







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Zapobieganie powikłaniom kostnym (złamania patologiczne, złamania kompresyjne kręgów, radioterapia lub operacja kości lub hiperkalcemia wywołana chorobą nowotworową) u dorosłych pacjentów z zaawansowanym procesem nowotworowym z przerzutami do kości.
Leczenie hiperkalcemii wywołanej chorobą nowotworową (ang. tumor-induced hypercalcaemia- TIH) u dorosłych pacjentów.
Dawkowanie i sposób podawania
Dla pacjentów z prawidłowym poziomem kreatyniny w surowicy przed rozpoczęciem leczenia (< 1,4 mg/dl lub < 124 mikromol/l), wzrost o 0,5 mg/dl lub 44 mikromol/l;
Dla pacjentów z podwyższonym poziomem kreatyniny w surowicy przed rozpoczęciem leczenia (> 1,4 mg/dl lub > 124 mikromol/l), wzrost o 1,0 mg/dl lub 88 mikromol/l.
W badaniach klinicznych wznawiano podawanie leku Zoledronic acid Juta tylko wtedy, gdy poziom kreatyniny powrócił do zakresu wartości wyjściowej z 10% odchyleniem (patrz punkt 4.4). Leczenie lekiem Zoledronic acid Juta należy wznowić, podając taką samą dawkę, jaką podano w chwili przerwania terapii.
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania i skuteczności monohydratu kwasu zoledronowego u dzieci w wieku od 1 roku do 17 lat. Aktualne dane przedstawiono w punkcie i 5.1, ale brak zaleceń dotyczących dawkowania.
Sposób podawania Podanie dożylne.
Produkt leczniczy Zoledronic acid Juta następnie rozcieńczony w 100 ml (patrz punkt 6.6), należy podawać w pojedynczej infuzji dożylnej trwającej przynajmniej 15 minut.
U pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek, zaleca się stosowanie zmniejszonych dawek leku Zoledronic acid Juta (patrz punkt „Dawkowanie” powyżej i punkt 4.4).
Sposób przygotowania zmniejszonych dawek leku Zoledronic acid Juta
Pobrać odpowiednią objętość koncentratu, zgodnie z ustalonym dawkowaniem:
4,4 ml dla dawki 3,5 mg
4,1 ml dla dawki 3,3 mg
3,8 ml dla dawki 3,0 mg
Instrukcja dotycząca rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6. Pobrany koncentrat należy następnie rozcieńczyć w 100 ml jałowego 0,9% roztworu chlorku sodu lub 5% w/v roztworze glukozy. Dawka produktu musi być podana w jednorazowej infuzji dożylnej, trwającej nie krócej niż 15 minut.
Nie wolno mieszać koncentratu produktu leczniczego Zoledronic acid Juta z roztworami do infuzji zawierającymi wapń lub inne kationy dwuwartościowe, takimi jak roztwór Ringera z dodatkiem mleczanu i należy go podawać jako pojedynczą dawkę dożylną przez oddzielną linię infuzyjną.
Przed i po podaniu produktu leczniczego Zoledronic acid Juta pacjenci muszą być odpowiednio nawodnieni.
Przeciwwskazania
Nadwrażliwość na substancję czynną, inne bisfosfoniany lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
Karmienie piersią (patrz punkt 4.6)
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
siła działania bisfosfonianu (większe ryzyko występuje po podaniu leków o dużej sile działania), droga podania (większe ryzyko występuje w przypadku podania pozajelitowego) i dawka skumulowana bifosfonianu.
rozpoznanie raka, choroby współistniejące (np. niedokrwistość, koagulopatia, zakażenia), palenie tytoniu.
leczenie skojarzone: chemioterapia, inhibitory angiogenezy (patrz punkt 4.5), radioterapia głowy i szyi, leczenie kortykosteroidami.
choroby zębów w przeszłości, nieodpowiednia higiena jamy ustnej, choroba przyzębia, inwazyjne zabiegi stomatologiczne (np. ekstrakcję zębów) i źle dopasowane protezy zębowe.
Należy zachęcać wszystkich pacjentów do należytego dbania o higienę jamy ustnej, przechodzenia rutynowych kontrolnych badań stomatologicznych i natychmiastowego zgłaszania wszelkich objawów w obrębie jamy ustnej, takich jak ruchomość zębów, ból lub obrzęk, lub brak gojenia się owrzodzenia albo obecność wydzieliny podczas leczenia produktem leczniczym Zoledronic acid Juta. W trakcie leczenia, inwazyjne zabiegi stomatologiczne należy wykonywać jedynie po starannym rozważeniu i unikać ich
przeprowadzania w terminie bliskim do podania monohydratu kwasu zoledronowego. Jeśli podczas terapii bisfosfonianami wystąpi martwica kości szczęki, przeprowadzenie zabiegów z zakresu chirurgii szczękowej może przyczynić się do nasilenia tego stanu. W przypadku pacjentów wymagających przeprowadzenia zabiegów stomatologicznych nie istnieją dane, które potwierdziłyby, że przerwanie leczenia bisfosfonianem zmniejsza ryzyko wystąpienia martwicy kości szczęki.
Plan postępowania z pacjentami, u których wystąpi martwica kości szczęki powinien zostać ustalony w ścisłej współpracy pomiędzy lekarzem prowadzącym a stomatologiem lub chirurgiem szczękowym posiadającym doświadczenie w leczeniu martwicy kości szczęki. Należy rozważyć czasowe przerwanie leczenia monohydratem kwasu zoledronowego, aż do ustąpienia tego stanu oraz zminimalizować czynniki ryzyka martwicy kości szczęki, o ile jest to możliwe.
Martwica kości innych miejsc anatomicznych
Podczas stosowania bisfosfonianów notowano martwicę kości przewodu słuchowego zewnętrznego, głównie związaną z długotrwałym leczeniem. Możliwe czynniki ryzyka martwicy kości przewodu słuchowego zewnętrznego obejmują stosowanie steroidów i chemioterapii i (lub) czynniki ryzyka miejscowe, takie jak zakażenie lub uraz. Możliwość wystąpienia martwicy kości przewodu słuchowego zewnętrznego należy rozważyć u pacjentów przyjmujących bisfosfoniany, u których występują objawy związane z uchem, w tym przewlekłe zakażenia ucha.
Ponadto, odnotowano sporadyczne przypadki martwicy kości w innych miejscach, w tym w biodrze i kości udowej, zgłaszane głównie u dorosłych pacjentów z rakiem leczonych lekiem Zoledronic acid Juta.
Ból mięśniowo-szkieletowy
Po wprowadzeniu leku Zoledronic acid Juta do obrotu, u pacjentów leczonych kwasem zoledronowym, odnotowywano przypadki silnego i sporadycznie powodującego niesprawność bólu kości, stawów i (lub) mięśni. Jednakże takie doniesienia nie były częste. Czas pojawienia się objawów może być różny od jednego dnia do kilku miesięcy po rozpoczęciu leczenia. U większości pacjentów objawy te ustępują po zakończeniu leczenia. Część pacjentów miała nawroty objawów po powtórnym rozpoczęciu leczenia lekiem Zoledronic acid Juta lub innym bisfosfonianem.
Nietypowe złamania kości udowej
Zgłaszano przypadki nietypowych złamań podkrętarzowych i trzonu kości udowej u osób stosujących bisfosfoniany, głównie u pacjentów długotrwale leczonych z powodu osteoporozy. Te poprzeczne lub krótkie, skośne złamania mogą pojawić się w dowolnym miejscu wzdłuż całej kości udowej, od miejsca zlokalizowanego tuż pod krętarzem mniejszym aż do okolicy nadkłykciowej. Do tego typu złamań dochodzi po minimalnym urazie lub bez urazu, a niektórzy pacjenci odczuwają ból uda lub ból w pachwinie. W badaniach obrazowych często na kilka tygodni lub miesięcy przed całkowitym złamaniem kości udowej widoczne są cechy złamań z przeciążenia. Złamania często występują obustronnie, dlatego u leczonych bisfosfonianami pacjentów, u których stwierdzono złamanie trzonu kości udowej, należy zbadać kość udową w drugiej kończynie. Zgłaszano również słabe gojenie się tych złamań. Na podstawie indywidualnej oceny stosunku korzyści do ryzyka u pacjentów, u których podejrzewa się nietypowe złamanie kości udowej, należy rozważyć odstawienie bisfosfonianów do czasu przeprowadzenia oceny.
Należy zalecić pacjentom zgłaszanie pojawienia się jakiegokolwiek bólu w obrębie uda, biodra lub pachwiny występującego w trakcie leczenia bisfosfonianami, a każdy pacjent zgłaszający się z takimi objawami powinien być zbadany pod względem obecności niecałkowitego złamania kości udowej.
Hipokalcemia
U pacjentów leczonych lekiem Zoledronic acid Juta zgłaszano występowanie hipokalcemii. Wtórnie do przypadków ciężkiej hipokalcemii zgłaszano występowanie arytmii serca i neurologicznych zdarzeń niepożądanych (w tym drgawek, niedoczulicy i tężyczki). Zgłaszano przypadki ciężkiej hipokalcemii wymagającej hospitalizacji. W niektórych przypadkach hipokalcemia może zagrażać życiu pacjenta (patrz punkt 4.8). Zaleca się ostrożność podczas stosowania leku Zoledronic acid Jutais z produktami leczniczymi, o których wiadomo, że powodują hipokalcemię, ponieważ mogą one mieć synergiczne działanie skutkujące ciężką hipokalcemią (patrz punkt 4.5). Przed rozpoczęciem leczenia lekiem Zoledronic acid Jutatherapy, należy oznaczyć poziom wapnia w surowicy i skorygować istniejącą hipokalcemię. Pacjenci powinni otrzymywać odpowiednią suplementację wapniem i witaminą D.
Sód
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na fiolkę (5 ml), to znaczy, że lek uznaje się za „wolny od sodu”.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
In vivo: hamowanie resorpcji kości przez osteoklasty, co zmienia mikrośrodowisko szpiku kostnego, powodując zmniejszenie podatności szpiku na wzrost komórek nowotworowych, działanie antyangiogenne i działanie przeciwbólowe.
In vitro: hamowanie proliferacji osteoblastów, bezpośrednie działanie cytostatyczne i proapoptotyczne dotyczące komórek nowotworowych, synergizm działania cytostatycznego z innymi lekami przeciwnowotworowymi, działanie przeciwadhezyjne i przeciwinwazyjne.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania oraz specjalistyczny sprzęt, służący do używania, podawania lub implantacji
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Zoledronic acid Juta
Jedna fiolka z 5 ml koncentratu zawiera 4 mg kwasu zoledronowego, co odpowiada 4,264 mg monohydratu kwasu zoledronowego.
Jeden ml koncentratu zawiera 0,8 mg kwasu zoledronowego (w postaci monohydratu). Substancje pomocnicze o znanym działaniu:
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na fiolkę (5 ml), tzn. lek uznaje się za
„wolny od sodu“.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Koncentrat do sporządzania roztworu do infuzji.
Klarowny i bezbarwny roztwór, niemal bez widocznych cząstek.
Lek Zoledronic acid Juta może być przepisywany i podawany pacjentom wyłącznie przez lekarzy mających doświadczenie w podawaniu dożylnych bisfosfonianów. Pacjenci leczeni produktem Zoledronic acid Juta powinni otrzymać ulotkę dla pacjenta oraz kartę przypominającą dla pacjenta.
Dawkowanie
Zapobieganie powikłaniom kostnym u pacjentów z zaawansowanym procesem nowotworowym z przerzutami do kości
Dorośli i osoby w podeszłym wieku
Zalecana dawka w zapobieganiu powikłaniom kostnym u pacjentów z zaawansowanymi nowotworami z przerzutami do kości wynosi 4 mg monohydratu kwasu zoledronowego co 3 do 4 tygodni.
Pacjenci powinni także otrzymywać doustną suplementację preparatami wapnia w ilości 500 mg/dobę oraz witaminą D w ilości 400 j.m./dobę.
Podejmując decyzję o leczeniu pacjentów z przerzutami do kości w celu zapobiegania powikłaniom kostnym, należy uwzględnić, że początek działania leku występuje po 2-3 miesiącach.
Leczenie TIH
Dorośli i osoby w podeszłym wieku
Zalecana dawka w leczeniu hiperkalcemii (poziom wapnia w surowicy z uwzględnieniem wapnia związanego z albuminami ≥ 12,0 mg/dl lub ≥ 3,0 mmol/l) to jednorazowa dawka 4 mg monohydratu kwasu zoledronowego.
Zaburzenia czynności nerek TIH:
Zastosowanie Zoledronic acid Juta u pacjentów z TIH oraz z ciężkimi zaburzeniami czynności nerek
można rozważyć wyłącznie po dokonaniu oceny ryzyka względem korzyści z leczenia. W badaniach klinicznych wyłączono z leczenia pacjentów, u których poziom kreatyniny w surowicy przekraczał 400 mikromol/l lub 4,5 mg/dl. Nie ma konieczności modyfikacji dawkowania u pacjentów z TIH, u których poziom kreatyniny w surowicy jest mniejszy niż 400 mikromol/l lub 4,5 mg/dl (patrz punkt 4.4).
Zapobieganie powikłaniom kostnym u pacjentów z zaawansowanym procesem nowotworowym z przerzutami do kości:
Rozpoczynając terapię lekiem Zoledronic acid Juta u pacjentów ze szpiczakiem mnogim lub przerzutami guzów litych do kości, należy oznaczyć pozio kreatyniny w surowicy i klirens kreatyniny (CLcr). Klirens kreatyniny oblicza się na podstawie poziomu kreatyniny w surowicy przy pomocy wzoru Cockcroft- Gaulta. Zoledronic acid Juta nie jest wskazany do stosowania u pacjentów z ciężkimi zaburzeniami czynności nerek przed rozpoczęciem terapii, rozumianymi w tej populacji jako wartość CLcr <30 ml/min. W badaniach klinicznych z lekiem Zoledronic acid Juta wyłączono z leczenia pacjentów, u których poziom kreatyniny w surowicy przekraczał 265 mikromol/l lub 3,0 mg/dl.
U pacjentów z przerzutami do kości z łagodnymi do umiarkowanych zaburzeniami czynności nerek, przed rozpoczęciem leczenia, definiowanymi w tej populacji na podstawie wartości CLcr 30 – 60 ml/min, zaleca się następujące dawkowanie Zoledronic acid Juta (patrz także punkt 4.4):
Klirens kreatyniny przed leczeniem (ml/min) | Zalecana dawka Zoledronic acid Juta* |
> 60 4,0 mg monohydrat kwasu zoledronowego | |
50–60 3,5 mg* monohydrat kwasu zoledronowego | |
40–49 3,3 mg* monohydrat kwasu zoledronowego | |
30–39 3,0 mg* monohydrat kwasu zoledronowego | |
* Dawki obliczono przyjmując, że docelowe wartości AUC wynoszą 0,66 (mg•h/l) (CLcr = 75 ml/min). Uważa się, że podanie mniejszych dawek pacjentom z zaburzeniami czynności nerek pozwoli osiągnąć takie same wartości AUC, jak u pacjentów z klirensem kreatyniny 75 ml/min.
Po wprowadzeniu leku Zoledronic acid Juta, przed podaniem każdej następnej dawki należy oznaczać poziom kreatyniny w surowicy, a leczenie należy przerwać, jeśli czynność nerek ulegnie pogorszeniu. W badaniach klinicznych pogorszenie czynności nerek definiowano w następujący sposób:
Ogólne
Przed podaniem leku Zoledronic acid Juta pacjenci muszą zostać zbadani, aby upewnić się, że są odpowiednio nawodnieni.
Należy unikać nadmiernego nawodnienia u pacjentów z ryzykiem wystąpienia niewydolności serca.
Po rozpoczęciu leczenia lekiem Zoledronic acid Juta należy dokładnie monitorować badane standardowo w hiperkalcemii parametry metaboliczne, takie jak: poziom wapnia, fosforanów i magnezu w surowicy. W przypadku wystąpienia hipokalcemii, hipofosfatemii lub hipomagnezemii, może być konieczne wprowadzenie krótkotrwałej terapii uzupełniającej. Pacjenci z nieleczoną hiperkalcemią mają z reguły w pewnym stopniu zaburzoną czynność nerek, dlatego u takich pacjentów należy rozważyć dokładne monitorowanie czynności nerek.
Pacjenci leczeni produktem leczniczym Zoledronic acid monohydratu nie powinni jednocześnie otrzymywać takich produktów leczniczych lub innych bisfosfonianów, ponieważ łączne skutki działania tych leków nie są znane.
Niewydolność nerek
Stan pacjentów z TIH i objawami pogorszenia czynności nerek należy odpowiednio ocenić, decydując, czy oczekiwana korzyść wynikająca z podawania leku Zoledronic acid Juta przewyższa możliwe ryzyko.
Podejmując decyzję o leczeniu pacjentów z przerzutami do kości w celu zapobieżenia powikłaniom kostnym, należy pamiętać, że początek działania leczniczego występuje po 2-3 miesiącach.
Stosowanie leku Zoledronic acid Juta związane jest z doniesieniami o występowaniu zaburzeń czynności nerek. Do czynników, które mogą zwiększać ryzyko pogorszenia czynności nerek należą: odwodnienie, zaburzenie czynności nerek przed rozpoczęciem leczenia, podawanie wielu cykli leku Zoledronic acid Juta i innych bisfosfonianów oraz zastosowanie innych produktów leczniczych o toksycznym działaniu na nerki. Pogorszenie czynności nerek jest rzadsze, chociaż możliwe po podaniu monohydratu kwasu zoledronowego w dawce 4 mg, w czasie 15 minut. Donoszono o pogorszeniu czynności nerek do
niewydolności nerek i konieczności przeprowadzenia dializ u pacjentów po dawce początkowej lub pojedynczej 4 mg monohydratu kwasu zoledronowego. Także, chociaż rzadziej, u niektórych pacjentów otrzymujących długotrwale w zalecanych dawkach lek Zoledronic acid Juta, w celu zapobieżenia powikłaniom kostnym, może wystąpić zwiększenie poziomu kreatyniny w surowicy.
Przed podaniem każdej kolejnej dawki leku Zoledronic acid Juta należy oznaczyć stężenie kreatyniny w surowicy. Rozpoczynając terapię u pacjentów z przerzutami do kości oraz łagodnymi do umiarkowanych zaburzeniami czynności nerek zaleca się podanie mniejszych dawek monohydratu kwasu zoledronowego. U pacjentów z oznakami pogorszenia czynności nerek podczas leczenia, należy odstawić lek Zoledronic acid Juta. Terapię lekiem Zoledronic acid Juta powinno się wznowić jedynie wówczas, gdy poziom kreatyniny w surowicy powróci do wartości wyjściowych z 10% odchyleniem.
Leczenie lekiem Zoledronic acid Juta należy wznowić, podając taką samą dawkę, jaką stosowano przed przerwaniem leczenia.
Ze względu na potencjalny wpływ monohydratu kwasu zoledronowego na czynność nerek, brak szczegółowych danych klinicznych dotyczących bezpieczeństwa jego stosowania u pacjentów z istniejącą ciężką niewydolnością nerek (w badaniach klinicznych określoną jako poziom kreatyniny w surowicy ≥ 400 mikromol/l lub ≥ 4,5 mg/dl dla pacjentów z TIH i ≥ 265 mikromol/l lub ≥ 3,0 mg/dl dla pacjentów z nowotworami i przerzutami do kości) oraz tylko ograniczone dane farmakokinetyczne dotyczące pacjentów z istniejącą ciężką niewydolnością nerek (klirens kreatyniny < 30 ml/min), nie zaleca się stosowania leku Zoledronic acid Juta u pacjentów z ciężkim zaburzeniem czynności nerek.
Niewydolność wątroby
Z uwagi na ograniczone dane kliniczne w grupie pacjentów z ciężkim zaburzeniem czynności wątroby, nie można podać specjalnych zaleceń dla tej grupy pacjentów.
Martwica kości
Martwica kości szczęki
Występowanie martwicy kości szczęki (ONJ, ang. osteonecrosis of the jaw) u pacjentów otrzymujących lek Zoledronic acid Juta zgłaszano niezbyt często w badaniach klinicznych. Dane z okresu po wprowadzeniu produktu do obrotu oraz z literatury fachowej wskazują na większą częstość występowania martwicy kości szczęki w zależności od rodzaju nowotworu złośliwego (zaawansowany rak piersi, szpiczak mnogi). W jednym z badań stwierdzono częstsze przypadki martwicy kości szczęki u pacjentów ze szpiczakiem w porównaniu z innym rodzajami nowotworów (patrz punkt 5.1).
Należy opóźnić rozpoczęcie leczenia lub nowego kursu terapii u pacjentów z niewygojonymi, otwartymi zmianami w obrębie tkanek miękkich jamy ustnej, z wyjątkiem sytuacji, które wymagają natychmiastowej pomocy lekarskiej. U pacjentów ze współistniejącymi czynnikami ryzyka, przed rozpoczęciem leczenia bifosfonianami zaleca się przeprowadzenie badania stomatologicznego i odpowiedniego zachowawczego leczenia stomatologicznego oraz indywidualnej oceny stosunku korzyści do ryzyka.
Dokonując oceny indywidualnego ryzyka wystąpienia ONJ należy wziąć pod uwagę następujące czynniki:
W badaniach klinicznych, w których lek Zoledronic acid Juta podawany był równocześnie z powszechnie stosowanymi lekami przeciwnowotworowymi, diuretykami, antybiotykami i lekami przeciwbólowymi, nie stwierdzono klinicznie istotnych interakcji. Monohydrat kwasu zoledronowego wiąże się w niewielkim stopniu z białkami osocza i nie hamuje aktywności enzymów ludzkiego cytochromu P450 in vitro (patrz punkt 5.2), ale nie przeprowadzono badań klinicznych dotyczących interakcji.
Zaleca się zachowanie ostrożności w przypadku podawania bisfosfonianów z antybiotykami z grupy aminoglikozydów, kalcytoniną lub pętlowymi lekami moczopędnymi, ponieważ łącznie mogą one wywoływać efekt addycyjny, w wyniku czego mniejszy poziom wapnia w surowicy utrzymuje się przez okres dłuższy niż wymagany (patrz punkt 4.4).
Wskazana jest ostrożność w czasie równoczesnego stosowania leku Zoledronic acid Juta z innymi potencjalnie nefrotoksycznymi produktami leczniczymi. Należy również zwrócić uwagę na możliwość powstania hipomagnezemii podczas leczenia.
U pacjentów ze szpiczakiem mnogim ryzyko pogorszenia czynności nerek może zwiększać się wtedy, gdy lek Zoledronic acid Juta podaje się w skojarzeniu z talidomidem.
Zaleca się zachowanie ostrożności podczas podawania leku Zoledronic acid Juta z lekami antyangiogennymi, ponieważ obserwowano zwiększenie częstości występowania martwicy kości szczęki u pacjentów leczonych jednocześnie tymi lekami.
Ciąża
Brak wystarczających danych dotyczących stosowania monohydratu kwasu zoledronowego u kobiet w ciąży. Badania nad wpływem monohydratu kwasu zoledronowego na płodność zwierząt wykazały toksyczny wpływ na reprodukcję (patrz punkt 5.3). Potencjalne zagrożenie dla człowieka nie jest znane. Leku Zoledronic acid Juta nie należy stosować w okresie ciąży. Kobietom w wieku rozrodczym należy doradzić unikanie zajścia w ciążę.
Karmienie piersią
Nie wiadomo, czy monohydrat kwasu zoledronowego przenika do mleka ludzkiego. Lek Zoledronic acid Juta jest przeciwwskazany do stosowania u kobiet karmiących piersią (patrz punkt 4.3).
Płodność
Monohydrat kwasu zoledronowego badano na szczurach w poszukiwaniu możliwych działań niepożądanych na płodność rodziców i pokolenia F1. Obserwowano nasilenie farmakologicznych działań monohydratu kwasu zoledronowego, co przypisywano hamującemu wpływowi na metabolizm wapnia w kościach, powodującemu hipokalcemię okołoporodową, skutek grupy bisfosfonianów, trudne porody, co spowodowało wcześniejsze zakończenie badania. Wyniki te wykluczyły możliwość definitywnego określenia wpływu monohydratu kwasu zoledronowego na płodność ludzi.
Działania niepożądane, takie jak zawroty głowy i senność mogą wywierać wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn, dlatego stosując lek Zoledronic acid Juta, należy zachować ostrożność podczas prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
W ciągu trzech dni po podaniu leku Zoledronic acid Juta często zgłaszano występowanie reakcji ostrej fazy z takimi objawami jak ból kości, gorączka, uczucie zmęczenia, bóle stawów, bóle mięśni, dreszcze i zapalenie stawów z obrzękiem; objawy te zazwyczaj ustępują w ciągu kilku dni (patrz opis wybranych działań niepożądanych).
Do ważnych zidentyfikowanych działań niepożądanych leku Zoledronic acid Juta stosowanego w zarejestrowanych wskazaniach należą: zaburzenia czynności nerek, martwica kości szczęki, reakcja ostrej fazy, hipokalcemia, migotanie przedsionków, anafilaksja choroba śródmiąższowa płuc. Częstość występowania każdego z tych zagrożeń przedstawiono w Tabeli 1.
Tabelaryczne zestawienie działań niepożądanych
Następujące działania niepożądane, wymienione w Tabeli 1, zebrano podczas przeprowadzania badań klinicznych oraz na podstawie zgłoszeń spontanicznych, podczas stosowania monohydratu kwasu zoledronowego w dawce 4 mg, głównie długookresowo:
Tabela 1
Działania niepożądane zostały sklasyfikowane zgodnie z częstością występowania, najczęściej występujące podano jako pierwsze. Zastosowano następującą skalę: Bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1 000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1 000), bardzo rzadko (< 1/10 000), częstość nieznana (nie można określić na podstawie dostępnych danych).
Zaburzenia krwi i układu chłonnego | |
Często: | Niedokrwistość |
Niezbyt często: | Trombocytopenia, leukopenia |
Rzadko: | Pancytopenia |
Zaburzenia układu immunologicznego | |
Niezbyt często: | Reakcja nadwrażliwości |
Rzadko: | Obrzęk naczynioruchowy |
Zaburzenia psychiczne | |
Niezbyt często: | Niepokój, zaburzenia snu |
Rzadko: | Splątanie |
Zaburzenia układu nerwowego Często: Niezbyt często: Bardzo rzadko: | Ból głowy Zawroty głowy, parestezje, zaburzenia smaku, niedoczulica, przeczulica, drżenie, senność Drgawki, niedoczulica i tężyczka (wtórne do hipokalcemii) |
Zaburzenia oka Często: Niezbyt często: Rzadko: Bardzo rzadko: | Zapalenie spojówek Niewyraźne widzenie, zapalenie twardówki i zapalenie oczodołu Zapalenie błony naczyniowej oka Zapalenie nadtwardówki |
Zaburzenia serca Niezbyt często: Rzadko: | Nadciśnienie, niedociśnienie, migotanie przedsionków, niedociśnienie prowadzące do omdleń lub zapaści krążeniowej Bradykardia, arytmia serca (wtórna do hipokalcemii) |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Niezbyt często: | Duszność, kaszel, skurcz oskrzeli |
Rzadko: | Śródmiąższowa choroba płuc |
Zaburzenia żołądkowo-jelitowe Często: Niezbyt często: | Nudności, wymioty, zmniejszony apetyt Biegunka, zaparcia, bóle brzucha, niestrawność, zapalenie błony śluzowej jamy ustnej, suchość błony śluzowej jamy ustnej |
Zaburzenia skóry i tkanki podskórnej Niezbyt często: | Świąd, wysypka (w tym wysypka rumieniowa i plamkowa), wzmożona potliwość |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często: Niezbyt często: Bardzo rzadko: | Ból kości, bóle mięśni, bóle stawów, uogólniony ból Skurcze mięśni, martwica kości szczęki Martwica kości zewnętrznego przewodu słuchowego (działanie niepożądane związane ze stosowaniem leków z grupy bisfosfonianów) i innych miejsc anatomicznych, w tym kości udowej i biodra |
Zaburzenia nerek i dróg moczowych | |
Często: Niezbyt często: Rzadko: | Zaburzenia czynności nerek Ostra niewydolność nerek, krwiomocz, białkomocz Nabyty zespół Fanconiego |
Zaburzenia ogólne i stany w miejscu podania | |
Często: Niezbyt często: Rzadko: | Gorączka, objawy grypopodobne (w tym zmęczenie, dreszcze, złe samopoczucie i zaczerwienienie) Osłabienie, obrzęki obwodowe, reakcje w miejscu podania (w tym ból, podrażnienie, obrzęk, stwardnienie), ból w klatce piersiowej, zwiększenie masy ciała, reakcja/wstrząs anafilaktyczny, pokrzywka Zapalenie stawów i obrzęk stawów jako objaw reakcji ostrej fazy |
Badania diagnostyczne Bardzo często: Często: Niezbyt często: Rzadko: | Hipofosfatemia Zwiększony poziom kreatyniny i mocznika we krwi, hipokalcemia Hipomagnezemia, hipokaliemia Hiperkaliemia, hipernatremia |
Opis wybranych działań niepożądanych
Zaburzenia czynności nerek
Stosowanie leku Zoledronic acid Juta, było związane ze zgłoszeniami występowania zaburzeń czynności nerek. W zbiorczej analizie danych bezpieczeństwa z badań rejestracyjnych leku Zoledronic acid Juta dotyczących zapobiegania zdarzeniom związanym z układem kostnym u pacjentów z zaawansowaną chorobą nowotworową obejmującą kości, częstość występowania zaburzeń czynności nerek, zdarzeń podejrzewanych o związek z lekiem Zoledronic acid Juta (działań niepożądanych) była następująca: szpiczak mnogi (3,2%), rak prostaty (3,1%), rak piersi (4,3%), guzy płuc i inne guzy lite (3,2%). Do czynników mogących zwiększać ryzyko pogorszenia czynności nerek należy odwodnienie, współistniejące zaburzenia czynności nerek, wielokrotne cykle leczenia lekiem Zoledronic acid Juta lub innymi bisfosfonianami, jak również jednoczesne stosowanie nefrotoksycznych produktów leczniczych lub krótszego czasu infuzji niż obecnie zalecany. U pacjentów, którzy przyjęli początkową dawkę lub pojedynczą dawkę wynoszącą 4 mg monohydratu kwasu zoledronowego, zgłaszano pogorszenie czynności nerek, progresję do niewydolności nerek lub do dializowania (patrz punkt 4.4).
Martwica kości szczęki
Zgłaszano przypadki martwicy kości (szczęki i żuchwy), przede wszystkim u pacjentów z rozpoznaniem raka, leczonych produktami leczniczymi hamującymi resorpcję kości, takimi jak lek Zoledronic acid Juta (patrz punkt 4.4). Wielu z tych pacjentów otrzymywało także chemioterapię oraz kortykosteroidy i stwierdzono u nich oznaki miejscowego zakażenia, w tym zapalenia szpiku. Większość tych doniesień dotyczy pacjentów z rozpoznaniem raka, po ekstrakcji zębów lub innych zabiegach stomatologicznych.
Migotanie przedsionków
W jednym randomizowanym, podwójnie ślepym, kontrolowanym badaniu trwającym 3 lata oceniano skuteczność i bezpieczeństwo stosowania monohydratu kwasu zoledronowego w dawce 5 mg podawanego raz na rok i porównywano go z placebo w leczeniu osteoporozy pomenopauzalnej (PMO), całkowita częstość występowania migotania przedsionków wyniosła 2,5% (96 spośród 3 862) i 1,9% (75 spośród 3 852) u pacjentów otrzymujących odpowiednio dawkę 5 mg monohydratu kwasu zoledronowego i placebo. Częstość ciężkich działań niepożądanych w postaci migotania przedsionków wyniosła 1,3% (51 spośród 3 862) i 0,6% (22 spośród 3 852) u pacjentów otrzymujących odpowiednio dawkę 5 mg monohydratu kwasu zoledronowego i placebo. Dysproporcji obserwowanej w tym badaniu
nie stwierdzano w innych badaniach z monohydratem kwasu zoledronowego, także w badaniach z lekiem Zoledronic acid Juta w dawce 4 mg podawanym co 3-4 tygodnie pacjentom onkologicznym. Mechanizm odpowiedzialny za zwiększoną częstość występowania migotania przedsionków w tym jednym badaniu klinicznym nie jest znany.
Reakcja ostrej fazy
To działanie niepożądane polega na występowaniu zespołu objawów, takich jak gorączka, bóle mięśni, ból głowy, bóle kończyn, nudności, wymioty, biegunka, bóle stawów i zapalenie stawów z obrzękiem. Czas wystąpienia reakcji ostrej fazy wynosi ≤ 3 dni po podaniu leku Zoledronic acid Juta w infuzji, a reakcję tę określa się również terminem objawy „grypopodobne” lub objawy „po podaniu dawki”.
Nietypowe złamania kości udowej
W okresie po wprowadzeniu produktu do obrotu zgłaszano następujące działania (rzadko): nietypowe złamania podkrętarzowe i trzonu kości udowej (działanie niepożądane leków należących do klasy bisfosfonianów).
Działania niepożądane leku związane z hipokalcemią
Hipokalcemia jest ważnym zidentyfikowanym ryzykiem związanym ze stosowaniem leku Zoledronic acid Juta w zatwierdzonych wskazaniach. Przegląd przypadków zgłaszanych podczas badań klinicznych oraz po wprowadzeniu leku do obrotu dostarczył wystarczających dowodów na istnienie związku między leczeniem lekiem Zoledronic acid Juta, zgłaszanymi zdarzeniami hipokalcemii, a wtórnym rozwojem arytmii serca. Ponadto, zebrano dowody na istnienie związku między hipokalcemią a wtórnymi zdarzeniami neurologicznymi zgłaszanymi w tych przypadkach, w tym drgawkami, niedoczulicą i tężyczką (patrz punkt 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem
Departament Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C PL-02 222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Doświadczenie kliniczne z ostrym przedawkowaniem leku Zoledronic acid Juta jest ograniczone. Zgłaszano niezamierzone podanie dawek wynoszących do 48 mg monohydratu kwasu zoledronowego. Należy dokładnie monitorować pacjentów, którzy otrzymali dawki większe niż zalecane (patrz punkt 4.2), ponieważ obserwowano zaburzenia czynności nerek (w tym niewydolność nerek) oraz nieprawidłowy poziom elektrolitów w surowicy (w tym wapnia, fosforu i magnezu). W przypadku hipokalcemii należy podać glukonian wapnia w infuzji, zależnie od wskazań klinicznych.
Grupa farmakoterapeutyczna: leki stosowane w leczeniu chorób kości, bisfosfoniany, kod ATC: M05BA08
Mechanizm działania
Monohydrat kwasu zoledronowego należy do klasy bisfosfonianów i działa głównie na tkankę kostną. Jest on inhibitorem resorpcji kości przez osteoklasty.
Działanie farmakodynamiczne
Selektywne działanie bisfosfonianów na tkankę kostną wynika z ich dużego powinowactwa do zmineralizowanej kości, ale dokładny mechanizm prowadzący do zaburzenia aktywności osteoklastów pozostaje nadal niewyjaśniony. W długookresowych badaniach na zwierzętach wykazano, że monohydrat kwasu zoledronowego hamuje resorpcję kości nie wpływając negatywnie na tworzenie, mineralizację oraz właściwości mechaniczne tkanki kostnej.
Poza silnym działaniem hamującym resorpcję kości, monohydrat kwasu zoledronowego ma także liczne właściwości przeciwnowotworowe, które mogą wpływać na jego ogólną skuteczność w leczeniu przerzutów nowotworowych do kości. W badaniach przedklinicznych wykazano następujące właściwości:
Wyniki badań klinicznych w zapobieganiu powikłaniom kostnym u pacjentów z zaawansowanym procesem nowotworowym z przerzutami do kości
W pierwszym, randomizowanym, kontrolowanym placebo badaniu, które prowadzono metodą podwójnie ślepą, porównywano monohydrat kwasu zoledronowego w dawce 4 mg z placebo w zapobieganiu powikłaniom kostnym (ang. skeletal related events-SRE) u pacjentów z rakiem prostaty. Monohydrat kwasu zoledronowego w dawce 4 mg znacząco zmniejszał ilość pacjentów, u których wystąpił co najmniej jeden epizod SRE, zwiększał medianę czasu do wystąpienia pierwszego incydentu SRE o ponad 5 miesięcy i zmniejszał ilość powikłań kostnych w ciągu roku na jednego pacjenta (ang. skeletal morbidity rate- SMR). Analiza przypadków wielokrotnych wykazała zmniejszenie o 36% ryzyka wystąpienia SRE u pacjentów otrzymujących monohydrat kwasu zoledronowego w dawce 4 mg w porównaniu z placebo. Pacjenci otrzymujący monohydrat kwasu zoledronowego w dawce 4 mg zgłaszali mniejszy wzrost bólu w porównaniu z pacjentami otrzymującymi placebo. Różnica osiągnęła istotność statystyczną w 3, 9, 21 i 24 miesiącu. U mniejszej liczby pacjentów otrzymujących monohydrat kwasu zoledronowego w dawce 4 mg wystąpiły patologiczne złamania. Wyniki leczenia były słabiej wyrażone u pacjentów z uszkodzeniami blastycznymi. Wyniki skuteczności przedstawiono w Tabeli 2.
W drugim badaniu obejmującym guzy lite inne niż rak sutka lub prostaty, monohydrat kwasu zoledronowego w dawce 4 mg istotnie zmniejszał liczbę pacjentów z SRE, zwiększał medianę czasu do wystąpienia pierwszego incydentu SRE o ponad 2 miesiące i zmniejszał SMR. Analiza przypadków wielokrotnych wykazała 30,7% zmniejszenie ryzyka wystąpienia SRE u pacjentów otrzymujących monohydrat kwasu zoledronowego w dawce 4 mg w porównaniu z placebo. Wyniki skuteczności przedstawiono w Tabeli 3.
Tabela 2: Wyniki skuteczności (pacjenci z rakiem prostaty leczeni hormonalnie)
Wszystkie SRE (+TIH) | Złamania * | Radioterapia kości | ||||
monohydrat kwasu zoledronowego 4 mg | Placebo | monohydrat kwasu zoledronowego 4 mg | Placebo | monohydrat kwasu zoledronowego 4 mg | Placebo | |
N | 214 | 208 | 214 | 208 | 214 | 208 |
Odsetek pacjentów z SRE (%) | 38 | 49 | 17 | 25 | 26 | 33 |
wartość p | 0,028 | 0,052 | 0,119 | |||
Mediana czasu do SRE (dni) | 488 | 321 | NR | NR | NR | 640 |
wartość p | 0,009 | 0,020 | 0,055 | |||
SMR | 0,77 | 1,47 | 0,20 | 0,45 | 0,42 | 0,89 |
wartość p | 0,005 | 0,023 | 0,060 | |||
Zmniejszenie ryzyka wielokrotnego wystąpienia SRE** (%) | 36 | - | NA | NA | NA | NA |
wartość p | 0,002 | NA | NA | |||
* Obejmuje złamania kręgów i inne złamania
** Obejmuje wszystkie powikłania kostne, ich całkowitą ilość jak również czas do wystąpienia każdego przypadku w czasie badania
NR Nie osiągnięto NA Nie dotyczy
Tabela 3: Wyniki skuteczności (guzy lite inne niż rak piersi lub prostaty)
Wszystkie SRE (+TIH) | Złamania * | Radioterapia kości | ||||
monohydrat kwasu zoledronowego 4 mg | Placebo | monohydrat kwasu zoledronowego 4 mg | Placebo | monohydrat kwasu zoledronowego 4 mg | Placebo | |
N | 257 | 250 | 257 | 250 | 257 | 250 |
Odsetek pacjentów z SRE (%) | 39 | 48 | 16 | 22 | 29 | 34 |
wartość p | 0,039 | 0,064 | 0,173 | |||
Mediana czasu do SRE (dni) | 236 | 155 | NR | NR | 424 | 307 |
wartość p | 0,009 | 0,020 | 0,079 | |||
SMR | 1,74 | 2,71 | 0,39 | 0,63 | 1,24 | 1,89 |
wartość p | 0,012 | 0,066 | 0,099 | |||
Zmniejszenie ryzyka wielokrotnego wystąpienia SRE** (%) | 30,7 | - | NA | NA | NA | NA |
wartość p | 0.003 | NA | NA | |||
* Obejmuje złamania kręgów i inne złamania
** Obejmuje wszystkie powikłania kostne, ich całkowitą ilość jak również czas do wystąpienia każdego przypadku w czasie badania
NR Nie osiągnięto NA Nie dotyczy
W trzecim randomizowanym badaniu fazy III, prowadzonym metodą podwójnie ślepą, w którym porównywano monohydrat kwasu zoledronowego w dawce 4 mg z pamidronianem w dawce 90 mg co 3 do 4 tygodni u pacjentów ze szpiczakiem mnogim lub rakiem sutka, u których wystąpiło co najmniej jedno uszkodzenie kości. Wyniki wskazują, że monohydrat kwasu zoledronowego w dawce 4 mg wykazywał skuteczność porównywalną z pamidronianem (Pam) w dawce 90 mg w zapobieganiu SRE. Analiza przypadków wielokrotnych wykazała 16% zmniejszenie ryzyka wystąpienia SRE u pacjentów otrzymujących monohydrat kwasu zoledronowego w dawce 4 mg w porównaniu do pacjentów otrzymujących pamidronian. Wyniki skuteczności przedstawiono w Tabeli 4.
Tabela 4: Wyniki skuteczności (pacjenci z rakiem piersi i szpiczakiem mnogim)
Wszystkie SRE (+TIH) | Złamania * | Radioterapia kości | ||||
monohydrat kwasu zoledronowego 4 mg | Pam 90 mg | monohydrat kwasu zoledronowego 4 mg | Pam 90 mg | monohydrat kwasu zoledronowego 4 mg | Pam 90 mg | |
N | 561 | 555 | 561 | 555 | 561 | 555 |
Odsetek pacjentów z SRE (%) | 48 | 52 | 37 | 39 | 19 | 24 |
wartość p | 0,198 | 0,653 | 0,037 | |||
Mediana czasu do SRE (dni) | 376 | 356 | NR | 714 | NR | NR |
wartość p | 0,151 | 0,672 | 0,026 | |||
SMR | 1,04 | 1,39 | 0,53 | 0,60 | 0,47 | 0,71 |
wartość p | 0,084 | 0,614 | 0,015 | |||
Zmniejszenie ryzyka wielokrotnego wystąpienia SRE** (%) | 16 | - | NA | NA | NA | NA |
wartość p | 0.030 | NA | NA | |||
* Obejmuje złamania kręgów i inne złamania
** Obejmuje wszystkie powikłania kostne, ich całkowitą ilość jak również czas do wystąpienia każdego przypadku w czasie badania
NR Nie osiągnięto NA Nie dotyczy
Monohydrat kwasu zoledronowego w dawce 4 mg był również badany w podwójnie ślepym, randomizowanym badaniu kontrolowanym placebo, z udziałem 228 pacjentek z udokumentowanymi przerzutami raka piersi do kości, w celu oceny wpływu monohydratu kwasu zoledronowego w dawce 4 mg na wskaźnik częstości występowania powikłań kostnych, obliczanych jako całkowita liczba powikłań kostnych (z wyjątkiem hiperkalcemii oraz po uwzględnieniu wcześniejszych złamań) podzielona przez całkowity okres ryzyka. Pacjentki otrzymywały monohydrat kwasu zoledronowego w dawce 4 mg lub placebo co cztery tygodnie przez jeden rok. Dokonano równomiernego przydziału pacjentek do grupy otrzymującej monohydrat kwasu zoledronowego lub do grupy placebo.
Częstość występowania powikłań kostnych (liczba powikłań/osoborok) wyniosła 0,628 dla monohydratu kwasu zoledronowego i 1,096 dla placebo. Odsetek pacjentek z co najmniej jednym powikłaniem kostnym (z wyjątkiem hiperkalcemii) wyniósł 29,8% w grupie otrzymującej monohydrat kwasu zoledronowego i 49,6% w grupie placebo (p=0,003). Mediana czasu do wystąpienia pierwszego powikłania kostnego nie została osiągnięta pod koniec badania w grupie otrzymującej monohydrat kwasu zoledronowego i była istotnie wydłużona w porównaniu do placebo (p=0,007). W analizie wieloczynnikowej monohydrat kwasu zoledronowego w dawce 4 mg zmniejszał ryzyko powikłań kostnych o 41% w porównaniu do placebo (współczynnik ryzyka = 0,59, p=0,019).
W grupie otrzymującej monohydrat kwasu zoledronowego obserwowano statystycznie istotną poprawę nasilenia bólu (ocenianego przy użyciu skali pomiaru bólu Brief Pain Inventory, BPI) po 4 tygodniach oraz w każdym kolejnym punkcie czasowym badania, w porównaniu z placebo (Rysunek 1). Wynik punktowy oceny bólu w grupie otrzymującej monohydrat kwasu zoledronowego utrzymywał się poniżej wartości wyjściowych, a zmniejszeniu nasilenia bólu towarzyszyła tendencja obniżenia wyniku punktowego dotyczącego zużycia leków przeciwbólowych.
Rysunek 1. Średnie zmiany w wyniku punktowym w skali BPI względem wartości początkowych. Statystycznie istotne różnice zaznaczono (*p< 0,05) dla porównania między grupami terapii (4 mg monohydratu kwasu zoledronowego w porównaniu do placebo)
Placebo ∆
Zoledronic acid
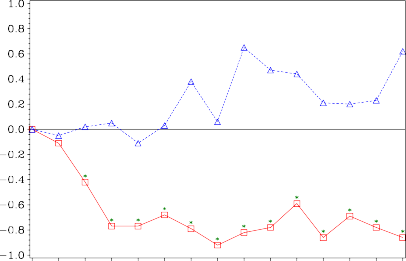









Średnia zmiana w wyniku BPI względem wartości wyjściowych
Czas badania (tygodnie)
Badanie CZOL446EUS122/SWOG
Głównym celem tego badania obserwacyjnego było oszacowanie skumulowanej częstości występowania martwicy kości szczęki po upływie 3 lat u pacjentów z rakiem i przerzutami do kości przyjmujących kwas zoledronowy. Terapię inhibitorem resorpcji kości, inne rodzaje terapii przeciwnowotworowej oraz opiekę stomatologiczną prowadzono zgodnie ze wskazaniami klinicznymi w celu zapewnienia jak najlepszej opieki zdrowotnej (opiekę akademickąa i środowiskową). Początkowe badanie stomatologiczne było zalecane, jednak nie było ono obowiązkowe.
Wśród 3491 pacjentów kwalifikujących się do oceny potwierdzono 87 przypadków rozpoznania martwicy kości szczęki. Szacowany skumulowany odsetek potwierdzonych przypadków martwicy kości szczęki po upływie 3 lat wyniósł 2,8% (95% CI: 2,3–3,5%) łącznie. Odsetek po 1. roku wyniósł 0,8%, a po 2. roku – 2,0%. Odsetek potwierdzonych przypadków martwicy kości szczęki po 3 latach był najwyższy wśród pacjentów ze szpiczakiem (4,3%), a najniższy wśród pacjentek z rakiem piersi (2,4%). Liczba potwierdzonych przypadków choroby była w sposób statystycznie istotny większa wśród pacjentów ze szpiczakiem mnogim (p=0,03) niż wśród pozostałych pacjentów z nowotworami złośliwymi łącznie.
Wyniki badań klinicznych w leczeniu TIH
Badania kliniczne monohydratu kwasu zoledronowego w hiperkalcemii wywołanej chorobą nowotworową (TIH) wykazały, że wpływa on na zmniejszenie poziomu wapnia w surowicy i obniżenie wydalania wapnia przez nerki. W badaniach I fazy dotyczących doboru dawki u pacjentów z łagodną do umiarkowanej hiperkalcemią wywołaną chorobą nowotworową (TIH), stwierdzono, że dawki skuteczne mieściły się w przybliżeniu w zakresie 1,2 do 2,5 mg.
W celu porównania skuteczności monohydratu kwasu zoledronowego w dawce 4 mg z pamidronianem w dawce 90 mg, połączono, poddając zaplanowanej analizie wyniki dwóch, podstawowych, wieloośrodkowych badań u pacjentów z hiperkalcemią wywołaną chorobą nowotworową (TIH).
Skorygowany względem albumin poziom wapnia w surowicy powracał szybciej do wartości prawidłowych w 4 dniu leczenia monohydratem kwasu zoledronowego i w dawce 8 mg oraz w 7 dniu leczenia monohydratem kwasu zoledronowego w dawkach 4 mg i 8 mg. Obserwowano następujący odsetek odpowiedzi:
Tabela 5: Odsetek całkowitych odpowiedzi uzyskanych w poszczególnych dniach leczenia, w połączonych badaniach dotyczących TIH
Dzień 4 | Dzień 7 | Dzień 10 | |
Monohydrat kwasu zoledronowego 4 mg (N=86) | 45,3% (p=0,104) | 82,6% (p=0,005)* | 88,4% (p=0,002)* |
Monohydrat kwasu zoledronowego 8 mg (N=90) | 55,6% (p=0,021)* | 83,3% (p=0,010)* | 86,7% (p=0,015)* |
Pamidronian 90 mg (N=99) | 33,3% | 63,6% | 69,7% |
* wartości p - porównanie z pamidronianem. | |||
Mediana czasu potrzebnego do uzyskania prawidłowego poziomu wapnia w surowicy (normokalcemii) wynosiła 4 dni. Mediana czasu do ponownego zwiększenia poziomu wapnia w surowicy z uwzględnieniem wapnia związanego z albuminami (≥ 2,9 mmol/l) wynosiła 30 do 40 dni w grupie pacjentów leczonych monohydratem kwasu zoledronowego i 17 dni w grupie otrzymującej pamidronian w dawce 90 mg (wartości p: 0,001 dla dawki 4 mg i 0,007 dla dawki 8 mg monohydraty kwasu zoledronowego). Nie było statystycznie istotnej różnicy między dwoma dawkami monohydratu kwasu zoledronowego.
W badaniach klinicznych, monohydrat kwasu zoledronowego w dawce 8 mg podano ponownie 69 pacjentom, u których doszło do ponownego zwiększenia poziomu wapnia lub którzy byli niewrażliwi na początkowe leczenie (monohydrat kwasu zoledronowego w dawce 4 mg, 8 mg lub pamidronian w dawce 90 mg). Wskaźnik odpowiedzi u tych pacjentów wynosił ok. 52%. Wyżej wymienieni pacjenci otrzymywali przy ponownym podaniu monohydratu kwasu zoledronowego tylko dawkę 8 mg, dlatego brak jest danych pozwalających na dokonanie porównania z dawką 4 mg.
W badaniach klinicznych wykonanych u pacjentów z hiperkalcemią wywołaną chorobą nowotworową (TIH) ogólny profil bezpieczeństwa (rodzaj i ciężkość działań niepożądanych) we wszystkich trzech leczonych grupach (monohydrat kwasu zoledronowego w dawce 4 mg i 8 mg oraz pamidronian w dawce 90 mg) był podobny.
Dzieci i młodzież
Wyniki badania klinicznego w leczeniu ciężkiej wrodzonej łamliwości kości u dzieci w wieku od 1 do 17 lat
Działanie monohydratu kwasu zoledronowego po podaniu dożylnym w leczeniu dzieci (w wieku od 1 do 17 lat) z ciężką wrodzoną łamliwością kości (typu I, III i IV) porównano z działaniem pamidronianu po podaniu dożylnym w jednym międzynarodowym, wieloośrodkowym, randomizowanym, otwartym badaniu z udziałem odpowiednio 74 i 76 pacjentów w każdej z grup terapeutycznych. Okres leczenia w tym badaniu wynosił 12 miesięcy i był poprzedzony 4 do 9 tygodniowym okresem kwalifikacji, podczas którego dzieci przyjmowały witaminę D i wapń cząsteczkowy przez co najmniej 2 tygodnie. W badaniu klinicznym, dzieciom w wieku od 1 do < 3 lat podawano 0,025 mg/kg monohydratu kwasu zoledronowego, (maksymalnie do 0,35 mg w pojedynczej dawce), co 3 miesiące. Pacjenci w wieku od 3 do 17 lat otrzymywali 0,05 mg/kg monohydratu kwasu zoledronowego, (maksymalnie do 0,83 mg w pojedynczej dawce), co 3 miesiące. Fazę rozszerzoną badania przeprowadzono w celu oceny długotrwałego bezpieczeństwa ogólnego i bezpieczeństwa dla nerek po podaniu monohydratu kwasu zoledronowego raz w roku lub dwa razy w roku przez dalsze 12 miesięcy dzieciom, które ukończyły 12- miesięczne leczenie monohydratem kwasu zoledronowego lub pamidronianem w badaniu głównym.
Pierwszorzędowym punktem końcowym badania była procentowa zmiana względem wartości wyjściowych w gęstości mineralnej kości kręgosłupa lędźwiowego (BMD) po 12 miesiącach leczenia. Szacunkowe wyniki badania nad BMD były podobne, ale badanie nie było wystarczająco dobrze zaprojektowane, aby wykazać przewagę skuteczności monohydratu kwasu zoledronowego. W szczególności, nie było oczywistych dowodów na skuteczność w przypadku wystąpienia złamania lub bólu. Działania niepożądane obejmujące złamania kości długich kończyn dolnych zgłaszano u około 24% (kość udowa) i 14% (kość piszczelowa) pacjentów leczonych monohydratem kwasu zoledronowego w stosunku do 12% i 5% pacjentów z ciężką wrodzoną łamliwością kości leczonych pamidronianem, bez względu na typ choroby i związek przyczynowy, ale liczba złamań była porównywalna u pacjentów leczonych monohydratem kwasu zoledronowego i pamidronianem: 43% (32/74) w stosunku do 41% (31/76). Interpretację ryzyka wystąpienia złamania utrudnia fakt, że u pacjentów z ciężką wrodzoną łamliwością kości złamania występują często, stanowiąc część procesu chorobowego.
Rodzaj działań niepożądanych obserwowany u tej grupy pacjentów był podobny do działań niepożądanych obserwowanych wcześniej u dorosłych z zaawansowanym rakiem kości (patrz punkt
4.8). Działania niepożądane przedstawione w Tabeli 6 pogrupowano według częstości występowania. Zastosowano następującą skalę: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często (≥ 1/1 000 do < 1/100), rzadko (≥ 1/10 000 do < 1/1 000), bardzo rzadko (< 1/10 000), częstość nieznana (nie może być określona na podstawie dostępnych danych).
Tabela 6: Działania niepożądane zaobserwowane u dzieci z ciężką wrodzoną łamliwością kości 1
Zaburzenia układu nerwowego | |
Często: | Ból głowy |
Zaburzenia serca | |
Często: | Tachykardia |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Często: | Nieżyt nosa i gardła |
Zaburzenia żołądkowo-jelitowe | |
Bardzo często: Często: | Wymioty, nudności Ból brzucha |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej Często: | Bóle kości, bóle stawów, bóle mięśni |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często: Często: | Gorączka, zmęczenie Reakcja ostrej fazy, ból |
Badania diagnostyczne | |
Bardzo często: Często: | Hipokalcemia Hipofosfatemia |
1 Działania niepożądane występujące z częstością < 5% oceniono pod względem medycznym i wykazano, że te przypadki są zgodne z ustalonym profilem bezpieczeństwa leku Zoledronic acid Juta (patrz punkt 4.8)
U dzieci z ciężką wrodzoną łamliwością kości monohydrat kwasu zoledronowego wydaje się wiązać z wyraźniejszym ryzykiem wystąpienia reakcji ostrej fazy, hipokalcemii i niewyjaśnionej tachykardii, w porównaniu do pamidronianu, ale ta różnica zmniejszała się po podaniu kolejnych wlewów.
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego monohydrat kwasu zoledronowego we wszystkich podgrupach populacji dzieci i młodzieży w leczeniu hiperkalcemii wywołanej chorobą nowotworową i zapobieganiu powikłaniom kostnym u pacjentów z zaawansowanym procesem nowotworowym z przerzutem do kości (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
W grupie 64 pacjentów z przerzutami nowotworowymi do kości, stosowano monohydrat kwasu zoledronowego w dawkach 2, 4, 8 i 16 mg w pojedynczej lub wielokrotnej 5-minutowej i 15-minutowej infuzji, uzyskując niezależnie od podanej dawki leku następujące dane farmakokinetyczne.
Po rozpoczęciu infuzji monohydratu kwasu zoledronowego, stężenie monohydratu kwasu zoledronowego w osoczu gwałtownie zwiększa się, osiągając stężenie maksymalne pod koniec infuzji. Następnie obserwuje się szybkie zmniejszenie stężenia leku do < 10% wartości maksymalnej po 4 godzinach i < 1% wartości maksymalnej po 24 godzinach. Następnie przez długi okres, do drugiej infuzji monohydratu kwasu zoledronowego w 28 dniu, obserwowano bardzo małe stężenia, nie przekraczające 0,1% wartości maksymalnej.
Eliminacja monohydratu kwasu zoledronowego z organizmu po podaniu dożylnym odbywa się trójfazowo: w formie szybkiego, dwufazowego usuwania leku z krążenia ogólnego z okresem półtrwania t1/2α wynoszącym 0,24 godziny i t ½ β 1,87 godziny, po którym następuje długa faza eliminacji z okresem półtrwania w końcowej fazie eliminacji t 1/2γ wynoszącym 146 godzin. Nie stwierdzono kumulacji monohydratu kwasu zoledronowego w osoczu po wielokrotnym podaniu co 28 dni. Monohydrat kwasu zoledronowego nie jest metabolizowany i wydala się przez nerki w formie niezmienionej. W ciągu
pierwszych 24 godzin, 39 ± 16% podanej dawki leku pojawia się w moczu, podczas gdy pozostała część wiąże się przede wszystkim z tkanką kostną.
Z kości lek uwalnia się bardzo powoli do krążenia ogólnego i jest wydalany przez nerki. Całkowity klirens leku wynosi 5,04 ± 2,5 l/godz. i jest niezależny od dawki, płci, wieku, rasy i masy ciała.
Przedłużenie czasu infuzji z 5 minut do 15 minut spowodowało zmniejszenie stężenia monohydratu kwasu zoledronowego pod koniec infuzji o 30%, ale nie miało wpływu na powierzchnię pola pod krzywą w układzie stężenie w osoczu względem czasu.
Tak jak w przypadku innych bisfosfonianów, zmienność międzyosobnicza parametrów farmakokinetycznych monohydratu kwasu zoledronowego była duża.
Brak jest danych farmakokinetycznych dla monohydratu kwasu zoledronowego w grupach pacjentów z hiperkalcemią lub z niewydolnością wątroby. Monohydrat kwasu zoledronowego nie hamuje aktywności enzymów ludzkiego cytochromu P450 in vitro i nie ulega biotransformacji. W badaniach na zwierzętach mniej niż 3% podanej dawki leku wydalało się z kałem, co wskazuje, że wątroba nie odgrywa istotnej roli w farmakokinetyce monohydratu kwasu zoledronowego.
Klirens nerkowy monohydratu kwasu zoledronowego był skorelowany z klirensem kreatyniny. Klirens nerkowy stanowi 75 ± 33% klirensu kreatyniny, którego średnia wartość u 64 badanych pacjentów z rakiem wynosiła 84 ± 29 ml/min (w zakresie od 22 do 143 ml/min). Analiza populacyjna wykazała, że u pacjentów z klirensem kreatyniny wynoszącym 20 ml/min (ciężkie zaburzenie czynności nerek) lub 50 ml/min (umiarkowane zaburzenie czynności nerek), przewidywany klirens monohydratu kwasu zoledronowego powinien wynosić odpowiednio 37% i 72% klirensu u pacjenta z klirensem kreatyniny wynoszącym 84 ml/min. U pacjentów z ciężką niewydolnością nerek (klirens kreatyniny < 30 ml/min) dostępne są tylko ograniczone dane farmakokinetyczne).
W badaniu in vitro monohydrat kwasu zoledronowego wykazywał słabe powinowactwo do elementów morfotycznych krwi ludzkiej, przy średnim stosunku stężeń krew/osocze wynoszącym 0,59 w zakresie stężeń od 30 ng/ml do 5000 ng/ml. Wiązanie z białkami osocza jest małe, a niezwiązana frakcja waha się od 60% przy stężeniu monohydratu kwasu zoledronowego 2 ng/ml do 77% przy stężeniu 2000 ng/ml.
Szczególne populacje pacjentów
Dzieci i młodzież
Ograniczone dane farmakokinetyczne u dzieci z ciężką wrodzoną łamliwością kości wskazują, że farmakokinetyka monohydratu kwasu zoledronowego u dzieci w wieku od 3 do 17 lat jest podobna jak u pacjentów dorosłych po podaniu podobnej dawki w mg/kg mc. Wiek, masa ciała, płeć i klirens kreatyniny wydają się nie mieć wpływu na ogólnoustrojową ekspozycję na monohydrat kwasu zoledronowego.
Toksyczność ostra
Największa pojedyncza dawka leku podawana dożylnie, która nie powodowała śmierci, wynosiła 10 mg/kg masy ciała u myszy i 0,6 mg/kg u szczurów.
Toksyczność długo- i krótkookresowa
Monohydrat kwasu zoledronowego podawany podskórnie szczurom i dożylnie psom w dawkach do 0,02 mg/kg mc. na dobę przez 4 tygodnie, był dobrze tolerowany. Podskórne podawanie monohydratu kwasu zoledronowego szczurom w dawce 0,001 mg/kg mc./dobę oraz dożylne psom w dawce 0,005 mg/kg mc. co 2-3 dni, w okresie czasu do 52 tygodni, było również dobrze tolerowane.
Najczęściej obserwowanym działaniem po podaniu wielokrotnym było zwiększenie pierwotnej warstwy gąbczastej przynasad kości długich u zwierząt w okresie wzrostu, w niemal wszystkich badanych dawkach. Zjawisko to jest wynikiem farmakologicznego działania związku polegającego na zahamowaniu procesu resorpcji kości.
W badaniach długookresowych, z wielokrotnym podawaniem pozajelitowym u zwierząt, margines bezpieczeństwa w odniesieniu do wpływu na nerki był wąski. Jednakże skumulowane wyniki badania
największych dawek niepowodujących działań niepożądanych (ang. No adverse event level - NOAEL) po podaniu jednorazowym (1,6 mg/kg mc.) i po podaniu wielokrotnym, w czasie do jednego miesiąca (0,06- 0,6 mg/kg mc./dobę), nie wskazywały na działanie na nerki przy dawkach równych lub przekraczających największą proponowaną dawkę terapeutyczną u ludzi. W badaniach narażenia długotrwałego w dawkach z przedziału największej proponowanej dawki terapeutycznej dla ludzi monohydrat kwasu zoledronowego działał toksycznie na inne narządy, w tym na przewód pokarmowy, wątrobę, śledzionę i płuca oraz w miejscu dożylnego podania.
Toksyczny wpływ na reprodukcję
Monohydrat kwasu zoledronowego podawany podskórnie szczurom w dawkach ≥ 0,2 mg/kg mc., wykazywał działanie teratogenne. Chociaż nie obserwowano działania teratogennego lub toksycznego na płód u królików, jednak stwierdzano toksyczne działanie u matek. U szczurów, po podaniu najniższej badanej dawki (0,01 mg/kg mc.) obserwowano dystocję.
Mutagenność i rakotwórczość
Przeprowadzone testy na mutagenność i rakotwórczość nie wykazały mutagennego ani rakotwórczego działania monohydratu kwasu zoledronowego.
Mannitol Cytrynian sodu
Woda do wstrzykiwań
W celu uniknięcia potencjalnych niezgodności, produktu leczniczego Zoledronic acid Juta, koncentrat należy rozcieńczyć 0,9% m/v roztworem chlorku sodu lub 5% m/v roztworem glukozy.
Nie wolno mieszać produktu leczniczego z roztworami do infuzji zawierającymi wapń lub inne kationy dwuwartościowe, takimi jak roztwór Ringera z dodatkiem mleczanu i należy go podawać jako osobny roztwór dożylny, przez oddzielną linię infuzyjną.
2 lata.
Z mikrobiologicznego punktu widzenia roztwór do infuzji należy natychmiast zużyć po rozcieńczeniu. Jeśli produkt leczniczy nie zostanie natychmiast zużyty, odpowiedzialność za czas i warunki jego przechowywania przed użyciem ponosi użytkownik, a roztwór jest trwały nie dłużej niż 24 godziny w temperaturze 2°C-8°C. Przed podaniem, schłodzony roztwór powinien osiągnąć temperaturę pokojową. Po rozcieńczeniu, chemiczna i fizyczna trwałość produktu leczniczego jest wykazana przez 24 godziny w temperaturze 2°C-8°C.
Brak specjalnych zaleceń dotyczących przechowywania tego produktu leczniczego. Warunki przechowywania odtworzonego produktu leczniczego, patrz punkt 6.3.
Fiolka: 5 ml bezbarwna fiolka szklana typu I z 20 mm korkiem z gumy butylowej pokrytym teflonem i 20 mm aluminiową uszczelką typu flip-off.
Opakowania jednostkowe zawierające 1 lub 4 fiolki.
Opakowania zbiorcze zawierające 10 fiolek (10 opakowań po 1 fiolce). Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Przed podaniem 5,0 ml koncentratu stanowiącego zawartość jednej fiolki lub odpowiednia ilość koncentratu pobrana z fiolki musi być rozcieńczona w 100 ml roztworu niezawierającego jonów wapnia (0,9% m/v roztwór chlorku sodu lub 5% m/v roztwór glukozy).
Dodatkowe informacje dotyczące postępowania z produktem leczniczym Zoledronic acid Juta, w tym wskazówki dotyczące przygotowania zmniejszonych dawek podano w punkcie 4.2.
Podczas przygotowywania infuzji należy przestrzegać zasad aseptyki. Produkt przeznaczony wyłącznie do jednorazowego użytku.
Należy używać wyłącznie przezroczystych roztworów, wolnych od cząstek i przebarwień.
Zaleca się pracownikom służby zdrowia niewrzucanie niewykorzystanych resztek produktu leczniczego Zoledronic acid Juta do kanalizacji.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Juta Pharma GmbH Gutenbergstr. 13
24941 Flensburg Niemcy
25719
23/04/2021







