu w surowicy dzięki łagodnemu działaniu przeciwmineralokortykosteroidowemu.
Stosowanie produktu leczniczego Asubtela w czasie ciąży jest przeciwwskazane.
Jeśli podczas stosowania produktu leczniczego Asubtela kobieta zajdzie w ciążę, należy natychmiast przerwać jego stosowanie. Szerokie badania epidemiologiczne nie wykazały, ani zwiększonego ryzyka wystąpienia wad wrodzonych u dzieci kobiet, które przed zajściem w ciążę stosowały złożone doustne produkty antykoncepcyjne, ani działania teratogennego, gdy produkty te były stosowane we wczesnym okresie ciąży.
Badania na zwierzętach wykazały występowanie działań niepożądanych podczas ciąży i karmienia piersią (patrz punkt 5.3). Na podstawie tych badań nie można wykluczyć działania niepożądanego związanego
z hormonalnym działaniem substancji czynnych. Jednakże, powszechne doświadczenie kliniczne
dotyczące stosowania złożonych doustnych produktów antykoncepcyjnych w czasie ciąży nie wskazuje na występowanie rzeczywistych działań niepożądanych u ludzi.
Dostępne dane dotyczące stosowania produktu leczniczego Asubtela w czasie ciąży są zbyt ograniczone aby móc wyciągnąć wnioski dotyczące szkodliwego działania produktu leczniczego Asubtela na ciążę, stan zdrowia płodu lub noworodka. Do chwili obecnej brak jest istotnych danych epidemiologicznych.
Podejmując decyzję o ponownym rozpoczęciu stosowania produktu leczniczego Asubtela należy wziąć pod uwagę, zwiększone ryzyko żylnej choroby zakrzepowo-zatorowej u kobiet w okresie poporodowym (patrz punkty 4.2 i 4.4).
Karmienie piersią
Złożone doustne produkty antykoncepcyjne mogą mieć wpływ na laktację, ponieważ mogą zmniejszać ilość oraz modyfikować skład pokarmu. Dlatego też, nie zaleca się stosowania złożonych doustnych produktów antykoncepcyjnych do czasu zakończenia karmienia piersią. Niewielkie ilości steroidowych produktów antykoncepcyjnych i (lub) ich metabolitów mogą przenikać do mleka kobiet stosujących złożone doustne produkty antykoncepcyjne. Takie ilości mogą mieć wpływ na dziecko.
Nie przeprowadzono badań nad wpływem produktu leczniczego na zdolność prowadzenia pojazdów i obsługiwania maszyn. U kobiet stosujących złożone doustne produkty antykoncepcyjne nie zaobserwowano wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Ciężkie działania niepożądane występujące u kobiet stosujących złożone doustne produkty antykoncepcyjne, patrz punkt 4.4.
Opis wybranych działań niepożądanych
U kobiet stosujących złożone hormonalne produkty antykoncepcyjne odnotowano zwiększone ryzyko zakrzepicy żył i tętnic oraz zdarzeń zakrzepowo-zatorowych, w tym zawału serca, udaru, przemijającego napadu niedokrwiennego, zakrzepicy żylnej oraz zatorowości płucnej, zostały one szerzej omówione
w punkcie 4.4.
U kobiet stosujących produkt leczniczy Asubtela zgłaszano następujące działania niepożądane:
W poniższej tabeli przedstawiono działania niepożadane zgodnie z klasyfikacją układów i narządów MedDRA (ang. MedDRA SOCs – MedDRA System Organ Class). Częstości są oparte na danych z badań klinicznych.
Układ i narząd | Częstość występowania działań niepożądanych | |||
Często | Niezbyt często | Rzadko | Częstość nieznana | |
≥1/100 do <1/10 | ≥1/1000 do <1/100 | ≥1/10000 do <1/1000) | (częstość nie może być określona na podstawie dostępnych danych) | |
Zaburzenia układu immunologicznego | Nadwrażliwość Astma | Zaostrzenie objawów wrodzonego lub nabytego obrzęku naczynioruchowego | ||
Zaburzenia psychiczne | Nastroje depresyjne | Zmniejszenie libido, zwiększenie libido | ||
Zaburzenia układu nerwowego | Bóle głowy | |||
Zaburzenia ucha i błędnika | Niedosłuch | |||
Zaburzenia naczyniowe | Migrena | Nadciśnienie tętnicze Niedociśnienie tętnicze | Żylna choroba zakrzepowo- zatorowa Tętnicze zaburzenia zakrzepowo- zatorowe | |
Zaburzenia żołądka i jelit | Nudności | Wymioty, biegunka | ||
Zaburzenia skóry i tkanki podskórnej | Trądzik Łysienie Egzema Świąd | Rumień guzowaty Rumień wielopostaciowy | ||
Zaburzenia układu rozrodczego i piersi | Zaburzenia miesiączkowania, Krwawienia śródcykliczne, Ból piersi Tkliwość piersi Białe upławy Kandydoza pochwy | Powiększenie piersi Zapalenie pochwy | Wydzielina z piersi | |
Zaburzenia ogólne i stany w miejscu podania | Zatrzymanie płynów Zmniejszenie masy ciała Zwiększenie masy ciała |
W celu opisania poszczególnych reakcji i ich synonimów oraz związanych z nimi stanów zastosowano najbardziej odpowiednią terminologię MedDRA.
U kobiet stosujących złożone doustne produkty antykoncepcyjne zgłoszono wymienione niżej ciężkie działania niepożądane, omówione w punkcie 4.4.
Częstość rozpoznawania raka piersi u kobiet stosujących złożone doustne produkty antykoncepcyjne jest nieznacznie zwiększona. Ponieważ rak piersi występuje rzadko u kobiet w wieku poniżej 40 lat, nadmierna liczba takich rozpoznań jest mała w porównaniu z ogólnym ryzykiem raka piersi. Związek przyczynowy ze stosowaniem złożonych doustnych produktów antykoncepcyjnych jest nieznany. W celu uzyskania dalszych informacji, patrz punkt 4.3 i 4.4.
Interakcje
Krwawienie śródcykliczne i (lub) nieskuteczność działania antykoncepcyjnego może być wynikiem interakcji innych produktów leczniczych (induktorów enzymatycznych) ze złożonymi doustnymi produktami antykoncepcyjnymi (patrz punkt 4.5).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, Al. Jerozolimskie 181C, 02-222 Warszawa, Tel.: + 48 22 49 21 301,
Fax: + 48 22 49 21 309, e-mail: ndl@urpl.gov.pl.
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie zgłoszono żadnego przypadku przedawkowania produktu leczniczego Asubtela. Na podstawie ogólnych doświadczeń dotyczących przedawkowania złożonych doustnych produktów antykoncepcyjnych, objawy, jakie mogą wówczas wystąpić są następujące: nudności, wymioty oraz, u młodych dziewcząt, niewielkie krwawienie z pochwy. Nie istnieje antidotum i należy zastosować leczenie objawowe.
Grupa farmakoterapeutyczna (ATC): progestageny i estrogeny, produkt złożony kod ATC: G03AA12
Indeks Pearl’a dla niepowodzenia metody: 0,11 (górna wartość obustronnej granicy przedziału ufności 95%: 0,60).
Całkowity wskaźnik Pearl’a (niepowodzenie metody + błąd pacjenta): 0,31 (górna wartość obustronnej granicy przedziału ufności 95%: 0,91).
Mechanizm działania
Działanie antykoncepcyjne produktu leczniczego Asubtela jest oparte na interakcji kilku czynników, z których najważniejszym jest hamowanie owulacji oraz zmiany w obrębie endometrium.
Działanie farmakodynamiczne
Produkt leczniczy Asubtela jest złożonym doustnym produktem antykoncepcyjnym zawierającym etynyloestradiol i progestagen: drospirenon. W dawkach terapeutycznych drospirenon wykazuje również działanie antyandrogenne oraz niewielkie działanie antymineralokortykosteroidowe. Nie ma właściwości estrogennych, glikokortykosteroidowych oraz antyglikokortykosteroidowych. Dzięki tym właściwościom, profil farmakologiczny drospirenonu jest bardzo zbliżony do naturalnego progesteronu.
Wyniki badań klinicznych wskazują, że niewielkie właściwości przeciwmineralokortykosteroidowe produktu leczniczego Asubtela powodują lekkie działanie przeciwmineralokortykosteroidowe.
Wchłanianie
Po podaniu doustnym drospirenon jest szybko i prawie całkowicie wchłaniany. Po pojedynczym podaniu maksymalne stężenie substancji czynnej w surowicy krwi występuje po upływie 1-2 godzin i wynosi około 38 ng/ml. Biodostępność wynosi 76-85 %. Jednoczesne spożycie pokarmu nie wpływa na biodostępność drospirenonu.
Dystrybucja
Po podaniu doustnym, stężenie drospirenonu w surowicy krwi zmniejsza się, a końcowy, okres półtrwania wynosi 31 godzin.
Drospirenon wiąże się albuminami surowicy krwi, ale nie wiąże się z globuliną wiążącą hormony płciowe (ang. Sex Hormone Binding Globulin, SGHB) i globuliną wiążącą kortykoidy (ang. Corticoid Binding Globulin, CBG). Tylko od 3 % do 5 % całkowitego stężenia substancji czynnej w surowicy występuje w stanie wolnym. Zwiększenie stężenia SHGB w surowicy krwi, spowodowane działaniem etynyloestradiolu, nie wpływa na wiązanie drospirenonu z białkami. Średnia pozorna objętość dystrybucji drospirenonu wynosi 3,7 ± 1,2 l/kg.
Metabolizm
Drospirenon po podaniu doustnym podlega szybkiemu metabolizmowi. Główne metabolity w osoczu to: kwasowa postać drospirenonu powstająca na skutek otwarcia pierścienia laktozowego oraz 3-siarczan 4,5- dihydrodrospirenon; uzyskiwany poprzez redukcję i następnie sulfatację. Oba związki powstają bez udziału układu cytochromu P450. Drospirenon podlega metabolizmowi oksydacyjnemu katalizowanemu przez CYP4A4. In vitro, Drospirenon w stopniu niewielkim do umiarkowanego, hamuje enzymy cytochromu P450: CYP1A1, CYP2C9, CYP2C19 i CYP3A4.
Wydalanie
Klirens metaboliczny drospirenonu w surowicy wynosi 1,5 ± 0,2 ml/min/kg. Jedynie śladowe ilości drospirenonu są wydalane w postaci niezmienionej. Metabolity drospirenonu są wydalane z kałem
i moczem w stosunku około 1,2 - 1,4. Okres półtrwania wydalania metabolitów z moczem i kałem wynosi około 40 godzin.
Warunki stanu stacjonarnego
Podczas cyklu stosowania produktu leczniczego, maksymalne stężenie drospirenonu w surowicy w stanie stacjonarnym wynoszące około 70 ng/ml występuje po około 8 dniach od rozpoczęcia stosowania.
Stężenie drospirenonu w surowicy charakteryzuje się kumulacją przy współczynniku równym
w przybliżeniu 3 (iloraz końcowego okresu półtrwania i odstępu pomiędzy stosowaniem kolejnych dawek).
Szczególne grupy pacjentów
Wpływ zaburzeń czynności nerek
W stanie stacjonarnym stężenie drospirenonu w surowicy u kobiet z łagodnymi zaburzeniami czynności nerek (klirens kreatyniny CLcr, 50-80 ml/min) było porównywalne ze stężeniem u kobiet z prawidłową czynnością nerek. Stężenie drospirenonu w surowicy było większe o około 37% u kobiet z umiarkowanymi zaburzeniami czynności nerek (CLcr, 30 - 50 ml/min) niż u kobiet z prawidłową czynnością nerek. Drospirenon był również dobrze tolerowany przez kobiety z łagodnymi lub umiarkowanymi zaburzeniami czynnościami nerek. Stosowanie drospirenonu nie wykazało żadnego wpływu na stężenie potasu w surowicy.
Wpływ zaburzeń czynności wątroby
U ochotników z umiarkowanymi zaburzeniami czynności wątroby po jednorazowym podaniu doustnym drospirenonu klirens został zmniejszony o 50 % w porównaniu z grupą osób z prawidłową czynnością wątroby. Obserwowane zmniejszenie klirensu drospirenonu w grupie ochotników z umiarkowanymi zaburzeniami czynności wątroby w porównaniu z grupą osób z prawidłową czynnością wątroby nie miało wpływu na powstanie istotnych różnic w stężeniu potasu w surowicy. Również w przypadku występującej jednocześnie cukrzycy oraz leczenia skojarzonego spironolaktonem (dwa czynniki zwiększające ryzyko hiperkaliemii) nie zaobserwowano zwiększenia stężenia potasu w surowicy krwi ponad górną granicę normy. Podsumowując, drospirenon jest dobrze tolerowany przez pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności wątroby (stopień B w skali Child-Pugh).
Grupy etniczne
Wykazano brak istotnych klinicznie różnic pod względem właściwości farmakokinetycznych drospirenonu oraz etynyloestradiolu pomiędzy kobietami rasy kaukaskiej i pochodzącymi z Japonii.
Etynyloestradiol
Wchłanianie
Etynyloestradiol jest szybko i całkowicie wchłaniany po podaniu doustnym. Po podaniu 30 µg, maksymalne stężenie produktu leczniczego w surowicy występuje po około 1-2 godzinach po podaniu i wynosi 100 pg/ml. Etynyloestradiol podlega efektowi pierwszego przejścia, który wykazuje dużą zmienność indywidualną. Całkowita biodostępność wynosi około 45%. U około 25% badanych osób następowało zmniejszenie dostępności biologicznej etynyloestradiolu po spożyciu pokarmu; w pozostałych przypadkach nie wykazano tego efektu.
Dystrybucja
Stężenie etynyloestradiolu w surowicy ulega zmniejszeniu w dwóch fazach; w końcowej fazie eliminacji okres półtrwania wynosi około 24 godzin. Pozorna objętość dystrybucji wynosi około 5 l/kg. Około 98,5% etynyloestradiolu podlega silnemu, nieswoistemu wiązaniu przez albuminę i indukuje zwiększenie stężenia SHGB i globuliny wiążącej kortykosteroidy (CBG). Podczas stosowania 30 µg etynyloestradiolu stężenie SHBG w osoczu zwiększa się z 70 do około 350 nmol/l. Śladowe ilości etynyloestradiolu przenikają do mleka matki (0,02 % dawki).
Metabolizm
Etynyloestradiol w znaczącym stopniu ulega efektowi pierwszego przejścia w wątrobie i jelicie. Etynyloestradiol jest głównie metabolizowany na drodze hydroksylacji pierścienia aromatycznego, jednak powstaje również wiele metabolitów hydroksylowanych i metylowanych, występujących w postaci wolnej oraz związanej z kwasem glukuronowym i siarkowym. Klirens metaboliczny etynyloestradiolu wynosi
5 ml/min/kg). In vitro, etynyloestradiol jest odwracalnym inhibitorem enzymów CYP2X19, CYP1A1 i CYP1A2 jak również nieodwracalnym inhibitorem CYP3A4/5, CYP2C8 i CYP2J2.
Wydalanie
Praktycznie nie stwierdza się wydalania etynyloestradiolu w postaci niezmienionej. Metabolity etynyloestradiolu są wydalane wraz z moczem i żółcią w stosunku 4:6. Okres półtrwania metabolitów w fazie wydalania wynosi ok. 1 dzień. Okres półtrwania eliminacji wynosi 20 godzin.
Warunki stanu stacjonarnego
Stan równowagi dynamicznej ustala się w drugiej połowie cyklu stosowania produktu leczniczego; kumulacja etynyloestradiolu w surowicy charakteryzuje się współczynnikiem równym w przybliżeniu 2,0- 2,3.
W badaniach na zwierzętach laboratoryjnych stwierdzono, że działanie drospirenonu i etynyloestradiolu nie wykraczało poza znane działania farmakologiczne. W szczególności, badania toksycznego wpływu na reprodukcję wykazały embriotoksyczne i toksyczne działanie na płód, u zwierząt postrzeganych jako gatunki swoiste. W przypadku zastosowania dawki drospirenonu przekraczającej dawkę stosowaną u osób przyjmujących produkt leczniczy Asubtela odnotowano wpływ na różnicowanie płciowe płodów u szczurów natomiast nie zaobserwowano tego działania u małp.
Laktoza jednowodna
Skrobia kukurydziana
Skrobia żelowana kukurydziana Krospowidon (typ A i typ B) Powidon K 30
Polisorbat 80 Magnezu stearynian
Otoczka tabletki:
Otoczka Opadry II 85F32450 Yellow o składzie: Alkohol poliwinylowy częściowo hydrolizowany Tytanu dwutlenek (E171)
Makrogol 3350 Talk
Żelaza tlenek żółty (E172)
Nie dotyczy.
3 lata.
Przechowywać w temperaturze poniżej 30oC.
Blister PVC/PVDC/Aluminium w tekturowym pudełku. Rodzaje opakowań:
6 x 21 tabletek powlekanych
13 x 21 tabletek powlekanych
Nie wszystkie rodzaje opakowań muszą znajdować się w obrocie.
Bez specjalnych wymagań.
Exeltis Poland Sp. z o.o. ul. Szamocka 8
01-748 Warszawa
Pozwolenie nr: 16628
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 17.03.2010 Data ostatniego przedłużenia pozwolenia: 11.01.2017
10.11.2022 r.








