







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Lutrate Depot, 3,75 mg, proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o przedłużonym uwalnianiu
Każda fiolka zawiera 3,75 mg leuproreliny octanu (co odpowiada 3,57 mg leuproreliny). l ml odtworzonej zawiesiny zawiera 1,875 mg leuproreliny octanu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o przedłużonym uwalnianiu Proszek: biały lub białawy proszek.
Rozpuszczalnik: przezroczysty, bezbarwny roztwór (pH 5,0 -7,0) bez widocznych cząstek.
Lutrate Depot jest wskazany w leczeniu paliatywnym zaawansowanego raka gruczołu krokowego.
Dawkowanie
Zazwyczaj zalecana dawka produktu leczniczego Lutrate Depot to 3,75 mg w postaci wstrzyknięcia o przedłużonym uwalnianiu, wykonywanego raz w miesiącu, i podawana w pojedynczym wstrzyknięciu domięśniowym co miesiąc.
.
Zastosowanie postaci leku o przedłużonym, kontrolowanym uwalnianiu zapewnia stałe uwalnianie leuproreliny octanu w ciągu miesiąca po podaniu produktu Lutrate Depot. Liofilizowany proszek należy odtworzyć i stosować w pojedynczym wstrzyknięciu domięśniowym, podawanym w odstępach co miesiąc. Należy unikać przypadkowego podania dotętniczego lub dożylnego. Proszek z mikrosferami, znajdujący się w fiolce produktu Lutrate Depot, należy odtworzyć bezpośrednio przed wstrzyknięciem domięśniowym. Jak w przypadku innych produktów leczniczych stosowanych długotrwale we wstrzyknięciach, należy okresowo zmieniać miejsce wstrzyknięcia.
Nie należy przerywać stosowania produktu Lutrate Depot, gdy wystąpi remisja choroby lub poprawa kliniczna. Należy monitorować reakcję pacjenta na leczenie produktem Lutrate Depot, oznaczając okresowo zarówno stężenie testosteronu w surowicy, jak i swoisty antygen sterczowy (PSA). W badaniach klinicznych wykazano, że u większości pacjentów niepoddanych wcześniej orchidektomii, stężenie testosteronu zwiększało się podczas 4 pierwszych dni leczenia, a następnie malało, osiągając wartości kastracyjne w ciągu 3 do 4 tygodni. Od tego momentu stężenie kastracyjne (zdefiniowane jako stężenie testosteronu mniejsze niż 0,5 ng/ml) utrzymywało się tak długo, jak długo stosowano leczenie.
Jeśli reakcja pacjenta na leczenie jest mniejsza od oczekiwanej, zaleca się upewnić, czy stężenia testosteronu w surowicy osiągnęły wartości kastracyjne lub utrzymują się na ich poziomie. Niekiedy w początkowym okresie leczenia występuje przemijające zwiększenie aktywności fosfatazy kwaśnej, które zazwyczaj po ok. 4 tygodniach leczenia powraca do wartości prawidłowych lub zbliżonych do normy.
Czas leczenia
Produkt Lutrate Depot jest stosowany w postaci wstrzyknięć domięśniowych podawanych raz w miesiącu.
Specjalne grupy pacjentów
Dzieci i młodzież:
Nie ustalono bezpieczeństwa stosowania i skuteczności produktu Lutrate Depot u dzieci i młodzieży. Z tego powodu nie zaleca się stosowania produktu Lutrate Depot do czasu uzyskania tych danych.
Niewydolność nerek lub wątroby:
Nie badano farmakokinetyki produktu Lutrate Depot u pacjentów z niewydolnością wątroby lub nerek.
Pacjenci w podeszłym wieku:
W badaniu klinicznym produktu Lutrate Depot średnia wieku osób badanych wynosiła 71,6 ± 9,2 lata. Tak więc dane odnoszące się do farmakokinetyki, skuteczności i bezpieczeństwa stosowania produktu Lutrate Depot dotyczą także tej populacji.
Sposób podawania
Produkt leczniczy Lutrate Depot powinien być przygotowywany, rekonstytuowany i podawany wyłącznie przez fachowy personel medyczny, który zna te procedury.
Lutrate Depot należy podawać wyłącznie domięśniowo. Nie stosować innych dróg podania. Jeśli produkt został omyłkowo podany podskórnie, należy uważnie monitorować stan pacjenta ze względu na brak danych, dotyczących innych niż domięśniowa dróg podania produktu Lutrate Depot.
Instrukcja dotycząca rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na analogi hormonu uwalniającego luteotropinę (LHRH) lub na którąkolwiek substancję pomocniczą produktu Lutrate Depot, wymienioną w punkcie 6.1. W piśmiennictwie medycznym znajdują się doniesienia o reakcjach anafilaktycznych po podaniu syntetycznego hormonu LHRH lub analogów agonistów LHRH.
Przebyta orchidektomia.
Nie należy stosować produktu Lutrate Depot w monoterapii u pacjentów z rakiem gruczołu krokowego, u których występuje ucisk na rdzeń kręgowy lub przerzuty do kręgosłupa.
Lutrate Depot nie jest wskazany do stosowania u kobiet.
Lutrate Depot nie jest wskazany do stosowania u dzieci i młodzieży.
W początkowym okresie leczenia produktem Lutrate Depot, tak jak podczas leczenia innymi agonistami LHRH, może wystąpić przemijające zwiększenie stężenia testosteronu. W niektórych przypadkach powoduje to wystąpienie charakterystycznych objawów lub nasilenie wzrostu guza,
prowadzące do zaostrzenia objawów raka gruczołu krokowego, które zazwyczaj zmniejszają się podczas kontynuacji leczenia. Charakterystyczne objawy („flare”) mogą występować w postaci układowych lub neurologicznych objawów (np. jako ból kości). Opisano także przypadki zaniku jąder i ginekomastii po stosowaniu innych agonistów LHRH.
Leczenie należy natychmiast przerwać, jeśli u pacjenta wystąpią objawy przedmiotowe i podmiotowe, sugerujące anafilaksję lub reakcję anafilaktyczną (duszność, astma, katar, obrzęk naczynioruchowy głośni, hipotonia, pokrzywka, wysypka, świąd lub śródmiąższowe zapalenie płuc). Przed rozpoczęciem leczenia należy poinformować o tym pacjentów, ostrzegając ich, że powinni przerwać leczenie i skonsultować się z lekarzem, jeśli wystąpią opisane powyżej objawy. Pacjentów, u których wystąpiła reakcja nadwrażliwości na leuprolid, należy uważnie monitorować. Nie powinni oni ponownie stosować produktu Lutrate Depot.
U pacjentów leczonych leuproreliny octanem obserwowano pojedyncze przypadki niedrożności cewki moczowej (z hematurią lub bez) i ucisku na rdzeń kręgowy lub przerzutów do kręgosłupa, mogących powodować porażenie, niekiedy z powikłaniami prowadzącymi do zgonu. Pacjentom z ryzykiem wystąpienia niedrożności cewki moczowej, ucisku na rdzeń kręgowy lub przerzutów do kręgosłupa należy w pierwszych tygodniach leczenia zapewnić ścisłą opiekę lekarską. U tych pacjentów należy rozważyć profilaktyczne leczenie antyandrogenami.
W razie wystąpienia urologicznych i neurologicznych powikłań należy wdrożyć odpowiednie leczenie.
U pacjentów leczonych agonistami GnRH jak np. leuprolidu octanem występuje zwiększone ryzyko depresji (która może być ciężka). Pacjenci powinni być o tym poinformowani i w przypadku wystąpienia objawów depresji odpowiednio leczeni.
W piśmiennictwie opisywano występowanie zmniejszenia gęstości mineralnej kości u mężczyzn po zabiegu orchidektomii lub leczonych agonistami LHRH. Włączenie antyandrogenu do schematu leczenia zmniejsza utratę masy kostnej, ale nasila ryzyko wystąpienia innych działań niepożądanych, takich jak zaburzenia krzepnięcia i obrzęki. Jeśli lek z grupy antyandrogenów jest stosowany przez dłuższy czas, należy zwrócić szczególną uwagę na przeciwwskazania i środki ostrożności związane z jego długotrwałym stosowaniem. Podczas leczenia leuproreliny octanem należy ściśle nadzorować pacjentów z ryzykiem wystąpienia osteoporozy lub wywiadem obciążonym osteoporozą.
Podczas stosowania leuproreliny octanu informowano o występowaniu zaburzeń czynności wątroby i żółtaczki przebiegającej ze zwiększeniem aktywności enzymów wątrobowych. Z tego powodu należy uważnie obserwować pacjentów i w razie konieczności wdrożyć odpowiednie postępowanie.
Należy kontrolować reakcję pacjenta na leczenie produktem Lutrate Depot, monitorując parametry kliniczne oraz oznaczając okresowo stężenie testosteronu i antygenu PSA w surowicy.
U pacjentów mogą wystąpić zmiany metaboliczne (np. nietolerancja glukozy lub zaostrzenie istniejącej cukrzycy), nadciśnienie tętnicze, zmiany masy ciała i zaburzenia sercowo-naczyniowe. Jak należy się spodziewać w odniesieniu do tej grupy leków, może wystąpić lub zaostrzyć się istniejąca wcześniej cukrzyca. Z tego powodu podczas leczenia produktem Lutrate Depot pacjenci z cukrzycą mogą wymagać częstszego kontrolowania stężenia glukozy we krwi. Przed rozpoczęciem leczenia oraz podczas prowadzenia deprywacji androgenowej należy starannie monitorować pacjentów z dużym ryzykiem wystąpienia choroby metabolicznej lub sercowo-naczyniowej. Leczenie leupropreliny octanem powoduje zahamowanie osi przysadka mózgowa-gruczoły płciowe. Podczas leczenia oraz po jego zakończeniu mogą ulec zmianie wyniki badań diagnostycznych czynności gonadotropowej przysadki oraz czynności gruczołów płciowych.
U pacjentów leczonych leuproreliny octanem informowano o występowaniu wydłużonego czasu protrombinowego.
Raportowano o występowaniu drgawek podczas stosowania leuproreliny octanu. Dotyczyło to pacjentów z wywiadem obciążonym występowaniem drgawek, padaczki, zaburzeń krążenia mózgowego, nieprawidłowości lub guzów ośrodkowego układu nerwowego oraz pacjentów leczonych jednocześnie lekami wywołującymi drgawki, takimi jak na przykład bupropion i selektywne inhibitory wychwytu zwrotnego serotoniny (SSRI). Opisywano także występowanie drgawek u pacjentów spoza tych grup.
Leuprolidu octan należy stosować z zachowaniem ostrożności w chorobie sercowo-naczyniowej (w tym w zastoinowej niewydolności serca), chorobie zakrzepowo-zatorowej, obrzękach, depresji i udarze przysadki mózgowej.
Należy zachować ostrożność, stosując leuprolidu octan u pacjentów z rozpoznanymi zaburzeniami krwawienia, trombocytopenią lub leczonych lekami przeciwzakrzepowymi.
Sportowcy powinni zachować ostrożność, ponieważ Lutrate Depot zawiera substancję, która może dawać pozytywny wynik badania podczas kontroli antydopingowej.
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) sodu na 1 fiolce, to znaczy lek uznaje się za „wolny od sodu”.
Leczenie przeciwandrogenowe może prowadzić do wydłużenia odstępu QT.
U pacjentów z czynnikami ryzyka wydłużenia odstępu QT w wywiadzie i u pacjentów otrzymujących równocześnie produkty lecznicze, które mogą wydłużać odstęp QT (patrz punkt 4.5), przed rozpoczęciem stosowania produktu leczniczego Lutrate Depot lekarze powinni ocenić stosunek korzyści do ryzyka, w tym możliwość wystąpienia częstoskurczu typu torsade de pointes.
Nie przeprowadzono badań dotyczących interakcji w fazie farmakokinetycznej leuproreliny octanu z innymi produktami leczniczymi. Ponieważ jednak leuproreliny octan jest peptydem rozkładanym przez peptydazę, a nie przez enzymy cytochromu P-450 oraz wiąże się z białkami osocza jedynie w 46%, nie należy spodziewać się interakcji farmakokinetycznych z innymi lekami.
Ponieważ leczenie przeciwandrogenowe może prowadzić do wydłużenia odstępu QT, należy uważnie przeanalizować równoczesne stosowanie produktu leczniczego Lutrate Depot z produktami leczniczymi, o których wiadomo, że wydłużają odstęp QT i które mogą wywoływać częstoskurcz typu torsade de pointes, takimi jak leki antyarytmiczne z klasy IA (np. chinidyna, dyzopiramid) lub klasy III (np. amiodaron, sotalol, dofetylid, ibutylid) oraz metadon, moksyfloksacyna, leki przeciwpsychotyczne itp. (patrz punkt 4.4).
Produkt Lutrate Depot 3,75 mg nie jest wskazany do stosowania u kobiet.
Leuproreliny octan, wstrzyknięty kobiecie w ciąży, może spowodować uszkodzenie płodu. Z tego powodu możliwe jest wystąpienie spontanicznego poronienia po jego podaniu w czasie ciąży.
Zdolność prowadzenia pojazdów i obsługiwania maszyn może ulec osłabieniu wskutek wystąpienia zaburzeń widzenia i zawrotów głowy.
Przedstawiony poniżej profil bezpieczeństwa produktu Lutrate Depot, o ile nie zaznaczono inaczej,
został opracowany na podstawie wyników badania klinicznego III fazy. W badaniu tym pacjentom z rakiem gruczołu krokowego podano domięśniowo w odstępach comiesięcznych 6 dawek produktu Lutrate Depot; okres leczenia i obserwacji po jego zakończeniu wynosił łącznie 26 tygodni.
Większość raportowanych działań niepożądanych związanych z leczeniem była spowodowana zahamowaniem stężenia testosteronu.
Najczęściej zgłaszanymi działaniami niepożądanymi po stosowaniu produktu Lutrate Depot były uderzenia gorąca, ból w miejscu wstrzyknięcia, podrażnienie w miejscu wstrzyknięcia, poty nocne i ból głowy.
Poniżej przedstawiono, zgodnie z klasyfikacją układów i narządów, działania niepożądane odnotowane podczas badań klinicznych. Wymieniono je według częstości występowania, zgodnie ze zmniejszającym się nasileniem (bardzo często: 1/10); często: l/100 do <l/10; niezbyt często: l/1000 do <l/100; rzadko: l/10 000 do <l/1000; bardzo rzadko: <l/10 000).
Tabela 1. Częstość występowania działań niepożądanych podczas leczenia produktem Lutrate Depot 3,75 mg
Klasyfikacja układów i narządów Częstość występowania: działania niepożądane |
Zaburzenia metabolizmu i odżywiania Często: Zwiększone łaknienie Niezbyt często: Jadłowstręt, hipercholesterolemia, hiperlipidemia |
Zaburzenia psychiczne Niezbyt często: Zaburzenia snu, bezsenność, zmniejszenie popędu seksualnego, zmiany nastroju i depresja* |
Zaburzenia układu nerwowego Często: Ból głowy Niezbyt często: Senność |
Zaburzenia ucha i błędnika Niezbyt często: Zawroty głowy |
Zaburzenia naczyń Bardzo często: Uderzenia gorąca |
Zaburzenia żołądka i jelit Niezbyt często: Ból w podbrzuszu, biegunka, nudności, wymioty |
Zaburzenia wątroby i dróg żółciowych Niezbyt często: Hiperbilirubinemia |
Zaburzenia skóry i tkanki podskórnej Często: Nadmierne pocenie, nocne poty, zimne poty Niezbyt często: Obrzęki wokół oczu, pokrzywka, świąd |
Zaburzenia mięśni szkieletowych i tkanki łącznej Często: Ból pleców Niezbyt często: Ból stawów, kurcze mięśni, ból kończyn |
Zaburzenia nerek i układu moczowego Niezbyt często: Zatrzymanie moczu, nietrzymanie moczu, częstomocz |
Zaburzenia układu rozrodczego i piersi Często: Zaburzenia erekcji Niezbyt często: Obrzęk sutków, tkliwość sutków, niemożność wytrysku |
Zaburzenia serca Nieznana: Wydłużenie odstępu QT (patrz punkty 4.4 i 4.5) |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia Nieznana: Śródmiąższowa choroba płuc |
Zaburzenia ogólne i stany w miejscu podania |
Często: Zmęczenie, astenia, gorączka, miejscowe reakcje niepożądane (patrz tab. 2) Niezbyt często: Osłabienie, uczucie gorąca i zimna, niepokój |
Badania diagnostyczne Niezbyt często: Zwiększenie aktywności AspAT, zwiększenie aktywności AlAT, zwiększenie stężenia bilirubiny, zwiększenie aktywności gamma- glutamylotransferazy (GGTP) |
*W badaniach post-marketingowych u osób długotrwale stosujących produkt leczniczy zmiany nastroju i depresja występowały często.
Pod względem ciężkości objawów 98% wszystkich działań niepożądanych związanych z leczeniem miało nasilenie łagodne do umiarkowanego. W 89% przypadków uderzenia gorąca określano jako łagodne, a w 9% - jako umiarkowane. Dwa przypadki uderzeń gorąca (0,2%) raportowano jako ciężkie.
Podczas badania odnotowano 35 miejscowych reakcji niepożądanych w miejscu wstrzyknięcia (LAR), zgłoszonych przez 29 pacjentów (18,1%).
Miejscowe działania niepożądane po stosowaniu produktu Lutrate Depot 3,75 mg są takie same, jak raportowane po domięśniowym wstrzyknięciu innych, podobnych produktów leczniczych. Najczęściej były to: ból w miejscu wstrzyknięcia, podrażnienie w miejscu wstrzyknięcia, dyskomfort w miejscu wstrzyknięcia, siniaki i rumień w miejscu wstrzyknięcia. Niezbyt często raportowano o wystąpieniu podrażnienia w miejscu wstrzyknięcia, obrzęku, rany i krwotoku (tab. 2).
Tabela 2. Odsetek pacjentów z miejscowymi działaniami niepożądanymi podczas leczenia produktem Lutrate Depot
Główna klasyfikacja układów i narządów* Zaburzenia ogólne i stany w miejscu podania | Pacjenci z miejscowymi działaniami niepożądanymi (%) |
Często Ból w miejscu wstrzyknięcia | 8,1 |
Podrażnienie w miejscu wstrzyknięcia | 4,4 |
Dyskomfort w miejscu wstrzyknięcia | 1,9 |
Rumień w miejscu wstrzyknięcia | 1,3 |
Siniaki w miejscu wstrzyknięcia | 1,3 |
Niezbyt często Podrażnienie w miejscu wstrzyknięcia Obrzęk w miejscu wstrzyknięcia | 0,6 0,6 |
Rana w miejscu wstrzyknięcia | 0,6 |
Krwotok w miejscu wstrzyknięcia | 0,6 |
*W odniesieniu do niektórych pacjentów występowała więcej niż 1 kategoria działań niepożądanych.
Nawracające miejscowe działania niepożądane związane z powtarzającymi się wstrzyknięciami produktu Lutrate Depot obejmowały: obrzęk (0,6%), ból (0,6%), siniaki (0,6%) i podrażnienie (0,6%). Zdarzenia te raportowano jako łagodne. Żaden pacjent nie przerwał leczenia z powodu miejscowych zdarzeń niepożądanych.
W badaniu klinicznym I fazy (CRO-02-43) przeprowadzonym z udziałem zdrowych ochotników, którym jednorazowo podano produkt Leuprolide Depot GP-Pharm 7,5 mg, odnotowano 1 przypadek stwardnienia w miejscu wstrzyknięcia.
Pozostałe zdarzenia niepożądane, które odnotowano podczas leczenia leuproreliny octanem obejmują: impotencję, zmniejszenie libido (oba przypadki jako farmakologiczne następstwo utraty testosteronu), obrzęki obwodowe, zator płucny, palpitacje, ból mięśni, osłabienie mięśni, dreszcze, duszność, zawroty głowy pochodzenia obwodowego, wysypkę, zaniki pamięci, zaburzenia widzenia i zaburzenia skórne. Po leczeniu zarówno krótko-, jak i długo działającymi analogami LHRH informowano o rzadko występujących przypadkach udaru przysadki u pacjentów z istniejącym wcześniej gruczolakiem przysadki. Rzadko odnotowano występowanie małopłytkowości i leukopenii.
Raportowano o zmianach w tolerancji glukozy.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C PL-02-222 Warszawa Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Brak danych klinicznych dotyczących ostrego przedawkowania produktu Lutrate Depot lub leuproreliny octanu. W badaniach klinicznych, w których pacjentom z rakiem gruczołu krokowego podawano codziennie, przez okres do 2 lat, leuproreliny octan w dawce wynoszącej aż 20 mg na dobę (podskórnie), nie występowały działania niepożądane inne niż te, które obserwowano po podaniu dawki 1 mg na dobę.
W badaniach na zwierzętach, którym podawano dawki przewyższające 500-krotnie zalecaną dawkę u człowieka, występowały: duszność, zmniejszona aktywność oraz podrażnienie w miejscu wstrzyknięcia.
Po przedawkowaniu należy ściśle monitorować stan pacjenta oraz wdrożyć leczenie objawowe i podtrzymujące.
Grupa farmakoterapeutyczna: leki stosowane w terapii hormonalnej. Hormony i ich pochodne. Analogi hormonu uwalniającego gonadotropinę; kod ATC: L02AE02.
Mechanizm działania
Nazwa chemiczna leuproreliny octanu to: 5-okso-L-prolilo-L-histydylo-L-tryptofylo-L-serylo-L- tyrozylo-D-leucylo-L-leucylo-L-arginylo-L-prolilo-etyloamid.
Leuproreliny octan nie wywiera działania farmakologicznego po podaniu doustnym ze względu na słabe przenikanie przez błony biologiczne i niemal całkowite unieczynnienie przez jelitowe enzymy proteolityczne.
Leuproreliny octan wykazuje właściwości silnego agonisty LHRH zarówno zastosowany
krótkotrwale, jak i leczeniu przerywanym, jednak analogi LHRH stosowane w sposób ciągły, niepulsacyjny, powodują zahamowanie wydzielania gonadotropin oraz steroidogenezy zachodzącej w jądrach.
Działanie farmakodynamiczne
Po związaniu się z receptorami LHRH w przysadce mózgowej leuproreliny octan wywołuje początkowo zwiększenie stężenia luteotropiny (LH) i folikulotropiny (FSH) we krwi, prowadząc do przemijającego zwiększenia stężeń testosteronu i dihydrotestosteronu. Jednak po 5 do 8 dniach od podania produktu leczniczego, analogi LHRH powodują zmniejszenie wrażliwości kompleksu receptorowego LHRH i (lub) zmniejszenie liczby receptorów w przednim płacie przysadki mózgowej. Z powodu zmniejszonej liczby receptorów na powierzchni komórek zmniejsza się pobudzenie komórek, a tym samym synteza i uwalnianie gonadotropin. W ten sposób po kilku tygodniach leczenia agonistą LHRH maleje wydzielanie LH i FSH. W wyniku tego komórki Leydiga w jądrach przestają wytwarzać testosteron, którego stężenie w surowicy w ciągu 2 do 4 tygodni po rozpoczęciu leczenia zmniejsza się do wartości kastracyjnych (mniejsze niż 0,5 ng/ml).
Skuteczność kliniczna i bezpieczeństwo stosowania
W otwartym, wieloośrodkowym badaniu klinicznym z zastosowaniem dawek wielokrotnych produktu Lutrate Depot uczestniczyło 160 pacjentów z rakiem gruczołu krokowego, u których nie stosowano wcześniej układowego leczenia przeciwnowotworowego, terapii hormonalnej stosowanej w leczeniu raka gruczołu krokowego, wcześniejszych zabiegów chirurgicznych dotyczących gruczołu krokowego ani orchidektomii. Celem badania była ocena skuteczności i bezpieczeństwa stosowania produktu Lutrate Depot u pacjentów z rakiem gruczołu krokowego, którzy mogli odnieść korzyści z deprywacji androgenów. Lutrate Depot stosowano domięśniowo w 6 dawkach podawanych co miesiąc.
Podczas trwającego 168 dni badania oznaczano w różnych dniach stężenie testosteronu. Jak było do przewidzenia, stężenie testosteronu w porównaniu do stężenia wyjściowego (4, 119 ± 1,341 ng/ml) zwiększyło się gwałtownie po pierwszym wstrzyknięciu, osiągając stężenie maksymalne (Cmax) trzeciego dnia po podaniu, wynoszące 6, 598 ± 2,2491 ng/ml. Po osiągnięciu wartości maksymalnych stężenie testosteronu zmniejszyło się, osiągając 21. dnia leczenia u 78,7% pacjentów wartości typowe dla kastracji medycznej (zdefiniowanej jako stężenia testosteronu mniejsze niż 0,5 ng/ml). Po 28 dniach leczenia poziom kastracyjny uzyskano u 96,8% pacjentów, przy czym u 73,1% z nich wystąpiły stężenia ≤0,2 ng/ml (ryc. 1).
Czas (dni)
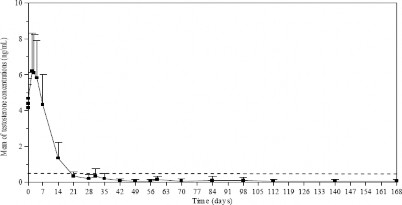
Średnie stężenie testosteronu (ng/mL)
Rycina 1. Średnie (±SD) stężenie testosteronu w osoczu podczas leczenia produktem Lutrate Depot 3,75 mg (6 wstrzyknięć domięśniowych podawanych raz w miesiącu)
Drugorzędowe punkty końcowe badania obejmowały oznaczanie aktywności LH i FSH oraz antygenu
PSA w surowicy. Średnie stężenie LH i FSH w surowicy zmniejszyło się poniżej wartości wyjściowych odpowiednio 14. i 4. dnia po pierwszym wstrzyknięciu produktu Lutrate Depot. Stężenia te utrzymywały się zdecydowanie poniżej wartości wyjściowych od 28. dnia badania do końca jego trwania. Podczas badania stopniowo zmniejszało się (w pierwszym miesiącu) średnie stężenie PSA w surowicy, a następnie utrzymywało się na stałym poziomie poniżej wartości wyjściowych do końca badania. Obserwowano jednak występowanie podczas badania znacznych różnic międzyosobniczych dotyczących stężenia PSA.
Częstość występowania zwiększenia stężenia testosteronu w odpowiedzi na kolejną iniekcję u przewlekle leczonego pacjenta wynosi 10,5%, jednocześnie częstość samoistnego zwiększenia stężenia testosteronu w czasie przewlekłego leczenia wynosi 11,8%. Nie zaobserwowano działań niepożądanych wskazujących klinicznie na objawy nagłego zwiększenia stężenia testosteronu (zatrzymanie moczu, ucisk rdzenia kręgowego, zaostrzenie bólu kości) u żadnego pacjenta, u którego zidentyfikowano spontaniczne zwiększenie stężenia testosteronu.
Wchłanianie
Po podaniu domięśniowym 3 dawek produktu Lutrate Depot w odstępach miesięcznych stężenie maksymalne w osoczu pacjentów z rakiem gruczołu krokowego (n=12) było podobne w każdym z 3 cykli leczenia. Po pierwszym wstrzyknięciu (dni 0-28) stężenie maksymalne (Cmax) wynosiło 13 145, 6
± 3 070,6 pg/ml. Mediana czasu do osiągnięcia Cmax (tmax) wynosiła 0,004 dnia, co odpowiada 0,96 h (przedział 0,96 do 4,08 h).
Dystrybucja
Nie badano dystrybucji produktu Lutrate Depot. Jednak u zdrowych ochotników płci męskiej, którym we wstrzyknięciu dożylnym podano 1 g leuproreliny octanu, objętość dystrybucji w stanie równowagi wynosiła 27 litrów. Wiązanie z białkami osocza ludzkiego in vitro wynosiło 43 do 49%.
Metabolizm
Nie badano metabolizmu produktu Lutrate Depot. Po wstrzyknięciu dożylnym 1 g leuproreliny octanu zdrowym ochotnikom płci męskiej średni klirens układowy wynosił 7,6 l/h, a okres półtrwania w końcowej fazie eliminacji – ok. 3 h (model dwukompartmentowy).
Przypuszczalnie leuprorelina jest metabolizowana do mniejszych, nieczynnych peptydów, które mogą być wydalane lub ulegać dalszemu rozpadowi.
Eliminacja
Nie badano eliminacji produktu Lutrate Depot. Jednak po podaniu 3 pacjentom leuproreliny octanu wydalone zostało z moczem w postaci niezmienionej oraz jako metabolit M-I mniej niż 5% podanej dawki.
Specjalne grupy pacjentów
Niewydolność nerek lub wątroby
Nie badano farmakokinetyki produktu u pacjentów z zaburzeniami czynności nerek lub wątroby.
Dane niekliniczne pochodzące z badań konwencjonalnych, dotyczących farmakologii bezpieczeństwa, toksyczności po podaniu wielokrotnym i genotoksyczności leuproreliny octanu, potwierdzają brak specjalnego ryzyka dla ludzi. Jak należało przypuszczać na podstawie znanych właściwości farmakologicznych produktu leczniczego, badania niekliniczne wykazały jego wpływ na reprodukcję, który był przemijający. W badaniach, dotyczących toksycznego wpływu na reprodukcję, leuproreliny octan nie wykazywał działania teratogennego. U królików obserwowano jednak działanie embriotoksyczne lub letalne.
Badania rakotwórczości przeprowadzone na szczurach, otrzymujących leuproreliny octan podskórnie (0,6 do 0,4 mg/kg/dobę), wykazały zależne od dawki zwiększenie częstości występowania gruczolaków przysadki. Ponadto obserwowano znamienne, lecz niezależne od dawki zwiększenie częstości występowania gruczolaków wyspowo-komórkowych trzustki u samic oraz gruczolaków wywodzących się z komórek śródmiąższowych jąder u samców, przy czym największy odsetek występował w grupie otrzymującej najmniejszą dawkę. Stosowanie leuproreliny octanu powodowało zahamowanie wzrostu różnych guzów zależnych od hormonów (guzy gruczołu krokowego u samców szczurów szczepów Noble i Dunning oraz wywołane przez dimetylobenzoantracen – DMBA nowotwory sutka u samic szczurów). Nie obserwowano takiego działania w badaniach rakotwórczości u myszy. Nie prowadzono badań dotyczących działania rakotwórczego produktu Lutrate Depot.
W badaniach in vitro oraz in vivo, dotyczących leuproreliny octanu, wykazano brak działania mutagennego. Nie badano działania mutagennego produktu Lutrate Depot.
Substancje pomocnicze proszku (fiolka):
Polisorbat 80
Mannitol (E 421) Karmeloza sodowa (E 466) Trietylu cytrynian
Polimer Poli-(DL-mleczano-ko-glikolowy (PLGA 50:50)
Substancje pomocnicze rozpuszczalnika (ampułkostrzykawka):
Mannitol (E 421)
Sodu wodorotlenek (do ustalenia pH) Kwas solny (do ustalenia pH)
Woda do wstrzykiwań
Ze względu na brak badań zgodności nie należy mieszać tego produktu z innymi produktami leczniczymi.
Do rekonstytucji proszku Lutrate Depot nie należy używać żadnego innego rozpuszczalnika, oprócz jałowego rozpuszczalnika znajdującego się w zestawie.
3 lata (w nieotwieranym opakowaniu).
Po rekonstytucji, poprzez dodanie rozpuszczalnika, należy niezwłocznie podać otrzymaną zawiesinę.
Przechowywać w temperaturze poniżej 25o C. Nie zamrażać. Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Opakowanie handlowe zawiera:
Sposób podawania:
Zawarty w fiolce produktu Lutrate Depot należy odtworzyć bezpośrednio przed pojedynczy wstrzyknięciem domięśniowym. Należy się upewnić, że procedura rekonstytucji odbywa się zgodnie z zasadami aseptyki.
Odtworzony produkt jest zawiesiną o mlecznobiałej barwie.
Używać wyłącznie substancji rozcieńczającej dołączonej do zestawu. Do rekonstytucji produktu leczniczego Lutrate Depot nie stosować żadnego innego rozpuszczalnika.
Produkt jest przeznaczony do jednorazowego wstrzyknięcia. Należy wyrzucić wszelkie pozostałości roztworu
Podczas rekonstytucji produktu leczniczego Lutrate Depot należy stosować się do poniższych zaleceń. Przeczytać uważnie przed podaniem produktu:
1 |
| Całkowicie usunąć zrywalne wieczko z górnej części fiolki, odsłaniając gumowy korek. Upewnić się, że żadne części zrywalnego wieczka nie pozostały na fiolce. |
2 |
| Umieścić fiolkę na stole, w pozycji pionowej. Oderwać osłonkę z blistra zawierającego łącznik fiolki (MIXJECT). Nie wyjmować łącznika fiolki z blistra. Umieścić blister z łącznikiem fiolki mocno na górnej części fiolki, przekłuwając fiolkę znajdującą się w całkowicie pionowej pozycji. Delikatnie docisnąć, aż poczuje się, że łącznik wskoczył na swoje miejsce. |
3 |
| Przymocować biały -uchwyt do strzykawki tak, aby go zatrzasnąć. Odkręcić gumową nasadkę strzykawki w kierunku przeciwnym do ruchu wskazówek zegara. Następnie zdjąć opakowanie blistrowe z systemu MIXJECT. |
4 |
| Podłączyć strzykawkę do adaptera ampułki, wkręcając ją zgodnie z ruchem wskazówek zegara w otwór z boku adaptera. Ostrożnie dokręcać strzykawkę do momentu, w którym przestanie się obracać, aby zapewnić szczelne połączenie. |
5 |
| Utrzymując strzykawkę i ampułkę w pozycji pionowej, powoli naciskać tłok, aby przenieść całość substancji rozcieńczającej do ampułki. |
6 |
| Gdy strzykawka wciąż jest połączona z ampułką, delikatnie wstrząsać ampułką przez około jedną minutę aż do uzyskania jednolitej mlecznobiałej zawiesiny. Aby uniknąć rozdzielenia zawiesiny, należy niezwłocznie przejść do następnych kroków. |
7 |
| Odwrócić system MIXJECT tak, aby ampułka znajdowała się na górze. Mocno chwycić system MIXJECT za strzykawkę i powoli odciągać tłok, aby wprowadzić przygotowany produkt do strzykawki. Część produktu może ulec zbryleniu lub przykleić się do ścianek ampułki. Jest to normalne zjawisko. |
8 |
| Odłączyć adapter ampułki od systemu MIXJECT połączonego ze strzykawką: Mocno chwycić strzykawkę i obrócić ampułkę (trzymając za plastikową nasadkę adaptera) w kierunku zgodnym z ruchem wskazówek zegara. |
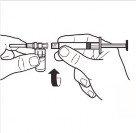
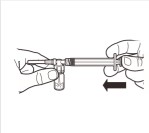
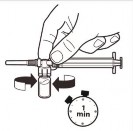
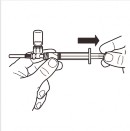
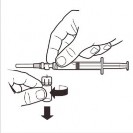
9 |
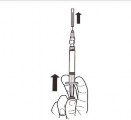
| Trzymać strzykawkę PIONOWO. Drugą ręką pociągnąć nasadkę igły do góry. Nacisnąć tłok tak, aby usunąć powietrze ze strzykawki. Strzykawka zawierająca produkt jest gotowa do natychmiastowego podania. |
10 |
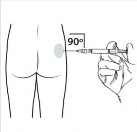
| Wykonać zastrzyk domięśniowy, wprowadzając igłę pod kątem 90 stopni w okolice pośladka. Upewnić się, że wstrzyknięta została cała ilość produktu. Miejsca wstrzyknięcia powinny być zmieniane. |
.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
GP-Pharm S.A.
Poligono Industrial Els Vinyets – Els Fogars, Sector 2 Carretera Comarcal C -244, Km. 22
08777 Sant Quintí de Mediona (Barcelona), Hiszpania
21174
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 02.05.2013 Data przedłużenia pozwolenia: 17.07.2017
28.12.2020







