







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
ml roztworu zawiera 0,2 mg nimodypiny (Nimodipinum). Substancje pomocnicze o znanym działaniu:
Każda butelka 50 ml zawiera 23 mg sodu i 10,0 g etanolu 96%. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
mg (czyli 10 ml roztworu do infuzji Nimotop S) na godzinę (około 30 μg/kg mc./godz). U pacjentów o masie ciała znacznie mniejszej niż 70 kg lub mających niestabilne ciśnienie tętnicze krwi, leczenie należy rozpoczynać od dawki 0,5 mg nimodypiny (czyli 2,5 ml roztworu do infuzji Nimotop S) na godzinę.
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Nimotop S, 0,2 mg/ml, roztwór do infuzji
Roztwór do infuzji
Profilaktyka i leczenie niedokrwiennych ubytków neurologicznych spowodowanych skurczem naczyń krwionośnych mózgu po krwotoku podpajęczynówkowym w następstwie pęknięcia tętniaka.
Dawkowanie
Jeżeli lekarz nie przepisze inaczej, zaleca się następujące dawkowanie:
Wlew dożylny:
Roztwór do infuzji Nimotop S podawany jest w ciągłym wlewie dożylnym.
Maksymalna dawka dobowa wynosi 48 mg nimodypiny (czyli 240 ml roztworu do infuzji Nimotop S). Na początku leczenia (przez pierwsze 2 godziny): 1 mg nimodypiny (czyli 5 ml roztworu do infuzji Nimotop S) na godzinę (około 15 μg/kg mc. /godz.).
Jeżeli dawka ta jest dobrze tolerowana przez pacjenta, szczególnie jeśli nie obserwuje się znacznego obniżenia ciśnienia tętniczego krwi, po dwóch godzinach dawkę zwiększa się do
U pacjentów, u których występują działania niepożądane, należy zmniejszyć dawkę lub nawet zaprzestać podawania leku. Ciężkie upośledzenie czynności wątroby, szczególnie marskość wątroby, może spowodować – w wyniku zmniejszenia efektu pierwszego przejścia i osłabienia metabolizmu - zwiększenie biodostępności nimodypiny. Działania terapeutyczne oraz niepożądane, np. obniżenie ciśnienia tętniczego krwi, mogą być znacznie nasilone. W takich przypadkach należy – w zależności od wartości ciśnienia tętniczego - zmniejszyć dawkę leku lub jeżeli jest to konieczne, rozważyć jego odstawienie.
Sposób podawania
Roztwór do infuzji Nimotop S należy podawać w ciągłym wlewie dożylnym przez wkłucie do żyły centralnej z zastosowaniem pompy infuzyjnej. Produkt należy podawać przez kranik trójkanałowy wraz z jednym z następujących roztworów do wlewów: 5% roztwór glukozy, 0,9% roztwór chlorku sodu, płyn Ringera z dodatkiem mleczanu, płyn Ringera z dodatkiem mleczanu i magnezu, roztwór dekstranu 40 lub 6% roztwór poli-(O-2-hydroksy-etylo)-skrobii (HAES). Roztwory należy zmieszać w stosunku 1:4, tj. 1 część roztworu do infuzji Nimotop S i 4 części odpowiedniego roztworu do wlewów.
Wraz z roztworem do infuzji Nimotop S można podawać również roztwór mannitolu, albuminę ludzką lub krew.
Polietylenowy cewnik doprowadzający nimodypinę i linię infuzyjną podawanego jednocześnie roztworu należy połączyć z cewnikiem żylnym przy pomocy kranika trójkanałowego.
Roztworu do infuzji Nimotop S nie należy dodawać do worka lub butelki z innymi roztworami do wlewów. Nie należy go także mieszać z roztworami innych leków.
Podawanie roztworu do infuzji Nimotop S należy kontynuować podczas stosowania znieczulenia, zabiegu chirurgicznego i angiografii.
Czas trwania leczenia
Zastosowanie profilaktyczne:
Leczenie za pomocą wlewu dożylnego należy rozpocząć nie później niż 4 dni po wystąpieniu krwotoku podpajęczynówkowego oraz kontynuować przez cały okres zwiększonego ryzyka skurczu naczyń, to znaczy przez 10-14 dni po krwotoku.
Jeśli w trakcie leczenia profilaktycznego istnieje konieczność ingerencji chirurgicznej w obrębie źródła krwotoku podawanie roztworu do infuzji Nimotop S należy kontynuować przez co najmniej 5 dni po zabiegu.
Po zakończeniu leczenia dożylnego zaleca się kontynuować leczenie, podając nimodypinę w postaci doustnej w dawce 6 x 60 mg na dobę, co 4 godziny, przez kolejnych 7 dni.
Zastosowanie lecznicze:
Jeżeli doszło do niedokrwiennych ubytków neurologicznych, spowodowanych skurczem naczyń krwionośnych po krwotoku podpajęczynówkowym, leczenie należy rozpocząć tak szybko, jak tylko jest to możliwe i prowadzić co najmniej przez 5 dni (maksymalnie przez 14 dni).
Po zakończeniu leczenia dożylnego zaleca się kontynuować leczenie, podając nimodypinę w postaci doustnej w dawce 6 x 60 mg na dobę, co 4 godziny, przez kolejnych 7 dni.
Jeżeli w trakcie leczenia nimodypiną jest przeprowadzany zabieg chirurgiczny, podawanie roztworu do infuzji Nimotop S należy kontynuować przez co najmniej 5 dni po zabiegu.
Szczególne grupy pacjentów
Nie określono bezpieczeństwa stosowania ani skuteczności nimodypiny u pacjentów w wieku poniżej 18 lat.
Nadwrażliwość na nimodypinę lub inne składniki produktu.
Mimo że nie wykazano zależności pomiędzy leczeniem produktem Nimotop S a zwiększeniem ciśnienia śródczaszkowego, zaleca się monitorowanie stanu pacjentów z podwyższonym ciśnieniem
śródczaszkowym lub ze zwiększoną zawartością wody w tkance mózgowej (uogólniony obrzęk mózgu).
Należy także zachować ostrożność podczas stosowania produktu u pacjentów ze znacznie obniżonym ciśnieniem tętniczym (ciśnienie skurczowe mniejsze niż 100 mm Hg).
U pacjentów z niestabilną dławicą piersiową lub którzy przebyli ostry zawał mięśnia sercowego w ciągu ostatnich 4 tygodni, należy rozważyć stosunek potencjalnego ryzyka (np. zmniejszony przepływ wieńcowy lub niedokrwienie mięśnia sercowego) do korzyści (np. poprawa perfuzji mózgowej) związanych ze stosowaniem produktu.
Dawka 10 ml/h tego leku podana osobie dorosłej o masie ciała 70 kg spowoduje ekspozycję na etanol wynoszącą 28 mg/kg/h, co może spowodować zwiększenie stężenia alkoholu we krwi (ang. blood alcohol concentration, BAC) o około 4 mg/100 ml. Dla porównania, u osoby dorosłej, po wypiciu kieliszka wina lub 500 ml piwa, stężenie alkoholu we krwi wyniesie prawdopodobnie około 50 mg/100 ml.
Jednoczesne podawanie z lekami zawierającymi, np. glikol propylenowy lub etanol może prowadzić do kumulacji etanolu i wywoływać działania niepożądane, w szczególności u małych dzieci o małej zdolności metabolicznej lub z niedojrzałością metaboliczną.
Ponieważ ten lek podaje się powoli przez ciągły wlew dożylny, działanie alkoholu może być zmniejszone.
Ta ilość etanolu może stanowić zagrożenie dla pacjentów z chorobą alkoholową i pacjentów z zaburzeniami metabolizmu alkoholu oraz musi być brana pod uwagę u kobiet w ciąży lub karmiących piersią, dzieci oraz pacjentów z grup wysokiego ryzyka np. z chorobą wątroby, padaczką. Alkohol zawarty w produkcie leczniczym może zmieniać działanie innych stosowanych jednocześnie leków (patrz pkt. 4.5).
Produkt leczniczy zawiera 23 mg sodu w 50 ml butelce, co odpowiada 1,15 % zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych. Podając roztwór do infuzji Nimotop S należy również wziąć pod uwagę zawartość sodu wchodzącego w skład roztworu do wlewu. Należy zapoznać się z charakterystyką produktu leczniczego stosowanego roztworu do wlewu w celu obliczenia całkowitej zawartości sodu w przygotowanym rozcieńczeniu produktu.
Należy wziąć to pod uwagę zalecając produkt leczniczy osobom stosującym dietę z małą ilością sodu.
Podawanie produktu z lekami mogącymi działać nefrotoksycznie (antybiotyki aminoglikozydowe, cefalosporyny, furosemid) może zaburzać czynność nerek. U pacjentów z niewydolnością nerek może nastąpić pogorszenie czynności nerek. W tych przypadkach należy monitorować parametry czynności nerek, a w sytuacji wystąpienia objawów ich niewydolności, należy rozważyć przerwanie leczenia.
Produkty lecznicze wpływające na działanie nimodypiny Fluoksetyna
Jednoczesne podanie nimodypiny i fluoksetyny prowadziło do zwiększenia w surowicy krwi stężenia nimodypiny w stanie stacjonarnym o około 50%. Ekspozycja na fluoksetynę wyraźnie się zmniejsza, zaś jej aktywnego metabolitu – norfluoksetyny - nie zmieniło się.
Nortryptylina
Jednoczesne podanie nimodypiny i nortryptyliny powodowało nieznaczne zmniejszenie ekspozycji na nimodypinę w stanie stacjonarnym; stężenie nortryptyliny nie zmienia się.
Wpływ nimodypiny na inne produkty lecznicze
Leki obniżające ciśnienie tętnicze
Jednoczesne podanie nimodypiny może nasilać hipotensyjne działanie leków przeciwnadciśnieniowych takich jak: leki moczopędne, leki blokujące receptory beta-adrenergiczne, inhibitory konwertazy angiotensyny, antagoniści receptorów A1, inni antagoniści wapnia, środki blokujące receptory alfa-adrenergiczne, inhibitory enzymu fosfodiesterazy 5 (PDE5), alfa-metylodopa. Jeśli konieczne jest zastosowanie nimodypiny w skojarzeniu z wymienionymi lekami, należy systematycznie kontrolować stan pacjenta.
Dożylne podawanie leków β-adrenolitycznych jednocześnie z nimodypiną może prowadzić do nasilenia działania inotropowego ujemnego i niewyrównanej niewydolności mięśnia sercowego.
Podawanie produktu z lekami mogącymi działać nefrotoksycznie (antybiotyki aminoglikozydowe, cefalosporyny, furosemid) może nasilić ujemny wpływ na czynność nerek. U pacjentów
z niewydolnością nerek może nastąpić pogorszenie czynności nerek. W tych przypadkach należy stale monitorować parametry czynności nerek, a w sytuacji wystąpienia objawów ich niewydolności, należy rozważyć przerwanie leczenia.
Zydowudyna
Badania przeprowadzone na małpach, którym podawano jednocześnie zydowudynę dożylnie
i nimodypinę w postaci wlewu skutkowało znaczącym zwiększeniem pola powierzchni pod krzywą zależności stężenia od czasu (AUC) dla zydowudyny podczas gdy wyraźnie zmniejszyła się objętość dystrybucji i klirens.
Inne formy interakcji
Ponieważ Nimotop S – roztwór do infuzji - zawiera 23,7% obj. alkoholu etylowego, należy wziąć pod uwagę możliwość interakcjach z lekami, których nie można podawać jednocześnie z etanolem (patrz pkt. 4.4).
Ciąża
Brak odpowiednich i dobrze kontrolowanych badań u kobiet w ciąży. W razie konieczności podania nimodypiny kobiecie w cięży należy ocenić stosunek korzyści do ryzyka, uwzględniając stan kliniczny pacjentki.
Laktacja
Wykazano, że nimodypina i jej metabolity przenikają do mleka kobiet karmiących w stężeniu odpowiadającym stężeniu w surowicy matki. Zaleca się zaprzestania karmienia piersią w trakcie terapii nimodypiną.
Płodność
W pojedynczych przypadkach zapłodnień in vitro zaobserwowano odwracalne zmiany biochemiczne w główce plemników pod wpływem antagonistów wapnia, które mogą skutkować zaburzeniami nasienia. Znaczenie tego odkrycia dla krótkotrwałego leczenia nie jest znane.
Nimodypina może zaburzać zdolność prowadzenia pojazdów i obsługiwania maszyn z powodu możliwości wystąpienia zawrotów głowy. W przypadku konieczności zastosowania leku w postaci roztworu do infuzji ten wpływ nie będzie miał znaczenia.
Związane z lekiem działania niepożądane, opisywane w badaniach klinicznych z zastosowaniem nimodypiny we wskazaniu „krwotok podpajęczynówkowy w następstwie tętniaka” podzielone wg
kategorii CIOMS III co do częstości zestawiono w poniższej tabeli (kontrolowane badania z placebo: nimodypina N=703; placebo N=692; niekontrolowane badania: nimodypina N=2496, stan na 31.08.2005). W zakresie każdej częstości, działania niepożadane przedstawiono zgodnie ze zmieniającym się nasileniem.
Częstości zdefiniowano w natępujący sposób: Bardzo często (≥ 1/10)
Często (≥ 1/100 do < 1/10)
Niezbyt często (≥ 1/1000 do < 1/100) Rzadko (≥ 1/10000 do < 1/1000) Bardzo rzadko (<1/10000)
Układ/narząd wg MedDRA | Często | Niezbyt często | Rzadko | Bardzo rzadko |
Zaburzenia krwi i układu chłonnego | ||||
małopłytkowość | ||||
Zaburzenia układu immunologicznego | ||||
reakcje alergiczne, wysypka | ||||
Zaburzenia układu nerwowego | ||||
ból głowy | ||||
Zaburzenia serca | ||||
tachykardia | bradykardia | |||
Zaburzenia naczyniowe | ||||
niedociśnienie tętnicze, rozszerzenie naczyń krwionośnych | ||||
Zaburzenia żołądka i jelit | ||||
nudności | niedrożność jelit | |||
Zaburzenia wątroby i dróg żółciowych | ||||
przemijające zwiększenie aktywności enzymów wątrobowych | ||||
Zaburzenia ogólne i stany w miejscu podania | ||||
reakcje w miejscu wlewu lub wstrzyknięcia, zakrzepowe zapalenie żył w miejscu wlewu | ||||
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C, 02-222 Warszawa,
Tel.: + 48 22 49 21 301,
Faks: + 48 22 49 21 309,
Strona internetowa: https://smz.ezdrowie.gov.pl
Objawy
Objawami zatrucia, występującego w wyniku ostrego przedawkowania, są: znaczne zmniejszenie ciśnienia tętniczego, tachykardia lub bradykardia a po podaniu doustnym zaburzenia żołądkowo- jelitowe i nudności.
Leczenie
W przypadku ostrego przedawkowania należy natychmiast odstawić Nimotop S. Należy wdrożyć leczenie objawowe oraz rozważyć płukanie żołądka i podanie węgla aktywowanego. Jeśli doszło do znacznego zmniejszenia ciśnienia tętniczego można podać dożylnie dopaminę lub noradrenalinę. Ponieważ brak specyficznego antidotum, należy wdrożyć leczenie objawowe w przypadku wystąpienia innych działań niepożądanych, zależnie od ich nasilenia.
Grupa farmakoterapeutyczna: wybiórczy antagoniści wapnia o dominującym działaniu naczyniowym; pochodne dihydropirydyny; Kod ATC: C08CA06
Nimodypina jest pochodną 1,4-dihydropirydyny, należącą do grupy antagonistów wapnia. Proces skurczu komórek mięśni gładkich zależy od jonów wapnia, które napływają do tych komórek w czasie depolaryzacji, w postaci wolnego prądu przezbłonowego. Nimodypina hamuje napływ jonów wapnia do tych komórek i w ten sposób hamuje skurcz mięśni gładkich ścian naczyniowych. W doświadczeniach na zwierzętach nimodypina wykazywała silniejsze działanie na tętnice mózgowe niż na naczynia tętnicze w innej lokalizacji. Prawdopodobnie ze względu na wysoce lipofilne właściwości przechodzi przez barierę krew-mózg. U pacjentów po krwotoku podpajęczynówkowym leczonych nimodypiną w płynie mózgowo-rdzeniowych stwierdzano stężenia na poziomie 12,5 ng/ml.
Nimodypina wykazuje szczególne działanie zapobiegające zwężeniu naczyń mózgowych oraz niedokrwieniu tkanki mózgowej. Może też zapobiegać lub eliminować efekt zwężenia naczyń wywoływany in vitro przez różne substancje wykazujące takie działanie (np. serotonina, prostaglandyny i histamina) a także przez krew i produkty jej rozpadu. Nimodypina ma także właściwości neurofarmakologiczne i psychofarmakologiczne.
Badania przeprowadzone z udziałem pacjentów z ostrymi zaburzeniami przepływu mózgowego krwi wykazały, że nimodypina rozszerza naczynia mózgowe i poprawia przepływ krwi. Wzrost przepływu jest większy w uszkodzonych i niedostatecznie ukrwionych obszarach mózgu, w porównaniu
z obszarami nie zmienionymi chorobowo. Niedokrwienne ubytki neurologiczne u pacjentów po krwotoku podpajęczynówkowym oraz śmiertelność są wyraźnie mniejsze po zastosowaniu nimodypiny.
Wchłanianie
Substancja czynna, nimodypina, podana doustnie, praktycznie całkowicie wchłania się z przewodu pokarmowego. Niezmieniona substancja czynna oraz jej wczesne metabolity wykrywane są w osoczu
krwi już po 10-15 minutach od przyjęcia tabletki. Maksymalne stężenie w surowicy (Cmax) po wielokrotnym doustnym podaniu dawek podzielonych (3 x 30 mg/dobę) wynosi 7,3-43,2 ng/ml u osób starszych i jest osiągane po 0,6-1,6 godziny (tmax). Podanie pojedynczej dawki 30 mg i 60 mg osobom młodym daje średnie stężenie w osoczu odpowiednio 16 ± 8 ng/ml i 31 ± 12 ng/ml. Cmax oraz AUC wzrastają proporcjonalnie do dawki, aż do najwyższych badanych dawek (90 mg).
Po dożylnym podaniu nimodypiny we wlewie ciągłym w dawce 0,03 mg/kg mc./godz. stężenie substancji czynnej w osoczu w stanie równowagi stacjonarnej wynosi 17,6-26,6 ng/ml. Po wstrzyknięciu dożylnym nimodypiny obserwuje się dwufazowy spadek stężenia nimodypiny w osoczu z okresem półtrwania 5-10 min. i ok. 60 min. Pozorna objętość dystrybucji (model dwukompartmentowy) po podaniu dożylnym wynosi 0,9 - 1,6 l/kg mc., zaś całkowity klirens (ogólnoustrojowy) 0,6 - 1,9 l/godz/kg mc.
Wiązanie z białkami oraz dystrybucja
Nimodypina wiąże się z białkami osocza w 97-99 %. W doświadczeniach na zwierzętach nimodypina znakowana radioaktywnym węglem 14C przenikała przez barierę łożyskową. Uważa się, że analogiczne zjawisko może występować u ludzi, choć brak jest na to dowodów empirycznych.
Wykazano, że nimodypina i (lub) jej metabolity występowały w mleku szczurów w stężeniach znacznie większych niż w osoczu karmiącej samicy.
Stężenia leku oznaczone w mleku kobiecym były podobne do odpowiednich stężeń w osoczu matki.
Po doustnym i dożylnym podaniu nimodypina występuje w płynie mózgowo - rdzeniowym w stężeniu około 0,5% stężenia stwierdzonego w osoczu. Odpowiada to w przybliżeniu stężeniu wolnej substancji w osoczu.
Metabolizm, eliminacja i wydalanie
Nimodypina jest metabolizowana przez enzym 3A4 cytochromu P450 do metabolitów, powstałych w wyniku dehydrogenacji pierścienia dihydropirydyny oraz oksydacyjnego rozszczepienia estrowego. Oksydacyjne rozszczepienie wiązania estrowego, hydroksylacja grup metylowych w pozycjach 2- i 6- oraz sprzęganie z kwasem glukuronowym są kolejnymi ważnymi etapami metabolizmu nimodypiny. Trzy pierwotne metabolity pojawiające się w osoczu krwi, nie wykazują żadnej aktywności lub mają aktywność nieistotną w sensie terapeutycznym.
Wpływ na enzymy wątrobowe - ich indukcja lub inhibicja - nie są znane. Z organizmu człowieka metabolity nimodypiny wydalają się w 50% przez nerki w moczu oraz w 30% z żółcią. Kinetyka eliminacji ma charakter liniowy. Okres półtrwania nimodypiny wynosi 1,1-1,7 godz. Końcowy okres półtrwania wynosi 5-10 godz. i nie ma znaczenia dla zalecanych przerw w podawaniu kolejnych dawek.
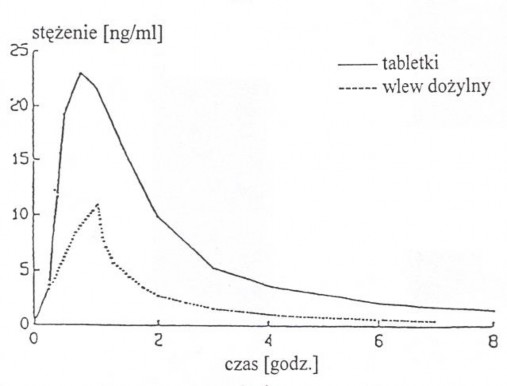
Krzywe średniego stężenia nimodypiny w osoczu po podaniu dawki 30 mg w postaci tabletki, a następnie wlewu dożylnego w dawce 0,015 mg/kg mc. przez godzinę (n=24 ochotników).
Biodostępność
Ze względu na znaczny efekt pierwszego przejścia (85-95 %) całkowita biodostępność nimodypiny wynosi 5-15 %.
Dane przedkliniczne uzyskane na podstawie standardowych badań toksyczności genotoksyczności, rakotwórczości i toksycznego wpływu na rozmnażanie po podaniu jednorazowym i wielokrotnym nie wykazały szczególnego zagrożenia dla ludzi. U ciężarnych samic szczurów dawki 30 mg/kg m.c./dobę hamowały rozwój płodu oraz skutkowały zmniejszeniem masy ciała płodu. Dawka 100 mg/kg m.c./dobę stanowiła dawkę śmiertelną dla zarodka. Nie zaobserwowano efektu teratogennego nimodypiny. U królików nie zaobserwowano efektu embriotoksycznego ani teratogennego w dawkach do 10 mg/kg m.c./dobę. W jednym przypadku badania noworodków szczura wykazały śmiertelność
i opóźnienie rozwoju fizycznego zaobserwowano po podawaniu dawek 10 mg/kg m.c./dobę i większych. Kolejne badania nie potwierdziły tych wyników.
Etanol 96%
Makrogol 400 Sodu cytrynian Kwas cytrynowy
Woda do wstrzykiwań
Roztwór zawiera 23,7% obj. alkoholu (200 mg w 1 ml roztworu do infuzji) i 17% makrogolu 400.
Ponieważ nimodypina jest adsorbowana przez polichlorek winylu (PVC), do podawania roztworu do infuzji Nimotop S należy używać aparatów, przewodów infuzyjnych oraz kateterów wykonanych wyłącznie z polietylenu (PE).
Mimo że nimodypina jest w niewielkim stopniu wrażliwa na światło, należy unikać bezpośredniej ekspozycji produktu na światło słoneczne. Jeżeli w trakcie prowadzenia wlewu nie można uniknąć ekspozycji produktu na światło słoneczne, należy używać strzykawek i przewodów w kolorach czarnym, brązowym, żółtym lub czerwonym lub osłaniać pompy infuzyjne i przewody folią nie przepuszczającą światła.
Roztwór można podawać bez osłony, jeżeli ekspozycja na światło sztuczne lub rozproszone światło dzienne jest krótsza niż 10 godzin.
4 lata
Przechowywać w temperaturze poniżej 25˚C, chronić od światła.
Butelka z oranżowego szkła 50 ml zamknięta korkiem z chlorobutylu umieszczona w tekturowym pudełku. Przewód infuzyjny z polietylenu (HDPE) łączący pompę infuzyjną z kranikiem trójkanałowym w torbie foliowo-papierowej.
Roztwór do infuzji Nimotop S należy podawać w ciągłym wlewie dożylnym przez wkłucie do żyły centralnej z zastosowaniem pompy infuzyjnej.
Bayer AG
Kaiser-Wilhelm-Allee 1
51373 Leverkusen Niemcy
Pozwolenie nr R/2746
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 01.08.1990 r. Data ostatniego przedłużenia pozwolenia: 29.07.2009 r.








