







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Dzieci i młodzież
Krwawienie u pacjentów z wrodzonym niedoborem fibrynogenu lub afibrynogenemią
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
1 g ludzkiego fibrynogenu w butelce o pojemności 100 ml z bezbarwnego szkła (typu II) zamkniętej korkiem z gumy bromobutylowej i aluminiowym odrywanym wieczkiem.
50 ml rozpuszczalnika (woda do wstrzykiwań) w fiolce o pojemności 50 ml z bezbarwnego szkła (typu II), zamkniętej korkiem z gumy halobutylowej i aluminiowym odrywanym wieczkiem.
1 urządzenie do rekonstytucji Octajet.
1 filtr cząstek stałych.
Szczególne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Przygotowany roztwór powinien być niemal bezbarwny i lekko opalizujący. Nie używać roztworów, które są mętne lub zawierają osad.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Fibryga, 1 g, proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań/ do infuzji.
Fibrynogen ludzki
Każda butelka produktu leczniczego Fibryga zawiera 1 g fibrynogenu ludzkiego. Po rekonstytucji w 50 ml wody do wstrzykiwań produkt Fibryga zawiera w przybliżeniu 20 mg/ml fibrynogenu ludzkiego.
Zawartość białka krzepliwego oznaczana jest zgodnie z monografią ludzkiego fibrynogenu podaną w Farmakopei Europejskiej.
Wyprodukowany z ludzkiego osocza pochodzącego od dawców.
Substancja pomocnicza o znanym działaniu: sód w ilości do 132 mg (5,8 mmol) na butelkę. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań/do infuzji. Biały lub bladożółty higroskopijny proszek lub krucha, zestalona masa.
Rozpuszczalnik jest bezbarwnym i przezroczystym roztworem.
Leczenie epizodów krwawienia i profilaktyka okołooperacyjna u pacjentów z wrodzoną hipo- lub afibrynogenemią ze skłonnością do krwawień.
Jako leczenie uzupełniające do leczenia niekontrolowanego ciężkiego krwotoku u pacjentów z hipofibrynogenemią nabytą w przebiegu interwencji chirurgicznej.
Leczenie powinno zostać rozpoczęte pod nadzorem lekarza doświadczonego w leczeniu zaburzeń krzepnięcia.
Dawkowanie
Dawkowanie i czas trwania terapii zastępczej zależą od stopnia ciężkości zaburzenia, lokalizacji i intensywności krwawienia oraz od stanu klinicznego pacjenta.
Należy oznaczać stężenie (czynnościowe) fibrynogenu w celu wyliczenia indywidualnego dawkowania oraz ilości i częstości podawanych dawek u każdego pacjenta poprzez regularne pomiary stężenia fibrynogenu w osoczu i ciągłe monitorowanie stanu klinicznego pacjenta i innych stosowanych terapii zastępczych.
W przypadku poważnych zabiegów chirurgicznych konieczne jest dokładne monitorowanie terapii zastępczej przez oznaczanie parametrów krzepnięcia.
W przypadku zabiegu chirurgicznego lub leczenia epizodu krwawienia dawkę dla młodzieży należy wyliczyć na podstawie podanego powyżej wzoru dla dorosłych, natomiast dawkę dla dzieci w wieku
<12 lat należy wyliczyć w następujący sposób:
Dawka (mg/kg masy ciała) = [Stężenie docelowe (g/l) - stężenie oznaczone (g/l)]
0,014 (g/l na mg/kg masy ciała)
Dalsze dawkowanie powinno być dostosowane do stanu klinicznego pacjenta i wyników testów laboratoryjnych.
Osoby w podeszłym wieku
Badania kliniczne produktu leczniczego Fibryga nie obejmowały pacjentów w wieku 65 lat i starszych w celu przedstawienia rozstrzygających dowodów, czy występowały u nich różnice w odpowiedzi na leczenie porównaniu do młodszych pacjentów.
Leczenie epizodów krwawienia należy prowadzić zgodnie z podanymi powyżej wzorami odpowiednio dla dorosłych/młodzieży i dzieci w celu osiągnięcia zalecanego docelowego stężenia fibrynogenu w osoczu wynoszącego 1 g/l. Stężenie to należy utrzymywać do czasu osiągnięcia hemostazy.
Krwawienie u pacjentów z nabytym niedoborem fibrynogenu Dorośli
Zwykle początkowo podaje się 1–2 g, a kolejne wlewy w razie potrzeby. W przypadku ciężkiego krwotoku, np. podczas poważnego zabiegu chirurgicznego, wymagana może być większa ilość fibrynogenu (4–8 g).
Dzieci i młodzież
Dawkowanie należy ustalić na podstawie masy ciała i wskazań klinicznych, ale zwykle wynosi 20– 30 mg/kg.
Sposób podawania
Infuzja lub wstrzyknięcie dożylne.
Produkt leczniczy Fibryga należy podawać powoli dożylnie przy zalecanej maksymalnej szybkości 5 ml na minutę u pacjentów z wrodzoną hipo- lub afibrynogenemią i przy zalecanej maksymalnej szybkości 10 ml na minutę u pacjentów z nabytym niedoborem fibrynogenu.
Instrukcja dotycząca rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Identyfikowalność
W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Zdarzenia zakrzepowo-zatorowe
Przy podawaniu ludzkiego fibrynogenu pacjentom z wrodzonym lub nabytym niedoborem istnieje ryzyko wystąpienia zakrzepicy, zwłaszcza przy dużej dawce leku lub jego wielokrotnym podawaniu. Pacjenci otrzymujący ludzki fibrynogen powinni być dokładnie obserwowani w kierunku przedmiotowych i podmiotowych objawów zakrzepicy.
W przypadku pacjentów z chorobą niedokrwienną serca lub zawałem mięśnia sercowego w wywiadzie, pacjentów z chorobą wątroby, pacjentów w okresie około- i pooperacyjnym,
u noworodków oraz u pacjentów z ryzykiem zdarzeń zakrzepowo-zatorowych lub rozsianego wykrzepiania wewnątrznaczyniowego, należy rozważyć możliwe korzyści z leczenia ludzkim fibrynogenem wobec ryzyka powikłań zakrzepowo-zatorowych. Należy postępować z ostrożnością i dokładnie monitorować stan pacjenta.
Nabyta hipofibrynogenemia jest związana z niskim stężeniem wszystkich czynników krzepnięcia (nie tylko fibrynogenu) oraz inhibitorów w osoczu, należy zatem rozważyć leczenie produktami krwiopochodnymi zawierającymi czynniki krzepnięcia. Konieczne jest staranne monitorowanie układu krzepnięcia.
Reakcje alergiczne i typu anafilaktycznego
W razie wystąpienia reakcji alergicznej lub typu anafilaktycznego, należy natychmiast przerwać wstrzyknięcie/infuzję. W razie wystąpienia wstrząsu anafilaktycznego należy zastosować odpowiednie standardowe postępowanie lecznicze.
Zawartość sodu
Ten produkt leczniczy zawiera sód w ilości do 132 mg na butelkę, co odpowiada 6,6% zalecanego przez WHO maksymalnego dziennego spożycia 2 g sodu przez dorosłych. Należy wziąć to pod uwagę u pacjentów kontrolujących zawartość sodu w diecie.
Bezpieczeństwo wirusologiczne
Standardowe środki ostrożności zapobiegające zakażeniom wynikającym ze stosowania produktów leczniczych przygotowanych z ludzkiej krwi lub osocza obejmują selekcję dawców, badania przesiewowe poszczególnych donacji i całych pul osocza w kierunku swoistych markerów zakażenia oraz wprowadzenie skutecznych etapów procesu wytwarzania w celu inaktywacji/usunięcia wirusów. Mimo to, w przypadku podawania produktów leczniczych przygotowanych z ludzkiej krwi lub osocza nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych. Dotyczy to również nieznanych lub pojawiających się od niedawna wirusów i innych patogenów.
Podejmowane środki ostrożności są uważane za skuteczne wobec wirusów otoczkowych, takich jak HIV, HBV i HCV, jak również wirusa bezotoczkowego HAV. Podejmowane środki ostrożności mogą mieć ograniczoną skuteczność wobec wirusów bezotoczkowych, takich jak parwowirus B19.
Zakażenie parwowirusem B19 może być poważne w przypadku kobiet w ciąży (zakażenie płodu) oraz osób z upośledzeniem odporności lub nasiloną erytropoezą (np. anemia hemolityczna).
W przypadku pacjentów przyjmujących regularnie/wielokrotnie produkty pochodzące z ludzkiego osocza należy rozważyć odpowiednie szczepienia (przeciwko WZW typu A i B).
Immunogenność
W przypadku terapii zastępczej czynnikami krzepnięcia przy występowaniu innych wrodzonych niedoborów obserwowano reakcje ze strony przeciwciał, jednakże aktualnie nie ma danych dotyczących koncentratu fibrynogenu.
Nie ma znanych interakcji produktów zawierających ludzki fibrynogen z innymi produktami leczniczymi.
Ciąża
W kontrolowanych badaniach klinicznych nie określono bezpieczeństwa stosowania produktu leczniczego Fibryga u kobiet w okresie ciąży. Doświadczenie kliniczne dotyczące stosowania produktów zawierających fibrynogen w leczeniu powikłań położniczych sugeruje, że nie należy oczekiwać działań szkodliwych dla przebiegu ciąży ani dla płodu czy noworodka. Nie przeprowadzono badań na zwierzętach dotyczących wpływu produktu leczniczego Fibryga na reprodukcję (patrz punkt 5.3). Ponieważ substancja czynna jest pochodzenia ludzkiego, jest katabolizowana w takich sam sposób, jak własne białko pacjenta. Nie oczekuje się, aby takie fizjologiczne składniki krwi ludzkiej wywierały niekorzystny wpływ na reprodukcję ani na płód.
Należy ocenić korzyści ze stosowania produktu leczniczego Fibryga podczas ciąży, biorąc pod uwagę to, że dostępne są dane kliniczne dotyczące stosowania koncentratów fibrynogenu, brakuje jednak danych pochodzących z kontrolowanych badań klinicznych.
Karmienie piersią
Nie wiadomo, czy produkt leczniczy Fibryga przenika do mleka ludzkiego. Jednak ze względu na właściwości substancji nie przewiduje się, by miała ona wpływ na karmione piersią noworodki/niemowlęta.
Z tego względu należy podjąć decyzję, czy stosowanie produktu leczniczego Fibryga jest wskazane podczas karmienia piersią, biorąc pod uwagę korzyści dla dziecka wynikające z karmienia piersią oraz korzyści dla matki wynikające z leczenia.
Płodność
Nie ma dostępnych danych dotyczących wpływu na płodność.
Produkt leczniczy Fibryga nie ma wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Nie ma wyczerpujących danych dotyczących częstości występowania działań niepożądanych w badaniach klinicznych tego produktu.
W badaniach klinicznych zgłoszono występowanie następujących działań niepożądanych: gorączka, wysypka polekowa, zapalenie żył i zakrzepica.
Następujące działania niepożądane zostały zgłoszone w związku ze stosowaniem produktu leczniczego Fibryga i innych koncentratów fibrynogenu:
Klasa układów i narządów MedDRA | Działania niepożądane | Częstość występowania* |
Zaburzenia układu immunologicznego: | Reakcje alergiczne i typu anafilaktycznego Reakcje skórne | Częstość nieznana |
Zaburzenia naczyniowe: | Zdarzenia zakrzepowo-zatorowe (w tym zawał mięśnia sercowego i zatorowość płucna) (patrz punkt 4.4) Zakrzepowe zapalenie żył | Częstość nieznana |
Zaburzenia ogólne i stany w miejscu podania: | Wzrost temperatury ciała (gorączka) | Częstość nieznana |
*Częstość jest nieznana, ponieważ nie można było jej obliczyć na podstawie dostępnych danych. Podczas badań klinicznych wystąpiły pojedyncze przypadki niewielkiej gorączki i reakcji skórnych. Reakcje alergiczne
i reakcje typu anafilaktycznego, zdarzenia zakrzepowo-zatorowe (w tym zawał mięśnia sercowego i zatorowość płucna) oraz zakrzepowe zapalenie żył to działania niepożądane wynikające ze stosowania tej grupy leków.
Informacje dotyczące bezpieczeństwa w odniesieniu do czynników zakaźnych, patrz punkt 4.4. Dzieci i młodzież:
Do analizy bezpieczeństwa stosowania w przypadku wrodzonego niedoboru fibrynogenu włączono dwadzieścioro sześcioro pacjentów w wieku od 1 do <18 lat, w tym 12 nastolatków w wieku od 12 do
<18 lat, 8 dzieci w wieku od 6 do <12 lat i 6 dzieci w wieku od 1 do <6 lat.
Nie stwierdzono różnic w zakresie ogólnego profilu bezpieczeństwa pomiędzy dorosłymi, młodzieżą i dziećmi.
Nie ma danych dotyczących stosowania produktu leczniczego Fibryga u dzieci z nabytym niedoborem fibrynogenu.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Wskazane jest regularne monitorowanie stężenia fibrynogenu w osoczu podczas leczenia, aby uniknąć przedawkowania (patrz punkt 4.2).
W przypadku przedawkowania zwiększone jest ryzyko powikłań zakrzepowo-zatorowych.
Grupa farmakoterapeutyczna: leki przeciwkrwotoczne, fibrynogen, kod ATC: B02BB01
Fibrynogen ludzki (czynnik krzepnięcia I), w obecności trombiny, aktywowanego czynnika krzepnięcia XIII (FXIIIa) oraz jonów wapnia jest przekształcany w stabilny i elastyczny, trójwymiarowy hemostatyczny skrzep fibryny.
Podanie fibrynogenu ludzkiego zapewnia wzrost stężenia fibrynogenu w osoczu i może czasowo ograniczać upośledzenie krzepnięcia u pacjentów z niedoborem fibrynogenu.
W prospektywnym badaniu farmakokinetycznym fazy II prowadzonym metodą otwartej próby z randomizacją i grupą kontrolną w dwóch grupach z zamianą grup leczenia obejmującym jedną
dawkę, obejmującym 22 pacjentów z wrodzonym niedoborem fibrynogenu (afibrynogenemia) (patrz punkt 5.2) oceniano także maksymalną twardość skrzepu (MCF) jako zastępczy wskaźnik skuteczności hemostatycznej (FORMA-01). MCF określano na podstawie tromboelastometrii (ROTEM). Dla każdego badanego MCF został określony przed (wartość wyjściowa) i jedną godzinę po podaniu pojedynczej dawki produktu Fibryga. Wartości MCF były znamiennie większe po podaniu produktu Fibryga w porównaniu do wartości wyjściowych (patrz tabela poniżej).
Tabela 1: Maksymalna twardość skrzepu MCF [mm] (populacja ITT), n=22
Punkt czasowy | Średnia ± SD | Mediana (zakres) |
Przed infuzją | 0 ± 0 | 0 (0–0) |
1 godz. po infuzji | 9,7 ± 3,0 | 10,0 (4,0–16,0) |
Średnia zmiana (analiza główna)* | 9,7 ± 3,0 | 10,0 (4,0–16,0) |
MCF = maksymalna twardość skrzepu; ITT = (populacja) zgodna z zamiarem leczenia.
*p < 0,0001 (95% przedział ufności: 8,37; 10,99)
Przeprowadzono prospektywne wieloośrodkowe badanie fazy III metodą otwartej próby bez grupy kontrolnej (FORMA-02) obejmujące 25 pacjentów z wrodzonym niedoborem fibrynogenu (afibrynogenemia lub hipofibrynogenemia) w wieku od 12 do 54 lat (6 nastolatków, 19 dorosłych). Badanie objęło leczenie 89 epizodów krwawienia i 12 zabiegów chirurgicznych. Stwierdzono znamienną zmianę wartości MCF ocenianej badaniem ROTEM i stężenia fibrynogenu w osoczu względem wartości wyjściowych. Mediana dawki produktu leczniczego Fibryga podawanej w ramach jednej infuzji w celu leczenia epizodów krwawienia wynosiła 57,5 mg/kg, a mediana dawki całkowitej wynosiła 59,4 mg/kg. Mediana dawki całkowitej produktu leczniczego Fibryga podawanej na jeden zabieg chirurgiczny wynosiła 85,8 mg/kg. Ogólna skuteczność hemostatyczna została potwierdzona (skuteczność dobra lub doskonała) w przypadku 98,9% leczonych epizodów krwawienia i 100% zabiegów chirurgicznych przez niezależną komisję orzekającą z zastosowaniem obiektywnego systemu oceny.
Przeprowadzono kolejne prospektywne wieloośrodkowe badanie fazy III metodą otwartej próby bez
grupy kontrolnej (FORMA-04) obejmujące 14 dzieci z wrodzonym niedoborem fibrynogenu (afibrynogenemia lub hipofibrynogenemia) w wieku od 1 do 10 lat (6 dzieci w wieku <6 lat i 8 dzieci w wieku od 6 do <12 lat). Badanie objęło leczenie 10 epizodów krwawienia i 3 zabiegów chirurgicznych, jak również farmakokinetykę dawki pojedynczej. Stwierdzono znamienną zmianę wartości MCF ocenianej w badaniu ROTEM i stężenia fibrynogenu w osoczu względem wartości wyjściowych. Mediana dawki produktu leczniczego Fibryga podawanej w ramach jednej infuzji
w celu leczenia epizodów krwawienia wynosiła 70,2 mg/kg, a mediana dawki całkowitej wynosiła 73,9 mg/kg. Mediana dawki całkowitej produktu leczniczego Fibryga podawanej na jeden zabieg chirurgiczny wynosiła 108 mg/kg. Ogólna skuteczność hemostatyczna została potwierdzona (skuteczność dobra lub doskonała) w przypadku 100% leczonych epizodów krwawienia i zabiegów chirurgicznych przez niezależną komisję orzekającą z zastosowaniem obiektywnego systemu oceny.
W prospektywnym, randomizowanym badaniu FORMA-05 z grupą kontrolną oceniano skuteczność hemostatyczną i bezpieczeństwo stosowania produktu leczniczego Fibryga w porównaniu
z krioprecypitatem jako źródłem suplementacji fibrynogenu u pacjentów z niedoborem fibrynogenu nabytym podczas zabiegu cytoredukcyjnego z powodu rozległego, złośliwego śluzaka rzekomego otrzewnej. W badaniu wzięło udział 43 pacjentów dorosłych w zbiorze analizy zgodnej z protokołem (Per Protocol, PP) – 21 pacjentów leczonych produktem leczniczym Fibryga oraz 22 pacjentów leczonych krioprecypitatem. Śródoperacyjną suplementację fibrynogenu prowadzono prewencyjnie (tj. po 60–90 minutach zabiegu chirurgicznego, gdy obserwowano nadmierną utratę krwi, ale zanim doszło do utraty 2 litrów krwi) produktem leczniczym Fibryga (4 g) lub krioprecypitatem (2 pule po 5 jednostek), powtarzając dawkowanie w razie potrzeby. Podczas 7,8 ± 1,7 godziny zabiegu chirurgicznego zastosowano odpowiednio 6,5 ± 3 g produktu leczniczego Fibryga (89 ± 39 mg/kg
mc.) oraz 4,1 ± 2,2 pule po 5 jednostek krioprecypitatu. W przypadku pacjentów leczonych produktem leczniczym Fibryga i krioprecypitatem mediana podanej śródoperacyjnie dawki KKCz wyniosła odpowiednio 1 jednostkę i 0,5 jednostki, a mediana dawki KKCz w ciągu pierwszych 24 godzin po operacji w obydwu grupach wyniosła 0 (patrz tabela poniżej). Podczas badania nie przetaczano świeżo mrożonego osocza ani koncentratów płytek krwi. Leczenie hemostatyczne polegające na suplementacji fibrynogenu zostało ocenione jako skuteczne w przypadku 100% zabiegów chirurgicznych w obydwu grupach przez niezależną komisję orzekającą z zastosowaniem obiektywnego systemu oceny.
Tabela 2: Transfuzja KKCz* [jednostki] śródoperacyjnie i w ciągu pierwszych 24 godzin po operacji (populacja PP)
Przedział czasowy | Grupa otrzymująca produkt Fibryga (n=21) Mediana (zakres) | Grupa otrzymująca krioprecypitat (n=22) Mediana (zakres) |
Śródoperacyjnie | 1 (0–4) | 0,5 (0–5) |
W ciągu pierwszych 24 godzin po operacji | 0 (0–2) | 0 (0–2) |
KKCz = koncentraty krwinek czerwonych; PP = zgodnie z protokołem*nie doszło do transfuzji innych allogenicznych produktów krwiopochodnych, takich jak świeżo mrożone osocze lub koncentraty płytek krwi
Dzieci i młodzież
W przypadku wrodzonego niedoboru fibrynogenu produkt leczniczy Fibryga podawany był w ramach dwóch badań klinicznych (FORMA-02 i FORMA-04) 20 pacjentom w wieku od 1 do <18 lat, w tym 6 nastolatkom w wieku od 12 do <18 lat, 8 dzieci w wieku od 6 do <12 lat i 6 dzieci w wieku od 1
do <6 lat. Skuteczność hemostatyczna została potwierdzona przez niezależną komisję orzekającą
w przypadku wszystkich leczonych epizodów krwawienia (10 epizodów krwawienia u nastolatków, 5 u dzieci w wieku od 6 do <12 lat oraz 5 u dzieci w wieku od 1 do <6 lat), a profilaktyka również została oceniona jako skuteczna w przypadku 4 zabiegów chirurgicznych przeprowadzonych u tych pacjentów (1 u nastolatków i 3 u dzieci w wieku od 1 do <6 lat).
Fibrynogen ludzki jest normalnym składnikiem ludzkiego osocza i działa jak fibrynogen endogenny.
Biologiczny okres półtrwania fibrynogenu w osoczu wynosi 3–4 dni. Produkt Fibryga jest podawany dożylnie i jest natychmiast dostępny w osoczu w stężeniu odpowiadającym podanej dawce.
W prospektywnym badaniu fazy II prowadzonym metodą otwartej próby z randomizacją i grupą kontrolną w dwóch grupach z zamianą grup leczenia, obejmującym 22 pacjentów z wrodzonym niedoborem fibrynogenu (afibrynogenemia) w wieku od 12 do 53 lat (6 nastolatków, 16 dorosłych) porównywano właściwości farmakokinetyczne jednej dawki produktu leczniczego Fibryga
z właściwościami innego dostępnego na rynku koncentratu fibrynogenu u tych samych pacjentów (FORMA-01). Każdy pacjent otrzymał jednorazową dawkę 70 mg/kg mc. produktu leczniczego Fibryga oraz produktu porównawczego. W punkcie początkowym oraz przez okres do 14 dni po infuzji pobierano próbki krwi w celu oznaczenia aktywności fibrynogenu. Parametry farmakokinetyczne produktu leczniczego Fibryga w analizie zgodnej z protokołem (PP) (n=21) są podsumowane w tabeli poniżej.
Tabela 3: Farmakokinetyczne parametry (n=21) aktywności fibrynogenu (populacja PP)
Parametr | Średnia ± SD | Zakres |
Okres półtrwania [godz.] | 75,9 ± 23,8 | 40,0–157,0 |
Cmax [mg/dl] | 139,0 ± 36,9 | 83,0–216,0 |
AUCnorm dla dawki 70 mg/kg mc. [mg*h/ml] | 113,7 ± 31,5 | 59,7–175,5 |
Klirens [ml/h/kg] | 0,67 ± 0,2 | 0,4–1,2 |
Średni czas przebywania [h] | 106,3 ± 30,9 | 58,7–205,5 |
Objętość dystrybucji w stanie równowagi [ml/kg] | 70,2 ± 29,9 | 36,9–149,1 |
*Jeden pacjent został wykluczony z populacji PP, ponieważ otrzymał<90% zaplanowanej dawki produktu Fibryga i produktu porównawczego.
Cmax = maksymalne stężenie w osoczu; AUCnorm = pole powierzchni pod krzywą znormalizowane względem podanej dawki; SD = odchylenie standardowe
Narastający odzysk in vivo (IVR) został określony na podstawie wartości stężenia oznaczonych
w okresie do 4 godzin po infuzji. Mediana narastającego IVR wynosiła 1,8 mg/dl (zakres 1,08–2,62 mg/dl) zwiększenia na mg/kg. Taka mediana IVR wskazuje, że dawka 70 mg/kg mc. zwiększy stężenie fibrynogenu w osoczu pacjenta o około 125 mg/dl.
Farmakokinetyka w specjalnych populacjach
Nie zaobserwowano statystycznie znamiennej różnicy w aktywności fibrynogenu pomiędzy pacjentami płci męskiej i żeńskiej.
Dzieci i młodzież
Dane farmakokinetyczne u młodzieży w wieku od 12 do poniżej 18 lat uzyskano w badaniu FORMA-02. W analizie PP zaobserwowano niewielką różnicę w zakresie okresu półtrwania
u młodzieży (n=5) i u dorosłych (n=16), wynoszącego odpowiednio 72,8 ± 16,5 godz. w porównaniu z 76,9 ± 26,1 godz. Klirens był niemal identyczny w obu grupach wiekowych, tzn. odpowiednio, 0,68 ± 0,18 ml/h/kg oraz 0,66 ± 0,21 ml/h/kg. Badania właściwości farmakokinetycznych produktu
leczniczego Fibryga kontynuowano w ramach badania FORMA-04 u 13 dzieci w wieku poniżej 12 lat z wrodzonym niedoborem fibrynogenu (afibrynogenemia). Każdy pacjent otrzymał pojedynczą dawkę dożylną 70 mg/kg produktu leczniczego Fibryga. Parametry farmakokinetyczne produktu leczniczego Fibryga podsumowano w tabeli poniżej. Mediana narastającego odzysku in vivo wynosiła 1,4 mg/dl (zakres, 1,3-2,1 mg/dl) zwiększenia na mg/kg.
Tabela 4: Parametry farmakokinetyczne (n=13) aktywności fibrynogenu
Parametr | Średnia ± SD | Zakres |
Okres półtrwania [godz.]* | 63,3 ± 12,0 | 45,6–91,6 |
Cmax [mg/dl] | 107,2 ± 16,8 | 93,0–154,0 |
AUCnorm dla dawki 70 mg/kg mc. [mg*h/ml]* | 92,0 ± 20,0 | 69,7–134,2 |
Klirens [ml/h/kg]* | 0,8 ± 0,2 | 0,5–1,0 |
Średni czas przebywania [h]* | 88,0 ± 16,8 | 63,6–126,7 |
Objętość dystrybucji w stanie równowagi [ml/kg]* | 67,6 ± 7,1 | 52,8–76,8 |
*Obliczono u 10 spośród 13 pacjentów z powodu niewystarczającej liczby oznaczalnych wartości u 3 pacjentów. IVR = odzysk in vivo; Cmax = maksymalne stężenie w osoczu; AUCnorm = pole powierzchni pod krzywą znormalizowane względem podanej dawki; SD = odchylenie standardowe
Bezpieczeństwo stosowania produktu leczniczego Fibryga zostało wykazane w kilku nieklinicznych badaniach farmakologicznych (działanie na układ krążenia, działanie trombogenne) oraz toksyczności (toksyczność ostra, tolerancja miejscowa). Wynikające z tych badań dane niekliniczne nie ujawniają szczególnego zagrożenia dla człowieka.
W teście zastoju żylnego (test Wesslera) nie wykazano trombogennego działania produktu Fibryga przy dawkach do 400 mg/kg masy ciała.
Proszek
L-argininy chlorowodorek Glicyna
Sodu chlorek
Sodu cytrynian dwuwodny
Rozpuszczalnik
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi.
2 lata
Wykazano stabilność chemiczną i fizyczną przygotowanego roztworu przez 24 godziny przy przechowywaniu w temperaturze pokojowej (do 25°C). Z mikrobiologicznego punktu widzenia produkt leczniczy należy użyć natychmiast po rekonstytucji. Jeżeli produkt leczniczy nie zostanie użyty natychmiast, użytkownik ponosi odpowiedzialność za czas i warunki jego przechowywania przed podaniem . Nie wolno zamrażać ani przechowywać w lodówce przygotowanego roztworu. Częściowo zużyte butelki należy usunąć.
Nie przechowywać w temperaturze powyżej 25C. Nie zamrażać. Przechowywać butelkę w opakowaniu zewnętrznym w celu ochrony przed światłem.
Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Każde tekturowe pudełko zawiera:
Instrukcje ogólne
Rekonstytucja
1. Zarówno proszek (Fibryga), jak i rozpuszczalnik (woda do wstrzykiwań) w nieotwartych opakowaniach ogrzać do temperatury pokojowej. Należy utrzymywać tę temperaturę przez cały proces rekonstytucji. Jeżeli do ogrzewania wykorzystywana jest łaźnia wodna, należy zachować ostrożność, by nie dopuścić do kontaktu wody z gumowym korkiem ani aluminiowym kapslem. Temperatura łaźni wodnej nie powinna przekraczać +37°C. |
2. Zdjąć wieczko z butelki z proszkiem (Fibryga) oraz fiolki z rozpuszczalnikiem, aby odsłonić centralną część korków infuzyjnych. Przetrzeć gumowe korki gazikiem z alkoholem i pozostawić do wyschnięcia. |
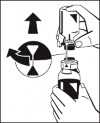
3. Oderwać pokrywę zewnętrznego opakowania urządzenia do rekonstytucji Octajet. Aby zachować jałowość pozostawić urządzenie Octajet w przezroczystym zewnętrznym opakowaniu. |
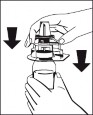
4. Wziąć urządzenie Octajet razem z zewnętrznym opakowaniem i odwrócić je do góry dnem nad butelką z proszkiem (Fibryga). Ustawić urządzenie, wciąż w opakowaniu zewnętrznym, nad środkiem butelki z proszkiem tak, aby zostały zablokowane zatrzaski iglicy przeznaczonej do produktu (bezbarwnej). Utrzymując urządzenie Octajet nad butelką z proszkiem, ostrożnie zdjąć z niego opakowanie zewnętrzne, uważając by nie dotknąć Ryc. 1 iglicy przeznaczonej do wody (niebieskiej) i pozostawić je ściśle połączone z butelką. (Ryc. 1) |
5. Trzymając mocno butelkę z proszkiem na poziomej powierzchni, odwrócić fiolkę z rozpuszczalnikiem do góry dnem i ustawić ją centralnie nad niebieską iglicą przeznaczoną do wody. Przebić pewnie niebieską plastikową iglicą urządzenia Octajet przez środek gumowego korka fiolki z rozpuszczalnikiem. (Ryc. 2) |
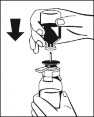
Ryc. 2
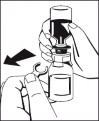
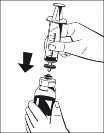
6. Usunąć pierścień oddzielajacy (Ryc. 3) i wcisnąć fiolkę z rozpuszczalnikiem w dół (Ryc. 4). Rozpuszczalnik przepłynie do butelki z proszkiem (Fibryga). |
Ryc. 3 Ryc. 4 7. Po zakończeniu przepływu rozpuszczalnika, wirować delikatnie butelką z produktem, aż proszek się całkowicie rozpuści. Nie wstrząsać butelką, aby uniknąć tworzenia się piany. Proszek powinien rozpuścić się całkowicie w ciągu 5 minut. Nie powinno to trwać dłużej niż 30 minut; Jeżeli po 30 minutach proszek nie będzie całkowicie rozpuszczony, usunąć produkt. |
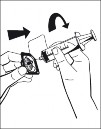
8. Przekręcić niebieskie złącze z fiolką po rozpuszczalniku (w dowolnym kierunku), tak aby wskaźniki położenia były w jednej linii i usunąć fiolkę po rozpuszczalniku razem z niebieską iglicą. (Ryc. 5) |
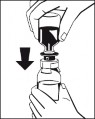
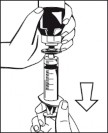
Ryc. 5 9. Podłączyć strzykawkę do filtra znajdującego się w zestawie (Ryc. 6) i połączyć filtr ze złączem Luer Lock urządzenia Octajet znajdującym się na butelce z proszkiem (Ryc. 7). Pobrać roztwór do strzykawki przez filtr. (Ryc. 8) |
Ryc. 6 Ryc. 7 Ryc. 8 10. Odłączyć napełnioną strzykawkę od filtra i usunąć pustą butelkę. |
Zaleca się dożylne podanie przygotowanego roztworu w temperaturze pokojowej przy użyciu standardowego zestawu do infuzji.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Octapharma (IP) SPRL Allée de la Recherche 65 1070 Anderlecht
Belgia
24152
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 26 lipca 2017 Data ostatniego przedłużenia pozwolenia:








