







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
zanik nabłonka rogówki i ścieńczenie rogówki,
martwica brodawek nerkowych,
martwica komórek wątroby i nacieki eozynofilowe makrofagów w zatokach wątrobowych.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Gefitinib Synthon, 250 mg, tabletki powlekane
Jedna tabletka zawiera 250 mg gefitynibu. Substancje pomocnicze o znanym działaniu:
jedna tabletka zawiera 163,5 mg laktozy (w postaci laktozy jednowodnej).
jedna tabletka zawiera 1,9 mg sodu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana.
Brązowe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem „Gefitinib Synthon 250” na jednej stronie tabletki.
Produkt Gefitinib Synthon jest wskazany w monoterapii u dorosłych pacjentów z niedrobnokomórkowym rakiem płuca (NDRP), miejscowo zaawansowanym lub z przerzutami, z aktywującą mutacją EGFR-TK (patrz punkt 4.4).
Leczenie produktem Gefitinib Synthon powinno być rozpoczynane i prowadzone przez lekarza posiadającego doświadczenie w stosowaniu leczenia przeciwnowotworowego.
Dawkowanie
Zalecana dawka to 250 mg (jedna tabletka) raz na dobę. W przypadku pominięcia dawki produktu, powinna ona zostać przyjęta tak szybko jak pacjent sobie o tym przypomni. Jeśli czas do zastosowania następnej dawki jest krótszy niż 12 godzin, pacjent nie powinien zażywać pominiętej dawki. Pacjent nie powinien zażywać podwójnej dawki (w tym samym czasie) w celu uzupełnienia pominiętej dawki.
Dzieci i młodzież
Bezpieczeństwo i skuteczność stosowania produktu Gefitinib Synthon u dzieci i młodzieży w wieku poniżej 18 lat nie zostały ustalone. Gefitynib nie ma zastosowania u dzieci i młodzieży we wskazaniu NDRP.
Zaburzenia czynności wątroby
U pacjentów z umiarkowaną do ciężkiej niewydolnością wątroby (stopień B lub C w skali Child- Pugh) z powodu marskości wątroby występuje zwiększone stężenie gefitynibu w osoczu. Należy dokładnie monitorować tych pacjentów ze względu na możliwość wystąpienia działań niepożądanych. Stężenie nie było zwiększone u pacjentów ze zwiększoną aktywnością
aminotransferazy asparaginowej (AspAT), fosfatazy alkalicznej lub bilirubiny z powodu przerzutów w wątrobie (patrz punkt 5.2).
Zaburzenia czynności nerek
Nie ma konieczności zmiany dawki u pacjentów z zaburzeniami czynności nerek, jeśli wartość klirensu kreatyniny 20 ml/min. Dane dotyczące pacjentów z klirensem kreatyniny 20 ml/min są ograniczone i zaleca się zachowanie ostrożności u tych pacjentów.
Pacjenci w podeszłym wieku
Nie ma konieczności dostosowania dawki w zależności od wieku pacjenta (patrz punkt 5.2).
Osoby z genotypem wolnego metabolizmu CYP2D6
Nie ma konieczności dostosowania dawki u osób z genotypem odpowiedzialnym za wolny metabolizm CYP2D6, jednak należy ściśle monitorować działania niepożądane u tych pacjentów (patrz punkt 5.2).
Dostosowanie dawki w zależności od toksyczności
U pacjentów źle tolerujących działania niepożądane leku takie, jak biegunka lub reakcje skórne zaleca się krótką przerwę w leczeniu (do 14 dni) i ponowne rozpoczęcie leczenia produktem Gefitinib Synthon
w dawce 250 mg (patrz punkt 4.8). U pacjentów, którzy nie tolerują leczenia po przerwie w terapii, należy zaprzestać stosowania gefitynibu i rozważyć inny sposób leczenia.
Sposób podawania
Tabletka może być zażywana doustnie z posiłkiem lub bez posiłku, codziennie, mniej więcej o tej samej porze dnia. Tabletkę można połknąć w całości popijając wodą, lub jeśli podawanie całej tabletki nie jest możliwe, tabletka może być podana po rozpuszczeniu w wodzie (niegazowanej). Nie należy używać innych płynów. Tabletkę należy wrzucić do szklanki w połowie wypełnionej wodą, nie należy kruszyć tabletki. Można raz na jakiś czas zamieszać, aż tabletka się rozpuści (może to trwać do
20 minut). Zawiesinę należy wypić niezwłocznie po przygotowaniu (tj. w ciągu 60 minut). Szklankę należy ponownie napełnić do połowy wodą i wypić. Roztwór można podawać także przez sondę nosowo-żołądkową lub gastrostomię.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą, wymienioną w punkcie 6.1.
Karmienie piersią (patrz punkt 4.6).
Rozważając zastosowanie produktu Gefitinib Synthon w leczeniu chorych na miejscowo zaawansowanego lub rozsianego NDRP jest istotnym, aby u wszystkich pacjentów podjąć próby badania w celu wykrycia obecności mutacji EGFR w tkance nowotworowej. Jeżeli materiał z guza nowotwrowego
jest niezdatny do oceny, wtedy można wykorzystać krążący DNA nowotworu (ang. circulating tumour DNA, ctDNA) uzyskany z krwi (osocza).
W celu uniknięcia fałszywie ujemnych lub fałszywie dodatnich wyników oznaczeń do określenia statusu mutacji EGFR w guzie nowotworowym lub na podstawie badania ctDNA należy stosować wyłącznie solidny, wiarygodny i czuły test(y) o udowodnionej przydatności (patrz punkt 5.1).
Choroba śródmiąższowa płuc (ChŚP)
U 1,3% pacjentów otrzymujących gefitynib zaobserwowano wystąpienie śródmiąższowej choroby płuc. Początek choroby może być nagły, w niektórych przypadkach nastąpił zgon pacjenta (patrz punkt 4.8). W razie nasilenia się u pacjenta takich objawów jak duszność, kaszel i gorączka, należy przerwać leczenie produktem gefitynib i natychmiast rozpocząć postępowanie diagnostyczne. Jeśli
rozpoznano śródmiąższową chorobę płuc, należy przerwać stosowanie produktu gefitynib i zastosować odpowiednie leczenie.
W badaniu farmakoepidemiologicznym przeprowadzonym w Japonii, u 3159 pacjentów
z niedrobnokomórkowym rakiem płuca otrzymujących gefitynib lub chemioterapię, obserwowanych przez 12 tygodni, stwierdzono następujące czynniki ryzyka wystąpienia choroby śródmiąższowej płuc niezależnie od tego, czy pacjent otrzymywał gefitynib czy chemioterapię: palenie tytoniu, zły stan
ogólny (PS 2), potwierdzone w tomografii komputerowej (CT) zmniejszenie ilości prawidłowego miąższu płucnego ( 50%), niedługi czas od rozpoznania NDRP ( 6 miesięcy), istniejąca wcześniej choroba śródmiąższowa płuc, starszy wiek ( 55 lat) i współistniejąca choroba serca. Zwiększone ryzyko wystąpienia ChŚP po zastosowaniu gefitynibu, w porównaniu z chemioterapią, występowało głównie podczas pierwszych 4 tygodni leczenia (skorygowany OR 3,8; 95% CI 1,9 do 7,7),
w późniejszym okresie względne ryzyko było mniejsze (skorygowany OR 2,5; 95% CI 1,1 do 5,8). Ryzyko zgonu u pacjentów, leczonych produktem gefitynib lub chemioterapią, u których wystąpiła ChŚP, było większe w grupie z następującymi czynnikami ryzyka: palenie tytoniu, potwierdzone
w tomografii komputerowej (CT) zmniejszenie ilości prawidłowego miąższu płucnego ( 50%), istniejąca wcześniej choroba śródmiąższowa płuc, starszy wiek ( 65 lat), znaczne obszary przyrośnięte do opłucnej ( 50 %).
Hepatotoksyczność i zaburzenia czynności wątroby
Obserwowano nieprawidłowe wyniki badań diagnostycznych oceniających czynność wątroby (w tym zwiększenie aktywności aminotransferazy alaninowej, aminotransferazy asparaginowej, stężenia bilirubiny), niezbyt często zmiany występowały jako zapalenie wątroby (patrz punkt 4.8). Istnieją pojedyncze doniesienia dotyczące występowania niewydolności wątroby prowadzącej w niektórych przypadkach do zgonu. Dlatego zaleca się okresową kontrolę czynności wątroby. Gefitynib należy stosować ostrożnie u pacjentów ze stwierdzonymi zmianami czynności wątroby o niewielkim lub umiarkowanym nasileniu. W przypadku znacznego zwiększenia aktywności enzymów wątrobowych należy rozważyć zaprzestanie leczenia produktem Gefitinib Synthon.
Wykazano, że zaburzenia czynności wątroby z powodu marskości wątroby prowadzą do zwiększenia stężenia gefitynibu w osoczu (patrz punkt 5.2).
Interakcje z innymi lekami
Substancje zwiększające aktywność izoenzymu CYP3A4, mogą zwiększać metabolizm gefitynibu i zmniejszać jego stężenie w osoczu. Dlatego należy unikać jednoczesnego stosowania substancji zwiększających aktywność izoenzymu CYP3A4 (np. fenytoiny, karbamazepiny, ryfampicyny, barbituranów i produktów ziołowych zawierających ziele dziurawca Hypericum perforatum), ponieważ mogą zmniejszać skuteczność leczenia (patrz punkt 4.5).
U niektórych pacjentów z wolnym metabolizmem CYP2D6, leczenie silnym inhibitorem enzymu CYP3A4 może spowodować zwiększenie stężenia gefitynibu w osoczu. Podczas rozpoczynania leczenia inhibitorem enzymu CYP3A4, pacjenta należy ściśle monitorować pod kątem wystąpienia działań niepożądanych związanych z gefitynibem (patrz punkt 4.5).
U niektórych pacjentów przyjmujących jednocześnie warfarynę oraz gefitynib stwierdzano krwawienia i (lub) zwiększone wartości międzynarodowego współczynnika znormalizowanego (INR) (patrz punkt 4.5). U pacjentów otrzymujących jednocześnie warfarynę i gefitynib, należy systematycznie kontrolować wartości INR lub czasu protrombinowego (PT).
Leki powodujące znaczne i długotrwałe zwiększenie pH soku żołądkowego, takie jak inhibitory pompy protonowej i antagoniści receptora H2, mogą zmniejszać biodostępność i stężenie gefitynibu w osoczu, a tym samym zmniejszać jego skuteczność. Leki zobojętniające podawane regularnie o tej samej porze dnia co gefitynib mogą mieć podobne działanie (patrz punkty 4.5 i 5.2).
Wyniki badania klinicznego fazy II z jednoczesnym zastosowaniem gefitynibu i winorelbiny wskazują, że gefitynib może nasilać neutropenię występującą po zastosowaniu winorelbiny.
Laktoza
Gefitinib Synthon zawiera laktozę. Lek nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, niedoborem laktazy (typu Lapp) lub zespołem złego wchłaniania glukozy-galaktozy.
Dalsze środki ostrożności
Należy poinformować pacjenta o konieczności niezwłocznego skontaktowania się z lekarzem w razie wystąpienia ciężkiej lub utrzymującej się biegunki, nudności, wymiotów lub braku łaknienia, ze względu
na możliwość odwodnienia.
W razie wystąpienia powyższych objawów należy zastosować właściwe leczenie (patrz punkt 4.8). Jeżeli u pacjenta występują nagłe objawy takie jak: zapalenie oka, łzawienie, nadwrażliwość na światło, niewyraźne widzenie, ból i (lub) zaczerwienienie oka, lub jeśli objawy te nasilą się, mogą być to objawy
zapalenia rogówki. W takim przypadku należy niezwłocznie skontaktować się z okulistą.
W przypadku stwierdzenia u pacjenta wrzodziejącego zapalenia rogówki, należy wstrzymać podawanie gefitynibu, a jeśli objawy nie ustąpią lub pojawią się ponownie po wznowieniu leczenia tym lekiem, należy rozważyć zaprzestanie leczenia gefitynibem.
W badaniu fazy I/II z zastosowaniem gefitynibu i leczenia napromienianiem u dzieci z nowo rozpoznanym glejakiem pnia mózgu lub po niecałkowitym usunięciu złośliwego glejaka nadnamiotowego, w grupie 45 pacjentów włączonych do badania stwierdzono 4 przypadki krwawienia do ośrodkowego układu nerwowego (w jednym przypadku nastąpił zgon). Kolejny przypadek krwawienia do ośrodkowego układu nerwowego stwierdzono u dziecka z wyściółczakiem, które brało udział w badaniu z zastosowaniem samego gefitynibu. Nie stwierdzono, aby u dorosłych pacjentów
z NDRP otrzymujących gefitynib istniało zwiększone ryzyko krwawienia do ośrodkowego układu nerwowego.
U pacjentów stosujących gefitynib obserwowano perforacje przewodu pokarmowego. W większości przypadków jest to związane z innymi znanymi czynnikami ryzyka, takimi jak: jednoczesne stosowanie steroidów lub produktów z grupy NLPZ, owrzodzenie przewodu pokarmowego
w wywiadzie, wiek, palenie tytoniu lub przerzuty do jelita w miejscu perforacji.
Gefitynib jest metabolizowany przez izoenzym CYP3A4 (przede wszystkim) i izoenzym CYP2D6 układu cytochromu P450.
Substancje czynne, które mogą zwiększać stężenie gefitynibu w osoczu
Wyniki badań in vitro wykazały, że gefitynib jest substratem dla glikoproteiny-p (Pgp). Dostępne dane nie wskazują, aby obserwacje in vitro miały znaczenie kliniczne.
Substancje, które hamują CYP3A4 mogą zmniejszać klirens gefitynibu. Jednoczesne stosowanie
z silnymi inhibitorami aktywności CYP3A4 (np. ketokonazolem, pozakonazolem, worykonazolem, inhibitorami proteaz, klarytromycyną, telitromycyną) może zwiększać stężenie gefitynibu.
Zwiększenie to może mieć znaczenie kliniczne, ponieważ działania niepożądane są zależne od dawki i ekspozycji. Zwiększenie może być jeszcze większe u osób z genotypem odpowiedzialnym za wolny metabolizm przez izoenzym CYP2D6. Wcześniejsze leczenie itrakonazolem (silnym inhibitorem CYP3A4) powodowało u zdrowych ochotników zwiększenie średniej powierzchni pola pod krzywą AUC o 80 %. W sytuacji jednoczesnego leczenia silnymi inhibitorami CYP3A4 należy ściśle obserwować pacjentów ze względu na możliwość wystąpienia działań niepożądanych po gefitynibie.
Brak danych klinicznych dotyczących jednoczesnego stosowania z inhibitorami CYP2D6, jednak silne inhibitory tego izoenzymu mogą powodować około 2-krotne zwiększenie stężenia gefitynibu u osób
z szybkim metabolizmem CYP2D6 (patrz punkt 5.2). Jeśli rozpoczyna się jednoczesne leczenie silnym inhibitorem CYP2D6, należy ściśle obserwować pacjenta pod kątem możliwości wystąpienia działań niepożądanych.
Substancje czynne, które mogą zmniejszać stężenie gefitynibu w osoczu
Substancje, które indukują aktywność CYP3A4 mogą zwiększać metabolizm i zmniejszać stężenie gefitynibu w osoczu i w ten sposób zmniejszyć skuteczność gefitynibu. Należy unikać jednoczesnego stosowania produktów leczniczych indukujących CYP3A4 (np. fenytoiny, karbamazepiny, ryfampicyny, barbituranów lub ziela dziurawca (Hypericum perforatum). Wcześniejsze leczenie ryfampicyną (silnie indukującą CYP3A4) powodowało u zdrowych ochotników zmniejszenie średniej powierzchni pola pod krzywą AUC o 83% (patrz punkt 4.4).
Leki, które powodują znaczne, trwałe zwiększenie pH soku żołądkowego mogą zmniejszać stężenie gefitynibu i tym samym zmniejszać skuteczność gefitynibu. Duże dawki krótkodziałających leków
zobojętniających mogą mieć podobne działanie, jeśli są stosowane regularnie w porze zbliżonej do czasu zażywania gefitynibu. Jednoczesne stosowanie gefitynibu i ranitydyny w dawkach powodujących trwałe zwiększenie pH soku żołądkowego 5 u zdrowych ochotników powodowało zmniejszenie średniej powierzchni pola pod krzywą AUC dla gefitynibu o 47% (patrz punkty
4.4 i 5.2).
Substancje czynne, których stężenie w osoczu może być zmienione przez gefitynib
Wyniki badań in vitro wykazały, że gefitynib w niewielkim stopniu hamuje izoenzym CYP2D6.
W badaniu klinicznym pacjentom podawano jednocześnie gefitynib i metoprolol (lek metabolizowany przez izoenzym CYP2D6). Zaobserwowano niewielkie zwiększenie ekspozycji na metoprolol (35%). Takie zwiększenie może mieć znaczenie w przypadku substancji o wąskim indeksie terapeutycznym, będących substratem izoenzymu CYP2D6. Kiedy rozważa się zastosowanie substratów dla izoenzymu CYP2D6 jednocześnie z gefitynibem, należy wziąć pod uwagę dostosowanie dawki substratu dla CYP2D6 zwłaszcza dla produktów o wąskim indeksie terapeutycznym.
Gefitynib in vitro hamuje białko transportujące BCRP, jednak znaczenie kliniczne tego działania jest nieznane.
Inne możliwe interakcje
U niektórych pacjentów przyjmujących jednocześnie warfarynę stwierdzano krwawienia
i (lub) zwiększone wartości międzynarodowego współczynnika znormalizowanego (INR) (patrz punkt 4.4).
Płodność
Pacjentki w wieku rozrodczym powinny być poinformowane o konieczności stosowania skutecznych metod antykoncepcji w trakcie leczenia.
Ciąża
Brak danych dotyczących stosowania gefitynibu u kobiet w ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Zagrożenie dla człowieka nie jest znane. Produktu Gefitinib Synthon nie należy stosować w ciąży, jeśli nie jest to bezwzględnie konieczne.
Karmienie piersią
Nie wiadomo czy gefitynib przenika do mleka karmiących kobiet. Gefitynib i jego metabolity kumulowały się w mleku karmiących samic szczura (patrz punkt 5.3). Gefitynib jest przeciwwskazany w okresie karmienia piersią i dlatego karmienie powinno być przerwane podczas leczenia gefitynibem (patrz punkt 4.3)
Podczas stosowania gefitynibu obserwowano występowanie osłabienia. Pacjenci, u których występuje ten objaw, powinni zachować ostrożność podczas prowadzenia pojazdów mechanicznych lub obsługiwania urządzeń mechanicznych.
Podsumowanie profilu bezpieczeństwa
Z łącznych danych pochodzących z badań klinicznych III fazy ISEL, INTEREST i IPASS (2462 pacjentów leczonych gefitynibem) wynika, że najczęściej zgłaszanymi działaniami niepożądanymi, stwierdzanymi u ponad 20% pacjentów, są biegunka i objawy skórne (w tym wysypka, trądzik, suchość skóry i świąd). Działania niepożądane występują zazwyczaj podczas
pierwszego miesiąca leczenia i najczęściej są odwracalne. U około 8% pacjentów wystąpiły ciężkie działania niepożądane (3. lub 4. stopnia według Wspólnej Skali Toksyczności - Common Toxicity Criteria, CTC), a u 3% pacjentów przerwano leczenie z powodu działań niepożądanych.
U 1,3% pacjentów wystąpiła choroba śródmiąższowa płuc, często ciężka (3. do 4. stopnia CTC).
Stwierdzono przypadki zgonu z powodu śródmiąższowej choroby płuc.
Tabelaryczne zestawienie działań niepożądanych
Profil bezpieczeństwa przedstawiony w tabeli 1 opiera się na danych z zastosowania gefitynibu w badaniach klinicznych i po dopuszczeniu go do obrotu. Działania niepożądane przedstawione w tabeli 1 zostały w miarę możliwości podzielone według częstości występowania na podstawie
łącznej analizy zgłoszeń porównywalnych działań niepożądanych z badań ISEL, INTEREST i IPASS (badania III fazy, 2462 pacjentów leczonych gefitynibem).
Częstość występowania działań niepożądanych została określona następująco: występujące bardzo często 1/10), występujące często ( 1/100, 1/10), występujące niezbyt często ( 1/1000, 1/100), występujące rzadko ( 1/10 000, 1/1000), występujące bardzo rzadko ( 1/10000), częstość nieznana (nie może być określona na podstawie dostępnych danych).
W każdej grupie, działania niepożądane zostały przedstawione w kolejności malejącej ciężkości.
Tabela 1 Działania niepożądane
Działania niepożądane podzielone według układów i narządów oraz częstości występowania | ||
Zaburzenia metabolizmu i odżywiania | Bardzo często | Brak łaknienia, łagodny do umiarkowanego (1. lub 2. stopnia CTC). |
Zaburzenia oka | Często | Zapalenie spojówek, zapalenie powiek, suchość oka*, zwykle łagodne (1. stopnia CTC). |
Niezbyt często | Nadżerka rogówki, odwracalna i czasami współistniejąca z nieprawidłowym wzrostem rzęs. | |
Zapalenie rogówki (0,12%). | ||
Zaburzenia naczyniowe | Często | Krwawienia takie jak krwawienie z nosa i krwiomocz. |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Często | Śródmiąższowa choroba płuc (1,3%), często ciężka (3.-4. stopnia według CTC). Stwierdzano przypadki zgonu. |
Zaburzenia żołądka i jelit | Bardzo często | Biegunka, głównie łagodna i umiarkowana (1. lub 2. stopnia według CTC). |
Wymioty, głównie łagodne i umiarkowane (1. lub 2. stopnia według CTC). | ||
Nudności, głównie łagodne (1. stopnia według CTC). | ||
Zapalenie jamy ustnej, głównie łagodne (1. stopnia według CTC). | ||
Często | Odwodnienie w związku z biegunką, nudnościami, wymiotami lub brakiem łaknienia. | |
Suchość w jamie ustnej*, przeważnie łagodna (1. stopnia według CTC). | ||
Niezbyt często | Zapalenie trzustki. | |
Perforacje przewodu pokarmowego. | ||
Zaburzenia wątroby i dróg żółciowych | Bardzo często | Zwiększenie aktywności aminotransferazy alaninowej, głównie łagodne i umiarkowane. |
Często | Zwiększenie aktywności aminotransferazy asparaginowej, głównie łagodne do umiarkowanego. | |
Zwiększenie stężenia bilirubiny całkowitej, głównie łagodne do umiarkowanego. | ||
Niezbyt często | Zapalenie wątroby** | |
Zaburzenia skóry i tkanki podskórnej | Bardzo często | Reakcje skórne, głównie łagodne lub umiarkowane (1. lub 2. stopnia według CTC), wysypka krostkowa, czasami swędząca z suchą skórą, w tym również pęknięcia skórne, na podłożu rumienia. |
Często | Zmiany w obrębie paznokci | |
Łysienie | ||
Reakcje alergiczne (1,1%), w tym obrzęk naczynioworuchowy i pokrzywka | ||
Rzadko | Zmiany pęcherzowe, w tym: martwica toksyczno-rozpływna naskórka, zespół Stevensa-Johnsona i rumień wielopostaciowy | |
Zapalenie naczyń skórnych | ||
Zaburzenia nerek i dróg moczowych | Często | Bezobjawowe zwiększenie stężenia kreatyniny w badaniach laboratoryjnych |
Białkomocz | ||
Zapalenie pęcherza moczowego | ||
Rzadko | Krwotoczne zapalenie pęcherza moczowego | |
Zaburzenia ogólne i stany w miejscu podania | Bardzo często | Osłabienie, przeważnie łagodne (1. stopnia według CTC). |
Często | Gorączka |
Częstość działań niepożądanych, które dotyczą nieprawidłowych wyników badań laboratoryjnych odnosi się do pacjentów ze zmianą o 2 lub więcej stopni w skali CTC dla poszczególnych badań.
* To działanie niepożądane może występować łącznie z suchością w innych miejscach (głównie skóry) po zastosowaniu gefitynibu.
** W pojedynczych doniesieniach opisano niewydolność wątroby, prowadzącą w niektórych przypadkach do zgonu.
Choroba śródmiąższowa płuc (ChŚP ang. Interstitial Lung Disease ILD)
W badaniu INTEREST częstość występowania zdarzeń niepożądanych zaliczanych do ChŚP wynosiła 1,4% (10 pacjentów) w grupie gefitynibu versus 1,1% (8 pacjentów) w grupie docetakselu. W jednym przypadku działania niepożądanego zaliczanego do ChŚP u pacjenta otrzymującego gefitynib,
wystąpił zgon.
W całej populacji badania ISEL częstość występowania zdarzeń niepożądanych zaliczanych do ChŚP wynosiła około 1%. Większość zdarzeń zgłaszanych jako ChŚP wystąpiło u pacjentów pochodzących z Azji. Częstość występowania ChŚP u pacjentów pochodzących z Azji otrzymujących gefitynib
i placebo wynosiła odpowiednio 3% i 4%. W jednym przypadku działania niepożądanego zaliczanego do ChŚP, u pacjenta otrzymującego placebo, nastąpił zgon.
W badaniu oceniającym bezpieczeństwo, wykonanym po wprowadzeniu produktu do obrotu w Japonii, częstość występowania ChŚP w grupie 3350 pacjentów leczonych gefitynibem
wynosiła 5,8%. Odsetek przypadków działań niepożądanych zaliczonych do ChŚP, w których wystąpił zgon wynosił 38,6%.
W otwartym badanu klinicznym III fazy (IPASS) z udziałem 1217 pacjentów pochodzenia azjatyckiego, w którym porównywano gefitynib i chemioterapię skojarzoną - karboplatyna/paklitaksel w leczeniu pierwszego rzutu u wybranych pacjentów z zaawansowanym NDRP, częstość występowania zdarzeń zaliczonych do ChŚP wynosiła 2,6% w grupie leczonej gefitynibem i 1,4% w grupie leczonej karboplatyną/paklitakselem.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie ma specyficznego leczenia w przypadku przedawkowania gefitynibu. Jednak w badaniach klinicznych I fazy ograniczona liczba pacjentów otrzymywała produkt w dawce dobowej do 1000 mg. Obserwowano zwiększenie częstości i ciężkości nasilenia niektórych działań niepożądanych, głównie biegunki i wysypek skórnych. Działania niepożądane występujące w związku z przedawkowaniem produktu powinny być leczone objawowo, szczególnie w przypadku ciężkiej biegunki należy zapewnić leczenie objawowe. W jednym badaniu klinicznym określona liczba pacjentów była leczona co tydzień dawkami od 1500 mg do 3500 mg. W tym badaniu ekspozycja na produkt gefitynib nie zwiększała się wraz ze zwiększaniem dawki, działania niepożądane były przeważnie łagodne do umiarkowanych i odpowiadały znanemu profilowi bezpieczeństwa produktu gefitynib.
Grupa farmakoterapeutyczna: inne leki przeciwnowotworowe, inhibitory kinazy proteinowej; kod ATC: L01XE02
Mechanizm działania i aktywność farmakodynamiczna
Naskórkowy czynnik wzrostu (ang. Epidermal Growth Factor EGF) i jego receptor (ang Epidermal Growth Factor Receptor EGFR [HER1; ErbB1]) zostały zidentyfikowane jako główne czynniki odpowiedzialne za proces wzrostu i podziału w komórkach prawidłowych i nowotworowych.
Aktywująca mutacja EGFR w komórce nowotworowej jest ważnym czynnikiem pobudzającym wzrost komórki, blokującym apoptozę, zwiększającym produkcję czynników angiogenezy i ułatwiającym proces powstawania przerzutów.
Gefitynib jest wybiórczym, małocząsteczkowym inhibitorem kinazy tyrozynowej receptora dla
naskórkowego czynnika wzrostu i jest skutecznym sposobem leczenia u pacjentów z nowotworem
z obecnością mutacji aktywującej domenę kinazy tyrozynowej EGFR, niezależnie od rzutu leczenia. Nie stwierdzono istotnego klinicznie działania u pacjentów z potwierdzonym brakiem mutacji EGFR w guzie.
Istnieją solidne dane wykazujące wrażliwość na gefitynib w przypadku częstych mutacji aktywujących EGFR (delecje w egzonie 19; mutacja L858R); na przykład w porównaniu gefitynibu do schematu chemioterapii dwulekowej iloraz ryzyk dla czasu wolnego od progresji - HR (95% CI) – wyniósł 0,489 (0,336; 0,710) [WJTOG3405]. Dane dotyczące odpowiedzi na leczenie gefitynibem u chorych, u których w tkance nowotworowej stwierdzono mniej częste mutacje, są rzadsze; dostępne dane wskazują, że mutacje G719X, L861Q oraz S7681 są mutacjami związanymi z wrażliwością na leczenie gefitynibem; natomiast mutacja punktowa T790M lub insercje w egzonie 20 związane są
z mechanizmami oporności na gefitynib.
Oporność
W przypadku większości guzów w NDRP z uwrażliwiającymi mutacjami kinazy EGFR wytworzy się oporność na leczenie produktem gefitynib, z medianą czasu do progresji choroby wynoszącą 1 rok. W około 60% przypadków oporność jest związana z wtórną mutacją T790M, w której inhibitory kinazy tyrozynowej EGFR skierowane przeciwko T790M mogą być stosowane jako leczenie kolejnego rzutu. Inne, potencjalne mechanizmy oporności, zgłoszone w związku z leczeniem produktami leczniczymi blokującymi przewodzenie sygnału dla EGFR, obejmują: przewodzenie sygnału innym szlakiem, np. poprzez amplifikację genów HER2 oraz MET oraz mutacje genu PIK3CA. Zmiana fenotypu na drobnokomórkowy rak płuca była obserwowano w 5-10% przypadków.
Krążący DNA komórek nowotworowych (ctDNA)
W badaniu IFUM status mutacji był badany w materiale z guza nowotworowego oraz w próbkach ctDNA pozyskanych z osocza przy użyciu zestawu testowego Therascreen EGFR RGQ PCR (Qiagen). Spośród 1060 pacjentów poddanych badaniu - ocena zarówno ctDNA jak i próbek z guza nowotworowego możliwa była u 652 pacjentów. Odsetek odpowiedzi obiektywnych (ORR) u pacjentów, u których stwierdzono dodatni wynik analizy na obecność mutacji w guzie nowotworowym i w ocenie ctDNA wyniósł 77% (95% CI: 66% do 86%), a u tych, u których stwierdzono dodatni wynik na obecność mutacji wyłącznie w guzie nowotworowym -60% (95% CI: 44% do 74%).
Tabela 2 Zestawienie wyjściowego statusu mutacji w odniesieniu do analizy próbek z guza nowotworowego oraz ctDNA u wszystkich poddanych badaniu pacjentów, u których możliwa była ocena z obu próbek.
Miara | Definicja | Odsetek IFUM % (CI) | IFUM N |
Czułość | Odsetek dodatnich wyników analizy ctDNA (M+) w obrębie dodatnich wyników analizy materiału z guza (M+) | 65,7 (55,8; 74,7) | 105 |
Swoistość | Odsetek ujemnych wyników analizy ctDNA (M-) w obrębie ujemnych wyników analizy materiału z guza (M-) | 99,8 (99,0; 100,0) | 547 |
Dane te są spójne z wynikami zaplanowanej a priori, analizy zwiadowczej obejmującej podgrupę chorych z Japonii włączonych do badania IPASS (Goto 2012). W tym badaniu do analizy mutacji EGFR przy użyciu zestawu EGFR Mutation Test Kit (DxS) wykorzystano ctDNA pozyskany
z surowicy, a nie z osocza (N=86). W badaniu tym czułość wyniosła 43,1%, a swoistość
wyniosła 100%.
Bezpieczeństwo i skuteczność kliniczna
Leczenie pierwszego rzutu
Randomizowane badanie kliniczne III fazy, w leczeniu pierwszego rzutu, IPASS, przeprowadzono z udziałem pacjentów pochodzących z Azji1 z zaawansowanym (stadium IIIB lub IV) NDRP o utkaniu gruczołowym, którzy w przeszłości palili papierosy w niewielkiej ilości (rzucili palenie ≥ 15 lat temu i palili < 10 paczko-lat) lub nigdy nie palili (patrz tabela 3).
1Chiny, Hong Kong, Indonezja, Japonia, Malezja, Filipiny, Singapur, Taiwan i Tajlandia.
Tabela 3 Ocena skuteczności gefitynibu w porównaniu do leczenia karboplatyną/paklitakselem w badaniua IPASS
Populacja | N | Odsetek odpowiedzi obiektywnych i przedział ufności 95% dla różnicy pomiędzy sposobami a | Pierwszorzędowy punkt końcowy (PFS) Czas przeżycia wolny od progresjia,b | Ogólne przeżycie abc |
Ogólna | 1217 | 43,0 % vs 32,2 % [5,3 %, 16,1 %] | ||
HR 0,74 [0,65, 0,85] 5,7 m vs 5,8 m p<0,0001 | HR 0,90 [0,79, 1,02] 18,8 m vs 17,4 m p=0,1087 | |||
Z mutacją EGFR | 261 | 71,2% vs 47,3 % [12,0 %, 34,9 %] | HR 1,00 [0,76, 1,33] 21,6 m vs 21,9 m | |
HR 0,48 [0,36, 0,64] 9,5 m vs 6,3 m p<0,0001 | ||||
Bez mutacji EGFR | 176 | 1,1 % vs 23,5 % [- 32,5 %, -13,3 %] | HR 1,18 [0,86, 1,63] 11,2 m vs 12,7 m | |
HR 2,85 [2,05, 3,98] 1,5 m vs 5,5 m p<0,0001 | ||||
Mutacja EFGR nieznana | 780 | 43,3 % vs 29,2% [7,3 %, 20,6 %] | HR 0,82 [0,70 tot 0,96] 18,9 m vs 17,2 m | |
HR 0,68 [0,58 tot 0,81] 6,6 m vs 5,8 m p<0,0001 |
a Przedstawiono wartości dla produktu gefitynib vs karboplatyna/paklitaksel.
b Mediana w miesiącach „m”. Liczby w nawiasach kwadratowych to przedział ufności 95% dla współczynnika ryzyka.
N Liczba zrandomizowanych pacjentów
HR Współczynnik ryzyka (współczynnik ryzyka 1 oznacza, że produkt gefitynib jest lepszy)
Wyniki oceny jakości życia różniły się w zależności od statusu mutacji EGFR. W grupie pacjentów
z mutacją EGFR, leczonych produktem gefitynib, u znacząco większej grupy pacjentów poprawiła się jakość życia i objawy raka płuca w porównaniu do grupy leczonej karboplatyną/paklitakselem
(patrz tabela 4).
Tabela 4 Wyniki badania jakości życia dla gefitynibu w porównaniu do leczenia karboksyplatyną/paklitakselem w badaniu IPASS
Populacja | N | Odsetek popraw w skali FACT-L QoLa | Odsetek popraw objawów w skali LCSa |
Ogólna | 1151 | (48,0% vs 40,8%) p=0,0148 | |
(51,5 % vs 48,5 %) p=0,3037 | |||
Z mutacją EGFR | 259 | (70,2% vs 44,5%) p<0,0001 | |
(75,6 % vs 53,9 %) p=0,0003 | |||
Bez mutacji EGFR | 169 | (14,6% vs 36,3%) p=0,0021 | (20,2 % vs 47,5 %) p=0,0002 |
Wyniki mierzone w skali TOI (Trial Outcome Index) potwierdzały wyniki uzyskane w skali FACT-L i LCS
a Przedstawiono wartości dla produktu gefitynib vs. karboplatyna/paklitaksel.
N - Liczba pacjentów, u których możliwe było przeprowadzenie analizy jakości życia. QoL – Jakość życia.
FACT-L – Czynnościowa ocena leczenia przeciwnowotworowego – płuca (ang. Functional Assessment Cancer Therapy –Lung)
LCS – Podskala oceny raka płuca (ang. Lung Cancer Subscale)
W badaniu IPASS - u nieleczonych wcześniej pacjentów z NDRP miejscowo zaawansowanym lub z przerzutami, z obecnością aktywującej mutacji kinazy tyrozynowej EFGR – wykazano, że zastosowanie produktu gefitynib przynosi większe korzyści w zakresie wskaźników takich, jak PFS, ORR, QoL, czy szybkość ustępowania objawów choroby z nieistotną różnicą w zakresie przeżycia całkowitego w porównaniu z karboplatyną/paklitakselem.
Pacjenci uprzednio leczeni
Przeprowadzono randomizowane badanie kliniczne III fazy INTEREST, u pacjentów z miejscowo zaawansowanym lub z przerzutami NDRP, którzy otrzymywali wcześniej leczenie związkami platyny.
W ogólnej populacji nie stwierdzono znamiennej statystycznie różnicy pomiędzy gefitynibem i docetakselem (75 mg/m2), jeśli chodzi o przeżycie całkowite, czas przeżycia wolny od progresji i odsetek odpowiedzi obiektywnych (patrz tabela 5).
Tabela 5 Wyniki badania skuteczności gefitynibu w porównaniu do leczenia docetakselem w badaniu INTEREST
Populacja | N | Odsetek odpowiedzi obiektywnych i przedział ufności 95% dla różnicy pomiędzy sposobami leczeniaa | Czas przeżycia wolny odab progresji | Pierwszorzędowy punkt końcowy Ogólne przeżycieab |
Ogólna | 1466 | 9,1 % vs 7,6 % [-1,5 %; 4,5 %] | HR 1,04 [0,93; 1,18] 2,2 m vs 2,7 m p=0,4658 | HR 1,020 [0,905; 1,150]c 7,6 m vs 8,0 m p=0,7332 |
Z mutacją EGFR | 44 | 42,1 % vs 21,1 % [-8,2 %; 46,0 %] | HR 0,16 [0,05; 0,49] 7,0 m vs 4,1 m p=0,0012 | HR 0,83 [0,41; 1,67] 14,2 m vs 16,6 m p=0,6043 |
Bez mutacji EGFR | 253 | 6,6 % vs 9,8 % [-10,5 %; 4,4 %] | HR 1,24 [0,94; 1,64] 1,7 m vs 2,6 m p=0,1353 | HR 1,02 [0,78; 1,33] 6,4 m vs 6,0 m p=0,9131 |
Azjacic | 323 | 19,7 % vs 8,7 % [3,1 %; 19,2 %] | HR 0,83 [0,64; 1,08] | HR 1,04 [0,80; 1,35] |
2,9 m vs 2,8 m p=0,1746 | 10,4 m vs 12,2 m p=0,7711 | |||
Nie Azjaci | 1143 | 6,2 % vs 7,3 % | HR 1,12 | HR 1,01 |
[-4,3 %; 2,0 %] | [0,98; 1,28] | [0,89; | ||
2,0 m vs 2,7 m | 1,14] | |||
p=0,1041 | 6,9 m vs 6,9 m | |||
p=0,9259 |
a Przedstawiono wartości dla gefitynibu vs. docetaksel
b Mediana w miesiącach. Liczby w nawiasach kwadratowych to przedział ufności 96% dla ogólnego przeżycia,
HR w ogólnej populacji, lub jeśli nie to przedział ufności 95% dla HR
c przedział ufności całkowicie poniżej zakładanego limitu non-inferiority 1,154 N Liczba zrandomizowanych pacjentów
HR Współczynnik ryzyka (współczynnik ryzyka 1 oznacza, że gefitynib jest lepszy)
Rysunek 1 i 2 Wyniki badań skuteczności w podgrupach pacjentów nie-Azjatów w badaniu INTEREST (N=liczba pacjentów)
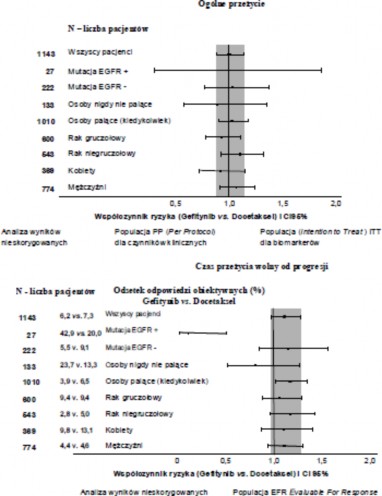
Randomizowane badanie kliniczne III fazy ISEL, było przeprowadzone u pacjentów
z zaawansowanym NDRP, którzy otrzymywali wcześniej 1 lub 2 kursy chemioterapii i nie tolerowali lub nie zareagowali na ostatni kurs leczenia. Gefitynib i najlepsze leczenie objawowe było porównywane z placebo i najlepszym leczeniem objawowym. W ogólnej populacji
gefitynib nie wydłużała przeżycia. Wyniki przeżycia były różne w zależności od palenia tytoniu i rasy
(patrz tabela 6).
Tabela 6 Wyniki skuteczności gefitynibu versus placebo w badaniu ISEL
Populacja | N | Odsetek odpowiedzi obiektywnych i przedział ufności 95% dla różnicy pomiędzy | Czas do niepowodzenia leczeniaab | Pierwszorzędowy punkt końcowy Ogólne przeżycieabc |
Ogólna | 1692 | 8,0% vs 1,3% [4,7%; 8,8%] | ||
HR 0,82 [0,73; 0,92] 3,0 m vs 2,6 m p=0,0006 | HR 0,89 [0,77; 1,02] 5,6 m vs 5,1 m p=0,0871 | |||
Obecna mutacja EGFR | 26 | 37,5% vs 0% [-15,1%; 61,4%] | HR NC NR vs 4,3 m | |
HR 0,79 [0,20; 3,12] 10,8 m vs 3,8 m p=0,7382 | ||||
Bez mutacji EGFR | 189 | 2,6% vs 0% [-5,6%; 7,3%] | ||
HR 1,10 [0,78; 1,56] 2,0 m vs 2,6 m p=0,5771 | HR 1,16 [0,79; 1,72] 3,7 m vs 5,9 m p=0,4449 | |||
Osoby nigdy nie paląc | 375 | 18,1% vs 0% [12,3%; 24,0 %] | ||
HR 0,55 [0,42; 0,72] 5,6 m vs 2,8 m p<0,0001 | HR 0,67 [0,49; 0,92] 8,9 m vs 6,1 m p=0,0124 | |||
Osoby palące (kiedykolwiek) | 1317 | 5,3 % vs 1,6% [1,4%; 5,7%] | ||
HR 0,89 [0,78; 1,01] 2,7 m vs 2,6 m p=0,0707 | HR 0,92 [0,79; 1,06] 5,0 m vs 4,9 m p=0,2420 | |||
Azjaci | 342 | 12,4% vs 2,1% [4,0%; 15,8%] | ||
HR 0,69 [0,52; 0,91] 4,4 m vs 2,2 m p=0,0084 | HR 0,66 [0,48; 0,91] 9,5 m vs 5,5 m p=0,0100 | |||
Nie-Azjaci | 1350 | 6,8% vs 1,0% [3,5%; 7,9%] | ||
HR 0,86 [0,76; 0,98] 2,9 m vs 2,7 m p=0,0197 | HR 0,92 [0,80; 1,07] 5,2 m vs 5,1 m p=0,2942 |
b
a Przedstawiona wartości dla gefitynibu versus placebo
Mediana jest podana w miesiącach. Liczby w nawiasach kwadratowych to przedział ufności 9c 5% dla HR
dla populacji ogólnej stratyfikowany test logarytmiczny rang, w innym wypadku model
proporcjonalnego ryzyka Coxa
d W grupie pacjentów pochodzenia azjatyckiego nie uwzględniono pacjentów pochodzących z Indii i odnoszonosię do rasy a nie do miejsca urodzenia
N - Liczba zrandomizowanych pacjentów
NC - współczynnik ryzyka ogólnego przeżycia nieliczony z powodu zbyt małej liczby zdarzeń NR – nie osiągnięto
HR Współczynnik ryzyka (współczynnik ryzyka 1 oznacza, że gefitynib jest lepszy)
Badanie IFUM było nieporównawczą, jednoramienną próbą wieloośrodkową przeprowadzoną na pacjentach rasy białej (n=106) z niedrobnokomórkowym rakiem płuca z aktywującą mutacją w genie dla EGFR. Celem badania było potwierdzenie, że skuteczność gefitynibu jest taka sama dla rasy białej i żółtej. Odsetek odpowiedzi obiektywnych (ORR) w ocenie badaczy wyniósł 70%, a mediana przeżycia bez progresji (PFS) 9,7 miesiąca. Wyniki te są podobne do uzyskanych w badaniu IPASS.
Status mutacji EGFR i charakterystyka kliniczna
W analizie badań klinicznych nad gefitynibem*, przeprowadzonej u 786 pacjentów rasy kaukaskiej, uwzględniającej różne czynniki, niezależnym czynnikiem klinicznym wskazującym na obecność mutacji EGFR okazał się być brak palenia tytoniu w wywiadzie, rak gruczołowy i płeć żeńska (patrz tabela 7). Guzy z mutacją EGFR częściej występują u pacjentów pochodzenia azjatyckiego.
Tabela 7 Podsumowanie wieloczynnikowej analizy regresji logistycznej w celu identyfikacji niezależnych czynników klinicznych wskazujących na obecność mutacji EGFR u 786 pacjentów
Czynniki wskazujące na obecność mutacji EGFR | Wartość p | Szansa mutacji EGFR | Wartość pozytywnie predestynująca (u 9,5% ogólnej populacji występuje mutacja EGFR (M+)) |
Palenie tytoniu | <0,0001 | 6,5 razy większa u osób, które nigdy nie paliły niż u osób, które kiedykolwiek paliły | u 28/70 (40%) osób nigdy nie palących M+ u 47/716 (7%) osób kiedykolwiek palących M+ |
Histologia | <0,0001 | 4,4 razy większa u osób z rakiem gruczołowym | u 63/396 (16%) pacjentów z rakiem gruczołowym występuje M+ u 12/390 (3%) pacjentów z rakiem niegruczołowym występuje M+ |
Płeć | 0,0397 | 1,7 razy większa u kobiet niż u mężczyzn | u 40/235 (17%) kobiet M+ u 35/551 (6%) mężczyzn M+ |
* badania kliniczne: INTEREST, ISEL, INTACT 1&2, IDEAL 1&2, INVITE
Wchłanianie
Po podaniu doustnym gefitynib jest umiarkowanie wolno wchłaniany, a maksymalne stężenie gefitynibu w osoczu występuje zazwyczaj po 3 do 7 godzinach od zażycia leku. Średnia bezwzględna biodostępność u pacjentów z nowotworem wynosi 59%. Pokarm nie wpływa istotnie na ekspozycję na gefitynib. W badaniu przeprowadzonym z udziałem zdrowych ochotników, gdy utrzymywano pH
soku żołądkowego powyżej 5, ekspozycja na gefitynib zmniejszyła się o 47%, prawdopodobnie ze względu na zaburzoną rozpuszczalność gefitynibu w żołądku (patrz punkt 4.4 i 4.5).
Dystrybucja
Średnia objętość dystrybucji gefitynibu w stanie stacjonarnym wynosi 1400 l, co wskazuje na znaczną dystrybucję do tkanek. Wiązanie z białkami osocza wynosi około 90%. Gefitynib wiąże się z albuminami osocza i kwaśną 1-glikoproteiną.
Dane z badań in vitro wskazują, że gefitynib jest substratem dla białka Pg-p odpowiedzialnego za transport przezbłonowy.
Metabolizm
Dane z badań in vitro wskazują, że izoenzym CYP3A4 i CYP2D6 są głównymi izoenzymami układu cytochromu P450 biorącymi udział w metabolizmie tlenowym gefitynibu.
Badania in vitro wykazały, że gefitynib ma ograniczony wpływ hamujący na izoenzym CYP2D6. W badaniach na zwierzętach gefitynib nie wykazuje działania pobudzającego enzymy ani istotnego działania hamującego (badania in vitro) na jakikolwiek inny enzym układu cytochromu P450.
U ludzi gefitynib jest intensywnie metabolizowany. W pełni określono pięć metabolitów znajdujących się w wydzielinach i 8 występujących w osoczu. Głównym zidentyfikowanym metabolitem jest
O-desmetylogefitynib. Ma on 14-krotnie słabsze działanie hamujące wzrost komórki powodowany przez pobudzenie EGFR, jak również nie wykazuje działania hamującego wzrost komórek guza
u myszy. Dlatego wydaje się, że nie bierze on udziału w istotnym klinicznie działaniu gefitynibu.
W badaniach in vitro wykazano, że O-desmetylogefitynib powstaje z udziałem CYP2D6. Rola CYP2D6 w przemianach metabolicznych gefitynibu była określona w badaniu klinicznym z udziałem zdrowych ochotników z określonym genotypem enzymu CYP2D6. U osób wolno metabolizujących nie stwierdzono wykrywalnych stężeń O-desmetylogefitynibu. Zakresy ekspozycji stwierdzane u osób wolno i szybko metabolizujących były szerokie i częściowo pokrywały się, jednak średnia ekspozycja na gefitynib była 2-krotnie większa u osób wolno metabolizujących. Większa średnia ekspozycja
u osób z nieaktywnym CYP2D6 może mieć znaczenie kliniczne, ponieważ działania niepożądane są zależne od dawki i ekspozycji.
Wydalanie
Gefitynib jest wydalany głównie w postaci zmetabolizowanej z kałem, mniej niż 4% podanej dawki jest wydalane przez nerki w postaci gefitynibu i jego metabolitów.
Całkowity klirens osoczowy gefitynibu wynosi mniej więcej 500 ml/min, a średni okres półtrwania u pacjentów z nowotworem wynosi 41 godzin. Podawanie gefitynibu raz na dobę powodowało 2- do 8-krotną kumulację, a typowa dla stanu stacjonarnego ekspozycja została osiągnięta po podaniu 7 do 10 dawek. W stanie stacjonarnym, stężenie w osoczu jest 2 do 3- krotnie większe, przez cały okres 24 godzin między kolejnymi dawkami.
Specjalne grupy pacjentów
W analizach uwzględniających dane farmakokinetyczne populacji pacjentów z nowotworem nie stwierdzono zależności między przewidywanym minimalnym stężeniem leku w stanie stacjonarnym a wiekiem, masą ciała, płcią, rasą lub wartością klirensu kreatyniny (powyżej 20 ml/min).
Zaburzenia czynności wątroby
W otwartym badaniu I fazy przeprowadzonym z udziałem pacjentów z łagodną, umiarkowaną i ciężką niewydolnością wątroby z powodu marskości (zgodnie z klasyfikacją Child-Pugh), po podaniu pojedynczej dawki gefitynibu 250 mg obserwowano zwiększenie ekspozycji we wszystkich grupach w porównaniu z ekspozycją u osób zdrowych. U osób z umiarkowaną i ciężką niewydolnością wątroby obserwowano średnio 3,1–krotne zwiększenie ekspozycji na gefitynib. U żadnego z pacjentów nie występował nowotwór, u wszystkich stwierdzano marskość, a u niektórych zapalenie wątroby. Zwiększenie ekspozycji może mieć znaczenie kliniczne, ponieważ działania niepożądane są zależne od dawki i ekspozycji na gefitynib.
Gefitynib był badany w grupie 41 pacjentów z litymi guzami nowotworowymi, którzy mieli prawidłową czynność wątroby albo umiarkowane lub ciężkie zaburzenia czynności wątroby (sklasyfikowane zgodnie z Common Toxicity Criteria, w oparciu o stwierdzone na początku badania wartości AspAT, fosfatazy alkalicznej i bilirubiny) z powodu przerzutów do wątroby. Wykazano, że gdy podawano gefitynib w dawce 250 mg na dobę, czas do osiągnięcia stanu stacjonarnego, całkowity klirens osoczowy (Cmaxss), ekspozycja na lek w stanie stacjonarnym (AUC24ss), były porównywalne w przypadku pacjentów z prawidłową czynnością wątroby i umiarkowanymi zaburzeniami czynności wątroby. Dane uzyskane od 4 pacjentów z ciężkimi
zaburzeniami czynności wątroby z powodu przerzutów sugerują, że ekspozycja w stanie stacjonarnym u tych pacjentów jest zbliżona do ekspozycji u osób z prawidłową czynnością wątroby.
Działania niepożądane, które nie występowały w badaniach klinicznych, natomiast były obserwowane u zwierząt po ekspozycji porównywalnej do ekspozycji klinicznej, i które mogą mieć znaczenie w praktyce klinicznej:
Dane z badań nieklinicznych (in vitro) wskazują, że gefitynib ma potencjalne działanie hamujące potencjału czynnościowego procesu repolaryzacji mięśnia serca (np. odcinek QT). Doświadczenie kliniczne nie wykazuje związku przyczynowego między wydłużeniem odstępu QT a gefitynibem.
U szczurów po dawce 20 mg/kg mc. na dobę obserwowano zmniejszenie płodności samic.
Wyniki opublikowanych badań wskazują, że genetycznie modyfikowane myszy, u których brak jest ekspresji EGFR, wykazują zaburzenia rozwojowe związane z niedojrzałością nabłonka w wielu narządach, w tym skórze, przewodzie pokarmowym i płucach. Gefitynib podawany szczurom
w okresie organogenezy w największej dawce (30 mg/kg mc. na dobę) nie wpływał na rozwój płodów, jednak u królików otrzymujących dawki 20 mg/kg na dobę i większe stwierdzono zmniejszoną masę ciała płodów. U żadnego z tych gatunków nie obserwowano występowania wad rozwojowych wywołanych przez podawany lek. Stosowany u szczurów w dawce 20 mg/kg na dobę w okresie ciąży i podczas porodu zmniejsza przeżycie nowo narodzonych szczurów.
Po doustnym podaniu, 14 dni po porodzie, karmiącym samicom szczura, znakowanego C-14 gefitynibu stwierdzano, że radioaktywność mleka była 11-19 razy większa niż krwi.
Gefitynib nie wykazuje działania genotoksycznego.
W trwającym dwa lata badaniu nad potencjalnym działaniem rakotwórczym przeprowadzonym na szczurach obserwowano małe, ale statystycznie znamienne zwiększenie częstości występowania gruczolaka wątroby u szczurów obu płci oraz naczyniakomięsaka krwionośnego węzłów krezki
u samic szczura tylko po największej zastosowanej dawce (10 mg/kg mc. na dobę). W trwającym dwa lata badaniu rakotwórczości u myszy obserwowano także występowanie gruczolaka wątroby, stwierdzono niewielkie zwiększenie częstości jego występowania u samców myszy po zastosowaniu średniej dawki i u myszy obu płci po zastosowaniu największej dawki. Działanie to osiągnęło poziom istotności statystycznej w grupie samic myszy, natomiast w grupie samców nie osiągnęło znamienności statystycznej. Dawki niewywołujące zmian u szczurów ani myszy były poza zakresem ekspozycji klinicznej.
Znaczenie kliniczne tych działań nie jest znane.
Wyniki przeprowadzonego in vitro badania nad fototoksycznością wskazują, że gefitynib może mieć działanie fototoksyczne.
Tabletka
Sodu laurylosiarczan Laktoza jednowodna Celuloza mikrokrystaliczna Powidon K-29/32 Kroskarmeloza sodowa Magnezu stearynian
Otoczka tabletki Alkohol poliwinylowy Makrogol 4000
Talk
Żelaza tlenek czerwony (E172) Żelaza tlenek żółty (E172) Żelaza tlenek czarny (E172)
Nie dotyczy.
3 lata
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
OPA/Aluminium/PVC/Aluminium blister z perforacją lub blister bez perforacji. Opakowanie zawiera 30 tabletek lub 30 x 1 tabletek.
Nie wszystkie rodzaje opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Synthon BV Microweg 22
6545 CM Nijmegen Holandia
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:








