







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO Berinert 2000
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER (-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO Berinert 2000
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację Ciąża
Wpływ na zdolność do prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Roztwór Berinert po rekonstytucji powinien być bezbarwny i przezroczysty do lekko opalizującego.
Po filtracji/opróżnieniu fiolki (patrz poniżej) produkt leczniczy po rekonstytucji przed podaniem powinien być
poddany ocenie wzrokowej; należy sprawdzić czy nie pojawiły się cząstki stałe
i czy nie nastąpiła zmiana zabarwienia.
Nie stosować mętnych roztworów i takich, które zawierają osad.
Podczas rekonstytucji i opróżniania fiolki muszą być zachowane aseptyczne warunki. Należy używać strzykawki
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER (-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU / DATA PRZEDŁUŻENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
2000 j.m.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
Berinert 3000
3000 j.m.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Substancja czynna: ludzki inhibitor C1-esterazy (pochodzący z ludzkiego osocza) do podawania podskórnego (s.c.)
W jednej fiolce Berinert 2000 znajduje się 2000 j.m. inhibitora C1-esterazy. W jednej fiolce Berinert 3000 znajduje się 3000 j.m. inhibitora C1-esterazy.
Aktywność ludzkiego inhibitora C1-esterazy jest wyrażona w jednostkach międzynarodowych (j.m.), które odnoszą się
do aktualnych standardów WHO dotyczących produktów inhibitora C1-esterazy.
Po rekonstytucji w 4 ml wody do wstrzykiwań Berinert 2000 zawiera 500 j.m./ml ludzkiego inhibitora C1-esterazy. Po rekonstytucji w 5.6 ml wody do wstrzykiwań Berinert 3000 zawiera 500 j.m./ml ludzkiego inhibitora C1-esterazy.
Zawartość białka całkowitego w roztworze po rekonstytucji produktu wynosi 65 mg/ml. Substancje pomocnicze o znanym działaniu:
Sód do 486 mg (około 21 mmol) na 100 ml roztworu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Berinert 2000
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
Berinert 3000
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Biały proszek.
Przeźroczysty, bezbarwny rozpuszczalnik.
Berinert do wstrzykiwań podskórnych jest wskazany do stosowania w zapobieganiu napadom nawracającego wrodzonego obrzęku naczynioruchowego (HAE) u młodzieży i pacjentów dorosłych z niedoborem inhibitora C1 - esterazy.
Berinert jest przeznaczony do samodzielnego podawania w postaci wstrzykiwań podskórnych. Pacjent lub jego opiekun powinni być przeszkoleni jak należy podawać Berinert.
Dawkowanie
Zalecana dawka:
Berinert s.c. 60 j.m./kg masy ciała dwa razy w tygodniu (co 3-4 dni)
Dzieci i młodzież
Dawkowanie u młodzieży jest takie samo jak u osób dorosłych.
Sposób podawania
Wyłącznie do podawania podskórnego
Instrukcje dotyczące rekonstytucji produktu leczniczego przed podaniem patrz punkt 6.6.
Proponowanym miejscem do wstrzyknięcia podskórnego produktu leczniczego Berinert jest okolica brzucha. W badaniach
klinicznych, Berinert był wstrzykiwany w pojedyncze miejsce.
Produkt po rekonstytucji powinien być podany we wstrzyknięciu podskórnym z szybkością tolerowaną przez pacjenta.
Pacjenci, którzy doświadczyli reakcji nadwrażliwości zagrażających życiu, włącznie z anafilaksją na produkty C1-INH lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Identyfikowalność
W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Reakcje nadwrażliwości
Jeżeli wystąpią ciężkie objawy alergiczne należy natychmiast zaprzestać podawania leku Berinert (tzn. przerwać wstrzykiwanie) i rozpocząć odpowiednią opiekę medyczną. W przypadku ostrego napadu HAE, należy rozpocząć leczenie dostosowane indywidualnie do pacjenta.
Zdarzenia zakrzepowo – zatorowe (TEE)
W przypadkach stosowania wysokich dawek C1-INH i.v. w profilaktyce lub leczeniu zespołu nieszczelności naczyń włosowatych przed, podczas i po zabiegach chirurgii serca z zastosowaniem krążenia zewnątrzustrojowego (niezatwierdzone wskazanie i dawkowanie) wystąpiły przypadki zakrzepicy.
Przy podawaniu podskórnych zalecanych dawek nie stwierdzono związku przyczynowego pomiędzy występowaniem TEE
a stosowaniem koncentratu C1-INH.
Bezpieczeństwo wirusowe
Standardowe postępowanie zabezpieczające przed przenoszeniem zakażeń poprzez podawanie produktów leczniczych otrzymywanych z krwi lub osocza ludzkiego obejmuje: selekcję dawców, badanie pojedynczych donacji i pul osocza na obecność markerów wirusów oraz zastosowanie skutecznych metod inaktywacji/usuwania wirusów podczas procesu wytwarzania. Pomimo tego, nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych po podaniu produktu leczniczego pochodzącego z krwi lub osocza ludzkiego. To ryzyko dotyczy nieznanych lub nowo odkrytych wirusów i innych czynników zakaźnych.
Podjęte środki zabezpieczające są uważane za skuteczne w stosunku do otoczkowych wirusów takich jak ludzki wirus niedoboru odporności (HIV), wirusowe zapalenie wątroby typu B (HBV), wirusowe zapalenie wątroby typu C (HCV) oraz wirusów bezotoczkowych HAV i parwowirusa B 19.
U pacjentów regularnie lub wielokrotnie otrzymujących produkty pochodzące z ludzkiego osocza należy rozważyć szczepienia przeciwko wirusowemu zapaleniu wątroby typu A i B.
Berinert 2000 zawiera mniej niż 1 mmol sodu (23 mg) na fiolkę, co oznacza, że jest uznany za „wolny od sodu”.
Berinert 3000 zawiera do 29 mg sodu na fiolkę, co odpowiada 1,5% zalecanej przez WHO maksymalnej 2 g dobowej dawki
spożycia sodu u osoby dorosłej.
Nie przeprowadzono badań dotyczących interakcji.
Istnieją ograniczone dane sugerujące brak zwiększonego ryzyka związanego ze stosowaniem produktów ludzkiego
inhibitora C1-esterazy u kobiet w ciąży. Ludzki inhibitor C1-esterazy jest fizjologicznym składnikiem ludzkiego osocza. Nie przeprowadzano badań w kierunku toksyczności Berinert w okresie ciąży i rozwoju płodowego u zwierząt. Nie należy spodziewać się wystąpienia działań niepożądanych związanych z płodnością oraz rozwojem przed- i pourodzeniowym u ludzi.
W trzech badaniach, które obejmowały 344 pacjentki, zebrano dane od 36 kobiet (50 ciąż) i nie stwierdzono żadnych działań niepożądanych związanych z leczeniem C1-INH przed, w trakcie lub po ciąży i kobiety urodziły zdrowe dzieci.
Karmienie piersią
Brak informacji dotyczących wydzielania produktu leczniczego Berinert do mleka matki, wpływu na niemowlę karmione piersią lub wpływu na wytwarzanie mleka.
Należy wziąć pod uwagę rozwojowe i zdrowotne korzyści wynikające z karmienia piersią, a także kliniczne zapotrzebowanie matki na lek Berinert oraz wszelkie potencjalne działania niepożądane karmionego piersią niemowlęcia przez matkę stosującą produkt leczniczy Berinert lub chorobę podstawową matki.
Płodność
Ludzki inhibitor C1-esterazy jest fizjologicznym składnikiem ludzkiego osocza. Nie przeprowadzono badań na zwierzętach produktu leczniczego Berinert dotyczących wpływu na reprodukcję i toksycznego wpływu na rozwój.
Berinert nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Informacje dotyczące działań niepożądanych zebrano w badaniu klinicznym 3 fazy (Badanie 3001) u pacjentów (n = 86) z HAE, którzy otrzymywali Berinert podskórnie. Zakwalifikowani pacjenci mogli również uczestniczyć w otwartym badaniu dodatkowym (Badanie 3002) przez maksymalnie 140 tygodni (n = 126). Częstość działań niepożądanych jest oparta na zdarzeniach związanych ze stosowaniem Berinert. Jest ona określona na pacjenta i klasyfikowana jako:
bardzo często: (≥ )1/10 często: (≥ 1/100 do< 1/10)
niezbyt często: (≥ 1/1 000 do < 1/100) rzadko: (≥ 1/10 000 do < 1/1 000) bardzo rzadko: (< 1/10 000)
Klasyfikacja układów i narządów MedDRA | Działania niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | Zapalenie nosogardzieli | Bardzo często |
Zaburzenia układu immunologicznego | Nadwrażliwość (nadwrażliwość, świąd, wysypka i pokrzywka) | Często |
Zaburzenia układu nerwowego | Zawroty głowy | Często |
Zaburzenia ogólne i stany w miejscu podania | Reakcje w miejscu wstrzyknięcia a | Bardzo często |
a Zasinienie w miejscu wstrzyknięcia, odczucie zimna w miejscu wstrzyknięcia, wyciek w miejscu wstrzyknięcia, rumień w miejscu wstrzyknięcia, krwiak w miejscu wstrzyknięcia, krwotok w miejscu wstrzyknięcia, stwardnienie w miejscu wstrzyknięcia, obrzęk w miejscu wstrzyknięcia , ból w miejscu wstrzyknięcia, świąd w miejscu wstrzyknięcia, wysypka w miejscu wstrzyknięcia, reakcje w miejscu wstrzyknięcia, blizna w miejscu wstrzyknięcia, opuchnięcie w miejscu wstrzyknięcia, pokrzywka w miejscu wstrzyknięcia, ciepło w miejscu wstrzyknięcia. | ||
Dzieci i młodzież
Profil bezpieczeństwa produktu leczniczego Berinert oceniano w podgrupie jedenastu pacjentów w wieku od 8 do <17 lat
w obu badaniach (Badanie 3001, Badanie 3002) i był zgodny z ogólnymi wynikami dotyczącymi bezpieczeństwa.
Inne szczególne grupy
Osoby w podeszłym wieku
Profil bezpieczeństwa produktu leczniczego Berinert oceniano w podgrupie dziesięciu pacjentów w wieku od 65 do 72 lat
w obu badaniach (Badanie 3001, Badanie 3002) i był zgodny z ogólnymi wynikami dotyczącymi bezpieczeństwa.
Możliwość przeniesienia czynników zakaźnych przedstawiono w punkcie 4.4.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie zgłoszono przypadków przedawkowania. Odpowiednie dawki do 117 j.m./kg s.c były podawane dwa razy w tygodniu,
w badaniu klinicznym z ustalonymi dawkami i były dobrze tolerowane.
Grupa farmakoterapeutyczna: Inne czynniki hematologiczne, Leki stosowane w dziedzicznym obrzęku naczynioworuchowym, C1-inhibitor, pochodna osocza
ATC kod: B06AC01
Inhibitor C1-esterazy jest osoczową glikoproteiną o masie cząsteczkowej 105 kDa zawierającą 40% węglowodanów. Stężenie w ludzkim osoczu wynosi około 240 mg/l. Poza osoczem inhibitor C1-esterazy wykrywany jest także w łożysku, komórkach wątroby, monocytach i płytkach krwi.
Inhibitor C1-esterazy należy do ludzkiego osoczowego układu inhibitorów proteaz serynowych (serpin) i działa jak inne białka tej grupy, takie jak: antytrombina III, alpha-2-antyplazmina, alpha-1- antytrypsyna i inne.
Mechanizm działania
W warunkach fizjologicznych inhibitor C1-esterazy blokuje klasyczną drogę układu dopełniacza poprzez inaktywację aktywnych enzymatycznie składników C1s i C1r. Aktywny enzym tworzy kompleks z inhibitorem w stechiometrycznym stosunku 1:1.
Poza tym inhibitor C1-esterazy stanowi najważniejszy inhibitor aktywatorów układu krzepnięcia poprzez hamowanie czynnika XIIa i jego fragmentów. Dodatkowo, oprócz alpha-2-makroglobuliny, jest głównym inhibitorem osoczowej kalikreiny.
Efekt leczniczy produktu Berinert we wrodzonym obrzęku naczynioruchowym polega na substytucji brakującej aktywności inhibitora C1-esterazy.
Skuteczność kliniczna i bezpieczeństwo
Skuteczność i bezpieczeństwo produktu leczniczego Berinert w rutynowej profilaktyce w celu zapobiegania napadom HAE wykazano w wieloośrodkowym, randomizowanym, podwójnie zaślepionym, kontrolowanym placebo, krzyżowym badaniu (Badanie 3001). W badaniu wzięło udział 90 osób dorosłych i młodzieży z objawowymi HAE typu I lub II. Mediana (zakres) wieku badanych wynosiła 40 (od 12 do 72) lat; w tym 60 kobiet i 30 mężczyzn. Pacjenci zostali losowo przydzieleni do grupy otrzymującej 60 IU/kg lub 40 IU/kg produktu leczniczego Berinert w jednym 16-tygodniowym okresie leczenia i placebo w drugim 16-tygodniowym okresie leczenia. Pacjenci stosowali samodzielnie produkt leczniczy Berinert lub placebo podskórnie 2 razy w tygodniu. Skuteczność oceniano przez ostatnie 14 tygodni każdego okresu leczenia. Kwalifikujący się pacjenci mogli również uczestniczyć w otwartym badaniu dodatkowym przez maksymalnie 140 tygodni (Badanie 3002). W przybliżeniu połowa osób zakwalifikowanych do przedłużonego badania uczestniczyła w badaniu 3001 (64/126, 50,8%), co przyczyniło się do podobieństwa między badanymi populacjami.
Badanie 3001:
Podanie podskórne dwa razy w tygodniu dawki 60 IU/kg lub 40 IU/kg produktu leczniczego Berinert powodowały istotną różnicę w znormalizowanej w czasie liczbie napadów HAE (wskaźnik napadów) w porównaniu z placebo (Tabela 1). Znormalizowana czasowo liczba napadów HAE u pacjentów, którym podawano dawkę 60 IU/kg, wynosiła 0,52 ataków na miesiąc w porównaniu do 4,03 ataków na miesiąc przyjmujących placebo (p <0,001). Znormalizowana czasowo liczba napadów HAE u pacjentów, którym podawano dawkę 40 IU/kg, wyniosła 1,19 ataków na miesiąc w porównaniu z 3,61 ataków na miesiąc przyjmujących placebo (p <0,001).
Tabela 1. Znormalizowana w czasie liczba ataków HAE (Liczba/Miesiąc)
60 IU/kg Sekwencja leczenia (N = 45) | 40 IU/kg Sekwencja leczenia (N = 45) | |||
Produkt leczniczy | Placebo | Produkt leczniczy | Placebo | |
n | 43 | 42 | 43 | 44 |
Średnia (SD) | 0,5 (0,8) | 4,0 (2,3) | 1,2 (2,3) | 3,6 (2,1) |
Min, Max | 0,0; 3,1 | 0,6: 11,3 | 0,0: 12,5 | 0,0; 8,9 |
Mediana | 0,3 | 3,8 | 0,3 | 3,8 |
LS Średnia (SE)* | 0,5 (0,3) | 4,0 (0,3) | 1,2 (0,3) | 3,6 (0,3) |
95% CI z LS Średniej* | (0,0, 1,0) | (3,5, 4,6) | (0,5, 1,9) | (3; 4,3) |
Różnica w leczeniu (biorąc pod uwagę pacjentów) | 60 IU/kg – Placebo | 40 IU/kg – Placebo | ||
LS Średnia* (95% CI) | -3,5 (-4,2: -2,8) | -2,4 (-3,4: -1,5) | ||
p-wartość* | < 0,001 | < 0,001 | ||
CI = przedział ufności; HAE = wrodzony obrzęk naczynioruchowy; N = liczba zrandomizowanych pacjentów; n = liczba pacjentów z danymi; LS = najmniejsze kwadraty.
* Z modelu mieszanego.
Mediana (centyl 25, 75) procentowego zmniejszenia znormalizowanej w czasie liczby napadów HAE w porównaniu z placebo wyniosła 95% (79, 100) dla dawki 60 IU/kg i 89% (70, 100) dla dawki 40 IU/kg produktu leczniczego Berinert wśród badanych z możliwymi do oceny danymi z obu okresów leczenia.
Odsetek osób z odpowiedzią (95% CI) z ≥50% zmniejszeniem znormalizowanej w czasie liczby napadów HAE na produkt leczniczy Berinert w porównaniu z placebo wyniósł 83% (73%, 90%). Dziewięćdziesiąt procent (90%) pacjentów otrzymujących dawkę 60 IU/kg odpowiedziało na leczenie oraz 76% pacjentów otrzymujących dawkę 40 IU/kg odpowiedziało na leczenie.
Siedemdziesiąt jeden procent (71%) pacjentów otrzymujących dawkę 60 IU/kg i 53% pacjentów otrzymujących dawkę 40 IU/kg miało ≥ 1 napad HAE na 4 tygodnie w grupie placebo i <1 napad HAE na 4 tygodnie w grupie otrzymujących produkt leczniczy Berinert.
W sumie 40% pacjentów otrzymujących dawkę 60 IU/kg i 38% pacjentów otrzymujących dawkę 40 IU/kg było bez ataków, a mediana częstości napadów HAE na miesiąc wynosiła 0,3 dla obu dawek.
Produkt leczniczy Berinert spowodował istotną różnicę w znormalizowanej w czasie liczbie stosowania leku doraźnie (wskaźnik użycia leku doraźnego) w porównaniu z placebo. Dawka 60 IU/kg skutkowała średnią szybkością zastosowania doraźnego leku wynoszącą 0,3 zastosowania na miesiąc w porównaniu z 3,9 zastosowaniami na miesiąc w przypadku placebo. Dawka 40 IU/kg spowodowała, że średni wskaźnik użycia leku doraźnie wyniósł 1,1 zastosowania na miesiąc, w porównaniu z 5,6 użycia na miesiąc w przypadku placebo.
Badanie 3002:
Wykazano długoterminowe bezpieczeństwo i skuteczność produktu leczniczego Berinert w rutynowej profilaktyce w celu zapobiegania napadom HAE w otwartym, randomizowanym parallel-arm badaniu. W badaniu oceniano 126 dorosłych i dzieci z objawowym HAE typu I lub II, w tym 64 pacjentów w grupie roll-over (z powtórnym udziałem badanych) z Badania 3001 i 62 pacjentów w grupie non-rollover (nie objętych powtórnym udziałem w badaniu). Mediana (zakres) wieku badanych wynosiła 41,0 (8-72) lat. Pacjenci z miesięcznym współczynnikiem ataków 4,3 na 3 miesiące przed przystąpieniem do badania byli włączani do badania i leczeni średnio przez 1,5 roku; 44 pacjentów (34,9%) miało ponad 2 lata ekspozycji. Średnia aktywność funkcjonalna C1-INH w stanie stacjonarnym wzrosła do 52,0% przy dawce 40 IU/kg i 66,6% przy dawce 60 IU/kg. Częstość występowania działań niepożądanych była niska i podobna w grupach dla obu dawek (odpowiednio 11,3 i 8,5 działań na pacjent/rok odpowiednio w grupie leczonej dawką 40 IU/kg i 60 IU/kg).
Odsetek osób z odpowiedzią (95% CI) z ≥ 50% zmniejszeniem znormalizowanej w czasie liczby napadów HAE przy zastosowaniu produktu leczniczego Berinert w stosunku do znormalizowanej w czasie liczby napadów HAE wykorzystanych do zakwalifikowania do udziału w Badaniu 3002 wyniósł 93,5% (84,6%; 97,5%) w grupie leczonej dawką 40 IU/kg i 91,7% (81,9%; 96,4%) w grupie leczonej dawką 60 IU/kg.
Odsetek pacjentów ze znormalizowaną w czasie częstością napadów HAE <1 napad HAE w okresie 4 tygodni wynosił 79,4% dla dawki 40 IU/kg i 85,7% dla dawki 60 IU/kg.
Odsetek pacjentów bez napadów HAE wyniósł 34,9% dla dawki 40 IU/kg i 44,4% dla dawki 60 IU/kg (przez cały czas trwania badania, przy maksymalnym czasie trwania ekspozycji> 2,5 roku). Spośród 23 pacjentów otrzymujących dawkę 60 IU/kg przez ponad 2 lata, 19 (83%) było bez ataków w okresie od 25 do 30 miesięcy leczenia.
Średnia znormalizowana w czasie liczba zastosowań doraźnych leku wynosiła 0,26 (0,572) zastosowań na miesiąc dla dawki 40 IU/kg i 0,31 (0,804) zastosowań na miesiąc dla dawki 60 IU/kg.
Dzieci i młodzież
Bezpieczeństwo i skuteczność produktu leczniczego Berinert oceniano w podgrupie 11 pacjentów w wieku od 8 do <17 lat, w randomizowanym, podwójnie zaślepionym, kontrolowanym placebo, krzyżowym, rutynowym badaniu profilaktycznym (Badanie 3001) oraz w randomizowanym, otwartym, aktywnie kontrolowanym badaniu (Badanie 3002). Wyniki analizy podgrup według wieku były zgodne z ogólnymi wynikami badania.
Charakterystyka farmakokinetyczna (PK) produktu leczniczego Berinert do podawania podskórnego opisano pierwotnie stosując PK dla populacji metodami zbiorczych danych z trzech badań klinicznych przeprowadzonych na osobach zdrowych i pacjentach z HAE.
Wchłanianie
Po zastosowaniu podskórnej dawki dwa razy w tygodniu Berinert jest wchłaniany powoli, z medianą (95% CI) czasu do osiągnięcia maksymalnego stężenia (tmax) około 59 godzin (23, 134 godziny). Oparty o medianę (95% CI) widoczny okres półtrwania w osoczu wynosi 69 godzin (24, 250 godzin), w stanie równowagi dla C1-INH jest oczekiwany w ciągu 3 tygodni od rozpoczęcia dawkowania. Średnie (95% CI) stężenie w stanie równowagi C1-INH wynosi 48 % (25,1; 102%) i jest oczekiwane po podskórnym dawkowaniu (s.c.) 2 razy w tygodniu 60 j.m./kg produktu leczniczego Berinert. Średnia (95% CI) względnej dostępności biologicznej Berinert po podaniu s.c. była w przybliżeniu określona na 43% (35,2; 50,2%).
Dystrybucja i eliminacja
Średnia w populacji (95%CI) klirensu i pozornej objętości dystrybucji produktu leczniczego Berinert została w przybliżeniu określona na 83 ml/godz. (72,7; 94,2 ml/godz.) i 4,33 l (3,51; 5,15 l). Klirens C1-INH został określony jako pozytywnie skorelowany z całkowitą masą ciała. PK w stanie równowagi po podaniu s.c. produktu leczniczego Berinert zostało rozpoznane jako niezależne od dawek pomiędzy 20-80 j.m./kg u pacjentów z HAE.
Nie przeprowadzono badań w celu określenia PK C1-INH w poszczególnych grupach pacjentów podzielonych ze względu na płeć, rasę, wiek, lub ze względu na występowanie niewydolności nerek lub wątroby.
Analiza populacji ze względu na wiek (8 do 72 lat) wykazała brak wpływu na PK C1-INH.
Dane nie kliniczne po podaniu dożylnym i podskórnym nie wykazały szczególnego zagrożenia toksyczności pojedynczych i powtarzanych dawek, dla tolerancji w miejscu podania i tworzenia się skrzepów u ludzi w oparciu o standardowe badania bezpieczeństwa farmakologicznego.
Nie przeprowadzono badań dotyczących właściwości rakotwórczych i toksycznego wpływu na reprodukcję,
Proszek:
Glicyna Sodu chlorek
Sodu cytrynian
Rozpuszczalnik:
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
36 miesięcy
Po rekonstytucji stabilność fizyko-chemiczna jest zachowana przez 48 godzin w temperaturze pokojowej (maks. 30ºC). Z mikrobiologicznego punktu widzenia oraz z uwagi na to, że Berinert nie zawiera środków konserwujących, produkt po rekonstytucji powinien być zużyty natychmiast. Jeżeli produkt nie został natychmiast podany, czas przechowywania w temperaturze pokojowej nie może przekroczyć 8 godzin. Produkt leczniczy po rekonstytucji powinien być przechowywany wyłącznie w fiolce.
Nie przechowywać w temperaturze powyżej 30 °C. Nie zamrażać.
Przechowywać fiolkę w opakowaniu zewnętrznym w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Opakowanie bezpośrednie:
Berinert 2000:
Proszek (2000 j.m.) w fiolce ze szkła typu II, zamkniętej korkiem z gumy bromobutylowej z niebieskim aluminiowym kapslem i szarym plastikowym wieczkiem typu flip-off.
4 ml rozpuszczalnika w fiolce ze szkła typu I, zamkniętej korkiem z (gumy chlorobutylowej lub bromobutylowej) z niebieskim aluminiowym kapslem i szarym plastikowym wieczkiem typu flip-off.
Berinert 3000:
Proszek (3000 j.m.) w fiolce ze szkła typu II, zamkniętej korkiem z gumy bromobutylowej z niebieskim aluminiowym kapslem i cytrynowym plastikowym wieczkiem typu flip-off.
5,6 ml rozpuszczalnika w fiolce ze szkła typu I, zamkniętej korkiem z (gumy chlorobutylowej lub bromobutylowej) z niebieskim aluminiowym kapslem i limonkowym plastikowym wieczkiem typu flip-off.
Dostępne opakowania:
Opakowanie zawiera:
1 fiolka z proszkiem
1 fiolka z rozpuszczalnikiem (Berinert 2000: 4 ml, Berinert 3000: 5,6 ml)
1 system do transferu 20/20 z filtrem
Zestaw do podawania (opakowanie wewnętrzne)
1 strzykawka jednorazowego użytku o pojemności (Berinert 2000: 5 ml, Berinert 3000: 10ml)
1 igła do podawania podskórnego 1 zestaw do wkłucia podskórnego, 2 waciki nasączone alkoholem,
1 plaster.
Opakowanie zawierające 5 x 2000 j.m. i 20 x 2000 j.m.
Opakowanie zawierające 5 x 3000 j.m. i 20 x 3000 j.m.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami.
Sposób podawania
Wskazówki ogólne
dołączonej do produktu.
Rekonstytucja
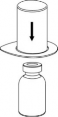
Doprowadzić rozpuszczalnik do temperatury pokojowej. Zdjąć z fiolek zawierających proszek i rozpuszczalnik plastikowe wieczka i przemyć korki w miejscach wkłucia aseptycznym roztworem. Po wyschnięciu otworzyć zestaw zawierający Mix2Vial.
1 | 1. Otworzyć opakowanie zawierające Mix2Vial poprzez usunięcie wieczka. Nie wyjmować Mix2Vial z blistra. |
2 | 2. Umieścić fiolkę z rozpuszczalnikiem na równej, czystej powierzchni i mocno przytrzymać. Nie wyjmując z blistra zestawu Mix2Vial nałożyć jego niebieską końcówkę z ostrzem na korek fiolki rozpuszczalnika i naciskając pionowo w dół przebić korek fiolki rozpuszczalnika. |
3 | 3. Przytrzymując krawędź zestawu Mix2Vial ostrożnie zdjąć blister pociągając go pionowo do góry. Należy zwrócić uwagę, aby zdjąć jedynie blister a nie cały zestaw Mix2Vial. |
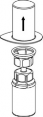
4 | 4. Umieścić fiolkę z proszkiem na równej i twardej powierzchni. Odwrócić do góry dnem fiolkę z rozpuszczalnikiem i dołączonym do niej zestawem Mix2Vial i wbić w korek fiolki z proszkiem, ruchem pionowo w dół, ostrze przezroczystego końca. Rozpuszczalnik samoczynnie zostanie przeniesiony do fiolki z proszkiem. |
5 | 5. Jedną ręką chwycić część zestawu Mix2Vial w fiolce zawierającej obecnie roztwór, drugą zaś ręką przytrzymać część łącznika od strony fiolki po rozpuszczalniku i ostrożnie odkręcając w przeciwnym kierunku do ruchu wskazówek zegara oddzielić od siebie obie części łącznika. Usunąć fiolkę po rozpuszczalniku z przyczepionym do niej niebieskim końcem zestawu Mix2Vial. |
6 | 6. Fiolkę z doczepionym przezroczystym końcem zestawu Mix2Vial, zawierającą roztwór, łagodnie mieszać ruchem wirowym do całkowitego rozpuszczenia leku. Nie wstrząsać. |
7 | 7. Nabrać powietrza do pustej, jałowej strzykawki. Stosować strzykawkę dołączoną do produktu. Trzymając fiolkę z produktem leczniczym pionowo korkiem do góry, przyłączyć strzykawkę do połączenia Luer Lock zestawu Mix2Vial przez przekręcenie zgodnie z ruchem wskazówek zegara. Wstrzyknąć powietrze do fiolki z produktem. |
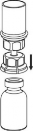
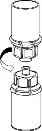

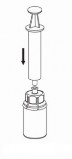

Pobieranie i sposób podawania
8. Przytrzymując tłok strzykawki odwrócić fiolkę wraz ze strzykawką do góry dnem i nabrać roztwór do strzykawki, powoli odciągając tłok. |
8 |
9. Po napełnieniu strzykawki roztworem, mocno uchwycić cylinder strzykawki (utrzymując strzykawkę tłokiem do dołu) odłączyć od niej przezroczysty koniec zestawu Mix2Vial poprzez odkręcenie w kierunku przeciwnym do ruchu wskazówek zegara. |
9 |
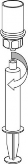
Podawanie
Produkt może być podawany za pomocą igły do wstrzykiwań podskórnych lub zestawu do wstrzyknięć podskórnych.
CSL Behring GmbH
Emil-von-Behring-Strasse 76
35041 Marburg
Niemcy
2000 j.m. - pozwolenie nr: 24613
3000 j.m. - pozwolenie nr: 24614
POZWOLENIA
2000 j.m. - Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 13.03.2018 r.
2000 j.m. - Data ostatniego przedłużenia pozwolenia: 13.02.2020 r.
3000 j.m. - Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:13.03.2018 r.
3000 j.m. - Data ostatniego przedłużenia pozwolenia: 13.02.2020 r.
12/2021







