







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
ml koncentratu zawiera 1 mg klofarabiny.
Jedna fiolka o objętości 20 ml zawiera 20 mg klofarabiny.
Substancja pomocnicza o znanym działaniu
Jedna fiolka o pojemności 20 ml zawiera 70,77 mg sodu (w postaci 180 mg sodu chlorku). Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
cyklach leczenia (patrz punkt 5.1). Zatem lekarz prowadzący leczenie pacjenta powinien dokonać oceny potencjalnych korzyści i ryzyka związanych z kontynuacją leczenia u pacjentów, którzy nie wykazują hematologicznej i (lub) klinicznej poprawy po 2 cyklach leczenia (patrz punkt 4.4).
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Regularnie badać pełną morfologię krwi i liczbę płytek krwi, częściej u pacjentów, u których wystąpiła cytopenia.
Czynność nerek i wątroby przed leczeniem, podczas aktywnego leczenia i po zakończeniu leczenia. Klofarabinę należy niezwłocznie odstawić, jeśli zostanie zaobserwowane znaczące zwiększenie stężenia kreatyniny, enzymów wątrobowych i(lub) bilirubiny.
Wartości parametrów układu oddechowego, ciśnienie krwi, równowaga płynów i masa ciała podczas leczenia i bezpośrednio po 5-dniowym okresie podawania klofarabiny.
Jeśli przewiduje się hiperurykemię (rozpad guza), należy rozważyć profilaktyczne stosowanie allopurynolu.
W celu zmniejszenia skutków rozpadu guza i innych zdarzeń pacjenci powinni otrzymywać płyny dożylne przez cały 5-dniowy okres leczenia klofarabiną.
Profilaktyczne podanie kortykosteroidów (np. hydrokortyzon w dawce 100 mg/m2 od 1. do
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Zahamowanie polimerazy DNA α powodujące zatrzymanie wydłużania łańcucha DNA i (lub) syntezy/naprawy DNA.
Zahamowanie reduktazy rybonukleotydowej wraz z obniżeniem puli komórkowych trifosforanów
deoksyrybonukleozydowych (dNTP).
Przerwanie integralności błony mitochondriów z uwolnieniem cytochromu C i innych czynników proapoptotycznych prowadzące do zaprogramowanej śmierci komórek nawet w przypadku limfocytów niedzielących się.
Brak blastów we krwi obwodowej lub dowodów
Szpik kostny M1 (≤ 5% blastów)
Poprawa liczby płytek w krwi obwodowej (płytki
Pacjenci, którzy spełnili wszystkie kryteria CR z wyjątkiem poprawy liczby płytek do > 100 x 109/l
Brak blastów we krwi obwodowej
Szpik kostny M2 (≥ 5% i ≤ 25% blastów) i pojawienie się komórek progenitorowych
Szpik M1, który nie kwalifikuje się do CR ani do CRp
(Liczba pacjentów z CR + Liczba pacjentów z CRp)
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Clofarabine Accord, 1 mg/ml, koncentrat do sporządzania roztworu do infuzji
Koncentrat do sporządzania roztworu do infuzji.
Klarowny, praktycznie bezbarwny roztwór bez widocznych cząstek, o pH od 4,5 do 7,5 i osmolarności od 270 do 310 mOsm/kg.
Leczenie ostrej białaczki limfoblastycznej (ALL) u dzieci i młodzieży z nawrotem lub oporną na leczenie chorobą po zastosowaniu przynajmniej dwóch wcześniejszych standardowych cykli
i w przypadku, gdy brak innych opcji pozwalających na przewidywanie długotrwałej odpowiedzi. Skuteczność i bezpieczeństwo stosowania oceniono w badaniach z udziałem pacjentów w wieku ≤ 21 lat podczas pierwszej diagnozy (patrz punkt 5.1).
Leczenie musi być rozpoczęte i nadzorowane przez lekarza posiadającego doświadczenie w leczeniu pacjentów z ostrą białaczką.
Dawkowanie
Dorośli (w tym osoby w podeszłym wieku)
Obecnie posiadane informacje są niewystarczające do ustalenia bezpieczeństwa stosowania i skuteczności klofarabiny u dorosłych pacjentów (patrz punkt 5.2).
Dzieci i młodzież
Dzieci i młodzież (≥ 1 roku życia)
Zalecana dawka w monoterapii wynosi 52 mg/m2 pc. podawana w infuzji dożylnej trwającej 2 godziny dziennie przez 5 kolejnych dni. Powierzchnię ciała należy obliczyć stosując rzeczywisty wzrost i masę ciała pacjenta przed rozpoczęciem każdego cyklu leczenia. Cykle leczenia należy powtarzać co 2 do 6 tygodni (licząc od pierwszego dnia poprzedniego cyklu) po przywróceniu normalnej hematopoezy (tj. bezwzględna liczba granulocytów obojętnochłonnych (ANC) ≥ 0,75 × 109/l) i powrotu do wyjściowych parametrów czynności narządów. Możliwe jest zmniejszenie dawki
o 25% u pacjentów, u których występuje duża toksyczność (patrz poniżej). Obecnie posiadane doświadczenie dotyczące pacjentów otrzymujących więcej niż 3 cykle leczenia jest ograniczone (patrz punkt 4.4).
U większości pacjentów reagujących na leczenie klofarabiną odpowiedź uzyskuje się po 1 lub
Dzieci o masie ciała mniejszej niż 20 kg
W celu zmniejszenia objawów niepokoju i drażliwości oraz w celu uniknięcia niepożądanego maksymalnego stężenia klofarabiny należy rozważyć wydłużenie czasu infuzji powyżej 2 godzin (patrz punkt 5.2).
Dzieci poniżej 1 roku życia
Brak danych dotyczących farmakokinetyki, bezpieczeństwa stosowania i skuteczności klofarabiny u niemowląt. Dlatego też bezpieczną i skuteczną dawkę zalecaną u pacjentów poniżej 1 roku życia należy dopiero ustalić.
Zmniejszenie dawki u pacjentów z toksycznością hematologiczną
Jeśli liczba ANC nie ulegnie poprawie w ciągu 6 tygodni od rozpoczęcia cyklu, w celu stwierdzenia choroby opornej na leczenie należy wykonać aspirację / biopsję szpiku kostnego. Jeśli oporna białaczka nie jest oczywista, wówczas po uzyskaniu liczby ANC ≥ 0,75 × 109/l zaleca się zmniejszenie dawki w kolejnym cyklu o 25%. Jeśli przez ponad 4 tygodnie od rozpoczęcia ostatniego cyklu ANC będzie mniejsze niż 0,5 × 109/l , zaleca się zmniejszenie dawki w kolejnym cyklu o 25%.
Zmniejszenie dawki u pacjentów z toksycznością niehematologiczną
Zakażenia
Jeśli wystąpi klinicznie istotne zakażenie, klofarabinę można odstawić aż do uzyskania klinicznej kontroli zakażenia. Wówczas leczenie można wznowić w pełnej dawce. W przypadku kolejnego klinicznie istotnego zakażenia, leczenie klofarabiną należy przerwać aż do uzyskania klinicznej kontroli zakażenia, a następnie można wznowić w dawce zmniejszonej o 25%.
Zdarzenia nie związane z zakażeniem
Jeśli wystąpi jedno lub kilka ciężkich objawów toksyczności [stopnia 3 według skali toksyczności amerykańskiego National Cancer Institute (NCI) Common Toxicity Criteria (CTC) oprócz nudności i wymiotów], leczenie należy opóźnić aż do powrotu objawów toksyczności do parametrów wyjściowych lub do stanu, gdy toksyczność nie jest już ciężka, a potencjalne korzyści wynikające
z kontynuacji leczenia klofarabiną przewyższają ryzyko. Zaleca się wówczas podawanie klofarabiny w dawce zmniejszonej o 25%.
Jeśli po raz drugi wystąpią objawy ciężkiej toksyczności, leczenie należy opóźnić aż do powrotu objawów toksyczności do parametrów wyjściowych lub do stanu, gdy takie działania toksyczne nie są już ciężkie, a potencjalne korzyści wynikające z kontynuacji leczenia klofarabiną przewyższają ryzyko. Zaleca się wówczas podawanie klofarabiny w dawce zmniejszonej o kolejne 25%.
Jeśli po raz trzeci wystąpią objawy ciężkiej toksyczności, które nie ustąpią w ciągu 14 dni (patrz powyższe wyjątki) lub toksyczność będzie zagrażała życiu lub spowoduje niesprawność (toksyczność stopnia 4. według amerykańskiego NCI CTC), leczenie klofarabiną należy przerwać (patrz punkt 4.4).
Szczególne grupy pacjentów
Zaburzenia czynności nerek
Dostępne, ograniczone dane wskazują, że klofarabina może kumulować się u pacjentów ze
zmniejszonym klirensem kreatyniny (patrz punkty 4.4 i 5.2). Klofarabina jest przeciwwskazana u pacjentów z ciężką niewydolnością nerek (patrz punkt 4.3) i należy ją stosować ostrożnie
u pacjentów z łagodną do umiarkowanej niewydolnością nerek (patrz punkt 4.4).
U pacjentów z umiarkowanymi zaburzeniami czynności nerek (klirens kreatyniny 30 − <60 ml/min) konieczne jest zmniejszenie dawki o połowę (patrz punkt 5.2).
Zaburzenia czynności wątroby
Nie ma doświadczenia w leczeniu pacjentów z zaburzeniami czynności wątroby (stężenie bilirubiny w surowicy > 1,5 x GGN i AspAT i AlAT > 5 x GGN), a wątroba jest potencjalnym narządem docelowym dla działania toksycznego. Dlatego też klofarabina jest przeciwwskazana u pacjentów
z ciężkimi zaburzeniami czynności wątroby (patrz punkt 4.3) i należy ją stosować ostrożnie
u pacjentów z łagodnymi do umiarkowanych zaburzeniami czynności wątroby (patrz punkt 4.4). Sposób podawania
Zalecaną dawkę należy podawać w infuzji dożylnej pomimo, że w badaniach klinicznych produkt leczniczy jest podawany przez cewnik założony do żyły centralnej. Produktu leczniczego
Clofarabine Accord nie wolno mieszać, ani podawać jednocześnie w tym samym zestawie do infuzji dożylnej z innymi lekami (patrz punkt 6.2). Instrukcja dotycząca filtrowania i rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Stosowanie u pacjentów z ciężką niewydolnością nerek lub ciężkimi zaburzeniami czynności wątroby. Karmienie piersią (patrz punkt 4.6).
Produkt Clofarabine Accord jest silnym lekiem przeciwnowotworowym powodującym potencjalnie istotne hematologiczne i niehematologiczne reakcje niepożądane (patrz punkt 4.8).
U pacjentów leczonych klofarabiną należy prowadzić ścisłą obserwację poniższych parametrów:
Zaburzenia krwi i układu chłonnego
Należy oczekiwać wystąpienia supresji szpiku kostnego. Supresja szpiku jest na ogół odwracalna i wydaje się, że przebiega w sposób zależny od dawki. U pacjentów leczonych klofarabiną
obserwowano przypadki ciężkiej supresji szpiku kostnego, również z neutropenią, niedokrwistością
i małopłytkowością. Zgłaszano przypadki, w tym przypadki śmiertelne krwotoku, w tym krwotoku do mózgu, krwotoku z przewodu pokarmowego i płuc. Występowanie większości z tych przypadków było związane z małopłytkowością (patrz punkt 4.8). W badaniach klinicznych na początku leczenia
u większości pacjentów stwierdzono pogorszenie parametrów hematologicznych jako objaw białaczki. Pacjenci z tej grupy są zagrożeni większym ryzykiem ciężkich zakażeń oportunistycznych, w tym ciężką posocznicą, mogącą prowadzić do zgonu, ze względu na uprzednie zaburzenia prowadzące do obniżenia odporności oraz długotrwałą neutropenię, która może być następstwem leczenia
klofarabiną. Należy obserwować stan pacjentów w celu wykrycia objawów podmiotowych i przedmiotowych zakażeń i w razie potrzeby bezzwłocznie rozpocząć leczenie.
Podczas leczenia klofarabiną opisywano przypadki zapalenia jelit, w tym zapalenie okrężnicy neutropeniczne lub wywołane C. difficile i zapalenie kątnicy. To powikłanie częściej występowało w ciągu 30 dni leczenia i w przypadku skojarzonego stosowania chemioterapii. Zapalenie jelit może
prowadzić do powikłań, takich jak martwica, perforacja lub posocznica i może zakończyć się zgonem (patrz punkt 4.8). Należy obserwować pacjentów w celu wykrycia objawów podmiotowych
i przedmiotowych zapalenia jelit.
Zaburzenia skóry i tkanki podskórnej
Zgłaszano występowanie zespołu Stevensa-Johnsona (SJS) i toksyczne martwicze oddzielanie się naskórka (TEN), w tym przypadki zakończone zgonem (patrz punkt 4.8). Podawanie klofarabiny należy przerwać w przypadku wystąpienia złuszczającej się lub pęcherzykowej wysypki albo
w przypadku podejrzewania SJS lub TEN.
Nowotwory łagodne i złośliwe (w tym torbiele i polipy) oraz zaburzenia układu immunologicznego Po podaniu klofarabiny następuje szybkie zmniejszenie liczby komórek białaczkowych we krwi obwodowej. U pacjentów leczonych klofarabiną należy prowadzić ocenę i obserwację objawów przedmiotowych i podmiotowych zespołu rozpadu guza i uwalniania cytokin (np. szybki oddech, częstoskurcz, niedociśnienie, obrzęk płuc), które mogą się przekształcić w uogólnioną odpowiedź zapalną (SIRS)/zespół przesiąkania włośniczek lub zaburzenie czynności narządów (patrz punkt 4.8).
3. dnia) może zapobiegać objawom przedmiotowym i podmiotowym zespołu ogólnoustrojowej reakcji zapalnej (SIRS) lub zespołu przesiąkania włośniczek.
Podawanie klofarabiny należy niezwłocznie przerwać w przypadku wystąpienia wczesnych objawów przedmiotowych lub podmiotowych SIRS/zespołu przesiąkania włośniczek lub poważnego zaburzenia czynności narządów i zastosować odpowiednie leczenie wspomagające. Ponadto należy przerwać leczenie klofarabiną, jeśli w ciągu 5 dni od podania wystąpi niedociśnienie tętnicze (niezależnie od przyczyny). Dalsze leczenie klofarabiną, generalnie w mniejszej dawce, można brać pod uwagę po ustabilizowaniu się stanu pacjenta i przywróceniu parametrów wyjściowych czynności narządów.
U większości pacjentów reagujących na leczenie klofarabiną odpowiedź uzyskuje się po 1 lub 2 cyklach leczenia (patrz punkt 5.1). Zatem lekarz prowadzący powinien dokonać oceny
potencjalnych korzyści i ryzyka związanego z kontynuacją leczenia u pacjentów, którzy nie wykazują hematologicznej i(lub) klinicznej poprawy po 2 cyklach leczenia.
Zaburzenia serca
Podczas leczenia klofarabiną pacjenci z chorobami serca oraz pacjenci przyjmujący leki wpływające na ciśnienie krwi lub czynność serca powinni być objęci ścisłą obserwacją (patrz punkty 4.5 i 4.8).
Zaburzenia nerek i dróg moczowych
Nie przeprowadzono badań klinicznych u dzieci i młodzieży z niewydolnością nerek (zdefiniowaną w badaniach klinicznych jako stężenie kreatyniny w surowicy ≥ 2 x GGN dla danego wieku),
a klofarabina jest wydalana głównie przez nerki. Dane farmakokinetyczne wskazują, że klofarabina może być kumulowana u pacjentów ze zmniejszonym klirensem kreatyniny (patrz punkt 5.2). Dlatego należy zachować ostrożność, stosując klofarabinę u pacjentów z łagodną lub umiarkowaną niewydolnością nerek (patrz punkt 4.2). Nie ustalono profilu bezpieczeństwa klofarabiny u pacjentów z ciężkimi zaburzeniami czynności nerek lub u pacjentów otrzymujących terapię nerkozastępczą (ang. Renal replacement therapy, RRT) (patrz punkt 4.3). Należy unikać, zwłaszcza podczas 5-dniowego leczenia klofarabiną, skojarzonego stosowania produktów leczniczych mogących mieć działania
nefrotoksyczne i wydalanych drogą wydzielania kanalikowego (np. niesteroidowe leki przeciwzapalne, amfoterycyna B, metotreksat, aminoglikozydy, pochodne platyny, foskarnet, pentamidyna, cyklosporyna, takrolimus, acyklowir i walgancyklowir); należy z wyboru stosować produkty lecznicze pozbawione działania nefrotoksycznego (patrz punkty 4.5 i 4.8). Niewydolność nerek lub ostra niewydolność nerek były obserwowane w wyniku zakażenia, posocznicy i zespołu rozpadu guza (patrz punkt 4.8). Pacjentów należy monitorować w celu wykrycia nefrotoksyczności i, jeżeli konieczne, należy przerwać podawanie klofarabiny.
Zaobserwowano, że częstość i nasilenie działań niepożądanych w szczególności takich jak zakażenia, zahamowanie czynności szpiku (neutropenia) i hepatotoksyczność zwiększa się podczas stosowania klofarabiny w leczeniu skojarzonym. W związku z tym należy uważnie monitorować pacjentów,
u których klofarabina stosowana jest w leczeniu skojarzonym.
U pacjentów leczonych klofarabiną mogą wystąpić wymioty i biegunka, dlatego należy doradzić odpowiednie postępowanie zapobiegające odwodnieniu. Należy pouczyć pacjentów, by zgłosili się do lekarza, jeśli wystąpią zawroty głowy, omdlenia lub zmniejszenie wydzielania moczu. Należy rozważyć profilaktyczne stosowanie przeciwwymiotnych produktów leczniczych.
Zaburzenia wątroby i dróg żółciowych
Brak doświadczenia w leczeniu pacjentów z zaburzeniami czynności wątroby (stężenie bilirubiny w surowicy > 1,5 x GGN plus AspAT i AlAT > 5 x GGN), a wątroba jest potencjalnym narządem docelowym dla działań toksycznych. Dlatego należy zachować ostrożność stosując klofarabinę
u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności wątroby (patrz punkty 4.2 lub 4.3). Jeśli tylko jest to możliwe, należy unikać równoczesnego podawania leków mogących mieć toksyczne działanie na wątrobę (patrz punkty 4.5 i 4.8). Jeśli wystąpi toksyczność hematologiczna
w postaci neutropenii 4. stopnia (ANC < 0,5 x 109/l) przez ≥ 4 tygodnie, w kolejnym cyklu należy zmniejszyć dawkę o 25%.
Klofarabinę należy odstawić, jeśli po raz trzeci wystąpią objawy ciężkiej toksyczności niehematologicznej (toksyczność 3. stopnia według NCI CTC), objawy ciężkiej toksyczności, które nie ustąpią w ciągu 14 dni (oprócz nudności i wymiotów) lub objawy toksyczności niehematologicznej i niezakaźnej zagrażające życiu lub powodujące niesprawność (toksyczność 4. stopnia według NCI CTC) (patrz punkt 4.2).
U pacjentów, którym wcześniej przeszczepiono macierzyste komórki krwiotwórcze (HSCT), po leczeniu klofarabiną (40 mg/m2) w skojarzeniu z etopozydem (100 mg/m2) i cyklofosfamidem (440 mg/m2) może wystąpić zwiększone ryzyko hepatotoksyczności sugerujące chorobę żylnookluzyjną wątroby. W okresie po wprowadzeniu produktu do obrotu, w następstwie leczenia
klofarabiną, u dzieci i młodzieży oraz pacjentów dorosłych, występowały ciężkie mające szkodliwy wpływ na wątrobę działania niepożądane choroby żylno-okluzyjnej (VOD), kończące się zgonem. W związku ze stosowaniem klofarabiny zgłaszano przypadki zapalenia wątroby i niewydolności wątroby, w tym zakończone zgonem (patrz punkt 4.8).
Większość pacjentów otrzymywała leczenie kondycjonujące obejmujące busulfan, melfalan i (lub) połączenie leczenia cyklofosfamidem i napromienianiem całego ciała. W łączonym badaniu klofarabiny fazy 1/2 opisywano ciężkie zdarzenia hepatotoksyczne u dzieci z ostrą białaczką nawracającą lub oporną na leczenie.
Obecnie dostępne są ograniczone dane dotyczące bezpieczeństwa stosowania i skuteczności klofarabiny podawanej dłużej niż podczas 3 cykli leczenia.
Ten produkt leczniczy zawiera 70,77 mg sodu (w postaci 180 mg sodu chlorku) na fiolkę, co odpowiada 3,54% zalecanej przez WHO maksymalnej dawki dobowej sodu dla osoby dorosłej. Maksymalna dawka dobowa tego produktu odpowiada 24,77 % zalecanej przez WHO maksymalnej dawki dobowej sodu dla osoby dorosłej. Uważa się, że produkt Clofarabine Accord zawiera dużo
sodu. Należy w szczególności brać to pod uwagę w przypadku pacjentów na diecie ograniczającej spożycie sodu.
Dotąd nie przeprowadzono żadnych badań interakcji. Jednak nie są znane żadne klinicznie istotne interakcje z innymi lekami lub badaniami laboratoryjnymi.
Klofarabina nie jest w wykrywalnym zakresie metabolizowana przez układ enzymów cytochromu P450 (CYP). Dlatego interakcja z czynnymi substancjami będącymi inhibitorami lub indukującymi enzymy cytochromu P450 jest mało prawdopodobna. Ponadto jest mało prawdopodobne, aby klofarabina była inhibitorem któregokolwiek z 5 głównych ludzkich izoform CYP (1A2, 2C9, 2C19, 2D6 i 3A4) lub, aby indukowała 2 z tych izoform (1A2 i 3A4) przy stężeniu w osoczu osiągniętym po infuzji dożylnej dawki 52 mg/m2/dobę. W związku z tym nie oczekuje się wpływu klofarabiny na metabolizm substancji czynnych, które są znanymi substratami tych enzymów.
Klofarabina jest głównie wydalana przez nerki. Należy unikać, zwłaszcza podczas 5-dniowego leczenia klofarabiną, skojarzonego stosowania produktów leczniczych mogących wywoływać działania nefrotoksyczne i wydalanych drogą wydzielania kanalikowego (np. niesteroidowe leki przeciwzapalne, amfoterycyna B, metotreksat, aminoglikozydy, pochodne platyny, foskarnet, pentamidyna, cyklosporyna, takrolimus, acyklowir i walgancyklowir); z wyboru należy stosować produkty lecznicze pozbawione działania nefrotoksycznego (patrz punkty 4.4, 4.8 i 5.2).
Wątroba jest potencjalnym narządem docelowym dla działania toksycznego. Z tego powodu należy unikać, o ile to możliwe, jednoczesnego stosowania produktów leczniczych mogących mieć działanie hepatotoksyczne (patrz punkty 4.4 i 4.8).
Klofarabina może mieć działanie hepatotoksyczne. Podczas leczenia klofarabiną pacjenci przyjmujący leki wpływające na ciśnienie krwi lub czynność serca powinni być objęci ścisłą obserwacją (patrz punkty 4.4 i 4.8).
Antykoncepcja u mężczyzn i kobiet
Ze względu na ryzyko genotoksyczności klofarabiny (patrz punkt 5.3), kobiety w wieku rozrodczym muszą stosować skuteczne metody antykoncepcji podczas leczenia klofarabiną oraz przez 6 miesięcy po zakończeniu leczenia.
Mężczyźni powinni stosować skuteczne metody antykoncepcji oraz należy ich poinformować, aby nie płodzili dziecka podczas leczenia klofarabiną oraz przez 3 miesiące po zakończeniu leczenia.
Ciąża
Brak danych dotyczących stosowania klofarabiny u kobiet w ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję, w tym działanie teratogenne (patrz punkt 5.3). Przypuszcza się, że klofarabina stosowana w okresie ciąży może wywoływać ciężkie wady wrodzone. Dlatego produktu leczniczego Clofarabine Accord nie wolno stosować w okresie ciąży, a szczególnie w czasie pierwszego trymestru, jeśli nie jest to bezwzględnie konieczne (tj. jedynie wówczas, gdy potencjalne korzyści matki przewyższają ryzyko dla płodu). Jeśli pacjentka zajdzie w ciążę podczas leczenia klofarabiną, należy ją poinformować o możliwym zagrożeniu dla płodu.
Karmienie piersią
Nie wiadomo, czy klofarabina lub jej metabolity są wydzielane z mlekiem kobiet. Przenikania klofarabiny do mleka nie badano na zwierzętach. Jednak w związku z możliwością wystąpienia poważnych reakcji niepożądanych u niemowląt karmionych piersią należy przerwać karmienie przed
rozpoczęciem leczenia, w trakcie leczenia i po zakończeniu leczenia produktem leczniczym Clofarabine Accord (patrz punkt 4.3).
Płodność
Uzależnione od dawki działania toksyczne na męskie narządy płciowe obserwowano u myszy, szczurów i psów, a działania toksyczne na żeńskie organy płciowe obserwowano u myszy (patrz punkt 5.3). Ponieważ nie jest znany wpływ leczenia klofarabiną na płodność u ludzi, lekarz powinien przedyskutować z pacjentem planowanie posiadania potomstwa.
Nie przeprowadzono badań nad wpływem klofarabiny na zdolność prowadzenia pojazdów
i obsługiwania maszyn. Jednak należy uprzedzić pacjentów, że podczas leczenia mogą odczuwać działania niepożądane takie jak zawroty głowy, uczucie pustki w głowie lub okresy omdleń i w takich okolicznościach nie powinni prowadzić pojazdów ani obsługiwać maszyn.
Podsumowanie profilu bezpieczeństwa
Niemal u wszystkich (98%) pacjentów wystąpiło co najmniej jedno działanie niepożądane uznane przez badacza jako związane ze stosowaniem klofarabiny. Najczęściej opisywano nudności
(61% pacjentów), wymioty (59%), gorączkę neutropeniczną (35%), ból głowy (24%), wysypkę
(21%), biegunkę (20%), świąd (20%), gorączkę (19%), zespół erytrodyzestezji
dłoniowo-podeszwowej (15%), przewlekłe zmęczenie (14%), lęk (12%), zapalenie błon śluzowych (11%) i uderzenia gorąca (11%). U 68 (59%) pacjentów wystąpiło co najmniej jedno ciężkie działanie niepożądane związane z klofarabiną. Po podaniu klofarabiny w dawce 52 mg/m2/dobę jeden pacjent odstąpił od leczenia ze względu na hiperbilirubinemię 4. stopnia, która została uznana za związaną ze stosowaniem klofarabiny. Trzech pacjentów zmarło wskutek następujących działań niepożądanych uznanych przez badacza jako związane z leczeniem klofarabiną: niewydolność oddechowa, uszkodzenie komórek wątrobowych i zespół przesiąkania włośniczek (jedna osoba), posocznica
i niewydolność wielonarządowa (jedna osoba) oraz wstrząs septyczny i niewydolność wielonarządowa (jedna osoba).
Tabelaryczne zestawienie działań niepożądanych
Przedstawione informacje opierają się na danych uzyskanych z badań klinicznych, podczas których 115 pacjentów (> 1 i ≤ 21 lat) z ALL lub z ostrą białaczką szpikową (AML) otrzymało przynajmniej jedną zalecaną dawkę klofarabiny 52 mg/m2 na dobę przez 5 dni.
Reakcje niepożądane wymieniono według klasyfikacji układów i narządów MedDRA i ich częstości [bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100, rzadko
(≥10 000 do <1/1 000) i bardzo rzadko (<1/10 000)] w tabeli poniżej. W poniższej tabeli, w kategorii częstości „nieznana” (nie może być określona na podstawie dostępnych danych) przedstawiono również działania niepożądane zgłaszane po wprowadzeniu produktu do obrotu. W obrębie każdej grupy częstości, reakcje niepożądane są przedstawione zgodnie ze zmniejszającym się nasileniem.
U pacjentów w zaawansowanym stadium ALL lub AML stan zdrowia mógł być czynnikiem zakłócającym powodując trudności w ocenie przyczyny działań niepożądanych w związku z różnymi objawami związanymi z podstawową chorobą, jej progresją i równoczesnym podawaniem wielu leków.
Reakcje niepożądane uznane za związane ze stosowaniem klofarabiny, występujące w badaniach klinicznych częściej niż w ≥ 1/1000 przypadków
(tj. > 1/115 pacjentów) oraz zgłaszane w raportach po wprowadzeniu produktu do obrotu | |
Zakażenia i zarażenia pasożytnicze | Często: Wstrząs septyczny*, posocznica, bakteriemia, zapalenie płuc, półpasiec, opryszczka, drożdżyca jamy ustnej Częstość występowania nieznana: Zapalenie okrężnicywywołane przez C. difficile |
Nowotwory łagodne i złośliwe (w tym torbiele i polipy) | Często: Zespół rozpadu guza* |
Zaburzenia krwi i układu chłonnego | Bardzo często: Gorączka neutropeniczna Często: Neutropenia |
Zaburzenia układu immunologicznego | Często: Nadwrażliwość |
Zaburzenia metabolizmu i odżywiania | Często: Jadłowstręt, zmniejszenie łaknienia, odwodnienie Częstość występowania nieznana: hiponatremia |
Zaburzenia psychiczne | Bardzo często: Lęk Często: Pobudzenie, niepokój, zmiany stanu psychicznego |
Zaburzenia układu nerwowego | Bardzo często: Ból głowy Często: Senność, neuropatia obwodowa, parestezje, zawroty głowy, drżenia mięśniowe |
Zaburzenia ucha i błędnika | Często: Niedosłuch |
Zaburzenia serca | Często: Wysięk osierdziowy*, częstoskurcz* |
Zaburzenia naczyniowe | Bardzo często: Uderzenia gorąca* Często: Niedociśnienie tętnicze*, zespół przesiąkania włośniczek, krwiak |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Często: Niewydolność oddechowa, krwawienia z nosa, duszność, przyspieszony oddech, kaszel |
Zaburzenia żołądka i jelit | Bardzo często: Wymioty, nudności, biegunka Często: Krwotok z ust, krwawienia z dziąseł, krwioplucie, ból brzucha, zapalenie błony śluzowej jamy ustnej, ból w nadbrzuszu, ból odbytu, owrzodzeniabłony śluzowej jamy ustnej Częstość występowania nieznana: Zapalenie trzustki, zwiększenie aktywności amylazy i lipazy w surowicy, zapalenie jelit, neutropeniczne zapalenie okrężnicy, zapalenie kątnicy. |
Zaburzenia wątroby i dróg żółciowych | Często: Hiperbilirubinemia, żółtaczka, choroba żylnookluzyjna wątroby, zwiększenie aktywności aminotransferazy alaninowej (AlAT)* i asparaginowej (AspAT)*, niewydolność wątroby Niezbyt często: Zapalenie wątroby |
Zaburzenia ogólne i stany w miejscu podania | Bardzo często: Zmęczenie, gorączka, zapalenie błon śluzowych Często: Niewydolność wielonarządowa, zespół zapalnej reakcji ogólnoustrojowej*, ból, dreszcze, drażliwość, obrzęki, obrzęki obwodowe, uczucie gorąca, poczucie choroby |
Zaburzenia skóry i tkanki podskórnej | Bardzo często: Zespół erytrodyzestezji dłoniowopodeszwowej, świąd Często: Wysypka grudkowo-plamista, wybroczyny, rumień, swędząca wysypka, złuszczanie skóry, uogólniona wysypka, łysienie, hiperpigmentacja skóry, uogólniony |
rumień, wysypka rumieniowa, suchość skóry, nadmierne wydzielanie potu Częstość występowania nieznana: Zespół Stevensa i Johnsona, toksyczne martwicze oddzielanie się naskórka | |
Zaburzenia mięśniowo-szkieletowe, tkanki łącznej i kości | Często: Ból kończyn, ból mięśni, ból kości, ból ściany klatki piersiowej, bóle stawów, ból szyi i pleców |
Zaburzenia nerek i dróg moczowych | Często: Krwiomocz* Często: Niewydolność nerek, ostra niewydolność nerek |
Badania diagnostyczne | Często: Zmniejszenie masy ciała |
Urazy, zatrucia i powikłania po zabiegach | Często: Uraz |
* patrz poniżej
** W tabeli przedstawiono wszystkie działania niepożądane, które wystąpiły co najmniej dwukrotnie (1,7%).
Opis wybranych działań niepożądanych
Zaburzenia krwi i układu chłonnego
Najczęstsze zaburzenia wyników badań hematologicznych obserwowane u pacjentów leczonych klofarabiną obejmowały niedokrwistość (83,3%; 95/114), leukopenię (87,7%; 100/114); limfopenię (82,3%; 93/113), neutropenię (63,7%; 72/113) i małopłytkowość (80,7%; 92/114). Większość tych działań niepożądanych miało nasilenie ≥ 3°.
Po wprowadzeniu produktu do obrotu zgłaszano przedłużone cytopenie, (małopłytkowość, niedokrwistość, neutropenia i leukopenia) oraz niewydolność szpiku kostnego. W początkowym okresie występowania małopłytkowości obserwowano przypadki krwawień. Zgłaszano przypadki, w tym przypadki śmiertelne krwotoku, w tym krwotoku do mózgu, krwotoku z przewodu pokarmowego i płuc (patrz punkt 4.4).
Zaburzenia naczyniowe
U 64 ze 115 pacjentów (55,7%) wystąpiło przynajmniej jedno zdarzenie niepożądane zaburzeń naczyń. U 23 ze 115 pacjentów wystąpiło zdarzenie zaburzenia naczyń, które uznano za związane z klofarabiną. Do najczęściej zgłaszanych należały nagłe zaczerwienienia twarzy (13 zdarzeń; nie miały dużego nasilenia) i niedociśnienie (5 zdarzeń; wszystkie uznano za poważne; patrz punkt 4.4). Jednak większość zdarzeń niedociśnienia zgłaszano u pacjentów z czynnikiem zakłócającym w postaci ciężkich zakażeń.
Zaburzenia serca
U 50% pacjentów wystąpiło przynajmniej jedno zdarzenie niepożądane zaburzeń serca. Jedenaście zdarzeń na 115 pacjentów uznano za związane z klofarabiną, żadne z nich nie było poważne,
a najczęściej opisywanym zaburzeniem kardiologicznym był częstoskurcz (35%) (patrz punkt 4.4); u 6,1% (7 ze 115) pacjentów uznano związek pomiędzy częstoskurczem i stosowaniem klofarabiny.
Większość opisywanych kardiologicznych zdarzeń niepożądanych wystąpiło w pierwszych 2 cyklach leczenia.
Wysięk osierdziowy i zapalenie osierdzia zgłoszono jako zdarzenia niepożądane u 9% (10/115) pacjentów. Trzy z nich oceniono później jako związane z klofarabiną: wysięk osierdziowy
(2 zdarzenia, z których 1 było poważne) i zapalenie osierdzia (1 zdarzenie; nie miało poważnego przebiegu). U większości pacjentów (8/10), wysięk osierdziowy i zapalenie osierdzia uznano za bezobjawowe i o małym znaczeniu klinicznym lub bez takiego znaczenia w ocenie echokardiograficznej. Jednak wysięk osierdziowy miał znaczenie kliniczne u 2 pacjentów, u których występowały pewne zaburzenia hemodynamiczne.
Zakażenia i zarażenia pasożytnicze
Przed rozpoczęciem leczenia klofarabiną u 48% pacjentów występowało jedno lub więcej zakażeń. Po
leczeniu klofarabiną u 83% pacjentów wystąpiło co najmniej 1 zakażenie, włączając zakażenie grzybicze, wirusowe i bakteryjne (patrz punkt 4.4). Za związane z klofarabiną uznano 21 zdarzeń (18,3%), spośród których zakażenie odcewnikowe (1 zdarzenie), posocznicę (2 zdarzenia) i wstrząs septyczny (2 zdarzenia; 1 pacjent zmarł (patrz informacje powyżej)) uznano za poważne.
Po wprowadzeniu produktu do obrotu zgłaszano infekcje bakteryjne, grzybicze i wirusowe, które mogły prowadzić do zgonu. Infekcje te mogą prowadzić do wstrząsu septycznego, niewydolności oddechowej, niewydolności nerek i (lub) niewydolności wielonarządowej.
Zaburzenia nerek i dróg moczowych
U 41% ze 115 pacjentów (35,7%) wystąpiło przynajmniej jedno zdarzenie niepożądane zaburzeń nerek i dróg moczowych. Zwiększone stężenie kreatyniny było najczęstszym objawem nefrotoksyczności u dzieci. U 8% pacjentów wystąpiło zwiększenie stężenia kreatyniny 3. lub 4. stopnia. Objawy nefrotoksyczności mogą się nasilać pod wpływem produktów leczniczych o działaniu nefrotoksycznym, rozpadu guza nowotworowego i rozpadu guza przebiegającego ze zwiększeniem stężenia kwasu moczowego (patrz punkt 4.3 i 4.4). Krwiomocz obserwowano u 13% pacjentów.
U 115 pacjentów odnotowano 4 zdarzenia niepożądane związane z nerkami, które uznano za związane z klofarabiną, żadne z nich nie miało ciężkiego przebiegu i obejmowały one krwiomocz (3
zdarzenia) i ostrą niewydolność nerek (1 zdarzenie) (patrz punkty 4.3 i 4.4).
Zaburzenia wątroby i dróg żółciowych
Wątroba jest potencjalnym organem docelowym dla działań toksycznych klofarabiny i u 25,2% pacjentów wystąpiło przynajmniej jedno zdarzenie niepożądane zaburzeń wątroby i dróg żółciowych (patrz punkty 4.3 i 4.4). Sześć zdarzeń uznano za związane z klofarabiną, z których za poważne uznano: ostre zapalenie pęcherzyka żółciowego (1 zdarzenie), kamicę żółciową (1 zdarzenie), uszkodzenie wątrobowokomórkowe (1 zdarzenie; pacjent zmarł (patrz powyżej)) i hiperbilirubinemię [1 zdarzenie; pacjent przerwał leczenie (patrz powyżej)]. Zgłoszono dwa przypadki (1,7%) choroby żylno-okluzyjnej wątroby (VOD) u dzieci, uznane jako związane z badanym lekiem.
Przypadki VOD zgłaszane u dzieci i dorosłych po wprowadzeniu produktu do obrotu były związane ze zgonem (patrz punkt 4.4).
Poza tym u 50 ze 113 pacjentów leczonych klofarabiną wystąpił co najmniej jeden przypadek dużego zwiększenia aktywności AlAT (stopień 3 lub wyższy wg skali CTC NCI w USA), u 36 ze
100 pacjentów wystąpiło zwiększenie aktywności AspAT, a u 15 ze 114 pacjentów zwiększone stężenie bilirubiny. Większość przypadków zwiększenia aktywności AlAT i AspAT wystąpiła
w ciągu 10 dni od podania klofarabiny, a w ciągu 15 dni wyniki wróciły do poziomu ≤ 2. stopnia. U pacjentów, dla których dostępne są dane z obserwacji kontrolnych, w większości przypadków w ciągu 10 dni zwiększone stężenie bilirubiny wróciło do poziomu ≤ 2. stopnia.
Uogólniona odpowiedź zapalna (SIRS) lub zespół przesiąkania włośniczek
Zespół ogólnoustrojowej reakcji zapalnej (SIRS), zespół przesiąkania włośniczek (objawy przedmiotowe i podmiotowe związane z uwalnianiem cytokiny, np. przyspieszenie oddychania, częstoskurcz, niedociśnienie tętnicze, obrzęk płuc) opisywano jako zdarzenie niepożądane u 5% (6 ze 115) dzieci (5 przypadków białaczki ALL i 1 AML) (patrz punkt 4.4). Opisano 13 przypadków zespołu rozpadu guza, zespołu przesiąkania włośniczek lub SIRS: 2 zdarzenia SIRS (oba uznano jako ciężkie) oraz 7 zdarzeń zespołu rozpadu guza (6 zdarzeń uznano za związane z leczeniem, z czego
3 zakwalifikowano jako ciężkie).
Przypadki zespołu przesiąkania włośniczek zgłaszane po wprowadzeniu klofarabiny do obrotu były związane ze zgonem (patrz punkt 4.4).
Zaburzenia żołądka i jelit
Podczas leczenia klofarabiną zgłaszano przypadki zapalenia jelit, w tym zapalenie okrężnicy neutropeniczne lub wywołane C. difficile i zapalenie kątnicy. Zapalenie jelit może prowadzić do powikłań, takich jak martwica, perforacja lub posocznica i może zakończyć się zgonem (patrz punkt 4.4).
Zaburzenia skóry i tkanki podskórnej
Zgłaszano występowanie zespołu Stevensa-Johnsona (SJS) i martwiczego toksycznego oddzielania się naskórka (TEN), w tym przypadki zakończone zgonem u pacjentów, którzy otrzymywali lub niedawno byli leczeni klofarabiną. Zgłaszano również inne stany związane ze złuszczaniem.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C, 02-222 Warszawa
tel.: + 48 22 49 21 301, faks: + 48 22 49 21 309
strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Objawy
Nie zgłoszono żadnego przypadku przedawkowania. Jednak oczekuje się, że objawy przedawkowania mogą obejmować nudności, wymioty, biegunkę i ciężką supresję szpiku kostnego. Dotąd największa podana dobowa dawka wynosiła 70 mg/m2 pc. przez 5 kolejnych dni (2 pacjentów pediatrycznych
z ALL). Objawy toksyczności obserwowane u tych pacjentów obejmowały wymioty, hiperbilirubinemię, podwyższone aktywności aminotransferaz i wysypkę grudkowo-plamkową.
Postępowanie w przedawkowaniu
Brak swoistego antidotum. Zaleca się natychmiastowe przerwanie leczenia, ścisłą obserwację i zastosowanie odpowiedniego leczenia wspomagającego.
Grupa farmakoterapeutyczna: leki przeciwnowotworowe, antymetabolity, kod ATC: L01BB06. Mechanizm działania
Klofarabina jest antymetabolitem nukleozydu puryn. Uważa się, że aktywność przeciwnowotworowa klofarabiny wynika z 3 mechanizmów:
Klofarabina musi najpierw przeniknąć lub zostać przetransportowana do komórek docelowych, gdzie za pośrednictwem kinaz wewnątrzkomórkowych ulega sekwencyjnej fosforylacji do mono- i difosforanów a na końcu do aktywnego koniugatu klofarabina 5’-trifosforanu. Klofarabina wykazuje duże powinowactwo do jednego z aktywujących enzymów fosforylujących – kinazy deoksycytydynowej, która przekracza powinowactwo naturalnego substratu deoksycytydyny.
Ponadto, klofarabina wykazuje większą oporność na rozkład w komórce przez deaminazę adenozynową i mniejszą podatność na rozpad fosforolityczny niż inne substancje aktywne w jej klasie, podczas gdy powinowactwo trifosforanów klofarabiny do polimerazy DNA α i reduktazy rybonukleotydowej jest podobne lub większe niż takie powinowactwo trifosforanów deoksyadenozyny.
Działanie farmakodynamiczne
Badania in vitro wykazały, że klofarabina hamuje wzrost komórek i wykazuje działanie cytotoksyczne wobec różnych szybko proliferujących linii komórek hematologicznych i litego guza. Klofarabina wykazywała również działanie wobec nieaktywnych limfocytów i makrofagów. Ponadto, w przypadku różnych ludzkich i mysich nowotworów przeszczepionych myszom klofarabina opóźniała wzrost nowotworu i w niektórych przypadkach powodowała regresję nowotworu.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność kliniczna: W celu umożliwienia systematycznej oceny odpowiedzi obserwowanych u pacjentów, odtajniony Panel Niezależnego Przeglądu Odpowiedzi (ang. Independent Response Review Panel, IRRP) określił następujące odpowiedzi na podstawie definicji przygotowanych przez Grupę Onkologii Dziecięcej:
CR = Całkowita remisja | Pacjenci, którzy spełniają wszystkie poniższe kryteria: występowania choroby pozaszpikowej ≥ 100 x 109/l i ANC ≥ 1,0 x 109/l) |
CRp = Całkowita remisja (CR) przy braku Całkowitej Poprawy Płytek | |
PR = Częściowa remisja | Pacjenci, którzy spełniają wszystkie poniższe kryteria: |
Współczynnik ogólnej remisji (OR) | ÷ Liczba pacjentów zakwalifikowanych, którzy otrzymali klofarabinę |
Bezpieczeństwo stosowania i skuteczność klofarabiny oceniono w otwartym badaniu I fazy bez grupy kontrolnej, z eskalacją dawek, z udziałem 25 dzieci z nawrotem/oporną białaczką (17 ALL; 8 AML), u których standardowe leczenie nie przyniosło poprawy lub dla których nie istniała inna terapia.
Podawanie rozpoczęto od dawki 11,25 zwiększając do 15, 30, 40, 52 i 70 mg/m2 pc./dobę w infuzji dożylnej przez 5 dni co 2 do 6 tygodni zależnie od toksyczności i odpowiedzi. Dziewięcioro
z siedemnastu pacjentów z białaczką ALL otrzymywało leczenie klofarabiną 52 mg/m2/dobę. Spośród 17 pacjentów z ALL, u 2 nastąpiła całkowita remisja (12%; CR), a u 2 częściowa remisja (12%; PR) po podawaniu różnych dawek. Podczas tego badania ograniczająca dawkę toksyczność obejmowała hiperbilirubinemię, podwyższone aktywności aminotransferaz i wysypkę grudkowo-plamkową występującą podczas podawania dawki 70 mg/m2 pc./dobę (2 pacjentów z ALL; patrz punkt 4.9).
Wieloośrodkowe otwarte badanie II fazy, bez grupy kontrolnej dotyczące klofarabiny zostało
przeprowadzone w celu oceny współczynnika ogólnej remisji (OR) u pacjentów, u których stosowano wcześniej liczne terapie (≤ 21 lat podczas pierwszej diagnozy) z nawrotem lub oporną ALL zdefiniowaną za pomocą klasyfikacji francusko-amerykańsko-angielskiej. Maksymalną tolerowaną dawkę klofarabiny, wskazaną podczas badania I fazy opisanego powyżej wynoszącą
52 mg/m2 pc./dobę, podawano w infuzji dożylnej przez 5 kolejnych dni co 2 do 6 tygodni. W poniższej tabeli podsumowano główne wyniki skuteczności w tym badaniu.
Pacjenci z ALL nie mogli być zakwalifikowani do bardziej intensywnego leczenia i musiało u nich dojść do drugiej lub kolejnej wznowy i (lub) nastąpiła oporność na leczenie, co oznaczało brak remisji po co najmniej dwóch poprzednio zastosowanych schematach leczenia. Przed zakwalifikowaniem do badania 58 z 61 pacjentów (95%) otrzymało od 2 do 4 różnych schematów leczenia indukującego
i 18/61 (30%) z tych pacjentów zostało poddanych przynajmniej 1 wcześniejszej transplantacji krwiotwórczych komórek macierzystych (ang. Haemopoetic stem cells transplantation − HSCT). Mediana wieku leczonych pacjentów (37 płci męskiej, 24 płci żeńskiej) wynosiła 12 lat.
Podanie klofarabiny spowodowało gwałtowne i szybkie zmniejszenie liczby komórek białaczkowych we krwi obwodowej u 31 z 33 pacjentów (94%), u których bezwzględną liczbę blastów określono na początku badania. U 12 pacjentów, którzy uzyskali ogólną remisję (CR + CRp) mediana czasu przeżycia wynosiła 66,6 tygodnia w granicznym dniu zbierania danych. Odpowiedź odnotowano
u pacjentów z różnymi immunofenotypami ALL, w tym z komórkami pre-B i komórkami T. Pomimo, że odsetek transplantacji nie stanowił punktu końcowego badania, 10/61 pacjentów (16%) otrzymało HSCT po leczeniu klofarabiną (3 po uzyskaniu CR, 2 po CRp, 3 po PR, 1 pacjenta, u którego uznano brak poprawy w związku z leczeniem według IRRP i 1, którego uznano za niekwalifikującego się do oceny według IRRP). Okresy trwania odpowiedzi są mylące u pacjentów, którzy otrzymali HSCT.
Wyniki skuteczności z badania głównego u pacjentów (≤ 21 lat podczas pierwszej diagnozy) z nawrotami lub oporną na leczenie ALL po podaniu przynajmniej dwóch wcześniejszych cykli | ||||
Kategoria odpowiedzi | Pacjenc i ITT* (n = 61) | Mediana czasu trwania remisji (tygodnie) (95% CI) | Mediana czasu do wystąpienia objawów (tygodnie)** (95% CI) | Mediana ogólnego przeżycia (tygodnie) (95% CI) |
Ogólna remisja | 12 | 32,0 | 38,2 | 69,5 |
(CR + CRp) | (20%) | (9,7 to 47,9) | (15,4 to 56,1) | (58,6 to -) |
CR | 7 | 47,9 | 56,1 | 72,4 |
(12%) | (6,1 to -) | (13,7 to -) | (66,6 to -) | |
CRp | 5 | 28,6 | 37,0 | 53,7 |
(8%) | (4,6 to 38,3) | (9,1 to 42) | (9,1 to -) | |
PR | 6 | 11,0 | 14,4 | 33,0 |
(10%) | (5,0 to -) | (7,0 to -) | (18,1 to -) | |
CR + CRp + PR | 18 | 21,5 | 28,7 | 66,6 |
(30%) | (7,6 to 47,9) | (13,7 to 56,1) | (42,0 to -) | |
Brak poprawy w | 33 | |||
związku z | (54%) | N/A | ||
leczeniem | 4,0 | 7,6 | ||
Brak | 10 | (3,4 to 5,1) | (6,7 to 12,6) | |
możliwości | (16%) | N/A | ||
oceny | ||||
Wszyscy | 61 | N/A | 5,4 | 12,9 |
pacjenci | (100%) | (4,0 to 6,1) | (7,9 to 18,1) | |
*ITT = populacja zgodna z zamiarem leczenia. ** Dane od pacjentów żyjących z remisją podczas ostatniej obserwacji ocenianych w danym punkcie czasu analizy. | ||||
Indywidualny okres remisji i dane dotyczące przeżywalności pacjentów, u których uzyskano całkowitą remisję (CR) lub całkowitą remisję bez pełnej normalizacji płytek (CRp) | |||
Najlepsza odpowiedź na leczenie | Czas do ogólnej remisji (OR) [tygodnie] | Długość okresu remisji [tygodnie] | Łączne przeżycie [tygodnie] |
Pacjenci, którzy nie otrzymali przeszczepu | |||
CR | 5,7 | 4,3 | 66,6 |
CR | 14,3 | 6,1 | 58,6 |
CR | 8,3 | 47,9 | 66,6 |
CRp | 4,6 | 4,6 | 9,1 |
CR | 3,3 | 58,6 | 72,4 |
CRp | 3,7 | 11,7 | 53,7 |
Pacjenci, którzy otrzymali przeszczep w okresie remisji* | |||
CRp | 8,4 | 11,6+ | 145,1+ |
CR | 4,1 | 9,0+ | 111,9+ |
CRp | 3,7 | 5,6+ | 42,0 |
CR | 7,6 | 3,7+ | 96,3+ |
Pacjenci, którzy otrzymali przeszczep po leczeniu alternatywnym lub wznowie choroby* | |||
CRp | 4,0 | 35,4 | 113,3+** |
CR | 4,0 | 9,7 | 89,4*** |
* Długość okresu remisji ustalana w chwili otrzymania przeszczepu ** Pacjent otrzymał przeszczep po zakończeniu leczenia alternatywnego *** Pacjent otrzymał przeszczep po wznowie choroby | |||
Referencyjny produkt leczniczy zawierający klofarabinę został dopuszczony do obrotu zgodnie
z procedurą dopuszczenia w wyjątkowych okolicznościach. Oznacza to, że ze względu na rzadkie występowanie choroby nie było możliwe uzyskanie pełnej informacji dotyczącej referencyjnego produktu leczniczego.
Europejska Agencja Leków dokona raz w roku przeglądu wszelkich nowych informacji i, w razie
konieczności, ChPL zostanie zaktualizowana zgodnie z ChPL referencyjnego produktu leczniczego.
Wchłanianie i dystrybucja
Farmakokinetykę klofarabiny badano u 40 pacjentów w wieku od 2 do 19 lat z nawrotem lub oporną ALL lub AML. Pacjenci zostali zakwalifikowani do jednego badania I fazy (n = 12) lub dwóch badań II fazy (n = 14 / n = 14) oceniających bezpieczeństwo stosowania i skuteczność i otrzymali wielokrotne dawki klofarabiny w infuzji dożylnej (patrz punkt 5.1).
Farmakokinetyka u pacjentów w wieku od 2 do 19 lat z nawrotem lub oporną ALL lub AML po podaniu wielokrotnych dawek klofarabiny w infuzji dożylnej | ||
Parametr | Oceny na podstawie analizy niekompartmentowej (n = 14 / n = 14) | Oceny oparte na innej analizie |
Dystrybucja: | ||
Objętość dystrybucji (stan stacjonarny) | 172 l/m2 pc | |
Wiązanie z białkami osocza | 47,1% | |
Albumina surowicy | 27,0% | |
Eliminacja: | ||
Okres półtrwania klofarabiny β | 5,2 godziny | |
Okres półtrwania trifosforanu klofarabiny | > 24 godziny | |
Klirens układowy | 28,8 l/godz./m2 pc | |
Klirens nerkowy | 10,8 l/godz./m2 pc | |
Dawka wydalana w moczu | 57% |
Analiza wieloczynnikowa wykazała, że farmakokinetyka klofarabiny zależy od masy ciała i pomimo, że wskazano na wpływ liczby białych krwinek (WBC) na farmakokinetykę klofarabiny, nie był on wystarczający do indywidualizacji schematu dawkowania u pacjenta na podstawie jego liczby białych krwinek. Infuzja dożylna klofarabiny w dawce 52 mg/m2 pc. zapewniała równoważną ekspozycję
w szerokim zakresie mas ciała. Jednak wartość Cmax jest odwrotnie proporcjonalna do masy ciała pacjenta, dlatego u małych dzieci wartość Cmax może być większa pod koniec infuzji niż
w przypadku typowego dziecka o masie ciała 40 kg, któremu podano taką samą dawkę klofarabiny na m2 pc. Zatem u dzieci o masie ciała < 20 kg należy rozważyć odpowiednio dłuższy czas trwania infuzji (patrz punkt 4.2).
Metabolizm i eliminacja
Klofarabina jest wydalana w wyniku połączonego wydalania przez nerki i wydalania poza nerkowego. Po 24 godzinach około 60% dawki jest wydalane w postaci niezmienionej w moczu. Wielkość klirensu klofarabiny wydaje się być większa niż wielkość filtracji kłębuszkowej wskazującą filtrację
i wydzielanie leku przez kanaliki nerkowe jako mechanizm eliminacji przez nerki. Jednak, ponieważ klofarabina nie jest w wykrywalnym zakresie metabolizowana przez układ enzymów cytochromu P450 (CYP), ścieżki eliminacji poza nerkowej pozostają obecnie nieznane.
Nie zauważono żadnych widocznych różnic w farmakokinetyce pomiędzy pacjentami z ALL i AML, ani pomiędzy pacjentami płci męskiej i żeńskiej.
W tej populacji nie ustalono żadnego związku pomiędzy ekspozycją na klofarabinę lub trifosforan klofarabiny a skutecznością i toksycznością.
Szczególne grupy pacjentów
Dorośli (> 21 i < 65 lat)
Obecnie posiadane informacje są niewystarczające do ustalenia bezpieczeństwa stosowania
i skuteczności klofarabiny u dorosłych pacjentów. Jednak farmakokinetykę klofarabiny, u dorosłych z nawrotem lub z oporną na leczenie AML po podaniu pojedynczej dawki klofarabiny wynoszącej 40 mg/m2 pc. w infuzji dożylnej trwającej 1 godzinę, porównywano z farmakokinetyką opisaną powyżej u pacjentów w wieku od 2 do 19 lat z nawrotem i z oporną na leczenie ALL lub AML po
podaniu klofarabiny w dawce 52 mg/m2 pc. w infuzji dożylnej trwającym 2 godziny przez 5 kolejnych
dni.
Osoby w podeszłym wieku (≥ 65 lat)
Obecnie posiadane informacje są niewystarczające do ustalenia bezpieczeństwa stosowania i skuteczności klofarabiny u pacjentów w wieku 65 lat i starszych.
Zaburzenia czynności nerek
Obecnie brak dostatecznych danych na temat farmakokinetyki klofarabiny dzieci i młodzieży
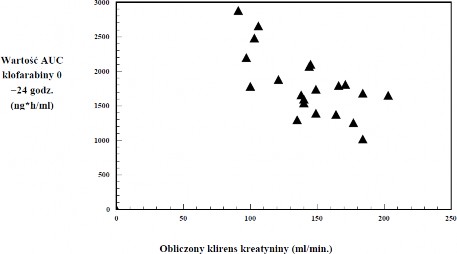
z obniżonym klirensem kreatyniny. Niemniej takie ograniczone dane wskazują, że klofarabina może gromadzić się u pacjentów z obniżonym klirensem kreatyniny (patrz wykres poniżej).
Dane farmakokinetyczne dotyczące populacji pacjentów dorosłych oraz dzieci i młodzieży wskazują, że u pacjentów ze stabilną umiarkowaną niewydolnością nerek (klirens kreatyniny 30 – < 60 ml/min) otrzymujących dawkę zmniejszoną o połowę, ekspozycja na klofarabinę jest podobna do tej
u pacjentów z prawidłową czynnością nerek otrzymujących dawkę standardową.
Wartość AUC0-24 godz klofarabiny według obliczonego wyjściowego klirensu kreatyniny
u pacjentów w wieku od 2 do 19 lat z nawrotem lub oporną ALL lub AML (n = 11 / n = 12) po podaniu wielokrotnych dawek klofarabiny w infuzji dożylnej (klirens kreatyniny obliczony za pomocą wzoru Schwartza)
Zaburzenia czynności wątroby
Brak doświadczenia w leczeniu pacjentów z zaburzeniami czynności wątroby (stężenie bilirubiny w surowicy > 1,5 x GGN plus AspAT i AlAT > 5 x GGN), a wątroba jest potencjalnym narządem docelowym dla działań toksycznych (patrz punkty 4.3 i 4.4).
Badania toksykologiczne klofarabiny prowadzone na myszach, szczurach i psach wykazały, że szybko proliferujące tkanki stanowiły podstawowe narządy docelowe dla działań toksycznych.
U szczurów obserwowano wpływ leku na serce zgodny z kardiomiopatią, który przyczyniał się do powstawania objawów niewydolności serca po ponownych cyklach leczenia. Częstość występowania takiej toksyczności zależała zarówno od wielkości podanej dawki klofarabiny jak i czasu trwania leczenia. Toksyczność obserwowano przy poziomach ekspozycji (Cmax) około 7 do 13 krotnie
(po 3 lub więcej cyklach leczenia) lub 16 do 35 krotnie (po jednym lub kilku cyklach leczenia) większych niż ekspozycja kliniczna. Minimalne działania obserwowane po mniejszych dawkach sugerują, że istnieje wartość progowa dla kardiotoksyczności, a nieliniowa farmakokinetyka osoczowa u szczurów może mieć znaczenie dla występowania obserwowanych działań. Potencjalne
zagrożenie dla człowieka nie jest znane.
U szczurów po podaniu dawki 3 do 5 krotnie większej niż kliniczna AUC po 6 cyklach leczenia klofarabiną obserwowano nefropatię kłębuszkową, która charakteryzowała się małym pogrubieniem kłębuszkowej błony podstawnej i jedynie niewielkim uszkodzeniem kanalików i nie towarzyszyły jej zmiany wyników badań biochemicznych surowicy.
Po długotrwałym podawaniu klofarabiny szczurom obserwowano jej wpływ na czynność wątroby. Zmiany te prawdopodobnie przedstawiają nałożenie się zmian zwyrodnieniowych i regeneracyjnych spowodowanych zastosowanymi cyklami leczenia i nie były związane z odchyleniami wyników badań biochemicznych surowicy. Histologiczne dowody na wywieranie przez lek wpływu na wątrobę obserwowano u psów po jednorazowym podaniu dużych dawek, lecz również nie towarzyszyły im zmiany w wynikach badań biochemicznych surowicy.
Zależną od wielkości dawki toksyczność na męskie organy płciowe obserwowano u myszy, szczurów i psów. Działania te obejmowały obustronne zwyrodnienie nabłonka kanalików nasiennych
z zatrzymanymi spermatydami i atrofią komórek śródmiąższowych u szczurów po podaniu nadmiernych dawek (150 mg/m2 pc. na dobę) oraz zwyrodnienie komórek najądrza i zwyrodnienie nabłonka kanalików nasiennych u psów po podaniu klinicznie istotnych dawek klofarabiny
(> 7,5 mg/m2 pc. na dobę).
U samic myszy po podaniu klofarabiny w jedynej zastosowanej dawce 225 mg/m2 pc. na dobę obserwowano opóźnioną atrofię jajników lub zwyrodnienie i apoptozę błony śluzowej macicy.
Klofarabina wykazywała działanie teratogenne u szczurów i królików. U szczurów otrzymujących klofarabinę w dawce stanowiącej około 2 do 3 krotność dawek klinicznych (54 mg/m2 pc. na dobę)
i u królików otrzymujących 12 mg/m2 pc. na dobę obserwowano zwiększenie strat po implantacji jaja, zmniejszoną masę płodów i zmniejszoną wielkość miotów wraz ze zwiększeniem liczby wad rozwojowych (ogólne zewnętrzne, tkanki miękkie) i zmian szkieletowych w tym opóźnione kostnienie). (Brak danych dotyczących ekspozycji królików). Za wartość progową dla toksyczności rozwojowej uznano 6 mg/m2 pc. na dobę u szczurów i 1,2 mg/m2 pc. na dobę u królików. Poziom niezauważalnych działań toksycznych u matki wynosił u szczurów 18 mg/m2 pc. na dobę, a u królików ponad 12 mg/m2 pc. na dobę. Nie prowadzono badań płodności.
Badania genotoksyczności wykazały, że klofarabina nie była mutagenna w teście mutacji powrotnych na bakteriach, lecz wywierała działanie klastogenne w nie-aktywowanych testach aberracji chromosomowej na komórkach jajnika chomika chińskiego (Chinese Hamster Ovary - CHO) i in vivo w teście mikrojąderkowym na szczurach.
Nie prowadzono badań działania rakotwórczego.
Sodu chlorek
Woda do wstrzykiwań
Produktu leczniczego nie wolno mieszać z innymi lekami poza podanymi w punkcie 6.6.
3 lata.
Po rozcieńczeniu
Rozcieńczony koncentrat zachowuje stabilność chemiczną i fizyczną przez 3 dni w temperaturze 2°C do 8°C i w temperaturze pokojowej (do 25°C).
Z mikrobiologicznego punktu widzenia produkt należy zużyć natychmiast. Jeśli produkt nie jest zastosowany natychmiast, użytkownik ponosi odpowiedzialność za dalszy okres oraz warunki przechowywania przed zastosowaniem. Przechowywanie zwykle nie powinno trwać dłużej niż 24 godziny w temperaturze 2°C do 8°C, jeśli rozcieńczenie zostało wykonane w kontrolowanych i zwalidowanych warunkach aseptycznych.
Nie zamrażać.
Warunki przechowywania produktu leczniczego po rozcieńczeniu, patrz punkt 6.3.
Fiolka ze szkła typu I, z korkiem z silikonowej gumy pokrytej teflonem, niebieskim kapslem typu flip- off i aluminiowym uszczelnieniem. Fiolki zawierają 20 ml koncentratu roztworu do infuzji i są umieszczone w tekturowych pudełkach.
Jedno tekturowe pudełko zawiera 1 fiolkę.
do stosowania
Specjalne środki ostrożności dotyczące przygotowania produktu leczniczego do stosowania
Przed podaniem produktu Clofarabine Accord 1 mg/ml koncentrat do sporządzania roztworu do infuzji, należy go rozcieńczyć. Należy go przefiltrować przez jałowy filtr strzykawkowy o średnicy porów 0,2 mikronów, a następnie rozcieńczyć roztworem chlorku sodu 9 mg/ml (0,9%) do wstrzykiwań, w workach lub butelkach nie-PVC, aby otrzymać łączną objętość zgodnie z podanym przykładem w tabeli poniżej. Jednak gotowa objętość rozcieńczenia może być różna, zależnie od stanu klinicznego pacjenta i decyzji lekarza. (Jeśli nie jest możliwe zastosowanie filtru strzykawkowego
o średnicy porów 0,2 mikronów, koncentrat należy wstępnie przefiltrować przez filtr o średnicy porów 5 mikronów, rozcieńczyć a następnie podawać przez filtr o średnicy porów 0,22 mikronów w zestawie do infuzji.)
Proponowany schemat rozcieńczeń w oparciu o zalecaną dawkę klofarabiny 52 mg/m2 pc na dobę | ||
Powierzchnia ciała (m2) | Koncentrat (ml)* | Łączna objętość po rozcieńczeniu |
≤ 1,44 | ≤ 74.9 | 100 ml |
1,45 do 2,40 | 75,4 do 124,8 | 150 ml |
2,41 do 2,50 | 125,3 do 130,0 | 200 ml |
*1 ml koncentratu zawiera 1 mg klofarabiny. Jedna fiolka o objętości 20 ml zawiera 20 mg klofarabiny. Dlatego w przypadku pacjentów o powierzchni ciała ≤ 0,38 m2, do przygotowania zalecanej dobowej dawki klofarabiny zostanie użyta część zawartości pojedynczej fiolki. Natomiast w przypadku pacjentów o powierzchni ciała > 0,38 m2 do przygotowania zalecanej dobowej dawki klofarabiny zostanie użyta zawartość od 1 do 7 fiolek. | ||
Rozcieńczony koncentrat powinien być klarownym i bezbarwnym roztworem. Przed podaniem należy go skontrolować wzrokowo w celu wykluczenia obecności cząstek stałych i zmiany zabarwienia.
Instrukcja podania
Należy przestrzegać procedur prawidłowego obchodzenia się z lekami przeciwnowotworowymi. Należy zachować ostrożność podczas obchodzenia się z lekami cytotoksycznymi.
Podczas obchodzenia się z produktem leczniczym Clofarabine Accord zaleca się stosowanie jednorazowych rękawiczek i ubrań ochronnych. W przypadku kontaktu z oczami, skórą lub błonami śluzowymi skażone miejsce należy natychmiast przemyć dużą ilością wody.
Kobiety w ciąży nie mogą mieć kontaktu z produktem leczniczym Clofarabine Accord. Usuwanie niewykorzystanego produktu
Clofarabine Accord jest przeznaczona do jednorazowego użycia. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
DOPUSZCZENIE DO OBROTU
Accord Healthcare Polska Sp. z o.o.
ul. Taśmowa 7
02-677 Warszawa
25718
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 27.01.2020
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
19.04.2023







