







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
Aprepitant Stada, 125 mg, kapsułki, twarde Aprepitant Stada, 80 mg, kapsułki, twarde Aprepitant Stada, 125 mg/80 mg, kapsułki, twarde
Każda kapsułka 125 mg zawiera 125 mg aprepitantu. Każda kapsułka 80 mg zawiera 80 mg aprepitantu.
Substancje pomocnicze o znanym działaniu
Każda kapsułka 125 mg zawiera 125 mg sacharozy i 0,00026 mmol (0,006 mg) sodu. Każda kapsułka 80 mg zawiera 80 mg sacharozy i 0,00022 mmol (0,005 mg) sodu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Kapsułka, twarda (kapsułka).
Kapsułki twarde 125 mg mają postać nieprzezroczystych, twardych kapsułek żelatynowych rozmiaru 1, z różowym wieczkiem i białym korpusem, z nadrukiem czarnym tuszem na korpusie „125mg”.
Kapsułki twarde 80 mg mają postać nieprzezroczystych, twardych kapsułek żelatynowych rozmiaru 2, z białym wieczkiem i białym korpusem, z nadrukiem czarnym tuszem na korpusie „80mg”.
Zapobieganie nudnościom i wymiotom związanym z przeciwnowotworową chemioterapią o wysokim i umiarkowanym ryzyku wymiotów u osób dorosłych i młodzieży w wieku od 12 lat.
Aprepitant Stada 125 mg/80 mg podaje się jako składnik leczenia skojarzonego (patrz punkt 4.2).
Dawkowanie
Dorośli
Aprepitant Stada podaje się przez 3 dni jako składnik schematu, który obejmuje kortykosteroid i antagonistę receptora 5-HT3.
Zalecana dawka to 125 mg doustnie raz na dobę, godzinę przed rozpoczęciem chemioterapii w dniu 1. i 80 mg doustnie raz na dobę w dniach 2. i 3. rano.
W zapobieganiu nudnościom i wymiotom związanym z emetogenną chemioterapią przeciwnowotworową u osób dorosłych zaleca się następujące schematy:
Schemat chemioterapii o wysokiej emetogenności
Dzień 1 | Dzień 2 | Dzień 3 | Dzień 4 | |
Aprepitant Stada | 125 mg doustnie | 80 mg doustnie | 80 mg doustnie | Brak |
Deksametazon | 12 mg doustnie | 8 mg doustnie | 8 mg doustnie | 8 mg doustnie |
Antagoniści | Standardowe | Brak | Brak | Brak |
receptorów 5-HT3 | dawki | |||
antagonistów | ||||
receptorów 5-HT3. | ||||
Należy zapoznać | ||||
się z zaleceniami | ||||
dotyczącymi | ||||
prawidłowego | ||||
dawkowania | ||||
podanymi w | ||||
drukach | ||||
informacyjnych | ||||
wybranego | ||||
antagonisty | ||||
receptora 5-HT3. |
Deksametazon należy podać 30 minut przed chemioterapią w dniu 1 oraz
rano w dniach 2. do 4. Dawka deksametazonu uwzględnia interakcje pomiędzy substancjami czynnymi.
Schemat chemioterapii o umiarkowanej emetogenności
Dzień 1 | Dzień 2 | Dzień 3 | |
Aprepitant Stada | 125 mg doustnie | 80 mg doustnie | 80 mg doustnie |
Deksametazon | 12 mg doustnie | Brak | Brak |
Antagoniści receptorów 5-HT3 | Standardowe dawki antagonistów receptorów 5-HT3. Należy zapoznać się z zaleceniami dotyczącymi prawidłowego dawkowania podanymi w drukach informacyjnych wybranego antagonisty receptora 5-HT3. | Brak | Brak |
Deksametazon należy podać 30 minut przed chemioterapią w dniu 1. Dawka deksametazonu uwzględnia interakcje pomiędzy substancjami czynnymi.
Dzieci i młodzież
Młodzież (w wieku od 12 do 17 lat włącznie)
Aprepitant Stada podaje się przez 3 dni w ramach schematu obejmującego antagonistę receptora 5- HT3. Zalecana dawka produktu leczniczego Aprepitant Stada w kapsułkach wynosi 125 mg doustnie w dniu 1. i 80 mg doustnie w dniach 2. i 3. Aprepitant Stada podaje się doustnie 1 godzinę przed rozpoczęciem chemioterapii w 1., 2. i 3. dniu. Jeśli w 2. i 3. dniu nie jest podawana chemioterapia, produkt leczniczy Aprepitant Stada podaje się rano. Patrz charakterystyka produktu leczniczego (ChPL) danego antagonisty receptora 5-HT3 w celu uzyskania informacji na temat dawkowania. Jeśli w skojarzeniu z produktem leczniczym Aprepitant Stada podawany jest kortykosteroid taki jak deksametazon, dawka kortykosteroidu powinna stanowić 50% zwykle stosowanej dawki (patrz punkty 4.5 i 5.1).
Nie wykazano bezpieczeństwa i skuteczności produktu leczniczego w kapsułkach 80 mg i 125 mg u dzieci w wieku poniżej 12 lat. Nie ma dostępnych danych.
Dane ogólne
Istnieją ograniczone dane dotyczące skuteczności produktu leczniczego Aprepitant Stada podczas stosowania w skojarzeniu z innymi kortykosteroidami i antagonistami receptora 5-HT3. Dodatkowe informacje dotyczące stosowania w skojarzeniu z kortykosteroidami znajdują się w punkcie 4.5.
Należy zapoznać się z ChPL stosowanych jednocześnie produktów leczniczych z grupy antagonistów receptora 5-HT3.
Szczególne grupy pacjentów
Pacjenci w podeszłym wieku (≥ 65 lat)
U osób w podeszłym wieku nie jest konieczne dostosowanie dawki (patrz punkt 5.2).
Płeć
Nie jest konieczne dostosowywanie dawki produktu leczniczego w zależności od płci pacjenta (patrz punkt 5.2).
Zaburzenia czynności nerek
Nie jest konieczna modyfikacja dawki u pacjentów z zaburzeniami czynności nerek lub u poddawanych hemodializie pacjentów ze schyłkową niewydolnością nerek (patrz punkt 5.2).
Zaburzenia czynności wątroby
U pacjentów z łagodnymi zaburzeniami czynności wątroby nie jest konieczna modyfikacja dawki. Istnieją ograniczone dane dotyczące pacjentów z umiarkowanymi zaburzeniami czynności wątroby, natomiast nie ma danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby.
Należy zachować ostrożność podczas stosowania aprepitantu w tej grupie pacjentów (patrz punkty 4.4 i 5.2).
Sposób podawania Podanie doustne
Kapsułki twarde należy połykać w całości.
Produkt leczniczy Aprepitant Stada można przyjmować niezależnie od posiłku.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Jednoczesne podawanie z pimozydem, terfenadyną, astemizolem lub cyzaprydem (patrz punkt 4.5).
Pacjenci z zaburzeniami czynności wątroby o nasileniu umiarkowanym do ciężkiego
Istnieją ograniczone dane dotyczące pacjentów z umiarkowanymi zaburzeniami czynności wątroby, natomiast nie ma danych dotyczących pacjentów z ciężkimi zaburzeniami czynności wątroby. Należy zachować ostrożność stosując Aprepitant Stada w tej grupie pacjentów (patrz punkt 5.2).
Interakcje związane z CYP3A4
Aprepitant Stada należy stosować ostrożnie u pacjentów przyjmujących jednocześnie doustne substancje czynne metabolizowane głównie przez cytochrom CYP3A4 oraz o wąskim zakresie terapeutycznym, takie jak cyklosporyna, takrolimus, syrolimus, ewerolimus, alfentanyl, pochodne alkaloidów sporyszu, fentanyl oraz chinidyna (patrz punkt 4.5). Ponadto należy zachować szczególną ostrożność podczas stosowania produktu leczniczego Aprepitant Stada w skojarzeniu z irynotekanem, ponieważ skojarzenie może prowadzić do nasilenia działania toksycznego.
Jednoczesne stosowanie z warfaryną (substrat CYP2C9)
U pacjentów przyjmujących przewlekle warfarynę należy ściśle monitorować wartość międzynarodowego współczynnika znormalizowanego (INR, ang. International Normalised Ratio) w okresie leczenia produktem leczniczym Aprepitant Stada i przez 14 dni po każdorazowej 3-dniowej terapii produktem leczniczym Aprepitant Stada (patrz punkt 4.5).
Jednoczesne stosowanie z hormonalnymi środkami antykoncepcyjnymi
Podczas stosowania i w ciągu 28 dni po podaniu produktu leczniczego Aprepitant Stada może dojść do obniżenia skuteczności hormonalnych środków antykoncepcyjnych. W trakcie leczenia produktem leczniczym Aprepitant Stada oraz przez 2 miesiące po przyjęciu ostatniej dawki produktu leczniczego Aprepitant Stada należy dodatkowo stosować alternatywne niehormonalne metody antykoncepcji (patrz punkt 4.5).
Substancje pomocnicze
Kapsułki produktu leczniczego Aprepitant Stada zawierają sacharozę. Tego produktu leczniczego nie należy stosować u pacjentów z rzadko występującą dziedziczną nietolerancją fruktozy, zespołem złego wchłaniania glukozy-galaktozy lub niedoborem sacharozy-izomaltazy.
Kapsułki produktu leczniczego Aprepitant Stada zawierają sód. Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na kapsułkę, to znaczy uznaje się go za „wolny od sodu".
Aprepitant (125 mg/80 mg) jest substratem, umiarkowanym inhibitorem oraz induktorem cytochromu CYP3A4. Aprepitant jest także induktorem cytochromu CYP2C9. Podczas leczenia produktem leczniczym Aprepitant Stada następuje zahamowanie cytochromu CYP3A4. Po zakończeniu leczenia Aprepitant Stada powoduje przemijające, łagodne pobudzenie aktywności cytochromów CYP2C9, CYP3A4 oraz procesu glukuronidacji. Wydaje się, że aprepitant nie wchodzi w interakcje z białkiem transportującym P-glikoproteiną, ponieważ nie stwierdzono interakcji aprepitantu z digoksyną.
Wpływ aprepitantu na farmakokinetykę innych substancji czynnych
Hamowanie cytochromu CYP3A4
Jako umiarkowany inhibitor cytochromu CYP3A4 aprepitant (125 mg/80 mg) może zwiększać w osoczu stężenia jednocześnie stosowanych substancji czynnych metabolizowanych przez cytochrom CYP3A4. Całkowita ekspozycja na podawane doustnie substraty cytochromu CYP3A4 może wzrosnąć nawet około 3-krotnie podczas 3-dniowego leczenia produktem leczniczym Aprepitant Stada; spodziewany jest mniejszy wpływ aprepitantu na stężenia substratów cytochromu CYP3A4 podawanych dożylnie. Produktu leczniczego Aprepitant Stada nie wolno stosować jednocześnie z pimozydem, terfenadyną, astemizolem lub cyzaprydem (patrz punkt 4.3). Hamowanie cytochromu CYP3A4 przez aprepitant może spowodować wzrost stężenia wymienionych substancji czynnych w osoczu, co może powodować poważne lub zagrażające życiu reakcje. Należy zachować ostrożność podczas jednoczesnego stosowania produktu leczniczego Aprepitant Stada i doustnych substancji czynnych metabolizowanych głównie przez cytochrom CYP3A4 oraz o wąskim indeksie terapeutycznym, takich jak cyklosporyna, takrolimus, syrolimus, ewerolimus, alfentanyl, diergotamina, ergotamina, fentanyl oraz chinidyna (patrz punkt 4.4).
Kortykosteroidy
Deksametazon: Podczas jednoczesnego stosowania produktu leczniczego Aprepitant Stada w schemacie 125 mg/80 mg, zwykle stosowaną, podawaną doustnie dawkę deksametazonu należy zmniejszyć o około 50%. Dawkę deksametazonu podawaną w badaniach klinicznych dotyczących nudności i wymiotów wywołanych przez chemioterapię wybrano z uwzględnieniem interakcji substancji czynnych (patrz punkt 4.2). Aprepitant podawany zgodnie ze schematem w dawce 125 mg w skojarzeniu z 20 mg deksametazonu doustnie w 1. dniu oraz podawany w dawce 80 mg na dobę, w skojarzeniu z 8 mg deksametazonu doustnie w dniach od 2. do 5., zwiększał AUC deksametazonu, który jest substratem cytochromu CYP3A4, 2,2-krotnie w 1. i 5. dniu.
Metyloprednizolon: Podczas jednoczesnego stosowania produktu leczniczego Aprepitant Stada w schemacie 125 mg/80 mg, zwykle stosowaną, podawaną dożylnie dawkę metyloprednizolonu należy zmniejszyć o około 25%, a zwykle stosowaną, podawaną doustnie dawkę metyloprednizolonu należy zmniejszyć o około 50%. Aprepitant podawany zgodnie ze schematem w dawce 125 mg, w skojarzeniu ze 125 mg metyloprednizolonu dożylnie w 1. dniu oraz podawany w dawce 80 mg na dobę, w skojarzeniu z 40 mg metyloprednizolonu doustnie w 2. i 3. dniu, zwiększał wartość AUC metyloprednizolonu, który jest substratem cytochromu CYP3A4, 1,3-krotnie w 1. dniu i 2,5-krotnie w
3. dniu.
Podczas długotrwałego stosowania metyloprednizolonu, jego AUC może zmniejszyć się w późniejszym okresie, w ciągu 2 tygodni po przyjęciu pierwszej dawki aprepitantu, ze względu na indukcję cytochromu CYP3A4 przez aprepitant. Można się spodziewać, że działanie to będzie silniejsze podczas stosowania metyloprednizolonu doustnie.
Chemioterapeutyczne produkty lecznicze
W badaniach farmakokinetycznych aprepitant podawany według schematu: w 1. dniu w dawce 125 mg, a w 2. i 3. dniu w dawce 80 mg na dobę, nie wpływał na farmakokinetykę docetakselu podawanego dożylnie w 1. dniu lub winorelbiny podawanej dożylnie w 1. lub 8. dniu. Ponieważ wpływ aprepitantu na farmakokinetykę substratów cytochromu CYP3A4 podawanych doustnie jest większy niż wpływ aprepitantu na farmakokinetykę substratów cytochromu CYP3A4 podawanych dożylnie, nie można wykluczyć interakcji z podawanymi doustnie cytostatykami metabolizowanymi głównie lub częściowo przez cytochrom CYP3A4 (np. etopozyd, winorelbina). Należy zachować ostrożność i dodatkowo monitorować pacjentów otrzymujących produkty lecznicze metabolizowane głównie lub częściowo przez cytochrom CYP3A4 (patrz punkt 4.4). Po wprowadzeniu produktu leczniczego do obrotu obserwowano występowanie objawów neurotoksyczności, które mogły być działaniem niepożądanym ifosfamidu, podczas jednoczesnego stosowania aprepitantu i ifosfamidu.
Immunosupresyjne produkty lecznicze
Podczas leczenia nudności i wymiotów wywołanych chemioterapią (ang. CINV, chemotherapy- induced nausea and vomiting) w schemacie 3-dniowym może dojść do przejściowego umiarkowanego zwiększenia, po którym następuje łagodny spadek ekspozycji na immunosupresyjne produkty lecznicze metabolizowane przez cytochrom CYP3A4 (np. cyklosporynę, takrolimus, ewerolimus i syrolimus). Biorąc pod uwagę krótki czas trwania 3-dniowego schematu i zależne od czasu ograniczone zmiany w ekspozycji, nie zaleca się zmniejszenia dawki immunosupresyjnych produktów leczniczych podczas 3 dni ich jednoczesnego stosowania z produktem leczniczym Aprepitant Stada.
Midazolam
Podczas jednoczesnego stosowania produktu leczniczego Aprepitant Stada (125 mg/80 mg) i midazolamu lub innych benzodiazepin metabolizowanych przez cytochrom CYP3A4 (alprazolam, triazolam) należy brać pod uwagę możliwość zwiększenia stężenia benzodiazepin w osoczu.
Aprepitant zwiększał AUC midazolamu, wrażliwego substratu cytochromu CYP3A4, 2,3-krotnie w 1. dniu oraz 3,3-krotnie w 5. dniu po jednoczesnym podaniu pojedynczej dawki doustnej 2 mg midazolamu w 1. i 5. dniu leczenia aprepitantem w dawce 125 mg w 1. dniu oraz 80 mg na dobę w dniach od 2. do 5.
W innym badaniu, w którym midazolam podawano dożylnie, aprepitant stosowano w dawce 125 mg w 1. dniu oraz 80 mg na dobę w 2. i 3. dniu. Midazolam podawano dożylnie w dawce 2 mg przed rozpoczęciem 3-dniowego schematu leczenia aprepitantem oraz w 4., 8. i 15. dniu. Aprepitant zwiększał AUC midazolamu o 25% w 4. dniu oraz zmniejszał AUC midazolamu o 19% w 8. dniu i o 4% w 15. dniu. Tych efektów nie uznano za klinicznie istotne.
W trzecim badaniu, w którym midazolam podawano dożylnie i doustnie, aprepitant stosowano w dawce 125 mg w 1. dniu oraz 80 mg na dobę w 2. i 3. dniu, razem z ondansetronem w dawce 32 mg w
1. dniu i deksametazonem w dawce 12 mg w 1. dniu oraz 8 mg w dniach od 2. do 4. Ta kombinacja (tj. aprepitant, ondansetron i deksametazon) zmniejszała wartość AUC midazolamu podanego
doustnie o 16% w 6. dniu, o 9% w 8. dniu, o 7% w 15. dniu oraz o 17% w 22. dniu. Tych efektów nie uznano za klinicznie istotne.
Zakończono dodatkowe badanie dotyczące dożylnego podawania midazolamu i aprepitantu. Podawano dożylnie 2 mg midazolamu 1 godzinę po podaniu doustnym aprepitantu w pojedynczej dawce 125 mg. AUC midazolamu w osoczu zwiększyło się 1,5-krotnie. Wpływ ten nie został uznany za klinicznie istotny.
Indukcja
Jako łagodny induktor cytochromów CYP2C9, CYP3A4 i procesu glukuronidacji, aprepitant może obniżać osoczowe stężenia substratów usuwanych tymi drogami w ciągu dwóch tygodni od rozpoczęcia leczenia. Działanie to może się uwidocznić dopiero po zakończeniu 3-dniowego leczenia produktem leczniczym Aprepitant Stada. Dla substratów cytochromów CYP2C9 i CYP3A4 pobudzenie aktywności jest przemijające, a maksymalny efekt osiągany jest w 3-5 dni po zakończeniu 3-dniowego leczenia produktem leczniczym Aprepitant Stada. Efekt ten utrzymuje się przez kilka dni, następnie powoli zmniejsza się, a do dwóch tygodni od zakończenia leczenia produktem leczniczym Aprepitant Stada nie jest już istotny klinicznie. Podczas stosowania aprepitantu w dawce 80 mg przez 7 dni doustnie zauważa się również łagodne pobudzenie glukuronidacji. Nie ma danych dotyczących wpływu na cytochromy CYP2C8 i CYP2C19. Należy zachować ostrożność podczas stosowania w tym czasie warfaryny, acenokumarolu, tolbutamidu, fenytoiny lub innych substancji czynnych, o których wiadomo, że są metabolizowane przez cytochrom CYP2C9.
Warfaryna
U pacjentów przewlekle przyjmujących warfarynę należy ściśle monitorować czas protrombinowy (INR) podczas leczenia produktem leczniczym Aprepitant Stada i przez 2 tygodnie po każdej 3- dniowej kuracji produktem leczniczym Aprepitant Stada podczas chemioterapii wywołującej nudności i wymioty (patrz punkt 4.4). Po podaniu aprepitantu w dawce 125 mg w 1. dniu oraz w dawce 80 mg na dobę w 2. i 3. dniu zdrowym ochotnikom w trakcie przewlekłej ustabilizowanej terapii warfaryną, nie stwierdzono wpływu aprepitantu na AUC warfaryny R(+) lub S(-) w osoczu w
3. dniu; niemniej jednak, stwierdzono obniżenie minimalnego stężenia warfaryny S(-) (substratu cytochromu CYP2C9) o 34%, czemu towarzyszyło obniżenie INR o 14% po 5 dniach od zakończenia leczenia aprepitantem.
Tolbutamid
Aprepitant podawany w dawce 125 mg w 1. dniu oraz w dawce 80 mg na dobę w 2. i 3. dniu, zmniejszał AUC tolbutamidu (substratu cytochromu CYP2C9) o 23% w 4. dniu, o 28% w 8. dniu oraz o 15% w 15. dniu, gdy tolbutamid podano doustnie w pojedynczej dawce 500 mg przed zastosowaniem 3-dniowego schematu podawania aprepitantu oraz w dniach 4., 8. i 15.
Hormonalne środki antykoncepcyjne
Podczas stosowania i w ciągu 28 dni po podaniu produktu leczniczego Aprepitant Stada może dojść do obniżenia skuteczności hormonalnych środków antykoncepcyjnych. W okresie stosowania produktu leczniczego Aprepitant Stada oraz przez 2 miesiące po przyjęciu ostatniej dawki produktu leczniczego Aprepitant Stada należy dodatkowo stosować alternatywne niehormonalne metody antykoncepcji.
W badaniu klinicznym pojedyncza dawka doustnego środka antykoncepcyjnego zawierającego etynyloestradiol i noretyndron podawana była w okresie od 1. do 21. dnia jednocześnie z aprepitantem, przyjmowanym w 8. dniu w dawce 125 mg, a w dniach 9. i 10. w dawce 80 mg na dobę, z ondansetronem podawanym dożylnie w 8. dniu w dawce 32 mg i deksametazonem podawanym doustnie w 8. dniu w dawce 12 mg oraz w dniach 9., 10. i 11. w dawce 8 mg na dobę. W badaniu tym, w okresie od 9. do 21. dnia, stwierdzono obniżenie najniższych wartości stężenia etynyloestradiolu o 64% i obniżenie najniższych wartości stężenia noretyndronu o 60%.
Antagoniści receptorów 5-HT3
W badaniach klinicznych interakcji produktów leczniczych, aprepitant nie miał istotnego klinicznie wpływu na farmakokinetykę ondansetronu, granisetronu lub hydrodolasetronu (czynnego metabolitu dolasetronu).
Wpływ innych produktów leczniczych na farmakokinetykę aprepitantu
Należy zachować ostrożność podczas jednoczesnego stosowania produktu leczniczego Aprepitant Stada z substancjami czynnymi będącymi inhibitorami aktywności CYP3A4 (np. ketokonazolem, itrakonazolem, worykonazolem, posakonazolem, klarytromycyną, telitromycyną, nefazodonem oraz inhibitorami proteazy), ponieważ przewiduje się, że jednoczesne stosowanie tych produktów leczniczych spowoduje kilkukrotny wzrost stężenia aprepitantu w osoczu (patrz punkt 4.4).
Należy unikać jednoczesnego podawania produktu leczniczego Aprepitant Stada z substancjami czynnymi silnie indukującymi aktywność CYP3A4 (np. ryfampicyną, fenytoiną, karbamazepiną, fenobarbitalem), ponieważ jednoczesne podawanie powoduje obniżenie stężenia aprepitantu w osoczu, co może prowadzić do zmniejszenia skuteczności produktu leczniczego Aprepitant Stada.
Nie zaleca się jednoczesnego podawania produktu leczniczego Aprepitant Stada i produktów ziołowych zawierających ziele dziurawca (Hypericum perforatum).
Ketokonazol
Po podaniu aprepitantu w pojedynczej dawce 125 mg w 5. dniu 10-dniowego leczenia ketokonazolem (który jest silnym inhibitorem cytochromu CYP3A4) w dawce 400 mg na dobę, AUC aprepitantu wzrosło około 5-krotnie, a średni okres półtrwania aprepitantu w fazie końcowej zwiększył się około 3-krotnie.
Ryfampicyna
Po podaniu aprepitantu w pojedynczej dawce 375 mg w 9. dniu 14-dniowego leczenia ryfampicyną (silnym induktorem cytochromu CYP3A4) w dawce 600 mg na dobę, AUC aprepitantu zmniejszyło się o 91%, a średni okres półtrwania w fazie końcowej aprepitantu zmniejszył się o 68%.
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono wyłącznie u dorosłych.
Antykoncepcja u mężczyzn i kobiet
Podczas stosowania i w ciągu 28 dni po podaniu produktu leczniczego Aprepitant Stada może dojść do obniżenia skuteczności hormonalnych środków antykoncepcyjnych. W okresie stosowania produktu leczniczego Aprepitant Stada oraz przez 2 miesiące po przyjęciu ostatniej dawki produktu leczniczego Aprepitant Stada należy dodatkowo stosować alternatywne niehormonalne metody antykoncepcji (patrz punkty 4.4 i 4.5).
Ciąża
Brak dostępnych danych dotyczących ekspozycji na aprepitant w okresie ciąży. Możliwy toksyczny wpływ aprepitantu na rozrodczość nie został w pełni określony, ponieważ w badaniach na zwierzętach nie można było uzyskać większej ekspozycji niż ekspozycja terapeutyczna u ludzi po podaniu dawki 125 mg/80 mg. Badania nie wykazują bezpośredniego lub pośredniego szkodliwego wpływu na przebieg ciąży, rozwój zarodka (płodu), przebieg porodu czy rozwój pourodzeniowy (patrz punkt 5.3). Potencjalny wpływ zmian regulacji neurokininowej na rozrodczość jest nieznany. Produktu leczniczego Aprepitant Stada nie należy stosować u kobiet w ciąży, o ile nie jest to bezwzględnie konieczne.
Karmienie piersią
Aprepitant przenika do mleka szczurów w okresie laktacji. Nie wiadomo, czy aprepitant przenika do mleka ludzkiego. Dlatego nie zaleca się karmienia piersią w okresie stosowania produktu leczniczego Aprepitant Stada.
Płodność
Możliwy wpływ aprepitantu na płodność nie został w pełni określony, ponieważ w badaniach na zwierzętach nie można było uzyskać większej ekspozycji niż ekspozycja terapeutyczna u ludzi. W badaniach tych, dotyczących płodności, nie wykazano bezpośredniego ani pośredniego szkodliwego wpływu na zdolność kojarzenia się w pary, płodność, rozwój zarodka/płodu oraz liczbę i ruchliwość plemników (patrz punkt 5.3).
Produkt leczniczy Aprepitant Stada może mieć niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów mechanicznych, rowerów i obsługiwania urządzeń mechanicznych w ruchu. Po przyjęciu produktu leczniczego Aprepitant Stada mogą wystąpić zawroty głowy i zmęczenie (patrz punkt 4.8).
Podsumowanie profilu bezpieczeństwa
Profil bezpieczeństwa aprepitantu oceniano u około 6500 osób dorosłych w ponad 50 badaniach klinicznych oraz u 184 pacjentów z grupy dzieci i młodzieży, w 2 głównych badaniach klinicznych.
Do najczęściej występujących działań niepożądanych, zgłaszanych z większą częstością u dorosłych leczonych wg schematu z aprepitantem w porównaniu z grupą pacjentów otrzymujących leczenie standardowe, u pacjentów otrzymujących chemioterapię o wysokim ryzyku wymiotów (HEC, ang. highly emetogenic chemotherapy) należały: czkawka (4,6% w porównaniu z 2,9%), zwiększenie aktywności aminotransferazy alaninowej (AlAT) (2,8% w porównaniu z 1,1%), niestrawność (2,6% w porównaniu z 2,0%), zaparcie (2,4% w porównaniu z 2,0%), ból głowy (2,0% w porównaniu z 1,8%) i zmniejszenie łaknienia (2,0% w porównaniu z 0,5%). Najczęstszym działaniem niepożądanym, zgłaszanym z większą częstością u pacjentów leczonych wg schematu z aprepitantem niż u pacjentów otrzymujących leczenie standardowe, u pacjentów otrzymujących chemioterapię o umiarkowanym ryzyku wymiotów (MEC, ang. moderately emetogenic chemotherapy), było zmęczenie (1,4% w porównaniu z 0,9%).
Do najczęściej zgłaszanych działań niepożądanych, obserwowanych z większą częstością u dzieci i młodzieży leczonych wg schematu z aprepitantem w porównaniu z grupą kontrolną, u pacjentów otrzymujących emetogenną chemioterapię przeciwnowotworową należały: czkawka (3,3% w porównaniu z 0,0%) i zaczerwienienie twarzy (1,1% w porównaniu z 0,0%).
Tabelaryczne zestawienie działań niepożądanych
Niżej wymienione działania niepożądane obserwowano w łącznej analizie badań dotyczących HEC oraz MEC z większą częstością dla aprepitantu, niż dla leczenia standardowego u pacjentów dorosłych i dzieci, lub po wprowadzeniu produktu do obrotu. Kategorie częstości występowania podane w tabeli zostały ustalone na podstawie badań z udziałem dorosłych; częstości obserwowane w badaniach z udziałem dzieci i młodzieży były podobne lub mniejsze, chyba że w tabeli wskazano inaczej. Niektóre działania niepożądane występujące u dorosłych z mniejszą częstością nie były obserwowane w badaniach z udziałem dzieci i młodzieży.
Częstości występowania są definiowane następująco: bardzo często (≥ 1/10), często (≥1/100 do
<1/10), niezbyt często (≥1/1 000 do < 1/100), rzadko (≥1/10 000 do < 1/1000), bardzo rzadko (<1/10 000), częstość nieznana (częstości nie można określić na podstawie dostępnych danych).
Klasa układu narządów | Działanie niepożądane | Częstość |
Zakażenia i zarażenia pasożytnicze | Zakażenia drożdżakowe, infekcje gronkowcowe | rzadko |
Zaburzenia krwi i układu chłonnego | Gorączka neutropeniczna, niedokrwistość | niezbyt często |
Zaburzenia układu odpornościowego | reakcje nadwrażliwości, w tym reakcje anafilaktyczne | częstość nieznana |
Zaburzenia metabolizmu i odżywiania | zmniejszony apetyt; | często |
nadmierne pragnienie | rzadko | |
Zaburzenia psychiczne | niepokój | niezbyt często |
dezorientacja, euforia | rzadko | |
Zaburzenia układu nerwowego | ból głowy | często |
zawroty głowy, senność | niezbyt często | |
zaburzenia funkcji poznawczych, letarg, zaburzenia smaku | rzadko | |
Zaburzenia oka | zapalenie spojówek | rzadko |
Zaburzenia ucha i błędnika | szumy uszne | rzadko |
Zaburzenia kardiologiczne | kołatanie serca | niezbyt często |
bradykardia, zaburzenia sercowo-naczyniowe | rzadko | |
Zaburzenia naczyniowe | nagłe zaczerwienienie skóry twarzy, nagłe uderzenia gorąca | niezbyt często |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | czkawka | często |
ból jamy ustnej i gardła, kichanie, kaszel, spływanie wydzieliny po tylnej ścianie gardła, podrażnienie gardła | rzadko | |
Zaburzenia żołądka i jelit | zaparcie, niestrawność | często |
kwaśne odbijanie, nudności†, wymioty†, refluks żołądkowo-przełykowy, ból brzucha, suchość jamy ustnej, wzdęcie | niezbyt często | |
perforujący wrzód dwunastnicy, zapalenie jamy ustnej, wzdęcie brzucha, twarde stolce, neutropeniczne zapalenie okrężnicy | rzadko | |
Zaburzenia skóry i tkanki podskórnej: | wysypka, trądzik | niezbyt często |
nadwrażliwość na światło, nadmierna potliwość, łojotok, zmiany skórne, swędząca wysypka, zespół Stevensa- Johnsona/toksyczne martwicze oddzielanie się naskórka | rzadko | |
świąd, pokrzywka | częstość nieznana | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | osłabienie mięśni, kurcze mięśni | rzadko |
Zaburzenia czynności nerek i układu moczowego | trudności w oddawaniu moczu | niezbyt często |
częstomocz | rzadko | |
Zaburzenia ogólne i stany w miejscu podania | zmęczenie | często |
astenia, złe samopoczucie | niezbyt często | |
obrzęki, dyskomfort w obrębie klatki piersiowej, zaburzenia chodu | rzadko | |
Badania diagnostyczne | wzrost aktywności AlAT | często |
wzrost aktywności AspAT, zwiększenie aktywności fosfatazy zasadowej | niezbyt często | |
obecność krwinek czerwonych w moczu, obniżenie stężenia sodu we krwi, zmniejszenie masy ciała, obniżenie liczby neutrofili, obecność glukozy w moczu, | rzadko |
wzmożone wydalanie moczu |
†Nudności i wymioty stanowiły parametry skuteczności w ciągu pierwszych 5 dni leczenia po chemioterapii i zgłaszane były jako działania niepożądane dopiero po tym okresie.
Opis wybranych działań niepożądanych
Profile działań niepożądanych u dorosłych podczas wielokrotnego stosowania w badaniach nad HEC oraz MEC przedłużonych do 6 dodatkowych cykli chemioterapii, były na ogół podobne do tych obserwowanych w 1. cyklu. W dodatkowym badaniu klinicznym z leczoną aktywnie grupą kontrolną obejmującym 1169 dorosłych pacjentów otrzymujących aprepitant i HEC profil działań niepożądanych był zasadniczo podobny do obserwowanego w innych badaniach z zastosowaniem HEC i aprepitantu.
U dorosłych pacjentów leczonych aprepitantem podczas nudności i wymiotów po zabiegach chirurgicznych (PONV, ang. postoperative nausea and vomiting) obserwowano dodatkowe działania niepożądane występujące z większą częstością niż w przypadku ondansetronu: ból w nadbrzuszu, nieprawidłowe zjawiska osłuchowe w przewodzie pokarmowym, zaparcie*, dyzartria, duszność, niedoczulica, bezsenność, zwężenie źrenicy, nudności, zaburzenia zmysłów, dyskomfort w żołądku, niedrożność przepuszczającą*, pogorszenie ostrości widzenia, świszczący oddech.
*Zgłaszano u pacjentów przyjmujących większe dawki aprepitantu.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel: + 48 22 49 21 301
Fax: + 48 22 49 21 309
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W przypadku przedawkowania należy przerwać stosowanie produktu leczniczego Aprepitant Stada i zastosować ogólne leczenie podtrzymujące oraz obserwację. Z uwagi na przeciwwymiotne działanie aprepitantu wywoływanie wymiotów poprzez podanie produktów leczniczych może nie być skuteczne.
Aprepitantu nie można usunąć metodą hemodializy.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Grupa farmakoterapeutyczna: leki przeciwwymiotne i przeciw nudnościom, inne leki przeciwwymiotne
Kod ATC: A04AD12
Aprepitant jest wybiórczym antagonistą o wysokim powinowactwie, działającym na ludzkie receptory neurokininowe 1 (NK1) substancji P.
3-dniowy schemat leczenia aprepitantem u dorosłych
W dwóch badaniach z randomizacją, z podwójnie ślepą próbą, obejmujących ogółem 1094 dorosłych pacjentów, którzy przyjmowali chemioterapię (w tym cisplatynę w dawce ≥ 70 mg/m2 p.c.), porównano działanie aprepitantu w skojarzeniu z ondansetronem i deksametazonem (patrz punkt 4.2) ze standardowym leczeniem (placebo plus ondansetron 32 mg dożylnie w 1. dniu plus deksametazon 20 mg doustnie w 1. dniu i 8 mg doustnie, dwa razy na dobę, w 2. i 4. dniu). Mimo że w badaniach klinicznych stosowano 32 mg ondansetronu dożylnie, dawka ta nie jest już zalecana. Należy zapoznać się z zaleceniami dotyczącymi odpowiedniego dawkowania podanymi w drukach informacyjnych wybranego antagonisty receptora 5-HT3.
Skuteczność leczenia określono na podstawie następującego kryterium złożonego: odpowiedź całkowita (określona jako brak wymiotów i niekorzystanie z terapii ratunkowej) głównie w 1. cyklu leczenia. Wyniki określono dla każdego badania osobno oraz dla 2 badań łącznie.
Tabela 1 przedstawia podsumowanie głównych wyników badań z łącznej analizy.
Tabela 1
Odsetek dorosłych pacjentów otrzymujących chemioterapię o wysokim ryzyku wymiotów, u których wystąpiła odpowiedź na leczenie, z podziałem na grupy leczenia i fazy – Cykl 1.
Schemat zawierający aprepitant (N= 521) † | Terapia standardowa (N= 524) † | Różnice* | ||
% | % | % | (95% CI) | |
WSKAŹNIKI ZBIORCZE | ||||
Całkowita odpowiedź (brak wymiotów i niekorzystanie z terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 67,7 | 47,8 | 19,9 | (14,0, 25,8) |
0-24 godziny | 86,0 | 73,2 | 12,7 | (7,9, 17,6) |
25-120 godzin | 71,5 | 51,2 | 20,3 | (14,5, 26,1) |
WSKAŹNIKI INDYWIDUALNE | ||||
Brak wymiotów (niezależnie od stosowania terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 71,9 | 49,7 | 22,2 | (16,4, 28,0) |
0-24 godziny | 86,8 | 74,0 | 12,7 | (8,0, 17,5) |
25-120 godzin | 76,2 | 53,5 | 22,6 | (17,0, 28,2) |
Brak istotnych nudności (maksymalny wynik w skali VAS [wzrokowa skala analogowa] < 25 mm w skali 0-100 mm] | ||||
W całym okresie (0-120 godzin) | 72,1 | 64,9 | 7,2 | (1,6, 12,8) |
25-120 godzin | 74,0 | 66,9 | 7,1 | (1,5, 12,6) |
* Przedziały ufności obliczono bez korekty względem płci i jednoczesnego stosowania chemioterapii, które uwzględniono w analizie pierwszorzędowej ilorazów szans i modelach logistycznych.
†W przypadku jednego pacjenta otrzymującego schemat z aprepitantem dane dotyczyły tylko fazy ostrej i został on wykluczony z analizy całościowej oraz analizy fazy opóźnionej; w przypadku jednego pacjenta w standardowym schemacie dane dotyczyły tylko fazy opóźnionej i został on wykluczony z analizy całościowej oraz analizy fazy ostrej.
Rycina 1 przedstawia szacunkowy czas do wystąpienia pierwszych wymiotów w analizie złożonej, w postaci krzywej Kaplana-Meiera.
Rycina 1
Odsetek dorosłych pacjentów otrzymujących chemioterapię o wysokim ryzyku wymiotów, u których nie występowały wymioty w zależności od czasu – Cykl 1
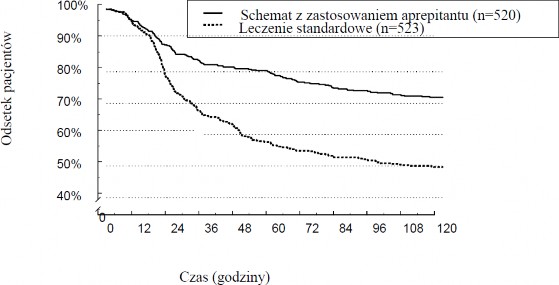
Statystycznie istotne różnice skuteczności obserwowano również w każdym z tych dwóch badań osobno.
W tych samych dwóch badaniach klinicznych, w fazie rozszerzonej, 851 dorosłych pacjentów kontynuowało leczenie przez okres do pięciu kolejnych cykli chemioterapii. We wszystkich cyklach skuteczność działania aprepitantu wyraźnie się utrzymywała.
W badaniu z randomizacją, z podwójnie ślepą próbą, które obejmowało ogółem 866 dorosłych pacjentów (864 kobiety, 2 mężczyzn) przyjmujących chemioterapię zawierającą cyklofosfamid w dawce 750-1500 mg/m2 p.c.; lub cyklofosfamid w dawce 500-1500 mg/m2 p.c. i doksorubicynę (< 60 mg/m2 p.c.) lub epirubicynę (< 100 mg/m2 p.c.), aprepitant w połączeniu ze schematem ondansetron/deksametazon (patrz punkt 4.2) porównano ze standardowym leczeniem (placebo plus ondansetron 8 mg doustnie (dwa razy w 1. dniu i co 12 godzin w 2. i 3. dniu) plus deksametazon 20 mg doustnie w 1. dniu).
Skuteczność leczenia określono na podstawie następującego kryterium złożonego: odpowiedź całkowita (określona jako brak wymiotów i niekorzystanie z terapii ratunkowej) głównie w 1. cyklu leczenia.
Tabela 2 przedstawia podsumowanie głównych wyników badań.
Tabela 2
Odsetek dorosłych pacjentów otrzymujących chemioterapię o umiarkowanym ryzyku wymiotów, u których wystąpiła odpowiedź na leczenie, z podziałem na grupy leczenia i fazy – Cykl 1.
Schemat zawierający aprepitant (N= 433) † | Terapia standardowa (N=424) | Różnice* | ||
% | % | % | (95% CI) | |
WSKAŹNIKI ZBIORCZE | ||||
Całkowita odpowiedź (brak wymiotów i niekorzystanie z terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 50,8 | 42,5 | 8,3 | (1,6, 15,0) |
0-24 godziny | 75,7 | 69,0 | 6,7 | (0,7, 12,7) |
25-120 godzin | 55,4 | 49,1 | 6,3 | (-0,4, 13,0) |
WSKAŹNIKI INDYWIDUALNE | ||||
Brak wymiotów (niezależnie od stosowania terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 75,7 | 58,7 | 17,0 | (10,8, 23,2) |
0-24 godziny | 87,5 | 77,3 | 10,2 | (5,1, 15,3) |
25-120 godzin | 80,8 | 69,1 | 11,7 | (5,9, 17,5) |
Brak istotnych nudności (maksymalny wynik w skali VAS [wzrokowa skala analogowa] < 25 mm w skali 0-100 mm] | ||||
W całym okresie (0-120 godzin) | 60,9 | 55,7 | 5,3 | (-1,3, 11,9) |
0-24 godziny | 79,5 | 78,3 | 1,3 | (-4,2, 6,8) |
25-120 godzin | 65,3 | 61,5 | 3,9 | (-2,6, 10,3) |
* Przedziały ufności obliczono bez korekty względem kategorii wiekowej (< 55 lat, ≥ 55 lat) i grupy badacza, które uwzględniono w analizie pierwszorzędowej ilorazów szans i modelach logistycznych.
†W przypadku jednego pacjenta otrzymującego schemat z aprepitantem dane dotyczyły tylko fazy ostrej i został on wykluczony z analizy całościowej oraz analizy fazy opóźnionej.
W tym samym badaniu klinicznym, w fazie rozszerzonej, 744 dorosłych pacjentów kontynuowało leczenie przez okres do trzech kolejnych cykli chemioterapii. We wszystkich cyklach skuteczność działania aprepitantu wyraźnie się utrzymywała.
W drugim wieloośrodkowym badaniu klinicznym z randomizacją, podwójnie ślepą próbą i prowadzonym na grupach równoległych, porównano schemat leczenia aprepitantem z leczeniem standardowym u 848 dorosłych pacjentów (652 kobiet, 196 mężczyzn) poddanych chemioterapii w schemacie zawierającym jakąkolwiek dożylną dawkę jednego z następujących produktów leczniczych: oksaliplatyny, karboplatyny, epirubicyny, idarubicyny, ifosfamidu, irynotekanu, daunorubicyny, doksorubicyny; cyklofosfamid podawany dożylnie (< 1500 mg/m2 p.c.); lub cytarabinę podawaną dożylnie (> 1 g/m2 p.c.). Aprepitant stosowano u pacjentów poddanych chemioterapii z powodu różnych typów nowotworów, z czego 52% stanowiły nowotwory piersi, 21% nowotwory przewodu pokarmowego, w tym rak okrężnicy i odbytnicy, 13% nowotwory płuc i 6% nowotwory ginekologiczne. Schemat leczenia aprepitantem w skojarzeniu z ondansetronem/deksametazonem (patrz punkt 4.2) porównano z leczeniem standardowym (placebo plus ondansetron 8 mg doustnie (dwa razy w 1. dniu, i co 12 godzin w 2. i 3. dniu) plus deksametazon 20 mg doustnie w 1. dniu).
Ocena skuteczności opierała się na ocenie następujących pierwszorzędowych i kluczowych drugorzędowych punktów końcowych: brak wymiotów w całym okresie (od 0 do 120 godzin po chemioterapii), ocena bezpieczeństwa i tolerancji aprepitantu w schemacie leczenia nudności i
wymiotów wywołanych chemioterapią (CINV, ang. chemotherapy-induced nausea and vomiting) i całkowita odpowiedź (określona jako brak wymiotów i niekorzystanie z terapii ratunkowej) w całym okresie (od 0 do 120 godzin po chemioterapii). Ponadto jako badawczy punkt końcowy oceniano odsetek przypadków braku istotnych nudności w całym okresie badania (od 0 do 120 godzin po chemioterapii) oraz w analizie post-hoc dla fazy ostrej i późnej.
Tabela 3 przedstawia podsumowanie głównych wyników badań.
Tabela 3
Odsetek dorosłych pacjentów otrzymujących chemioterapię o umiarkowanym ryzyku wymiotów, u których wystąpiła odpowiedź na leczenie, z podziałem na grupy leczenia i fazy w Badaniu 2 – Cykl 1.
Schemat zawierający aprepitant (N= 425) | Terapia standardowa (N=406) | Różnice* | ||
% | % | % | (95% CI) | |
Całkowita odpowiedź (brak wymiotów i niekorzystanie z terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 68,7 | 56,3 | 12,4 | (5,9, 18,9) |
0-24 godziny | 89,2 | 80,3 | 8,9 | (4,0, 13,8) |
25-120 godzin | 70,8 | 60,9 | 9,9 | (3,5, 16,3) |
Brak wymiotów (niezależnie od stosowania terapii ratunkowej) | ||||
W całym okresie (0-120 godzin) | 76,2 | 62,1 | 14,1 | (7,9, 20,3) |
0-24 godziny | 92,0 | 83,7 | 8,3 | (3,9, 12,7) |
25-120 godzin | 77,9 | 66,8 | 11,1 | (5,1, 17,1) |
Brak istotnych nudności (maksymalny wynik w skali VAS [wzrokowa skala analogowa] < 25 mm w skali 0-100 mm] | ||||
W całym okresie (0-120 godzin) | 73,6 | 66,4 | 7,2 | (1,0, 13,4) |
0-24 godziny | 90,9 | 86,3 | 4,6 | (0,2, 9,0) |
25-120 godzin | 74,9 | 69,5 | 5,4 | (-0,7, 11,5) |
*Przedziały ufności obliczono bez korekty względem płci i regionu, które uwzględniono w analizie pierwszorzędowej przy użyciu modeli logistycznych.
Korzyść ze stosowania leczenia skojarzonego z zastosowaniem aprepitantu, obserwowana w pełnej populacji badania, związana była przede wszystkim z wynikami obserwowanymi u pacjentów ze słabą kontrolą objawów podczas stosowania standardowego schematu leczenia, na przykład u kobiet, pomimo, że liczbowo wyniki te były lepsze niezależnie od wieku, rodzaju nowotworu czy płci pacjenta. Pełną odpowiedź na leczenie zawierające aprepitant i leczenie standardowe uzyskano odpowiednio u 209/324 (65%) i 161/320 (50%) kobiet oraz u 83/101 (82%) i 68/87 (78%) mężczyzn.
Dzieci i młodzież
W badaniu klinicznym z randomizacją, podwójnie ślepą próbą i grupą kontrolną otrzymującą aktywne leczenie z udziałem 302 pacjentów z grupy dzieci i młodzieży (w wieku od 6 miesięcy do 17 lat) otrzymujących chemioterapię o umiarkowanym lub wysokim ryzyku wymiotów, schemat leczenia aprepitantem porównywano z grupą kontrolną w zapobieganiu CINV. Skuteczność aprepitantu oceniano w pojedynczym cyklu (Cykl 1). Pacjenci mieli możliwość otrzymywania aprepitantu w kolejnych niezaślepionych cyklach (opcjonalne cykle 2-6); jednakże w tych dodatkowych cyklach nie oceniano skuteczności. Schemat leczenia aprepitantem u młodzieży w wieku od 12 do 17 lat (n=47) składał się z doustnego podawania kapsułki aprepitantu w dawce 125 mg w 1. dniu oraz 80 mg w 2. i
3. dniu, w skojarzeniu z ondansetronem w 1. dniu. Schemat leczenia aprepitantem u dzieci w wieku
od 6 miesięcy do poniżej 12 lat (n=105) składał się z aprepitantu w postaci proszku do sporządzania zawiesiny doustnej w dawce 3,0 mg/kg m.c. (do 125 mg) doustnie w 1. dniu oraz 2,0 mg/kg m.c. (do 80 mg) ustnie w 2. i 3. dniu, w skojarzeniu z ondansetronem w 1. dniu. Schemat leczenia w grupie kontrolnej u młodzieży w wieku od 12 do 17 lat (n=48) i dzieci w wieku od 6 miesięcy do poniżej 12 lat (n=102) składał się z placebo dla aprepitantu w 1., 2. i 3. dniu, w skojarzeniu z ondansetronem w
1. dniu. Odpowiednio aprepitant lub placebo i ondansetron były podawane na 1 godzinę i 30 minut przed rozpoczęciem chemioterapii. Dożylne podanie deksametazonu było dozwolone jako element leczenia przeciwwymiotnego u dzieci i młodzieży w obu grupach wiekowych, w zależności od decyzji lekarza. U dzieci i młodzieży przyjmujących aprepitant wymagane było obniżenie dawki deksametazonu (o 50%). Obniżenie dawki nie było wymagane u dzieci i młodzieży w grupie kontrolnej. Wśród dzieci i młodzieży 29% pacjentów leczonych aprepitantem oraz 28% pacjentów w grupie kontrolnej podawano deksametazon jako składnik schematu leczenia w Cyklu 1.
Działanie przeciwwymiotne aprepitantu oceniano w okresie 5 dni (120 godzin) po rozpoczęciu chemioterapii w 1. dniu. Pierwszorzędowym punktem końcowym była całkowita odpowiedź w fazie opóźnionej (25 do 120 godzin po rozpoczęciu chemioterapii) w Cyklu 1. Tabela 4 przedstawia podsumowanie głównych wyników badania.
Tabela 4
Liczba (%) pacjentów z grupy dzieci i młodzieży z całkowitą odpowiedzią i brakiem wymiotów z podziałem na grupy leczenia i fazy – Cykl 1 (populacja wyłoniona w oparciu o zamiar leczenia; ang.: intent-to-treat, ITT)
Schemat zawierający aprepitant n/m (%) | Schemat kontrolny n/m (%) | |
GŁÓWNY PUNKT KOŃCOWY | ||
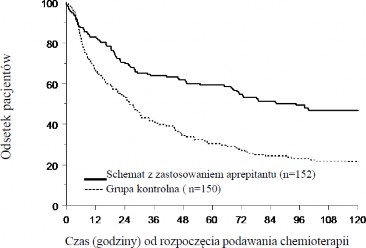
Odpowiedź całkowita* – Faza opóźniona | 77/152 (50,7)† | 39/150 (26,0) |
INNE OKREŚLONE WCZEŚNIEJ PUNKTY KOŃCOWE | ||
Odpowiedź całkowita* – Faza ostra | 101/152 (66,4)‡ | 78/150 (52,0) |
Odpowiedź całkowita* – Obie fazy | 61/152 (40,1)† | 30/150 (20,0) |
Brak wymiotów§ – Obie fazy | 71/152 (46,7)† | 32/150 (21,3) |
*Odpowiedź całkowita = brak wymiotów, nieefektywnych wymiotów i nudności oraz niekorzystanie z leków ratunkowych. †p < 0,01 w porównaniu do schematu kontrolnego †p < 0,05 w porównaniu do schematu kontrolnego §Brak wymiotów = brak wymiotów, nieefektywnych wymiotów i nudności n/m = liczba pacjentów z pożądaną odpowiedzią/liczba pacjentów włączonych do badania w danym punkcie czasowym. Faza ostra: 0 do 24 godzin po rozpoczęciu chemioterapii. Faza opóźniona: 25 do 120 godzin po rozpoczęciu chemioterapii. Obie fazy: 0 do 120 godzin po rozpoczęciu chemioterapii. | ||
Szacunkowy czas do wystąpienia pierwszych wymiotów od rozpoczęcia chemioterapii był dłuższy w przypadku schematu obejmującego aprepitant (szacunkowy średni czas do wystąpienia pierwszych wymiotów wynosił 94,5 godziny) w porównaniu z grupą kontrolną (szacunkowy średni czas do wystąpienia pierwszych wymiotów wynosił 26,0 godzin), co zostało przedstawione w postaci krzywej Kaplana-Meiera na Rycinie 2.
Rycina 2
Czas do wystąpienia pierwszych wymiotów od rozpoczęcia podawania chemioterapii – u dzieci i młodzieży w obu fazach Cyklu 1. (populacja wyłoniona w oparciu o zamiar leczenia; ang.: intent-to- treat, ITT)
Analiza skuteczności w podgrupach w Cyklu 1. wykazała, że niezależnie od kategorii wiekowej, płci, stosowania deksametazonu w celu zapobiegania wymiotom oraz ryzyka działania wymiotnego chemioterapii, stosowanie schematu z aprepitantem pozwoliło na lepsze opanowanie objawów w porównaniu z grupą kontrolną w odniesieniu do punktów końcowych odpowiedzi całkowitej.
Aprepitant charakteryzuje się farmakokinetyką nieliniową. Zarówno klirens, jak i bezwzględna dostępność biologiczna obniżają się wraz ze zwiększeniem dawki.
Wchłanianie
Średnia bezwzględna biodostępność aprepitantu po podaniu doustnym wynosi 67% dla kapsułek 80 mg oraz 59% dla kapsułek 125 mg. Średnie stężenie maksymalne aprepitantu w osoczu (Cmax) występowało po około 4 godzinach (tmax). Po podaniu kapsułki doustnie ze standardowym śniadaniem o wartości energetycznej około 800 kcal stwierdzono zwiększenie AUC aprepitantu o maksymalnie 40%. Ten wzrost nie jest uważany za istotny klinicznie.
Farmakokinetyka aprepitantu jest nieliniowa w całym zakresie dawek stosowanych klinicznie. U młodych zdrowych osób dorosłych zwiększenie wartości AUC0-∞ była o 26% większa niż wynikałoby to z proporcji dawek pojedynczych 80 mg i 125 mg, przyjmowanych po jedzeniu.
Po podaniu doustnym pojedynczej dawki 125 mg aprepitantu w 1. dniu oraz 80 mg raz na dobę w 2. i
3. dniu, AUC0-24h (średnia±odchylenie standardowe) wynosiło odpowiednio 19,6±2,5 μg∙h/ml oraz 21,2±6,3 μg∙h/ml w 1. i 3. dniu. Wartość Cmax wyniosła odpowiednio 1,6±0,36 µg/ml oraz 1,4±0,22 µg/ml w 1. i 3. dniu.
Dystrybucja
Aprepitant w znacznym stopniu wiąże się z białkami osocza, średnio w 97%. Średnia geometryczna pozornej objętości dystrybucji w stanie równowagi (Vdss) wynosi u ludzi około 66 litrów.
Metabolizm
Aprepitant jest w znacznym stopniu metabolizowany. U zdrowych młodych osób aprepitant stanowi około 19% aktywności promieniotwórczej w osoczu w okresie 72 godzin, po wstrzyknięciu pojedynczej dawki 100 mg [14C]-fosaprepitantu, prekursora aprepitantu, co oznacza, że w osoczu występuje duża ilość metabolitów. W osoczu krwi ludzkiej zidentyfikowano dwanaście metabolitów
aprepitantu. Metabolizm aprepitantu w znacznym stopniu następuje poprzez utlenianie pierścienia morfolinowego i jego łańcuchów bocznych, a metabolity, które powstają, mają słabe działanie farmakologiczne. W badaniach in vitro z zastosowaniem mikrosomów wątroby ludzkiej stwierdzono, że aprepitant jest metabolizowany głównie z udziałem cytochromu CYP3A4 i w mniejszym stopniu przez cytochromy CYP1A2 oraz CYP2C19.
Eliminacja
Aprepitant nie jest wydalany w moczu w postaci niezmienionej. Metabolity są wydalane w moczu oraz poprzez wydzielanie z żółcią, do stolca. Po podaniu dożylnym pojedynczej dawki 100 mg [14C]- fosaprepitantu, prekursora aprepitantu osobom zdrowym, 57% aktywności promieniotwórczej stwierdzano w moczu, a 45% w stolcu.
Klirens osoczowy aprepitantu zależy od dawki; przy zwiększeniu dawki zmniejsza się i w zakresie dawek terapeutycznych wynosi od 60 do 72 ml/min. Okres półtrwania w fazie końcowej wynosi od 9 do 13 godzin.
Farmakokinetyka w specjalnych grupach pacjentów
Pacjenci w podeszłym wieku: Po podaniu doustnym pojedynczej dawki 125 mg aprepitantu w dniu 1. i dawek 80 mg raz dziennie w dniach od 2 do 5, wartość AUC0-24hr dla aprepitantu była o 21% większa w dniu 1. i o 36% wyższa w dniu 5. u osób w podeszłym wieku (≥ 65 lat), w porównaniu do młodszych dorosłych. Wartość Cmax była o 10% wyższa w 1. dniu i o 24% wyższa w 5. dniu u osób w podeszłym wieku w porównaniu do młodszych dorosłych. Uznano, że różnice te nie są istotne klinicznie.
U osób w podeszłym wieku nie jest wymagane obniżenie dawki produktu leczniczego Aprepitant Stada.
Płeć: Po podaniu doustnym aprepitantu w dawce pojedynczej 125 mg wartość Cmax jest o 16% większa u kobiet niż u mężczyzn. Okres półtrwania aprepitantu u kobiet jest o 25% krótszy w porównaniu z mężczyznami, a wartość tmax jest zbliżona u kobiet i u mężczyzn. Uznano, że różnice te nie są istotne klinicznie.
Nie jest wymagana modyfikacja dawki produktu leczniczego Aprepitant Stada w zależności od płci pacjenta.
Zaburzenia czynności wątroby: Łagodne zaburzenie czynności wątroby (klasa A wg Child-Pugh) nie wpływa na parametry farmakokinetyczne aprepitantu w stopniu istotnym klinicznie. U pacjentów z łagodnymi zaburzeniami czynności wątroby nie jest konieczna modyfikacja dawki. Na podstawie dostępnych danych nie można wyciągać wniosków na temat wpływu umiarkowanych zaburzeń czynności wątroby (klasa B wg Child-Pugh) na parametry farmakokinetyczne aprepitantu. Brak danych klinicznych lub dotyczących farmakokinetyki aprepitantu u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasa C wg Child-Pugh).
Zaburzenia czynności nerek: Pacjentom z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny
< 30 ml/min.) oraz pacjentom ze schyłkową niewydolnością nerek (ESRD, ang. end stage renal disease) wymagającym hemodializy podawano aprepitant w dawce pojedynczej 240 mg.
U pacjentów z ciężkimi zaburzeniami czynności nerek wartość AUC0--∞ dla całkowitej puli aprepitantu (w formie niezwiązanej oraz związanej z białkami) obniżyła się o 21%, a wartość Cmax obniżyła się o 32% w porównaniu z osobami zdrowymi. U poddawanych hemodializie pacjentów z ESRD wartość AUC0-∞ dla całej puli aprepitantu obniżyła się o 42%, a wartość Cmax – o 32%. Ze względu na umiarkowane obniżenie stopnia wiązania się aprepitantu z białkami u pacjentów z chorobami nerek, AUC aprepitantu niezwiązanego, czynnego farmakodynamicznie nie było znamiennie różne u pacjentów z zaburzeniami czynności nerek, w porównaniu z osobami zdrowymi. Hemodializa przeprowadzona po 4 lub 48 godzinach po przyjęciu dawki produktu leczniczego nie miała istotnego wpływu na parametry farmakokinetyczne aprepitantu. W dializacie stwierdzano poniżej 0,2% dawki.
Nie jest wymagana modyfikacja dawki produktu leczniczego Aprepitant Stada u pacjentów z upośledzeniem czynności nerek ani u pacjentów z ESRD poddawanych hemodializie.
Dzieci i młodzież: W czasie 3-dniowego schematu podawania aprepitantu w postaci kapsułek (125/80/80 mg) u młodzieży (w wieku od 12 do 17 lat) wartość AUC0-24h wyniosła powyżej 17 μg∙hr/ml w 1. dniu ze stężeniem (Cmin) pod koniec 2. i 3. dnia powyżej 0,4 μg/ml u większości pacjentów. Średnie stężenie maksymalne w osoczu (Cmax) wynosiło około 1,3 μg/ml w 1. dniu i występowało po około 4 godzinach. W czasie 3-dniowego schematu podawania aprepitantu w postaci proszku do sporządzania zawiesiny doustnej (3/2/2 mg/kg m.c.) u pacjentów w wieku od 6 miesięcy do poniżej 12 lat wartość AUC0-24h wynosiła powyżej 17 μg∙hr/ml w 1. dniu ze stężeniem (Cmin) pod koniec 2. i 3. dnia powyżej 0,1 μg/ml u większości pacjentów.
Mediana stężenia maksymalnego w osoczu (Cmax) wynosiła około 1,2 μg/ml w 1. dniu i występowała po 5 do 7 godzin.
Analiza farmakokinetyki populacyjnej aprepitantu u dzieci i młodzieży (w wieku od 6 miesięcy do 17 lat) wskazuje, że płeć i rasa nie mają znaczącego klinicznie wpływu na farmakokinetykę aprepitantu.
Związek pomiędzy stężeniem i działaniem
Przeprowadzono badania metodą pozytonowej tomografii emisyjnej (PET, ang. positron emission tomography) z zastosowaniem wysoce swoistego znacznika wiążącego się z receptorem NK1. W badaniach tych uczestniczyli młodzi zdrowi mężczyźni. Wykazano, że aprepitant przenika do mózgu i łączy się z receptorami NK1 w sposób zależny od dawki oraz od stężenia w osoczu. Przewiduje się, że stężenia aprepitantu w osoczu, które występują po 3 dniach stosowania produktu leczniczego Aprepitant Stada, powodują zajęcie ponad 95% receptorów NK1 w mózgu.
Dane przedkliniczne, wynikające z konwencjonalnych badań toksyczności, genotoksyczności, potencjału rakotwórczego oraz toksycznego wpływu na rozród i rozwój potomstwa po podaniu dawki pojedynczej i dawek wielokrotnych nie ujawniają szczególnego zagrożenia dla człowieka.
Niemniej jednak należy zauważyć, że ekspozycja układowa u gryzoni była podobna, a nawet niższa niż ekspozycja u pacjentów stosujących produkt leczniczy w dawkach terapeutycznych 125 mg/80 mg. W szczególności, chociaż przy ekspozycji, jaka występuje u ludzi, nie stwierdzono niekorzystnego wpływu produktu leczniczego w badaniach nad reprodukcją, jednak na podstawie obserwacji zwierząt z taką ekspozycją na produkt leczniczy nie można właściwie określić ryzyka stosowania produktu leczniczego u ludzi.
W badaniu toksyczności u młodych, prowadzonym na szczurach od 10. do 63. dnia po urodzeniu, aprepitant w dawce od 250 mg/kg m.c. dwa razy na dobę u samic powodował przedwczesne otwarcie pochwy, oraz w dawce od 10 mg/kg m.c. dwa razy na dobę u samców opóźnienie w separacji napletka. Nie stwierdzono granic narażenia o znaczeniu klinicznym. Nie stwierdzono zależnego od leczenia wpływu na kojarzenie się zwierząt w pary, płodność, przeżywalność zarodków/płodów ani zmian patologicznych w narządach rozrodczych. W badaniu toksyczności u młodych, prowadzonym na psach od 14. do 42. dnia po urodzeniu, obserwowano obniżenie masy jąder oraz wielkości komórek Leydiga u samców po podawaniu 6 mg/kg m.c. na dobę, a także zwiększenie masy macicy, przerost macicy i szyjki macicy oraz obrzęk tkanek pochwy u samic po zastosowaniu produktu leczniczego w dawkach od 4 mg/kg m.c. na dobę. Nie stwierdzono granic narażenia o znaczeniu klinicznym dla aprepitantu. W leczeniu krótkotrwałym zgodnym z zalecanym schematem dawkowania jest mało prawdopodobne, aby dane te miały znaczenie kliniczne.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
1 blister aluminiowy zawierający jedną kapsułkę 125 mg.
5 blistrów aluminiowych zawierających po jednej kapsułce 125 mg.
Produkt leczniczy Aprepitant Stada, 80 mg, kapsułki, twarde dostępny jest w następujących wielkościach opakowań:
1 blister aluminiowy zawierający jedną kapsułkę 80 mg,
opakowanie na dwudniowe leczenie zawierające dwie kapsułki po 80 mg.
5 blistrów aluminiowych zawierających po jednej kapsułce 80 mg.
Produkt leczniczy Aprepitant Stada, 125 mg/80 mg, kapsułki twarde dostępny jest w następujących wielkościach opakowań:
opakowanie na trzydniowe leczenie zawierające jedną kapsułkę 125 mg i dwie kapsułki po 80 mg. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Zawartość kapsułki Hypromeloza 2910
Poloksamer 407 Sacharoza
Celuloza mikrokrystaliczna
Osłonka kapsułki (125 mg) Żelatyna
Sodu laurylosiarczan (E 487) Tytanu dwutlenek (E 171) Żelaza tlenek czerwony (E 172)
Osłonka kapsułki (80 mg) Żelatyna
Sodu laurylosiarczan (E 487) Tytanu dwutlenek (E 171)
Czarny tusz do nadruku Szelak
Żelaza tlenek czarny (E 172) Glikol propylenowy (E 1520)
Nie dotyczy.
30 miesięcy
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Produkt leczniczy Aprepitant Stada pakowany jest w tekturowe pudełka zawierające odpowiednią liczbę blistrów z folii OPA/Aluminium/PVC/Aluminium wraz z ulotką informacyjną.
Produkt leczniczy Aprepitant Stada, 125 mg, kapsułki, twarde dostępny jest w następujących wielkościach opakowań:
Brak specjalnych wymagań dotyczących usuwania.
STADA Arzneimittel AG Stadastrasse 2-18
61118 Bad Vilbel Niemcy
Aprepitant Stada, 125 mg, kapsułki, twarde: pozwolenie nr Aprepitant Stada, 80 mg, kapsułki, twarde: pozwolenie nr Aprepitant Stada, 125 mg/80 mg kapsułki, twarde: pozwolenie nr
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:








