







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
w celu profilaktycznego leczenia wrodzonego niedoboru czynnika XIII oraz
w postępowaniu okołooperacyjnym w celu leczenia krwawień podczas zabiegów chirurgicznych u pacjentów z wrodzonym niedoborem czynnika XIII.
Dawkowanie i sposób podawania
40 jednostek międzynarodowych (j.m.) na kilogram masy ciała.
Szybkość infuzji nie powinna być większa niż 4 ml na minutę
Kolejne dawki
Dawkowanie powinno być ustalone w oparciu o bieżący poziom aktywności FXIII, dawki należy podawać w odstępach 28-dniowych (4 tygodnie) w celu utrzymania minimalnej aktywności F XIII na poziomie ok. 5 do 20%.
Zalecane dostosowanie dawkowania o +/- 5 j.m. na kg masy ciała należy obliczyć biorąc pod uwagę
minimalny poziom aktywności F XIII jak pokazano w tabeli nr 1 oraz kliniczny stan pacjenta.
Dostosowywanie dawkowania powinno być prowadzone w oparciu o specyficzny, czuły test stosowany do oznaczania poziomu F XIII. Przykłady dostosowywania przy użyciu standardowego testu aktywności Berichrom zostały przedstawione w poniższej tabeli nr 1.
Tabela 1 : Dostosowanie dawkowania przy użyciu testu aktywności Berichrom
Minimalny poziom aktywności czynnika XIII(%)
Zmiana dawkowania
Jeden poziom minimalny < 5%
Zwiększenie o 5 jednostek na kg.
Poziom minimalny 5% do 20%
Brak zmian
Dwa poziomy minimalne > 20%
Zmniejszenie o 5 jednostek na kg.
Jeden poziom minimalny > 25%
Zmniejszenie o 5 jednostek na kg.
Aktywność wyrażona w jednostkach jest oznaczana przy użyciu testu aktywności Berichrom, odnoszącego się do aktualnego Międzynarodowego Standardu dla Osoczowego XIII Czynnika Krzepnięcia Krwi. W związku z tym jednostka ta odpowiada Jednostce Międzynarodowej.
Profilaktyka przedooperacyjna.
Po ostatniej dawce zastosowanej w rutynowej profilaktyce, w przypadku planowanego zabiegu operacyjnego:
Pomiędzy 21 a 28 dniem od ostatniej dawki - należy podać pacjentowi pełną dawkę bezpośrednio przed zabiegiem chirurgicznym, a kolejna dawka profilaktyczna powinna zostać podana 28 dni później.
Pomiędzy 8 a 21 dniem od ostatniej dawki- dodatkowa dawka (pełna lub częściowa) może zostać podana przed zabiegiem chirurgicznym. Dawka powinna być uzależniona od poziomu aktywności FXIII pacjenta, jego stanu klinicznego i powinna być dobrana zgodnie z okresem półtrwania produktu leczniczego Cluvot.
W ciągu 7 dni od ostatniej dawki – dodatkowe podanie może nie być potrzebne.
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
strzykawka jednorazowego użytku o pojemności 5 ml 1 zestaw do wkłucia
waciki nasączone alkoholem 1 niejałowy plaster
strzykawka jednorazowego użytku o pojemności 20 ml 1 zestaw do wkłucia
waciki nasączone alkoholem 1 niejałowy plaster
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
Cluvot 250 j.m. proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań / infuzji
Cluvot 1250 j.m. proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań / infuzji
Substancja czynna: Cluvot jest oczyszczonym koncentratem ludzkiego XIII czynnika krzepnięcia krwi
(FXIII) pochodzącego z osocza. Produkt występuje w postaci białego proszku.
Każda fiolka zawiera nominalnie 250 lub 1250 j.m. ludzkiego XIII czynnika krzepnięcia krwi pochodzącego z osocza. Po rekonstytucji odpowiednio w 4 i 20 ml wody do wstrzykiwań Cluvot zawiera w przybliżeniu 62,5 j.m./ml ludzkiego XIII czynnika krzepnięcia krwi (250 j.m./4 ml i 1250 j.m./ 20ml) .
Swoista aktywność produktu leczniczego Cluvot wynosi w przybliżeniu 6-10 j.m./ mg białka. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań/infuzji. Biały proszek i przezroczysty, bezbarwny rozpuszczalnik.
Cluvot jest wskazany do stosowania u pacjentów dorosłych, dzieci i młodzieży
Dawkowanie
1 ml odpowiada w przybliżeniu 62,5 j.m., a 100 j.m. jest równoważne odpowiednio 1,6 ml.
Ważne:
Ilość potrzebna do podania i częstość podawania powinny być zawsze dostosowane do skuteczności klinicznej u poszczególnych pacjentów.
Dawkowanie
Dawkowanie powinno być indywidualnie dobrane w zależności od masy ciała, wyników badań laboratoryjnych oraz stanu klinicznego pacjenta.
Rutynowy schemat dawkowania w profilaktyce Dawka początkowa
Dostosowanie dawkowania może być różne od dawkowania zalecanego i powinno być indywidualnie dobrane w oparciu o poziom aktywności F XIII i stan kliniczny pacjenta. Wszyscy pacjenci powinni być ściśle monitorowani podczas i po zabiegu chirurgicznym.
W związku z powyższym zaleca się monitorowanie wzrostu poziomu aktywności F XIII poprzez przeprowadzenie testu czynnika XIII. W przypadku poważnych zabiegów chirurgicznych oraz dużych krwawień celem powinno być osiągnięcie poziomu zbliżonego do prawidłowych wartości (osoby zdrowe: 70%-140%).
Dzieci i młodzież
Dawkowanie i sposób podawania u dzieci i młodzieży jest oparte o masę ciała i w związku z tym ogólnie nie odbiega od wytycznych dla dorosłych. Dawkowanie i/lub częstość podawania dla każdego pacjenta powinna zawsze być dobrana w oparciu o skuteczność kliniczną i poziom aktywności FXIII.(Patrz także punkt 5.1 oraz 5.2).
Pacjenci w podeszłym wieku
Dawkowanie i sposób podawania u pacjentów w podeszłym wieku (> 65 lat) nie zostały udokumentowane
w badaniach klinicznych.
Sposób podawania
Po rekonstytucji roztwór powinien być klarowny lub lekko opalizujący. Przed podaniem, przygotowany roztwór powinien być ogrzany do temperatury pokojowej lub temperatury ciała. Dożylne wstrzyknięcie lub wlew należy wykonywać powoli do oddzielnego zestawu do wstrzykiwań/ infuzji (dostarczonego z produktem), z szybkością komfortową dla pacjenta. Szybkość wstrzyknięcia lub infuzji nie powinna przekraczać około 4 ml na minutę.
Należy obserwować pacjenta pod kątem wystąpienia jakichkolwiek reakcji natychmiastowych. Jeżeli wystąpią jakiekolwiek reakcje, które mogą być związane z podawaniem produktu leczniczego Cluvot, szybkość infuzji należy zmniejszyć lub przerwać podawanie, w zależności od stanu klinicznego pacjenta.
W celu zapoznania się z instrukcją przeprowadzenia rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na substancję czynna lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
W przypadku pacjentów ze stwierdzoną nadwrażliwością na produkt leczniczy (z objawami takimi, jak uogólniona pokrzywka, wysypka, spadek ciśnienia tętniczego, duszność), leki przeciwhistaminowe i kortykosteroidy mogą być podawane profilaktycznie.
Po zastosowaniu produktu leczniczego Cluvot możliwe jest wystąpienie reakcji nadwrażliwości typu alergicznego. Jeżeli wystąpią objawy nadwrażliwości (takie, jak wysypka, uogólniona pokrzywka, ucisk w klatce piersiowej, świszczący oddech, hipotensja lub anafilaksja), należy natychmiast przerwać infuzję produktu leczniczego Cluvot. W przypadku wstrząsu należy zastosować aktualne standardowe procedury obowiązujące w leczeniu wstrząsu.
W przypadkach świeżej zakrzepicy należy zachować ostrożność z powodu fibryno-stabilizacyjnego efektu
FXIII.
Immunogenność
Tworzenie przeciwciał neutralizujących (inhibitorów) przeciwko F XIII został stwierdzony u pacjentów przyjmujących Cluvot. W związku z tym pacjenci powinni być monitorowani pod kątem możliwego wytworzenia przeciwciał neutralizujących. Obecność przeciwciał neutralizujących (inhibitorów) może przejawiać się jako niewłaściwa odpowiedź na leczenie. Jeżeli nie zostaną osiągnięte oczekiwane poziomy aktywności osoczowego F XIII, lub pojawi się krwawienie podczas stosowanej profilaktyki, należy zmierzyć stężenie przeciwciał neutralizujących.
Informacja dla pacjentów na diecie niskosodowej
Cluvot zawiera od 124,4 do 195,4 mg (5,41 do 8,50 mmol) sodu na dawkę (40 j.m. /masę ciała - przy średniej masie ciała 70 kg), jeśli podano rekomendowaną dawkę (2800 j.m. = 44.8 ml) . Należy wziąć to pod uwagę u pacjentów będących na kontrolowanej diecie niskosodowej.
Bezpieczeństwo wirusowe
Zastosowano standardowe postępowanie zabezpieczające przed przenoszeniem zakażeń poprzez podawanie produktów leczniczych otrzymywanych z krwi lub osocza ludzkiego, do którego należy: selekcja dawców, badanie pojedynczych donacji i pul osocza na obecność markerów wirusów i zastosowanie skutecznych metod inaktywacji/usuwania wirusów w czasie procesu wytwarzania. Pomimo tego, nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych po podaniu produktu leczniczego pochodzącego z krwi lub osocza ludzkiego. To ryzyko dotyczy nieznanych lub nowo odkrytych wirusów i innych czynników zakaźnych.
Podjęte środki zabezpieczające są uważane za skuteczne w stosunku do otoczkowych wirusów takich jak: HIV, HBV, HCV oraz wirusów bezotoczkowych HAV i parwowirusa B 19.
Zdecydowanie zaleca się, aby przy każdym podawaniu pacjentowi, odnotować nazwę oraz numer serii produktu leczniczego, aby móc powiązać pacjenta z daną serią leku.
Należy rozważyć odpowiednie szczepienie (przeciw wirusowemu zapaleniu wątroby typu A i B) u pacjentów
otrzymujących regularnie/okresowo produkty pochodzące z osocza ludzkiego.
Nie przeprowadzono badań dotyczących interakcji.
Ciąża
Ograniczone dane dotyczące klinicznego stosowania Cluvot w ciąży nie wykazały żadnych negatywnych skutków związanych z ciążą oraz rozwojem embrionalnym i poporodowym. W przypadku konieczności może być rozważone zastosowanie Cluvot w czasie ciąży.
Karmienie piersią
Nie jest znany stopień przenikania Cluvot do ludzkiego mleka, jednak zważywszy jego wysoką masę cząsteczkową prawdopodobieństwo wydzielania z mlekiem matki jest bardzo niskie i z powodu jego białkowego charakteru absorpcja nienaruszonych cząstek przez noworodka jest także mało prawdopodobna. W związku z tym Cluvot może być stosowany podczas karmienia piersią.
Płodność
Brak danych dotyczących wpływu Cluvot na płodność.
Nie prowadzono badań dotyczących wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Poniższe działania niepożądane są oparte o doświadczenia po wprowadzeniu produktu leczniczego na rynek. Tabelaryczne zestawienie działań niepożądanych
Poniższa tabela przedstawia działania niepożądane zgodnie z klasyfikacją układów i narządów MedDRA. Częstość występowania przedstawia się w następujący sposób: bardzo często:( ≥ 1/10); często: (≥ 1/100 do< 1/10); niezbyt często:( ≥ 1/1 000 do < 1/100); rzadko:( ≥ 1/10 000 do < 1/1 000); bardzo rzadko:( < 1/10 000)
Klasyfikacja układów i Narządów MedDRA | Działanie niepożądane | Częstość |
Zaburzenia układu immunologicznego | Reakcje alergiczno- anafilaktyczne (jak uogólniona pokrzywka, wysypka, obniżenie ciśnienia krwi, duszność) | Rzadko |
Rozwój inhibitorów FXIII | Bardzo rzadko | |
Zaburzenia ogólne i stany w miejscu podania leku | Wzrost temperatury | Rzadko |
Jeżeli pojawią się reakcje alergiczno- anafilaktyczne, podawanie leku Cluvot powinno być natychmiast przerwane, i powinno zostać rozpoczęte odpowiednie leczenie. Powinny zostać zastosowane aktualne standardy medyczne stosowane w leczeniu wstrząsu.
Dzieci i młodzież
Profil bezpieczeństwa dla dzieci i młodzieży nie różni się od zaobserwowanego w badaniach klinicznych
u dorosłych.
Możliwość przeniesienia czynników zakaźnych przedstawiono w punkcie 4.4. Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie odnotowano przypadków przedawkowania.
Grupa farmakoterapeutyczna: Środki przeciwkrwotoczne Kod ATC: B02B D07
Czynnik XIII łączy grypę aminową lizyny z glutaminą przez użycie jego funkcji enzymatycznej prowadzącej do krzyżowego połączenia cząstek fibryny.
Krzyżowe łączenie i stabilizacja fibryny umożliwia penetrację fibroblastów i wspomaga proces gojenia się
rany.
Dzieci i młodzież
W badaniach klinicznych obejmujących pacjentów poniżej 18. roku życia z wrodzonym niedoborem F XIII profilaktyczne podawanie Cluvot co 28 dni było skuteczne w podtrzymywaniu minimalnych poziomów aktywności F XIII w zakresie od 5% do 20%.
Dystrybucja
Produkt jest podawany dożylnie, w związku z tym jest natychmiast biodostępny, skutkując uzyskaniem stężenia w osoczu odpowiadającego podanej dawce.
Eliminacja
U pacjentów z wrodzonym nidoborem F XIII biologiczny okres półtrwania został określony jako 6,6 ± 2,29 dni (średnia ± SD). Cluvot jest metabolizowany w taki sam sposób jak endogenny XIII czynnik krzepnięcia.
Przegląd parametrów farmakokinetycznych (populacja dorosłych / 18 lat i starsi) został przedstawiony
w poniższej tabeli:
Parametry | Mediana (min-maks) |
AUC ss,0-inf (jednostki x godz/ml) | 182,9 (133,5-300,2) |
Css,max(Jednostki/ml)* | 0,9 (0,6-1,2) |
Css,min(Jednostki/ml)* | 0,07 (0,0-0,16) |
Tmax (godz.) | 1,2 (0,7-4,2) |
Okres półtrwania (dni) | 7,8 (3,1-11,02) |
CL (ml/godz/kg) | 0,22 (0,13-0,30) |
Vss (ml/kg) | 49,4 (31,.65-62,91) |
MRT (dni) | 11,7 (5,7-17,02) |
AUC ss,0-inf=obszar pod krzywą stężenia w osoczu w czasie od 0 do nieskończoności w stanie równowagi
*100% aktywności odpowiada 1 jednostce/ ml Css,max : maksymalne stężenie w stanie równowagi Css,min: minimalne stężenie w stanie równowagi Tmax : czas do maksymalnego stężenia
CL : klirens
Vss: objętość dystrybucji w stanie równowagi
MRT: średni czas trwania Dzieci i młodzież
W badaniach klinicznych dotyczących stężenia koncentratu czynnika XIII (ludzkiego) obejmujących 188 pacjentów, z których 117 było < 18. roku życia w czasie rozpoczynania badań (1 miesiąc do < 2 lat, n= 17; 2 do < 12 lat, n= 62; 12 do < 16 lat, n=30; 17 do 18 lat, n=8). W badaniach farmakokinetycznych PK 2002,
5 z 14 pacjentów w przedziale wiekowym od 2 do < 18 lat( 2-11 lat, n=3; 12-16 lat, n=2; 17-18 lat, n=0). U pacjentów młodszych niż 16 lat okres półtrwania był krótszy i szybszy klirens (okres półtrwania: 5,7 ± 1,00 dni; klirens: 0,291 ± 0,12 ml/godz/kg) w porównaniu do osób dorosłych (okres półtrwania: 7,1 ± 2,74 dni; klirens: 0,22 ± 0,07 ml/godz/kg).
U dzieci produkt ma krótszy okres półtrwania i szybszy klirens niż u dorosłych. Ponieważ we wszystkich grupach wiekowych dawkowanie jest ustalane indywidualnie w oparciu o masę ciała pacjenta i minimalne poziomy aktywności F XIII, nie ma potrzeby ustlania dawkowania w zależności od wieku.
Białko zawarte w Cluvot pochodzi z ludzkiego osocza i odgrywa taką samą rolę jak białka ludzkiego osocza.
Badania toksycznośći przeprowadzone na zwierzętach zarówno po pojedyńczej dawce, jak i podaniu wielkrotnym nie wykazały potencjalnej toksyczności Cluvot.
Nie przeprowadzano badań nad rozrodem i rozwojem embrionalno-płodowym.
Proszek:
Albumina ludzka Glukoza jednowodna Sodu chlorek
Sodu wodorotlenek (do ustalenia pH)
Rozpuszczalnik:
Woda do wstrzykiwań
Produktu leczniczego Cluvot nie wolno mieszać z innymi produktami leczniczymi, rozcieńczalnikami lub rozpuszczalnikami z wyjątkiem tych, wymienionych w punkcie 6.6 oraz powinien być on podawany za pomocą oddzielnych zestawów infuzyjnych.
3 lata.
Nie stosować po upływie daty ważności podanej na etykiecie i opakowaniu zewnętrznym.
Stabilność chemiczna i fizyczna roztworu po rekonstytucji została wykazana przez 24 godziny w temperaturze
≤ 25°C. Z mikrobiologicznego punktu widzenia, produkt powinien zostać natychmiast zużyty. Jeżeli nie zostanie zużyty natychmiast, przechowywanie w temperaturze pokojowej nie może przekraczać 4 godzin. Roztworu po rekonstytucji nie przechowywać w lodówce ani nie zamrażać.
Przechowywać w lodówce (2˚C – 8˚C) Nie zamrażać.
Przechowywać fiolkę w opakowaniu zewnętrznym w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Fiolka 250 j.m.:
Proszek: fiolka z bezbarwnego szkła, zamknięta gumowym korkiem (z gumy bromobutylowej), aluminiowym
wieczkiem i plastikowym dyskiem.
Rozpuszczalnik (woda do wstrzykiwań): fiolka z bezbarwnego szkła.
Fiolka 1250 j.m.
Proszek: fiolka z bezbarwnego szkła, zamknięta gumowym korkiem (z gumy bromobutylowej), aluminiowym
wieczkiem i plastikowym dyskiem.
Rozpuszczalnik (woda do wstrzykiwań): fiolka z bezbarwnego szkła
Dostępne opakowania:
Opakowanie z produktem leczniczym w dawce 250 j.m.:
1 fiolka z proszkiem
1 fiolka z 4 ml wody do wstrzykiwań
1 system do transferu 20/20 z filtrem (Mix2Vial)
Zestaw do podawania (opakowanie wewnętrzne):
Opakowanie z produktem leczniczym w dawce 1250 j.m.:
1 fiolka z proszkiem
1 fiolka z 20 ml wody do wstrzykiwań
1 system do transferu 20/20 z filtrem (Mix2Vial)
Zestaw do podawania (opakowanie wewnętrzne):
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
stosowania
Wskazówki ogólne
Roztwór powinien być klarowny lub lekko opalizujący. Po filtracji/pobraniu zawartości fiolki (patrz poniżej) produkt po rekonstytucji przed podaniem powinien być poddany wizualnej ocenie; należy sprawdzić czy nie pojawiły się żadne zanieczyszczenia i czy nie nastąpiła zmiana zabarwienia.
Nie stosować mętnych roztworów i takich, które zawierają płatki lub cząstki. Rekonstytucja i pobieranie z fiolki muszą być przeprowadzone w warunkach aseptycznych.
Rekonstytucja

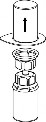
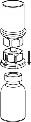
Doprowadzić rozpuszczalnik do temperatury pokojowej. Zdjąć z fiolek zawierających proszek i rozpuszczalnik plastikowe wieczka i przemyć korki aseptycznym roztworem. Po wyschnięciu otworzyć system zawierający łącznik Mix2Vial.
1 | 1. Otworzyć opakowanie zawierające Mix2Vial poprzez usunięcie wieczka. Nie wyjmować Mix2Vial z blistra. |
2 | 2. Umieścić fiolkę z rozpuszczalnikiem na równej, czystej powierzchni i mocno przytrzymać. Nie wyjmując z blistra systemu Mix2Vial nałożyć jego niebieską końcówkę z ostrzem na korek fiolki rozpuszczalnika i naciskając pionowo w dół przebić korek fiolki rozpuszczalnika. |
3 | 3. Przytrzymując krawędź systemu Mix2Vial ostrożnie zdjąć blister pociągając go pionowo do góry. Należy zwrócić uwagę aby zdjąć jedynie blister a nie cały system Mix2Vial. |
4 | 4. Umieścić fiolkę z proszkiem na równej i twardej powierzchni. Odwrócić do góry dnem fiolkę z rozpuszczalnikiem i dołączonym do niej systemem Mix2Vial i wbić w korek fiolki z proszkiem, ruchem pionowo w dół, ostrze przezroczystego końca. Rozpuszczalnik samoczynnie zostanie przeniesiony do fiolki z proszkiem. |
5 | 5. Jedną ręką chwycić część systemu Mix2Vial w fiolce zawierającej obecnie roztwór, drugą zaś ręką przytrzymać część łącznika od strony fiolki po rozpuszczalniku i ostrożnie odkręcając oddzielić od siebie obie części łącznika. Usunąć fiolkę po rozpuszczalniku z przyczepionym do niej niebieskim końcem systemu Mix2Vial. |
6 | 6. Fiolkę z doczepionym przezroczystym końcem systemu Mix2Vial, zawierającą roztwór, łagodnie mieszać ruchem wirowym do całkowitego rozpuszczenia leku. Nie wstrząsać. |
7 | 7. Nabrać powietrza do pustej, jałowej strzykawki. Trzymając fiolkę z produktem leczniczym pionowo korkiem do góry, przyłączyć strzykawkę do połączenia Luer Lock systemu Mix2Vial. Wstrzyknąć powietrze do fiolki z produktem. |
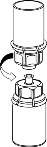
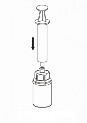
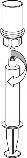
Pobieranie i sposób podawania
8 | 8. Przytrzymując tłok strzykawki odwrócić fiolkę wraz ze strzykawką do góry dnem i nabrać roztwór do strzykawki, powoli odciągając tłok. |
9 | 9. Po napełnieniu strzykawki roztworem, mocno uchwycić cylinder strzykawki (utrzymując strzykawkę tłokiem do dołu) odłączyć od niej przezroczysty koniec systemu Mix2Vial |
Należy zachować ostrożność, aby krew nie dostała się do strzykawki wypełnionej produktem leczniczym, gdyż istnieje ryzyko, że krew w strzykawce może ulec wykrzepianiu i skrzepliny fibryny mogłyby zostać podane pacjentowi.
Roztwór po rekonstytucji powinien być podany do oddzielnych zestawów do wstrzykiwań/ infuzji (dostarczonych z produktem), w powolnym wstrzyknięciu dożylnym, z szybkością nie przekraczającą 4 ml na minutę.
Wszelkie niezużyte produkty lecznicze oraz ich pozostałości powinny zostać usunięte zgodnie z lokalnymi wymaganiami.
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg
Niemcy
Cluvot 250 j.m.: 21950
Cluvot 1250 j.m.: 21951
PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 23.06.2014
Data ostatniego przedłużenia pozwolenia: 22.07.2019
PRODUKTU LECZNICZEGO
27.08.2021







