







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Szczególne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Druniler, 80 mg, tabletki powlekane
Każda tabletka powlekana zawiera 80 mg febuksostatu (Febuxostatum) w postaci febuksostatu
półwodnego.
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 72,68 mg laktozy (w postaci laktozy jednowodnej).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana
Jasnożółta lub żółta tabletka powlekana w kształcie kapsułki, z wytłoczonym symbolem „80” na jednej stronie i gładka na drugiej stronie, o wymiarach 16,5 mm x 7,0 mm.
Produkt leczniczy Druniler jest wskazany w leczeniu przewlekłej hiperurykemii w chorobach,
w których wystąpiło już odkładanie się złogów moczanowych (w tym guzki dnawe i (lub) zapalenie
stawów dnawe czynne lub w wywiadzie).
Produkt leczniczy Druniler jest wskazany do stosowania u dorosłych.
Dawkowanie
Zalecana doustna dawka produktu Druniler to 80 mg raz na dobę, niezależnie od posiłków. Jeśli po upływie 2-4 tygodni leczenia stężenie kwasu moczowego w surowicy krwi wynosi >6 mg/dl
(357 μmol/l), można rozważyć zastosowanie produktu leczniczego Druniler w dawce 120 mg raz na
dobę.
Działanie produktu Druniler jest na tyle szybkie, że umożliwia kontrolę stężenia kwasu moczowego w surowicy po 2 tygodniach. Celem terapeutycznym jest zmniejszenie i utrzymanie stężenia kwasu moczowego w surowicy krwi poniżej 6 mg/dl (357 μmol/l).
Zaleca się zapobieganie zaostrzeniom dny moczanowej przez co najmniej 6 miesięcy (patrz punkt
4.4).
Osoby w podeszłym wieku
Modyfikacja dawki u osób w podeszłym wieku nie jest konieczna (patrz punkt 5.2).
Zaburzenia czynności nerek
Nie oceniono w pełni skuteczności i bezpieczeństwa stosowania u pacjentów z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny <30 ml/min, patrz punkt 5.2).
U pacjentów z lekkimi lub umiarkowanymi zaburzeniami czynności nerek modyfikacja dawki nie jest konieczna.
Zaburzenia czynności wątroby
Nie badano skuteczności i bezpieczeństwa stosowania febuksostatu u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasa C wg skali Childa-Pugha).
Zalecana dawka u pacjentów z lekkimi zaburzeniami czynności wątroby wynosi 80 mg. Dostępne są ograniczone dane dotyczące pacjentów z umiarkowanymi zaburzeniami czynności wątroby.
Dzieci i młodzież
Nie ustalono bezpieczeństwa stosowania i skuteczności febuksostatu u dzieci i młodzieży w wieku
poniżej 18 lat. Brak dostępnych danych.
Sposób stosowania Podanie doustne.
Produkt leczniczy Druniler należy przyjmować doustnie, z jedzeniem lub niezależnie od posiłków.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną
w punkcie 6.1 (patrz także punkt 4.8).
Zaburzenia sercowo-naczyniowe
Należy unikać stosowania febuksostatu u pacjentów z istniejącymi uprzednio ciężkimi chorobami układu sercowo-naczyniowego (np. zawał serca, udar mózgu lub niestabilna dławica piersiowa), chyba że inne opcje leczenia nie są odpowiednie. Większą liczbowo częstość zgłaszanych przez badacza zdarzeń sercowo-naczyniowych APTC (punkty końcowe określone zgodnie z Anti Platelet Trialists’ Collaboration [APTC], obejmujące zgon z przyczyn sercowo-naczyniowych, niezakończony zgonem zawał mięśnia sercowego, niezakończony zgonem udar mózgu) zaobserwowano w łącznej grupie pacjentów otrzymujących febuksostat w porównaniu z grupą otrzymującą allopurynol w ramach badań APEX i FACT (1,3 vs. 0,3 zdarzenia na 100 pacjentolat), ale nie w badaniu CONFIRMS (szczegółowy opis badań, patrz punkt 5.1). Częstość zgłaszanych przez badacza zdarzeń sercowo- naczyniowych APTC w połączonych badaniach 3 fazy (APEX, FACT i CONFIRMS) wyniosła 0,7 vs. 0,6 zdarzenia na 100 pacjentolat. W długotrwałych badaniach rozszerzonych częstość zgłaszanych przez badacza zdarzeń sercowo-naczyniowych wyniosła odpowiednio 1,2 vs. 0,6 zdarzenia na 100 pacjentolat dla febuksostatu i dla allopurynolu. Nie stwierdzono statystycznie znaczących różnic ani związku przyczynowo-skutkowego ze stosowaniem febuksostatu. Rozpoznanymi czynnikami ryzyka u tych pacjentów były miażdżyca i (lub) zawał mięśnia sercowego albo zastoinowa niewydolność serca w wywiadzie. W badaniu porejestracyjnym CARES (patrz punkt 5.1, aby zapoznać się ze szczegółami badania) odsetek występowania ciężkich zdarzeń sercowo-naczyniowych (MACE – ang, Major Adverse Cardiac Events) był porównywalny u pacjentów leczonych febuksostatem oraz u pacjentów leczonych allopurynolem (HR 1,03; 95% CI 0.87-1,23), ale zaobserwowano wyższy odsetek zgonów z przyczyn sercowo-naczyniowych (odpowiednio 4,3% vs. 3,2% pacjentów, HR 1,34; 95% CI 1,03-1,73).
Alergia/nadwrażliwość na produkt leczniczy
Po wprowadzeniu febuksostatu do obrotu odnotowano rzadkie przypadki ciężkich reakcji alergicznych/nadwrażliwości, w tym zagrażającego życiu zespołu Stevensa-Johnsona, toksycznego martwiczego oddzielania się naskórka oraz ostrych reakcji anafilaktycznych/wstrząsu. W większości przypadków reakcje te występowały w pierwszym miesiącu leczenia febuksostatem. U niektórych pacjentów (nie u wszystkich) występowały zaburzenia czynności nerek i (lub) wcześniejsza nadwrażliwość na allopurynol. Ciężkie reakcje nadwrażliwości, w tym reakcje z eozynofilią
i objawami ogólnoustrojowymi (ang. Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS) wiązały się w niektórych przypadkach z gorączką, zaburzeniami układu krwiotwórczego, zaburzeniami czynności nerek lub wątroby.
Pacjentów należy poinformować o przedmiotowych i podmiotowych objawach reakcji alergicznych/ nadwrażliwości i ściśle kontrolować ich wystąpienie (patrz punkt 4.8). W razie wystąpienia ciężkich reakcji alergicznych/nadwrażliwości (w tym zespołu Stevensa-Johnsona) leczenie febuksostatem należy natychmiast przerwać, gdyż wczesne odstawienie daje lepsze rokowanie. U pacjentów,
u których wystąpiła reakcja alergiczna/nadwrażliwości (w tym zespół Stevensa-Johnsona i ostra
reakcja anafilaktyczna/wstrząs), nie wolno nigdy ponownie stosować febuksostatu.
Ostre napady dny (zaostrzenia dny moczanowej)
Leczenia febuksostatem nie należy rozpoczynać do czasu całkowitego ustąpienia ostrego ataku dny moczanowej. Na początku leczenia możliwe jest zaostrzenie dny moczanowej w wyniku zmian stężenia kwasu moczowego w surowicy na skutek uwalniania moczanu ze złogów w tkankach (patrz punkty 4.8 i 5.1). Podczas rozpoczynania leczenia febuksostatem zaleca się profilaktyczne podawanie NLPZ lub kolchicyny przez co najmniej 6 miesięcy w celu zapobiegania zaostrzeniom dny moczanowej (patrz punkt 4.2).
W razie zaostrzenia dny moczanowej w trakcie stosowania febuksostatu, leczenia nie należy przerywać. Zaostrzenie dny można równolegle leczyć w sposób odpowiedni dla danego pacjenta. Ciągłe leczenie febuksostatem zmniejsza częstość i nasilenie zaostrzeń dny.
Odkładanie się złogów ksantyny
U pacjentów ze znacznie przyspieszonym wytwarzaniem moczanu (np. z nowotworem złośliwym i w trakcie leczenia przeciwnowotworowego, z zespołem Lescha-Nyhana) bezwzględne stężenie ksantyny w moczu może w rzadkich przypadkach zwiększyć się w stopniu umożliwiającym odkładanie się jej złogów w drogach moczowych.
Ze względu na brak doświadczeń dotyczących febuksostatu, nie zaleca się stosowania febuksostatu u pacjentów z zespołem Lescha-Nyhana.
Merkaptopuryna/azatiopryna
Nie zaleca się stosowania febuksostatu u pacjentów jednocześnie leczonych merkaptopuryną lub azatiopryną, gdyż hamowanie aktywności oksydazy ksantynowej przez febuksostat może spowodować zwiększenie stężenia tych leków w osoczu, prowadzące do ciężkich działań toksycznych. Nie przeprowadzono badań interakcji u ludzi.
Jeśli nie można uniknąć jednoczesnego stosowania, zalecane jest zmniejszenie dawki merkaptopuryny lub azatiopryny. Na podstawie modelowania i analizy symulacyjnej danych z nieklinicznego badania na szczurach zaleca się, aby podczas jednoczesnego stosowania z febuksostatem dawkę merkaptopuryny lub azatiopryny zmniejszyć do 20% wcześniej przepisanej dawki lub mniejszej
w celu uniknięcia możliwych reakcji hematologicznych (patrz punkt 4.5).
Należy ściśle kontrolować stan pacjentów, a dawkę merkaptopuryny lub azatiopryny dostosowywać następnie na podstawie oceny odpowiedzi na leczenie i ewentualnego wystąpienia objawów działania toksycznego.
Pacjenci po przeszczepieniu narządu
Nie zaleca się stosowania febuksostatu u biorców przeszczepów ze względu na brak doświadczenia
w tej grupie pacjentów (patrz punkt 5.1).
Teofilina
Jednoczesne podanie febuksostatu w dawce 80 mg i pojedynczej dawki 400 mg teofiliny nie wykazało
u zdrowych ochotników jakiejkolwiek interakcji farmakokinetycznej (patrz punkt 4.5). Febuksostat w dawce 80 mg można stosować u pacjentów leczonych jednocześnie teofiliną bez ryzyka zwiększenia stężenia teofiliny w osoczu.
Brak dostępnych danych w odniesieniu do febuksostatu w dawce 120 mg.
Zaburzenia czynności wątroby
W trakcie połączonych badań klinicznych fazy 3 obserwowano u pacjentów leczonych febuksostatem
niewielkie zmiany wyników badań czynności wątroby (5,0%). Badanie czynności wątroby jest zalecane przed rozpoczęciem leczenia febuksostatem, a następnie okresowo zgodnie z oceną kliniczną (patrz punkt 5.1).
Zaburzenia tarczycy
U pacjentów długotrwale leczonych febuksostatem (5,5%) w ramach długofalowych otwartych badań rozszerzonych zaobserwowano zwiększone stężenie TSH (>5,5 μIU/ml). U pacjentów z zaburzeniami czynności tarczycy konieczne jest zachowanie ostrożności podczas stosowania febuksostatu (patrz punkt 5.1).
Laktoza
Produkt Druniler zawiera laktozę. Nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy- galaktozy.
Sód
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na tabletkę, to znaczy uznaje się go za
„wolny od sodu”.
Merkaptopuryna i azatiopryna
Ze względu na mechanizm działania hamującego aktywność oksydazy ksantynowej (XO) przez febuksostat, nie zaleca się jednoczesnego stosowania. Hamowanie aktywności XO przez febuksostat może być przyczyną zwiększenia stężenia tych substancji czynnych w osoczu, prowadzącego do toksyczności.
Nie przeprowadzono u ludzi badań interakcji febuksostatu z produktami leczniczymi (z wyjątkiem
teofiliny) metabolizowanymi przez XO.
Modelowanie i analiza symulacyjna danych z nieklinicznego badania na szczurach wskazuje że w przypadku jednoczesnego stosowania z febuksostatem dawka merkaptopuryny lub azatiopryny powinna zostać zmniejszona do 20% wcześniej przepisanej dawki lub mniejszej (patrz punkty 4.4 i 5.3).
Nie przeprowadzono badań interakcji febuksostatu z innymi lekami stosowanymi w chemioterapii. Brak dostępnych danych dotyczących bezpieczeństwa stosowania febuksostatu w trakcie leczenia cytotoksycznego.
Rozyglitazon/substraty CYP2C8
W warunkach in vitro wykazano, że febuksostat jest słabym inhibitorem CYP2C8. W badaniu
z udziałem zdrowych osób jednoczesne podanie febuksostatu w dawce 120 mg raz na dobę
i rozyglitazonu w pojedynczej doustnej dawce 4 mg nie wpływało na farmakokinetykę rozyglitazonu ani jego metabolitu N-demetylorozyglitazonu, co wskazuje, że febuksostat nie jest in vivo inhibitorem CYP2C8. W związku z tym jednoczesne podawanie febuksostatu i rozyglitazonu lub innych substratów CYP2C8 nie wymaga jakiejkolwiek modyfikacji dawkowania.
Teofilina
Przeprowadzono badanie interakcji z zastosowaniem febuksostatu u zdrowych osób w celu oceny, czy hamowanie aktywności XO może spowodować zwiększenie stężenia teofiliny we krwi, tak jak zgłaszano w przypadku innych inhibitorów XO. Wyniki badania wykazały, że jednoczesne podanie febuksostatu w dawce 80 mg raz na dobę i teofiliny w pojedynczej dawce 400 mg nie wpływa na farmakokinetykę lub bezpieczeństwo stosowania teofiliny. Dlatego jednoczesne stosowanie febuksostatu w dawce 80 mg i teofiliny nie wymaga szczególnej ostrożności. Brak dostępnych danych w odniesieniu do febuksostatu w dawce 120 mg.
Naproksen i inne inhibitory glukuronidacji
Metabolizm febuksostatu zależy od UDP-glukuronylotransferaz (UGT). Produkty lecznicze, które
hamują glukuronidację, takie jak NLPZ i probenecyd, mogą teoretycznie wpływać na eliminację
febuksostatu. U zdrowych osób jednoczesne stosowanie febuksostatu i naproksenu w dawce 250 mg dwa razy na dobę wiązało się ze zwiększoną ekspozycją na febuksostat (Cmax 28%, AUC 41% i t1/2 26%). W badaniach klinicznych stosowanie naproksenu lub innych NLPZ/inhibitorów COX-2 nie wiązało się z żadnym klinicznie istotnym zwiększeniem częstości działań niepożądanych.
Febuksostat można stosować jednocześnie z naproksenem bez konieczności modyfikacji dawki żadnego z tych produktów leczniczych.
Induktory glukuronidacji
Leki silnie pobudzające aktywność enzymów UGT mogą powodować nasilenie metabolizmu
i zmniejszać skuteczność febuksostatu. Dlatego zaleca się kontrolowanie stężenia kwasu moczowego w surowicy przez 1-2 tygodnie po rozpoczęciu leczenia silnym lekiem indukującym glukuronidację. Odwrotnie, odstawienie produktu leczniczego indukującego glukuronidację może spowodować zwiększenie stężenia febuksostatu w osoczu.
Kolchicyna/indometacyna/hydrochlorotiazyd/warfaryna
Febuksostat można podawać razem z kolchicyną lub indometacyną bez konieczności modyfikowania dawki febuksostatu lub jednocześnie stosowanej substancji czynnej.
Nie jest konieczna modyfikacja dawki febuksostatu stosowanego jednocześnie z hydrochlorotiazydem. Nie jest konieczna modyfikacja dawki warfaryny stosowanej jednocześnie z febuksostatem.
Stosowanie febuksostatu (80 mg lub 120 mg raz na dobę) z warfaryną u zdrowych osób nie wpływało
na farmakokinetykę warfaryny. Jednocześnie stosowany febuksostat nie wpływał również na wartość INR ani aktywność czynnika VII.
Dezypramina/substraty CYP2D6
Wykazano, że w warunkach in vitro febuksostat jest słabym inhibitorem CYP2D6. W badaniu
u zdrowych osób podanie 120 mg febuksostatu raz na dobę powodowało zwiększenie średnio o 22% wartości AUC dla dezypraminy (substratu CYP2D6), co wskazuje na możliwość słabego hamującego działania febuksostatu in vivo na aktywność CYP2D6. Dlatego jednoczesne podawanie febuksostatu z innymi substratami CYP2D6 nie powinno wymagać modyfikacji dawki którejkolwiek z tych substancji.
Leki zobojętniające kwas solny w żołądku
Wykazano, że jednoczesne przyjęcie leku zobojętniającego zawierającego wodorotlenek magnezu
i wodorotlenek glinu opóźnia wchłanianie febuksostatu (o około 1 godziny) i powoduje zmniejszenie o 32% wartości Cmax bez znaczącej zmiany AUC. Dlatego febuksostat można przyjmować niezależnie od stosowania leków zobojętniających.
Ciąża
Bardzo nieliczne dane dotyczące stosowania febuksostatu w okresie ciąży nie wskazują na jakikolwiek niepożądany wpływ na przebieg ciąży lub zdrowie płodu i (lub) noworodka. Badania na zwierzętach nie wykazują bezpośredniego lub pośredniego szkodliwego wpływu na przebieg ciąży, rozwój zarodka i (lub) płodu ani na przebieg porodu (patrz punkt 5.3). Potencjalne zagrożenie dla człowieka nie jest znane. Febuksostatu nie należy stosować w okresie ciąży.
Karmienie piersią
Nie wiadomo, czy febuksostat przenika do mleka kobiecego. Badania na zwierzętach wykazały przenikanie tej substancji czynnej do mleka i zaburzenia rozwoju karmionych młodych. Nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią. Febuksostatu nie należy stosować w okresie karmienia piersią.
Płodność
W badaniach reprodukcji u zwierząt nie wykazano, aby febuksostat podawany w dawkach do
48 mg/kg mc. na dobę wykazywał zależny od dawki niekorzystny wpływ na płodność (patrz punkt
5.3). Wpływ febuksostatu na płodność u ludzi nie jest znany.
Podczas stosowania febuksostatu zgłaszano senność, zawroty głowy, odczucie mrowienia
i niewyraźne widzenie. Pacjenci powinni zachować ostrożność przed podjęciem prowadzenia pojazdów, obsługiwania maszyn lub wykonywania niebezpiecznych czynności do czasu upewnienia się, że produkt Druniler nie wpływa niekorzystnie na ich sprawność.
Podsumowanie profilu bezpieczeństwa
Najczęściej zgłaszanymi działaniami niepożądanymi u pacjentów z dną moczanową, odnotowanymi w badaniach klinicznych (4072 pacjentów, którzy otrzymali co najmniej jedną dawkę z zakresu od 10 mg do 300 mg) oraz po wprowadzeniu febuksostatu do obrotu były: zaostrzenie objawów dny, zaburzenia czynności wątroby, biegunka, nudności, ból głowy, wysypka i obrzęk. Nasilenie tych
działań było przeważnie lekkie lub umiarkowane. Po wprowadzeniu febuksostatu do obrotu zgłaszano rzadkie przypadki ciężkich reakcji nadwrażliwości, niektóre z nich związane z objawami ogólnoustrojowymi, oraz rzadkie przypadki nagłego zgonu sercowego.
Tabelaryczne zestawienie działań niepożądanych
Niżej wymieniono częste (1/100 do <1/10), niezbyt częste (1/1000 do <1/100) oraz rzadkie (1/10 000 do <1/1000) działania niepożądane występujące u pacjentów leczonych febuksostatem.
W obrębie każdej z grup o danej częstości działania niepożądane wymieniono zgodnie ze zmniejszającym się nasileniem.
Tabela 1: Działania niepożądane notowane w trakcie badań klinicznych fazy 3, długotrwałych badań rozszerzonych i po wprowadzeniu febuksostatu do obrotu
Zaburzenia krwi i układu chłonnego | Rzadko Pancytopenia, małopłytkowość, agranulocytoza* |
Zaburzenia układu immunologicznego | Rzadko Reakcja anafilaktyczna*, nadwrażliwość na lek* |
Zaburzenia endokrynologiczne | Niezbyt często Zwiększona aktywność TSH we krwi |
Zaburzenia oka | Rzadko Niewyraźne widzenie |
Zaburzenia metabolizmu i odżywiania | Często*** Zaostrzenie objawów dny Niezbyt często Cukrzyca, hiperlipidemia, zmniejszenie łaknienia, zwiększenie masy ciała Rzadko Zmniejszenie masy ciała, zwiększenie łaknienia, jadłowstręt |
Zaburzenia psychiczne | Niezbyt często Zmniejszony popęd płciowy, bezsenność Rzadko Nerwowość |
Zaburzenia układu nerwowego | Często Ból głowy Niezbyt często Zawroty głowy, parestezje, niedowład połowiczy, senność, zmiana smaku, niedoczulica, osłabienie węchu |
Zaburzenia ucha i błędnika | Rzadko Szum w uszach |
Zaburzenia serca | Niezbyt często Migotanie przedsionków, kołatanie serca, nieprawidłowy zapis EKG Rzadko Nagły zgon sercowy* |
Zaburzenia naczyniowe | Niezbyt często Nadciśnienie tętnicze, zaczerwienienie skóry, uderzenia gorąca |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Niezbyt często Duszność, zapalenie oskrzeli, zakażenie górnych dróg oddechowych, kaszel |
Zaburzenia żołądka i jelit | Często Biegunka**, nudności Niezbyt często Ból brzucha, rozdęcie brzucha, refluks żołądkowo- przełykowy, wymioty, suchość w jamie ustnej, niestrawność, zaparcie, częste oddawanie stolca, wzdęcia z oddawaniem gazów, odczucie dyskomfortu w przewodzie pokarmowym Rzadko Zapalenie trzustki, owrzodzenie jamy ustnej |
Zaburzenia wątroby i dróg żółciowych | Często Nieprawidłowa czynność wątroby** Niezbyt często Kamica żółciowa Rzadko Zapalenie wątroby, żółtaczka*, uszkodzenie wątroby* |
Zaburzenia skóry i tkanki podskórnej | Często Wysypka (w tym różne rodzaje rzadziej występujących wysypek, patrz niżej) Niezbyt często Zapalenie skóry, pokrzywka, świąd, przebarwienie skóry, uszkodzenie skóry, wybroczyny, wysypka plamkowa, wysypka grudkowo-plamkowa, wysypka grudkowa Rzadko Toksyczne martwicze oddzielanie się naskórka*, zespół Stevensa-Johnsona*, obrzęk naczynioruchowy*, reakcja polekowa z eozynofilią i objawami układowymi*, uogólniona wysypka (ciężka)*, rumień, wysypka złuszczająca, wysypka grudkowa, wysypka pęcherzykowa, wysypka krostkowa, swędząca wysypka*, wysypka rumieniowata, wysypka odropodobna, łysienie, nadmierne pocenie się |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | Niezbyt często Ból stawów, zapalenie stawów, ból mięśni, ból mięśniowo- kostny, osłabienie mięśni, skurcze mięśni, nadmierne napięcie mięśni, zapalenie kaletki Rzadko Rabdomioliza*, sztywność stawów, sztywność mięśniowo- kostna |
Zaburzenia nerek i dróg moczowych | Niezbyt często Niewydolność nerek, kamica nerkowa, krwiomocz, częstomocz, białkomocz Rzadko |
Cewkowo-śródmiąższowe zapalenie nerek*, nagła potrzeba oddania moczu | |
Zaburzenia układu rozrodczego i piersi | Niezbyt często Zaburzenia wzwodu |
Zaburzenia ogólne i stany w miejscu podania | Często Obrzęk Niezbyt często Zmęczenie, ból w klatce piersiowej, odczucie dyskomfortu w klatce piersiowej Rzadko Nadmierne pragnienie |
Badania diagnostyczne | Niezbyt często Zwiększenie aktywności amylazy we krwi, zmniejszenie liczby płytek krwi, zmniejszenie liczby krwinek białych, zmniejszenie liczby limfocytów, zwiększenie stężenia kreatyny we krwi, zwiększenie stężenia kreatyniny we krwi, zmniejszenie stężenia hemoglobiny, zwiększenie stężenia mocznika we krwi, zwiększenie stężenia triglicerydów we krwi, zwiększenie stężenia cholesterolu we krwi, zmniejszenie hematokrytu, zwiększenie aktywności LDH we krwi, zwiększenie stężenia potasu we krwi Rzadko Zwiększenie stężenia glukozy we krwi, wydłużony czas kaolinowo-kefalinowy, zmniejszenie liczby krwinek czerwonych, zwiększenie aktywności fosfatazy zasadowej, zwiększenie aktywności kinazy kreatynowej* |
* Działania niepożądane odnotowane w okresie po wprowadzeniu febuksostatu do obrotu
** Wywołana leczeniem biegunka niezakaźna i nieprawidłowe wyniki badań czynności wątroby w łączonych badaniach 3 fazy występują częściej u pacjentów leczonych jednocześnie kolchicyną.
*** Częstość przypadków zaostrzenia dny moczanowej w poszczególnych randomizowanych, kontrolowanych badaniach 3 fazy, patrz punkt 5.1.
Opis wybranych działań niepożądanych
Po wprowadzeniu febuksostatu do obrotu odnotowano rzadkie przypadki ciężkich reakcji nadwrażliwości na tę substancję, w tym zespołu Stevensa-Johnsona, toksycznego martwiczego oddzielania się naskórka i reakcji anafilaktycznych/wstrząsu. Zespół Stevensa-Johnsona i toksyczne martwicze oddzielanie się naskórka charakteryzują się nasilającą się wysypką skórną z powstawaniem pęcherzy lub uszkodzeniem błon śluzowych i podrażnieniem oczu. Razem z reakcjami nadwrażliwości na febuksostat mogą wystąpić następujące objawy: reakcje skórne w postaci naciekowych zmian grudkowo-plamkowych, uogólnionych lub złuszczających wysypek, uszkodzenie skóry, obrzęk twarzy, gorączka, nieprawidłowe parametry hematologiczne (takie jak małopłytkowość i eozynofilia) oraz zmiany obejmujące pojedyncze narządy lub wielonarządowe (wątroba i nerki, w tym cewkowo- śródmiąższowe zapalenie nerek), patrz punkt 4.4.
Wkrótce po rozpoczęciu leczenia i podczas pierwszych miesięcy leczenia często obserwowano objawy zaostrzenia dny moczanowej, ale później częstość zaostrzeń objawów dny zmniejszała się z czasem.
Zaleca się stosowanie leczenia profilaktycznego przeciw dnie (patrz punkty 4.2 i 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301, faks: + 48 22 49 21 309, strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W razie przedawkowania należy zastosować leczenie objawowe i podtrzymujące.
Grupa farmakoterapeutyczna: leki przeciw dnie, leki hamujące biosyntezę kwasu moczowego. Kod ATC: M04AA03
Mechanizm działania
Kwas moczowy jest końcowym produktem metabolizmu puryn u ludzi i jest wytwarzany w kaskadzie hipoksantyna ksantyna kwas moczowy. Oba etapy tych przemian katalizowane są przez oksydazę ksantynową (XO). Febuksostat jest pochodną 2-aryltiazolu. Działanie lecznicze polegające na zmniejszeniu stężenia kwasu moczowego w surowicy wywiera przez wybiórcze hamowanie aktywności XO. Febuksostat jest silnym, niepurynowym, wybiórczym inhibitorem XO (NP-SIXO) ze stałą Ki w warunkach in vitro poniżej jednego nanomola. Wykazano, że febuksostat silnie hamuje zarówno utlenione, jak i zredukowane postacie XO. W stężeniach terapeutycznych febuksostat nie hamuje innych enzymów uczestniczących w metabolizmie puryn lub pirymidyn, tzn. deaminazy guaninowej, fosforybozylotransferazy hipoksantynowo-guaninowej, fosforybozylotransferazy orotanowej, dekarboksylazy monofosforanu orotydyny lub fosforylazy nukleozydów purynowych.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność kliniczną febuksostatu wykazano w trzech głównych badaniach klinicznych 3 fazy (dwa główne badania APEX i FACT oraz dodatkowe badanie CONFIRMS opisane niżej), które przeprowadzono z udziałem 4101 pacjentów z hiperurykemią i dną moczanową. W każdym
z głównych badań 3 fazy febuksostat wykazywał większą niż allopurynol zdolność zmniejszania
i utrzymywania stężenia kwasu moczowego w surowicy. Pierwszorzędowym punktem końcowym skuteczności w badaniach APEX i FACT był odsetek pacjentów, u których stężenie kwasu moczowego w surowicy w ostatnich 3 comiesięcznych pomiarach było poniżej 6,0 mg/dl
(357 μmol/l). W dodatkowym badaniu 3 fazy CONFIRMS, którego wyniki zostały udostępnione po uzyskaniu pierwszego pozwolenia na dopuszczenie do obrotu febuksostatu, pierwszorzędowym punktem końcowym skuteczności był odsetek pacjentów, u których stężenie kwasu moczowego
w surowicy było mniejsze niż 6,0 mg/dl podczas wizyty końcowej. Do badań nie włączono pacjentów
po przeszczepieniu narządu (patrz punkt 4.4).
Badanie APEX
APEX (Allopurinol and Placebo-Controlled Efficacy Study of Febuxostat) było randomizowanym, wieloośrodkowym, trwającym 28 tygodni badaniem klinicznym 3 fazy z podwójnie ślepą próbą.
Tysiąc siedemdziesięciu dwóch (1072) pacjentów przydzielono losowo do następujących grup: placebo (n=134), febuksostat w dawce 80 mg raz na dobę (n=267), febuksostat w dawce 120 mg raz na dobę (n=269), febuksostat w dawce 240 mg raz na dobę (n=134) lub allopurynol w dawce 300 mg raz na dobę (n=258) u pacjentów z wyjściowym stężeniem kreatyniny w surowicy ≤1,5 mg/dl albo
w dawce 100 mg raz na dobę (n=10) u pacjentów z wyjściowym stężeniem kreatyniny w surowicy
>1,5 mg/dl, ale ≤2,0 mg/dl. Dawką do oceny bezpieczeństwa febuksostatu było 240 mg (2-krotność największej zalecanej dawki).
Badanie APEX wykazało statystycznie istotną przewagę zarówno febuksostatu w dawce 80 mg raz na
dobę, jak i febuksostatu w dawce 120 mg raz na dobę nad allopurynolem w standardowych dawkach
300 mg (n=258) / 100 mg (n=10) w odniesieniu do zmniejszania stężenia kwasu moczowego
w surowicy poniżej 6 mg/dl (357 μmol/l), patrz tabela 2 i rycina 1.
Badanie FACT
FACT (Febuxostat Allopurinol Controlled Trial) było randomizowanym, wieloośrodkowym, trwającym 52 tygodnie badaniem 3 fazy z podwójnie ślepą próbą. Siedmiuset sześćdziesięciu (760) pacjentów przydzielono losowo do grup otrzymujących raz na dobę: febuksostat w dawce 80 mg (n=256), febuksostat w dawce 120 mg (n=251) lub allopurynol w dawce 300 mg (n=253).
Badanie FACT wykazało statystycznie istotną przewagę febuksostatu zarówno w dawce 80 mg raz na
dobę, jak i w dawce 120 mg raz na dobę nad allopurynolem w standardowej dawce 300 mg
w odniesieniu do zmniejszania i utrzymania stężenia kwasu moczowego w surowicy poniżej 6 mg/dl (357 μmol/l).
W tabeli 2 podsumowano wyniki dla pierwszorzędowego punktu końcowego.
Tabela 2
Odsetek pacjentów ze stężeniem kwasu moczowego w surowicy <6,0 mg/dl (357 μmol/l) Ostatnie trzy comiesięczne wizyty
Badanie | Febuksostat 80 mg QD | Febuksostat 120 mg QD | Allopurynol 300 mg / 100 mg QD1 |
APEX | 48% * | 65%*, # | 22% |
(28 tygodni) | (n=262) | (n=269) | (n=268) |
FACT | 53%* | 62%* | 21% |
(52 tygodnie) | (n=255) | (n=250) | (n=251) |
Łączne wyniki | 51%* | 63%*, # | 22% |
(n=517) | (n=519) | (n=519) | |
1 analiza łącznych wyników uzyskanych u osób otrzymujących 100 mg QD (n=10: pacjenci ze stężeniem kreatyniny w surowicy >1,5 i ≤2,0 mg/dl) lub 300 mg QD (n=509) * p <0,001 vs. allopurynol, # p <0,001 vs. 80 mg | |||
Febuksostat szybko i trwale zmniejszał stężenie kwasu moczowego w surowicy. Zmniejszenie stężenia kwasu moczowego w surowicy do wartości <6,0 mg/dl (357 μmol/l) stwierdzano do wizyty w 2. tygodniu, a następnie stężenie to utrzymywało się przez cały okres leczenia. Średnie stężenie kwasu moczowego w surowicy w czasie u pacjentów każdej z grup terapeutycznych w dwóch głównych badaniach klinicznych 3. fazy przedstawiono na rycinie 1.
Rycina 1 Średnie stężenie kwasu moczowego w surowicy – połączone wyniki z głównych badań 3. fazy
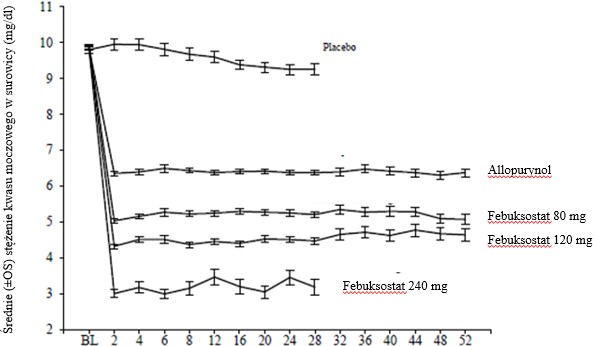
Tydzień
BL = wartości początkowe OS = odchylenie standardowe
Uwaga: 509 pacjentów otrzymywało allopurynol w dawce 300 mg raz na dobę, a 10 pacjentów ze
stężeniem kreatyniny w surowicy w zakresie >1,5 mg/dl i < 2,0 mg/dl dawkę 100 mg na dobę (10 z 268 pacjentów w badaniu APEX). Zastosowano febuksostat w dawce 240 mg w celu oceny bezpieczeństwa stosowania dawki dwukrotnie większej od zalecanej maksymalnej dawki.
Badanie CONFIRMS
To randomizowane, kontrolowane, 26-tygodniowe badanie 3.fazy miało na celu ocenę bezpieczeństwa stosowania i skuteczności febuksostatu w dawce 40 mg i 80 mg u pacjentów z dną moczanową
i hiperurykemią w porównaniu z allopurynolem w dawce 300 mg lub 200 mg. Dwa tysiące sześćdziesięciu dziewięciu (2269) pacjentów przydzielono losowo do grup otrzymujących raz na dobę: febuksostat w dawce 40 mg (n=757), febuksostat w dawce 80 mg (n=756) lub allopurynol w dawce 300 albo 200 mg (n=756). U co najmniej 65% pacjentów występowały łagodne do umiarkowanych zaburzenia czynności nerek (klirens kreatyniny 30-89 ml/min). Przez 26 tygodni trwania badania obowiązkowa była profilaktyka zaostrzenia dny moczanowej.
Odsetek pacjentów ze stężeniem kwasu moczowego w surowicy <6,0 mg/dl (357 μmol/l) podczas wizyty końcowej wynosił, odpowiednio, 45% w grupie otrzymującej febuksostat w dawce 40 mg, 67% w grupie otrzymującej febuksostat w dawce 80 mg i 42% w grupie otrzymującej allopurynol w dawce 300 lub 200 mg.
Pierwszorzędowy punkt końcowy w podgrupie pacjentów z zaburzeniami czynności nerek
W badaniu APEX oceniano skuteczność u 40 pacjentów z zaburzeniami czynności nerek (tzn.
z początkowym stężeniem kreatyniny w surowicy >1,5 mg/dl i ≤2,0 mg/dl). Pacjentom z zaburzeniami czynności nerek przydzielonym do grupy otrzymującej allopurynol ograniczono dawkę do 100 mg raz na dobę. Pierwszorzędowy punkt końcowy osiągnięto u 44% pacjentów z grupy otrzymującej febuksostat w dawce 80 mg na dobę, 45% pacjentów otrzymujących febuksostat w dawce 120 mg na dobę i 60% pacjentów otrzymujących febuksostat w dawce 240 mg na dobę w porównaniu z 0%
w grupie otrzymującej allopurynol w dawce 100 mg na dobę i grupie placebo.
Nie odnotowano klinicznie istotnych różnic w procentowym zmniejszeniu stężenia kwasu moczowego
w surowicy u osób zdrowych, bez względu na ich czynność nerek. Zmniejszenie wyniosło 58%
w grupie z prawidłową czynnością nerek i 55% w grupie z ciężkimi zaburzeniami czynności nerek.
W badaniu CONFIRMS dokonano prospektywnej analizy wyników uzyskanych u pacjentów z dną moczanową i zaburzeniami czynności nerek. Wykazano znacząco większą skuteczność febuksostatu w zmniejszaniu stężenia kwasu moczowego w surowicy do <6,0 mg/dl w porównaniu z allopurynolem w dawce 300 mg lub 200 mg u pacjentów z dną moczanową i łagodnymi do umiarkowanych zaburzeniami czynności nerek (65% badanych pacjentów).
Pierwszorzędowy punkt końcowy w podgrupie pacjentów ze stężeniem kwasu moczowego w surowicy
≥10 mg/dl
U około 40% pacjentów (połączone wyniki badań APEX i FACT) początkowe stężenie kwasu moczowego w surowicy wynosiło ≥10 mg/dl. W tej podgrupie pierwszorzędowy punkt końcowy (stężenie kwasu moczowego w surowicy <6,0 mg/dl podczas ostatnich trzech wizyt) osiągnięto u 41% pacjentów otrzymujących febuksostat w dawce 80 mg na dobę), u 48% pacjentów otrzymujących febuksostat w dawce 120 mg na dobę) i u 66% pacjentów otrzymujących febuksostat w dawce 240 mg na dobę w porównaniu z 9% w grupie otrzymującej allopurynol w dawce 300 mg lub 100 mg na dobę i 0% w grupie otrzymującej placebo.
W badaniu CONFIRMS odsetek pacjentów z początkowym stężeniem kwasu moczowego w surowicy
≥10 mg/dl, u których osiągnięto pierwszorzędowy punkt końcowy (stężenie kwasu moczowego
w surowicy <6,0 mg/dl podczas wizyty końcowej), otrzymujących febuksostat w dawce 40 mg raz na dobę wyniósł 27% (66/249), otrzymujących febuksostat w dawce 80 mg raz na dobę 49% (125/254), a otrzymujących allopurynol w dawce 300 mg lub 200 mg raz na dobę 31% (72/230).
Wyniki kliniczne: odsetek pacjentów wymagających leczenia z powodu zaostrzenia dny moczanowej
Badanie APEX
Podczas 8-tygodniowego okresu stosowania profilaktyki u większego odsetka pacjentów z grupy otrzymującej febuksostat w dawce 120 mg (36%) konieczne było leczenie z powodu zaostrzenia dny moczanowej niż u pacjentów z grupy otrzymującej febuksostat w dawce 80 mg (28%), allopurynol 300 mg (23%) i placebo (20%). Zaostrzenie następowało po zakończeniu okresu leczenia profilaktycznego i zmniejszało się stopniowo z czasem. Od 46% do 55% pacjentów otrzymało leczenie z powodu zaostrzenia dny moczanowej od tygodnia 8. do 28. Zaostrzenie dny moczanowej w ostatnich 4 tygodniach badania (tygodnie 24-28) odnotowano u 15% pacjentów otrzymujących febuksostat w dawce 80 mg, 120 mg, u 14% pacjentów otrzymujących allopurynol w dawce 300 mg i u 20% pacjentów otrzymujących placebo.
Badanie FACT
W trakcie 8-tygodniowego okresu stosowania profilaktyki u większego odsetka pacjentów z grupy otrzymującej febuksostat w dawce 120 mg (36%) konieczne było leczenie z powodu zaostrzenia dny moczanowej niż zarówno u pacjentów z grupy otrzymującej febuksostat w dawce 80 mg (22%), jak
i otrzymujących allopurynol w dawce 300 mg (21%). Po zakończeniu 8-tygodniowego okresu leczenia profilaktycznego następowało zaostrzenie i zmniejszało się stopniowo z czasem (64% i 70% pacjentów otrzymało leczenie z powodu zaostrzenia dny moczanowej od 8. do 52. tygodnia).
Zaostrzenie dny moczanowej w ciągu ostatnich 4 tygodni badania (tygodnie 49-52) zaobserwowano u 6-8% pacjentów otrzymujących febuksostat w dawce 80 mg, 120 mg i 11% pacjentów otrzymujących allopurynol w dawce 300 mg.
Odsetek pacjentów wymagających leczenia z powodu zaostrzenia dny moczanowej (badania APEX
i FACT) był liczebnie mniejszy w grupach, które osiągnęły średnie stężenie kwasu moczowego
w surowicy po rozpoczęciu badania <6,0 mg/dl, <5,0 mg/dl lub <4,0 mg/dl w porównaniu z grupą, która osiągnęła średnie stężenie kwasu moczowego w surowicy po rozpoczęciu badania ≥6,0 mg/dl w trakcie ostatnich 32 tygodni okresu leczenia (odstępy czasowe tygodnie 20-24 do tygodni 49-52).
Podczas badania CONFIRMS odsetek pacjentów wymagających leczenia z powodu zaostrzenia dny moczanowej (dzień 1. przez 6 miesięcy) wynosił odpowiednio 31% i 25% w grupie otrzymującej febuksostat i allopurynol. Nie odnotowano różnic w odsetku pacjentów wymagających leczenia
z powodu zaostrzenia dny moczanowej między grupami, którym podawano febuksostat w dawkach 40 mg i 80 mg.
Długotrwałe, otwarte badania rozszerzające
Badanie EXCEL (C02-021)
Badanie EXCEL było trwającym 3 lata, otwartym, wieloośrodkowym, kontrolowanym allopurynolem rozszerzającym badaniem 3.fazy oceniającym bezpieczeństwo stosowania obejmującym pacjentów, którzy ukończyli główne badanie 3.fazy (APEX lub FACT). Do badania włączono 1086 pacjentów otrzymujących raz na dobę: febuksostat w dawce 80 mg (n=649), febuksostat w dawce 120 mg (n=292) i allopurynol w dawce 300 mg lub 100 mg [n=145]. U około 69% pacjentów zmiana leczenia w celu osiągnięcia finalnego stabilnego leczenia nie była konieczna. Z badania wyłączono pacjentów ze stężeniem kwasu moczowego w surowicy <6,0 mg/dl podczas 3 kolejnych badań.
Stężenie kwasu moczowego w surowicy utrzymywało się na stałym poziomie w czasie całego badania
(tzn. u 91% i 93% pacjentów otrzymujących początkowo febuksostat odpowiednio w dawce 80 mg i 120 mg stężenie kwasu moczowego w surowicy w 36. miesiącu wynosiło <6,0 mg/dl).
Dane z trzech lat leczenia wykazały zmniejszenie częstości zaostrzeń dny moczanowej, a leczenie z powodu zaostrzenia konieczne było u mniej niż 4% pacjentów (tzn. ponad 96% pacjentów nie wymagało leczenia w związku z zaostrzeniem) w miesiącach 16-24 oraz 30-36.
U 46% i 38% pacjentów otrzymujących raz na dobę stałą dawkę podtrzymującą febuksostatu 80 mg lub 120 mg odnotowano podczas wizyty końcowej całkowity zanik wyczuwalnych guzków dnawych obserwowanych na początku leczenia.
Badanie FOCUS (TMX-01-005) było 5-letnim, otwartym, wieloośrodkowym, rozszerzającym badaniem 2. fazy dotyczącym bezpieczeństwa stosowania febuksostatu z udziałem pacjentów, którzy ukończyli 4-tygodniowe badanie TMX-00-004 z podwójnie ślepą próbą. Do badania włączono 116 pacjentów, którym podawano początkowo febuksostat w dawce 80 mg raz na dobę. U 62% pacjentów dostosowanie dawki w celu utrzymania stężenia kwasu moczowego w surowicy <6,0 mg/dl nie było konieczne, a 38% pacjentów wymagało modyfikacji dawki w celu osiągnięcia końcowej stabilnej dawki.
U ponad 80% (81-100%) pacjentów stężenie kwasu moczowego w surowicy podczas końcowej wizyty kontrolnej wyniosło <6,0 mg/dl (357 μmol/l) po podaniu każdej dawki febuksostatu.
Podczas klinicznych badań 3. fazy obserwowano niewielkie nieprawidłowości w testach czynności wątroby u pacjentów leczonych febuksostatem (5%). Podobne wyniki zgłaszano po zastosowaniu allopurynolu (4,2%), patrz punkt 4.4. Zwiększone stężenie TSH (>5,5 μIU/ml) odnotowano
u pacjentów leczonych przez długi okres febuksostatem (5,5%) i u pacjentów leczonych allopurynolem (5,8%) w długotrwałych otwartych badaniach rozszerzonych (patrz punkt 4.4).
Długotrwałe badania po wprowadzeniu do obrotu
Badanie CARES było wieloośrodkowym, randomizowanym badaniem z podwójnie ślepą próbą prowadzonym w celu wykazania równoważności, porównującym zdarzenia sercowo-naczyniowe występujące przy stosowaniu febuksostatu i przy stosowaniu allopurynolu u pacjentów z dną moczanową i ciężkimi chorobami układu sercowo-naczyniowego w wywiadzie, obejmującymi: zawał mięśnia sercowego, hospitalizację z powodu niestabilnej dławicy piersiowej, zabiegi rewaskularyzacji naczyń serca lub mózgu, udar, hospitalizację z powodu przemijającego ataku niedokrwiennego, chorobę naczyń obwodowych lub cukrzycę z oznakami powikłań mikro- lub makronaczyniowych. W celu osiągnięcia stężenia kwasu moczowego w surowicy mniejszego niż 6,0 mg/dl, dawkę febuksostatu podawano stopniowo, od 40 mg do 80 mg (niezależnie od czynności nerek) a dawkę allopurynolu zwiększano stopniowo o 100 mg w zakresie od 300 mg do 600 mg u pacjentów z prawidłową funkcją nerek i łagodnymi zaburzeniami czynności nerek oraz w zakresie dawek od 200 mg do 400 mg u pacjentów z umiarkowanymi zaburzeniami czynności nerek. Pierwszorzędowym punktem końcowym w badaniu CARES był czas do wystąpienia pierwszego ciężkiego zdarzenia niepożądanego sercowo-naczyniowego (MACE), złożonego z niezakończonego zgonem zawału mięśnia sercowego, niezakończonego zgonem udaru, zgonu z przyczyn sercowo-naczyniowych i
niestabilnej dławicy piersiowej z nagłą koniecznością rewaskularyzacji wieńcowej. Punkty końcowe (pierwszorzędowy i drugorzędowy) były analizowane w populacji zgodnej z zaplanowanym leczeniem (ITT, ang. intention-to-treat analysis) włączając wszystkie osoby przydzielone przez randomizację, które otrzymały przynajmniej jedną dawkę produktu w badaniu z podwójnie ślepą próbą. Ogółem 56,6% pacjentów zakończyło badanie przed czasem i 45% pacjentów nie stawiło się na wszystkie zaplanowane w badaniu wizyty. W sumie obserwowano 6190 pacjentów średnio przez medianę 32 miesięcy, przy ekspozycji trwającej średnio 728 dni w grupie pacjentów leczonych febuksostatem (n=3098) i 719 dni w grupie otrzymującej allopurynol (n=3092). Pierwszorzędowy punkt końcowy MACE wystąpił z podobną częstością w grupie otrzymującej febuksostat i grupie otrzymującej allopurynol (odpowiednio, 10,8% vs. 10,4% pacjentów; ryzyko względne [HR – hazard ratio] 1,03; obustronny powtórzony 95 % przedział ufności [CI – confidence interval] 0,87- 1,23). W analizie poszczególnych składowych MACE odsetek zgonów z przyczyn sercowo-naczyniowych był wyższy w związku ze stosowaniem febuksostatu, niż w przypadku stosowania allopurynolu (4,3% vs. 3,2% pacjentów, HR 1,34; 95% CI 1,03–1,73). Odsetek innych składowych MACE był porównywalny w grupie otrzymującej febuksostat i grupie otrzymującej allopurynol, tj. niezakończony zgonem zawał mięśnia sercowego (3,6% vs. 3,8% pacjentów; HR 0,93; 95% CI 0,72-1,21), niezakończony zgonem
udar (2,3% vs. 2,3% pacjentów; HR 1,01; 95% CI 0,73-1,41) i nagła rewaskularyzacja z powodu
niestabilnej dławicy piersiowej (1,6% vs. 1,8% pacjentów, HR 0,86; 95% CI 0,59-1,26). Odsetek śmiertelności ze wszystkich powodów był również wyższy dla febuksostatu niż dla allopurynolu (7,8% vs. 6,4% pacjentów; HR 1,22 95% CI 1,01-1,47), co było spowodowane głównie przez wyższy odsetek zgonów z przyczyn sercowo-naczyniowych w tej grupie (patrz punkt 4.4). Odsetek potwierdzonych hospitalizacji z powodu niewydolności serca, przyjęć do szpitala z powodu zaburzeń rytmu serca niezwiązanych z niedokrwieniem, żylnymi incydentami zakrzepowo-zatorowymi i hospitalizacji z powodu przemijającego ataku niedokrwiennego, był porównywalny dla febuksostatu i allopurynolu.
U zdrowych osób maksymalne stężenie febuksostatu w osoczu (Cmax) oraz pole powierzchni pod krzywą zależności jego stężenia w osoczu od czasu (AUC) po jednorazowym i wielokrotnym podaniu dawek 10 mg do 120 mg zwiększa się proporcjonalnie do dawki. Po podaniu dawek 120 mg i 300 mg zwiększenie wartości AUC dla febuksostatu było większe niż proporcjonalne. Podawanie dawek od 10 mg do 240 mg nie powodowało znaczącej kumulacji febuksostatu. Pozorny okres półtrwania febuksostatu w fazie eliminacji (t1/2) wynosi około 5 do 8 godzin.
Farmakokinetyczne i farmakodynamiczne analizy populacyjne przeprowadzono u 211 pacjentów z hiperurykemią i dną moczanową, leczonych febuksostatem w dawce 40-240 mg raz na dobę.
Oceniane w tej analizie parametry farmakokinetyczne były zasadniczo zgodne z danymi uzyskanymi u osób zdrowych, co wskazuje, że zdrowi uczestnicy są reprezentatywni dla oceny farmakokinetyki lub farmakodynamiki w populacji pacjentów z dną moczanową.
Wchłanianie
Febuksostat jest szybko (tmax = 1,0-1,5 godziny) i dobrze wchłaniany (≥84%). Po jednorazowym lub wielokrotnym doustnym podaniu dawek 80 i 120 mg raz na dobę febuksostat osiąga maksymalne stężenie (Cmax) odpowiednio około 2,8-3,2 µg/ml i 5,0-5,3 µg/ml. Nie oceniano bezwzględnej biodostępności febuksostatu uwalnianego z tabletki.
Po wielokrotnym podawaniu febuksostatu w dawce 80 mg raz na dobę lub w pojedynczej dawce 120 mg z bogatotłuszczowym posiłkiem wartość Cmax zmniejszyła się, odpowiednio o 49% i 38%, a wartość AUC odpowiednio o 18% i 16%. Wielokrotne dawki 80 mg nie spowodowały klinicznie
istotnej zmiany procentowego zmniejszenia stężenia kwasu moczowego w surowicy. Z tego względu febuksostat można przyjmować niezależnie od posiłków.
Dystrybucja
Pozorna objętość dystrybucji w stanie stacjonarnym febuksostatu (Vss /F) po doustnym podaniu dawek 10 do 300 mg mieści się w zakresie od 29 do 75 l. Febuksostat wiąże się z białkami osocza (głównie
z albuminą) w około 99,2% i wartość ta pozostaje stała w zakresie stężeń uzyskanych po podaniu
dawki 80 mg i dawki 120 mg. Wiązanie się czynnych metabolitów febuksostatu z białkami osocza wynosi od około 82% do 91%.
Metabolizm
Febuksostat jest w znacznym stopniu metabolizowany w procesie sprzęgania z udziałem transferazy urydylodifosfoglukuronowej (UDPGT) oraz w procesie oksydacji z udziałem enzymów układu cytochromu P450 (CYP). Zidentyfikowano cztery farmakologicznie czynne metabolity hydroksylowane, z których trzy obecne są w ludzkim osoczu. Badania in vitro z zastosowaniem mikrosomów ludzkiej wątroby wykazały, że w procesie oksydacji udział biorą głównie izoenzymy CYP1A1, CYP1A2, CYP2C8 lub CYP2C9, a w powstaniu glukuronidu febuksostatu uczestniczą głównie UGT 1A1, 1A8 i 1A9.
Wydalanie
Febuksostat eliminowany jest z organizmu drogą wątrobową i nerkową. Po podaniu doustnym znakowanego 14C febuksostatu w dawce 80 mg, w moczu wykryto około 49% dawki w postaci niezmienionego febuksostatu (3%), acylowego glukuronidu febuksostatu (30%), znane metabolity po oksydacji oraz ich sprzężone pochodne (13%), a także inne nieznane metabolity (3%). Ponadto około 45% dawki wykryto w kale w postaci niezmienionego febuksostatu (12%), acylowego glukuronidu febuksostatu (1%), znane metabolity po oksydacji oraz ich sprzężone pochodne (25%) oraz inne nieznane metabolity (7%).
Zaburzenia czynności nerek
Po wielokrotnym podaniu febuksostatu w dawce 80 mg jego wartość Cmax nie zmieniła się u pacjentów z lekkimi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek w porównaniu z pacjentami z prawidłową czynnością nerek. Średnia całkowita wartość AUC dla febuksostatu zwiększyła się około 1,8-krotnie z 7,5 μgh/ml u pacjentów z prawidłową czynnością nerek do 13,2 μgh/ml u pacjentów z ciężkimi zaburzeniami czynności nerek. Wartości Cmax i AUC czynnych metabolitów febuksostatu zwiększyły się odpowiednio 2- i 4-krotnie. U pacjentów z lekkimi lub umiarkowanymi zaburzeniami czynności nerek modyfikacja dawki nie jest konieczna.
Zaburzenia czynności wątroby
Po wielokrotnym podaniu febuksostatu w dawce 80 mg wartość Cmax i AUC febuksostatu oraz jego metabolitów nie zmieniła się znacząco u pacjentów z lekkimi (klasa A wg Childa-Pugha) lub umiarkowanymi (klasa B wg Childa-Pugha) zaburzeniami czynności wątroby w porównaniu
z pacjentami z prawidłową czynnością wątroby. Nie przeprowadzono badań dotyczących pacjentów
z ciężkimi (klasa C wg Childa-Pugha) zaburzeniami czynności wątroby.
Wiek
Nie stwierdzono, aby wielokrotne dawki doustne febuksostatu powodowały znaczące zmiany wartości AUC dla febuksostatu lub jego metabolitów u osób w podeszłym wieku w porównaniu z młodszymi zdrowymi osobami.
Płeć
Po wielokrotnym podaniu febuksostatu wartości Cmax i AUC u kobiet były większe odpowiednio o 24% i 12% niż u mężczyzn. Jednak skorygowane wobec masy ciała wartości tych parametrów u pacjentów obu płci były zbliżone. Modyfikacja dawki w zależności od płci nie jest konieczna.
Działania toksyczne w trakcie badań nieklinicznych obserwowano zazwyczaj przy ekspozycji na febuksostat większej niż maksymalna ekspozycja u ludzi.
Modelowanie farmakokinetyczne i symulacja danych z badania na szczurach wskazuje że
w przypadku jednoczesnego stosowania z febuksostatem dawka merkaptopuryny lub azatiopryny w warunkach klinicznych powinna zostać zmniejszona do 20% wcześniej przepisanej dawki lub mniejszej w celu uniknięcia możliwych reakcji hematologicznych (patrz punkty 4.4 i 4.5).
Działanie rakotwórcze, genotoksyczne, zaburzenie płodności
U samców szczura stwierdzono statystycznie istotne zwiększenie częstości nowotworów pęcherza moczowego (brodawczak i rak z komórek nabłonka przejściowego) w połączeniu z kamieniami ksantynowymi tylko u zwierząt otrzymujących duże dawki, odpowiadające około 11-krotnej ekspozycji u ludzi. Nie nastąpiło znaczące zwiększenie częstości innych rodzajów nowotworów
u samców lub samic myszy lub szczura. Obserwacje te uznaje się za skutek specyficznego dla gatunku metabolizmu puryny oraz składu moczu. Nie mają one znaczenia w praktyce klinicznej.
Standardowa seria badań genotoksyczności nie ujawniła żadnych biologicznie istotnych działań
genotoksycznych febuksostatu.
Stwierdzono, że febuksostat w dawkach doustnych do 48 mg/kg mc. na dobę nie wpływa na płodność
ani na zdolności rozrodcze samców i samic szczura.
Nie dowiedziono, aby febuksostat zaburzał płodność, działał teratogenie lub szkodliwie na płód.
U szczurów duże dawki (prowadzące do ekspozycji odpowiadającej 4,3-krotnej ekspozycji u ludzi) powodowały u matek działanie toksyczne ze zmniejszeniem wskaźnika odstawienia młodych od piersi i ograniczeniem rozwoju potomstwa. Badania działania teratogennego na ciężarnych samicach szczura i ciężarnych samicach królika po ekspozycji większej odpowiednio około 4,3-krotnie i około
13-krotnie od ekspozycji u ludzi, nie ujawniły działań teratogennych.
Laktoza jednowodna Celuloza mikrokrystaliczna Hydroksypropyloceluloza Kroskarmeloza sodowa
Krzemionka koloidalna bezwodna Magnezu stearynian
Otoczka
Alkohol poliwinylowy Talk
Tytanu dwutlenek (E 171) Makrogol 3350
Kwasu metakrylowego i etylu akrylanu kopolimer (1:1), typ A
Żelaza tlenek żółty (E 172) Sodu wodorowęglan
Nie dotyczy.
3 lata
Bez specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Blistry z folii Aluminium/OPA/Aluminium/PVC lub Aluminium/PVC/PE/PVDC w tekturowym
pudełku.
Wielkość opakowań: 14, 28, 42, 56, 84 i 98 tabletek powlekanych. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z
lokalnymi przepisami.
DO OBROTU
Sandoz GmbH Biochemiestrasse 10
6250 Kundl, Austria
Pozwolenie nr 24728
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 15.05.2018 r.
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
05.04.2020 r.








