







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
nowo rozpoznaną przewlekłą białaczką szpikową (ang. chronic myelogenous leukemia, CML) z chromosomem Philadelphia (ang. Philadelphia chromosome positive, Ph+) w fazie przewlekłej (ang. chronic phase, CP),
przewlekłą białaczką szpikową w fazie przewlekłej, w fazie akceleracji lub w fazie przełomu blastycznego w przypadku oporności lub nietolerancji na uprzednie leczenie, w tym leczenie imatynibem,
ostrą białaczką limfoblastyczną (ang. acute lymphoblastic leukaemia, ALL) z chromosomem Philadelphia (Ph+) oraz z limfoblastyczną postacią przełomu blastycznego CML, w przypadku oporności lub nietolerancji wcześniejszej terapii.
Dazatynib jest wskazany do leczenia dzieci i młodzieży z:
nowo rozpoznaną przewlekłą białaczką szpikową z chromosomem Philadelphia w fazie przewlekłej (ang. Philadelphia chromosome-positive chronic myelogenous leukaemia in chronic phase, Ph+ CML CP) lub Ph+ CML CP w przypadku oporności lub nietolerancji na uprzednie leczenie, w tym leczenie imatynibem.
nowo rozpoznaną Ph+ ALL w skojarzeniu z chemioterapią.
Dawkowanie i sposób podawania
Przerwać leczenie do czasu, gdy ANC ≥ 1,0 x 109/l oraz liczba płytek krwi
Ponownie rozpocząć leczenie w pierwotnej dawce.
W przypadku spadku liczby płytek krwi
Sprawdzić, czy cytopenia jest związana z białaczką (aspiracja szpiku kostnego lub biopsja)
Jeśli cytopenia nie jest związana z białaczką, należy wstrzymać leczenie aż do czasu, gdy ANC ≥ 1,0 x 109/l i liczba płytek krwi ≥ 20 x 109/l oraz ponownie rozpocząć leczenie w pierwotnej dawce początkowej.
W przypadku nawrotu cytopenii, powtórzyć punkt 1 i ponownie rozpocząć leczenie w mniejszej dawce, 100 mg raz na dobę (drugi epizod) lub 80 mg raz na dobę (trzeci epizod).
Jeśli cytopenia związana jest z białaczką, należy rozważyć zwiększenie dawki do 180 mg raz na dobę
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Dasatinib Alvogen, 20 mg, tabletki powlekane Dasatinib Alvogen, 50 mg, tabletki powlekane Dasatinib Alvogen, 70 mg, tabletki powlekane Dasatinib Alvogen, 80 mg, tabletki powlekane Dasatinib Alvogen, 100 mg, tabletki powlekane Dasatinib Alvogen, 140 mg, tabletki powlekane
Dasatinib Alvogen, 20 mg, tabletki powlekane
Każda tabletka powlekana zawiera 20 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 27 mg laktozy jednowodnej i około 0,0161 mg sodu.
Dasatinib Alvogen, 50 mg, tabletki powlekane
Każda tabletka powlekana zawiera 50 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 67,5 mg laktozy jednowodnej i około 0,0403 mg sodu.
Dasatinib Alvogen, 70 mg, tabletki powlekane
Każda tabletka powlekana zawiera 70 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 94,5 mg laktozy jednowodnej i około 0,0564 mg sodu.
Dasatinib Alvogen, 80 mg, tabletki powlekane
Każda tabletka powlekana zawiera 80 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 108 mg laktozy jednowodnej i około 0,0645 mg sodu.
Dasatinib Alvogen, 100 mg, tabletki powlekane
Każda tabletka powlekana zawiera 100 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 135,0 mg laktozy jednowodnej i około 0,0806 mg sodu.
Dasatinib Alvogen, 140 mg, tabletki powlekane
Każda tabletka powlekana zawiera 140 mg dazatynibu (w postaci dazatynibu bezwodnego).
Substancja pomocnicza o znanym działaniu
Każda tabletka powlekana zawiera 189 mg laktozy jednowodnej i około 0,1129 mg sodu. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana (tabletka).
Dasatinib Alvogen, 20 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, okrągłe tabletki powlekane, z wytłoczonym napisem "DAS" na jednej stronie oraz napisem "20" na drugiej stronie, o przybliżonym wymiarze 5,6 mm.
Dasatinib Alvogen, 50 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, owalne tabletki powlekane, z wytłoczonym napisem "DAS" na jednej stronie oraz napisem "50" na drugiej stronie, o przybliżonych wymiarach 10,.9 x 5,8 mm.
Dasatinib Alvogen, 70 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, okrągłe tabletki powlekane, z wytłoczonym napisem "DAS" na jednej stronie oraz napisem "70" na drugiej stronie, o przybliżonym wymiarze 8,8 mm.
Dasatinib Alvogen, 80 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, trójkątne tabletki powlekane, z wytłoczonym napisem "DAS" na jednej stronie oraz napisem "80" na drugiej stronie, o przybliżonych wymiarach 10,4 x 10,1 mm.
Dasatinib Alvogen, 100 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, owalne tabletki powlekane, z wytłoczonym napisem
"DAS" na jednej stronie oraz napisem "100" na drugiej stronie, o przybliżonych wymiarach 14,9 x 7,2 mm.
Dasatinib Alvogen, 140 mg, tabletki powlekane
Białe lub białawe, dwustronnie wypukłe, okrągłe tabletki powlekane, z wytłoczonym napisem "DAS" na jednej stronie oraz napisem "140" na drugiej stronie, o przybliżonym wymiarze 11,2 mm.
Dazatynib jest wskazany do leczenia dorosłych pacjentów z:
Leczenie powinno być rozpoczęte przez lekarza doświadczonego w rozpoznawaniu i leczeniu białaczki.
Dawkowanie
Dorośli pacjenci
Zalecana dawka początkowa dazatynibu w fazie przewlekłej CML wynosi 100 mg raz na dobę.
Zalecana dawka początkowa dazatynibu w fazie akceleracji mieloblastycznej lub limfoblastycznej postaci przełomu blastycznego (faza zaawansowana) CML, lub w ostrej białaczce limfoblastycznej z chromosomem Philadelphia (Ph+ ALL) wynosi 140 mg raz na dobę (patrz punkt 4.4).
Dzieci i młodzież (Ph+ CML CP i Ph+ ALL)
Dawkowanie u dzieci i młodzieży ustala się w zależności od masy ciała (patrz Tabela 1). Dazatynib jest podawany doustnie raz na dobę w postaci tabletek powlekanych albo w postaci proszku do sporządzania zawiesiny doustnej. Dawkę należy przeliczać co 3 miesiące z uwzględnieniem zmian masy ciała lub częściej, o ile jest to konieczne. Nie zaleca się stosowania tabletek u pacjentów ważących mniej niż 10 kg; u tych pacjentów należy stosować proszek do sporządzania zawiesiny doustnej. Zaleca się zwiększenie lub zmniejszenie dawki w zależności od indywidualnej odpowiedzi pacjenta na leczenie i tolerancji. Nie ma doświadczenia w leczeniu dazatynibem dzieci w wieku poniżej 1 roku.
Dazatynib w postaci tabletek powlekanych i dazatynib w postaci proszku do sporządzania zawiesiny doustnej nie są biorównoważne. Pacjenci, którzy są w stanie połykać tabletki i chcą zmienić stosowanie dazatynibu w postaci proszku do sporządzania zawiesiny doustnej na stosowanie dazatynibu w tabletkach lub pacjenci, którzy nie są w stanie połykać tabletek i chcą zamienić tabletki na zawiesinę doustną, mogą to zrobić pod warunkiem, że będą przestrzegać prawidłowych zaleceń dotyczących dawkowania danej postaci farmaceutycznej.
Zalecane początkowe dawkowanie dobowe dazatynibu w tabletkach u dzieci i młodzieży przedstawiono w Tabeli 1.
Tabela 1.: Dawkowanie dazatynibu w tabletkach u dzieci i młodzieży z Ph+ CML CP lub Ph+ ALL
Masa ciała (kg)a | Dawka dobowa (mg) |
10 do mniej niż 20 kg | 40 mg |
20 do mniej niż 30 kg | 60 mg |
30 do mniej niż 45 kg | 70 mg |
co najmniej 45 kg | 100 mg |
aNie zaleca się stosowania tabletek u pacjentów o masie ciała mniejszej niż 10 kg; u tych pacjentów należy stosować proszek do sporządzania zawiesiny doustnej.
Czas trwania leczenia
W badaniach klinicznych, leczenie z użyciem dazatynibu u dorosłych z Ph+ CML CP, w fazie akceleracji, z mieloblastyczną lub limfoblastyczną postacią przełomu blastycznego (faza zaawansowana) CML lub Ph+ ALL oraz u dzieci i młodzieży z Ph+ CML CP prowadzono do czasu progresji choroby lub wystąpienia nietolerancji leczenia przez pacjenta. Nie badano wpływu zaprzestania leczenia po osiągnięciu cytogenetycznej lub molekularnej odpowiedzi [w tym pełnej odpowiedzi cytogenetycznej (ang. complete cytogenetic response, CCyR), większej odpowiedzi molekularnej (ang. major molecular response, MMR i MR 4.5] na odległe skutki choroby.
W badaniach klinicznych, leczenie z użyciem dazatynibu u dzieci i młodzieży z Ph+ ALL stosowano w sposób ciągły, w uzupełnieniu do kolejnych bloków chemioterapii podstawowej, maksymalnie przez dwa lata. U pacjentów, którzy przechodzą następnie przeszczepienie komórek macierzystych, dazatynib można podawać dodatkowo przez rok po przeszczepieniu.
Dazatynib jest dostępny w postaci tabletek powlekanych o mocy 20 mg, 50 mg, 70 mg, 80 mg, 100 mg i 140 mg oraz proszku do sporządzania zawiesiny doustnej (10 mg/ml po przygotowaniu zawiesiny), aby umożliwić podanie zalecanej dawki. Zaleca się zwiększenie lub zmniejszenie dawki w zależności od odpowiedzi pacjenta na leczenie i tolerancji.
Zwiększanie dawki
W badaniach klinicznych obejmujących pacjentów dorosłych z CML oraz z Ph+ ALL zezwalano na zwiększenie dawki do 140 mg raz na dobę (faza przewlekła CML) lub do 180 mg raz na dobę
(faza zaawansowana CML lub Ph+ ALL) u pacjentów, u których nie uzyskano odpowiedzi hematologicznej lub cytogenetycznej stosując zalecaną dawkę początkową.
Następujące zwiększanie dawkowania, które przedstawiono w Tabeli 2., zalecane jest u dzieci i młodzieży z Ph+ CML CP, u których nie uzyskano odpowiedzi hematologicznej, cytogenetycznej i molekularnej w zalecanych punktach w czasie, zgodnie z aktualnymi wytycznymi terapeutycznymi a którzy tolerowali leczenie.
Tabela 2.: Zwiększanie dawki u dzieci i młodzieży z Ph+ CML-CP
Dawka (maksymalna dawka na dobę) | ||
Dawka początkowa | Zwiększanie dawki | |
Tabletki | 40 mg 60 mg 70 mg 100 mg | 50 mg 70 mg 90 mg 120 mg |
Nie zaleca się zwiększania dawki u dzieci i młodzieży z Ph+ ALL, gdyż u tych pacjentów dazatynib podaje się w skojarzeniu z chemioterapią.
Zmiana dawkowania ze względu na działania niepożądane Zahamowanie czynności szpiku kostnego
W przypadku wystąpienia zahamowania czynności szpiku kostnego podczas badań klinicznych stosowano przerwy w dawkowaniu, zmniejszenie dawki lub zakończenie terapii. Stosowano także, w zależności od wskazań, przetoczenia masy czerwonokrwinkowej oraz przetoczenia płytek krwi. U pacjentów z przedłużającym się zahamowaniem czynności szpiku kostnego stosowano hematopoetyczny czynnik wzrostu.
W Tabeli 3. podsumowano wytyczne dotyczące zmiany dawkowania u dorosłych a w Tabeli 4. u dzieci i młodzieży z Ph+ CML CP. Wytyczne dotyczące dzieci i młodzieży z Ph+ ALL leczonych w skojarzeniu z chemioterapią znajdują się w oddzielnym akapicie pod tabelami.
Tabela 3.: Dostosowanie dawki w przypadku neutropenii i małopłytkowości u dorosłych
Dorośli z CML w fazie przewlekłej (dawka początkowa 100 mg raz na dobę) | ANC<0,5x109/l i (lub) liczba płytek < 50 x 109/l | ≥50xl09/l. < 25 x 109/l oraz (lub) ANC < 0,5 x 109/l utrzymujących się przez > 7 dni, powtórzyć postępowanie podane w punkcie 1 i włączyć leczenie w mniejszej dawce, 80 mg raz na dobę w przypadku drugiego epizodu. W przypadku trzeciego epizodu należy zmniejszyć dawkę do 50 mg raz na dobę (u nowo zdiagnozowanych pacjentów) lub zaprzestać leczenia u pacjentów z opornością lub nietolerancją na wcześniejsze leczenie zawierające imatynib. |
Dorośli z CML w fazie akceleracji lub fazie przełomu blastycznego i Ph+ ALL (dawka początkowa 140 mg raz na dobę) | ANC<0,5x109/l i (lub) liczba płytek krwi < 10 x 109/l |
ANC (ang. absolute neutrophil count): bezwzględna liczba granulocytów obojętnochłonnych
Tabela 4.: Dostosowanie dawki w przypadku neutropenii i małopłytkowości u dzieci i młodzieży z Ph+ CML CP
≥ 1,0 x 109/l i liczba płytek krwi ≥ 75 x109/l oraz ponownie rozpocząć leczenie w pierwotnej dawce początkowej lub w zmniejszonej dawce . mniejszej dawce. | Dawka | (maksymalna dawka dobowa) | ||
Pierwotna dawka początkowa | Zmniejszenie dawki o jeden poziom | Zmniejszenie dawki o dwa poziomy | ||
Tabletki | 40 mg 60 mg 70 mg 100 mg | 20 mg 40 mg 60 mg 80 mg | * 20 mg 50 mg 70 mg | |
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Klinicznie istotne interakcje
Dazatynib jest substratem i inhibitorem cytochromu P450 (CYP)3A4. Dlatego istnieje możliwość wystąpienia interakcji z innymi równocześnie stosowanymi produktami leczniczymi metabolizowanymi głównie przez CYP3A4 lub takimi, które wpływają na jego aktywność (patrz punkt 4.5).
Jednoczesne stosowanie dazatynibu i innych produktów leczniczych lub substancji, które silnie hamują CYP3A4 (np. ketokonazolu, itrakonazolu, erytromycyny, klarytromycyny, rytonawiru, telitromycyny, soku grejpfrutowego) może zwiększać ekspozycję na dazatynib. Dlatego, nie zaleca
się stosowania silnych inhibitorów CYP3A4 u pacjentów przyjmujących dazatynib (patrz punkt 4.5).
Jednoczesne stosowanie dazatynibu i leków, które indukują CYP3A4 (np. deksametazonu, fenytoiny, karbamazepiny, ryfampicyny, fenobarbitalu lub produktów roślinnych zawierających ziela dziurawca [Hypericumperforatum]) może znacznie zmniejszyć ekspozycję na dazatynib, zwiększając potencjalnie ryzyko niepowodzenia terapeutycznego. Dlatego u pacjentów otrzymujących dazatynib należy stosować inne, alternatywne produkty lecznicze, o słabszym działaniu indukującym CYP3A4 (patrz punkt 4.5).
Jednoczesne stosowanie dazatynibu i substratów CYP3A4 może zwiększyć ekspozycję na substrat CYP3A4. Dlatego należy zachować ostrożność stosując dazatynib równocześnie z substratami CYP3A4 o wąskim przedziale terapeutycznym, takimi jak astemizol, terfenadyna, cisapryd, pimozyd, chinidyna, beprydyl lub alkaloidy sporyszu (ergotamina, dihydroergotamina) (patrz punkt 4.5).
Jednoczesne stosowanie dazatynibu i antagonistów receptora H2 (np. famotydyny), inhibitorów pompy protonowej (np. omeprazolu), lub wodorotlenku aluminium/wodorotlenku magnezu może zmniejszyć ekspozycję na dazatynib. Dlatego, nie zaleca się stosowania antagonistów receptora H2 i inhibitorów pompy protonowej, a leki zawierające wodorotlenek aluminium/wodorotlenek magnezu powinny być podawane do 2 godzin przed lub w 2 godziny po podaniu dazatynibu (patrz punkt 4.5).
Szczególne populacje
Na podstawie wyników badań farmakokinetycznych po jednorazowym podaniu, pacjenci
z zaburzeniami czynności wątroby w stopniu łagodnym, umiarkowanym lub ciężkim mogą otrzymywać zalecaną dawkę początkową (patrz punkt 5.2). Ze względu na ograniczenia tego badania, zaleca się zachowanie ostrożności podczas podawania dazatynibu pacjentom
z zaburzeniami czynności wątroby. Ważne działania niepożądane
Zahamowanie czynności szpiku kostnego
Stosowanie dazatynibu wiąże się z wystąpieniem niedokrwistości, neutropenii oraz małopłytkowości. Do ich wystąpienia dochodzi wcześniej i częściej u pacjentów z CML w fazie zaawansowanej lub z Ph+ ALL, niż u pacjentów z CML w fazie przewlekłej. U dorosłych pacjentów z CML w fazie zaawansowanej lub z Ph+ ALL leczonych dazatynibem w monoterapii badanie morfologii krwi z rozmazem (ang. complete blood counts, CBCs) należy wykonywać co tydzień w trakcie pierwszych 2 miesięcy leczenia, a następnie co miesiąc lub zgodnie ze wskazaniami klinicznymi. U dorosłych oraz u dzieci i młodzieży z CML w fazie przewlekłej badanie morfologii krwi z rozmazem należy wykonywać co 2 tygodnie przez 12 tygodni, a następnie co 3 miesiące lub zgodnie ze wskazaniami klinicznymi. Zahamowanie czynności szpiku kostnego jest zasadniczo odwracalne i ustępowało po tymczasowym wstrzymaniu podawania dazatynibu lub po zmniejszeniu dawki. U dzieci i młodzieży z Ph+ ALL leczonych dazatynibem w skojarzeniu z chemioterapią badanie CBC należy wykonywać przed rozpoczęciem każdego bloku chemioterapii, a także zgodnie ze wskazaniami klinicznymi. W trakcie konsolidacyjnych bloków chemioterapii badanie CBC należy wykonywać co 2 dni aż do czasu ustąpienia objawów (patrz punkty 4.2 i 4.8).
Krwawienie
U pacjentów z CML w fazie przewlekłej (n=548), krwawienie stopnia 3. lub 4. wystąpiło u 5 pacjentów (1%) otrzymujących dazatynib. W badaniach klinicznych u pacjentów z CML w fazie zaawansowanej, otrzymujących zalecaną dawkę dazatynibu (n=304), ciężkie krwawienie w ośrodkowym układzie nerwowym (OUN) wystąpiło u 1% pacjentów. W jednym przypadku zakończyło się ono zgonem, a związane było z małopłytkowością stopnia 4. według kryteriów CTC (ang. Common Toxicity Criteria). Krwawienie z przewodu pokarmowego stopnia 3. lub 4. wystąpiło u 6% pacjentów z CML w fazie zaawansowanej i w większości przypadków konieczne było przerwanie leczenia i podanie preparatów krwi. Inne krwawienia stopnia 3. lub 4. wystąpiły u 2% pacjentów z CML w fazie zaawansowanej. Większość działań niepożądanych związanych z krwawieniem u tych pacjentów związanych było zazwyczaj z małopłytkowością stopnia 3. lub 4.
(patrz punkt 4.8). Ponadto, badania z zastosowaniem płytek krwi w warunkach in vitro i in vivo
sugerują, że stosowanie dazatynibu wpływa w odwracalny sposób na aktywację płytek.
Należy zachować ostrożność u pacjentów, u których konieczne jest stosowanie produktów leczniczych hamujących czynność płytek krwi lub przeciwzakrzepowych.
Retencja płynów
Dazatynib powoduje retencję płynów. W badaniu III fazy u pacjentów z nowo rozpoznaną CML w fazie przewlekłej, retencję płynów stopnia 3. lub 4. obserwowano u 13 pacjentów (5%) w badanej grupie stosującej dazatynib i u 2 pacjentów (1%) w badanej grupie stosującej imatynib po co najmniej 60 miesiącach obserwacji (patrz punkt 4.8). Spośród wszystkich pacjentów z CML w fazie zaawansowanej leczonych dazatynibem, znacznie nasilona retencja płynów wystąpiła u 32 pacjentów (6%) otrzymujących dazatynib w zalecanej dawce (n=548). W badaniach klinicznych u pacjentów z CML w fazie zaawansowanej lub Ph+ ALL otrzymujących dazatynib w zalecanej dawce (n=304), retencję płynów stopnia 3. lub 4. obserwowano u 8% pacjentów, włącznie z wysiękiem w jamie opłucnej stopnia 3. lub 4. u 7% oraz do osierdzia u 1% pacjentów. Wśród tych pacjentów, niekardiogenny obrzęk płuc stopnia 3. lub 4. i nadciśnienie płucne obserwowano u 1% pacjentów.
U pacjentów z objawami przypominającymi wysięk w jamie opłucnej, takimi jak duszność oraz suchy kaszel, należy wykonać zdjęcie radiologiczne klatki piersiowej. W przypadku wysięku w jamie opłucnej stopnia 3. lub 4. konieczny może być drenaż jamy opłucnej i podanie tlenu.
Działania niepożądane związane z retencją płynów były zazwyczaj leczone objawowo, lekami moczopędnymi i krótkimi kursami glikokortykosteroidów (patrz punkty 4.2 i 4.8). U pacjentów w wieku 65 lat i starszych wystąpienie wysięku opłucnowego, duszności, kaszlu, wysięku osierdziowego i zastoinowej niewydolności serca jest bardziej prawdopodobne niż u młodszych pacjentów i dlatego należy ich dokładni monitorować.
Tętnicze nadciśnienie płucne (TNP)
TNP (przedwłośniczkowe tętnicze nadciśnienie płucne potwierdzone poprzez cewnikowanie prawej komory i przedsionka serca) zgłaszano w związku z leczeniem dazatynibem (patrz punkt 4.8). W tych przypadkach TNP było zgłaszane po rozpoczęciu leczenia dazatynibem, w tym po ponad roku leczenia.
Przed rozpoczęciem leczenia dazatynibem należy ocenić, czy u pacjentów nie występują objawy podstawowej choroby sercowo-płucnej. U każdego pacjenta z objawami choroby serca należy wykonać badanie echokardiograficzne na początku leczenia oraz rozważyć jego wykonanie u pacjentów z czynnikami ryzyka choroby serca lub płuc. U pacjentów, u których po rozpoczęciu leczenia wystąpiła duszność i zmęczenie, należy ocenić czynniki etiologiczne, w tym wysięk w jamie opłucnej, obrzęk płuc, niedokrwistość lub nacieki w płucach. Zgodnie z zaleceniami dotyczącymi postępowania w przypadku niehematologicznych działań niepożądanych (patrz punkt 4.2) należy zmniejszyć dawkę dazatynibu lub przerwać leczenie podczas przeprowadzania oceny. W przypadku niestwierdzenia innej przyczyny albo braku poprawy po przerwaniu leczenia lub zmniejszeniu dawki leku należy rozważyć rozpoznanie TNP. Sposób diagnozowania powinien być zgodny z wytycznymi dotyczącymi standardowego postępowania. W przypadku potwierdzenia TNP, należy na stałe zaprzestać leczenia dazatynibem. Dalszą obserwację należy przeprowadzać zgodnie z wytycznymi dotyczącymi standardowego postępowania. U pacjentów z TNP leczonych dazatynibem obserwowano poprawę parametrów hemodynamicznych i klinicznych po zaprzestaniu leczenia tym lekiem.
Wydłużenie odstępu QT
Z badań in vitro wynika, że dazatynib może wydłużać czas repolaryzacji komór serca (odstęp QT) (patrz punkt 5.3). W badaniu III fazy u pacjentów z nowo rozpoznaną CML w fazie przewlekłej, obejmującym 258 pacjentów leczonych dazatynibem i 258 pacjentów leczonych imatynibem z co najmniej 60-miesięczną obserwacją, wydłużenie odcinka QTc jako działanie niepożądane stwierdzono u 1 pacjenta (<1%) w każdej z grup. Mediana zmian QTcF w porównaniu z wartościami wyjściowymi wynosiła 3,0 ms u pacjentów leczonych dazatynibem w porównaniu do 8,2 ms u pacjentów leczonych imatynibem. U jednego pacjenta (<1%) w każdej z grup stwierdzono QTcF > 500 ms. W badaniach klinicznych II fazy przeprowadzonych u 865 chorych na białaczkę, leczonych dazatynibem, średnia zmiana odcinka QTc w porównaniu z wartościami wyjściowymi, obliczonego zgodnie z wzorem Fridericia (QTcF) wynosiła 4 - 6 ms; górne 95% przedziału ufności
dla wszystkich średnich zmian w porównaniu z wartościami wyjściowymi wynosił < 7 ms (patrz punkt 4.8). U 15 (1%) z 2182 pacjentów z opornością lub nietolerancją na wcześniejsze leczenie imatynibem, którzy otrzymywali dazatynib w badaniach klinicznych zgłoszono wydłużenie QTc jako działanie niepożądane. U dwudziestu jeden z tych pacjentów (1%) obserwowano wydłużenie QTcF > 500 ms.
U pacjentów, którzy mają wydłużony odstęp QTc, lub u których może dojść do jego wydłużenia należy stosować dazatynib z ostrożnością. Pacjenci ci to osoby z hipokalemią lub hipomagnezemią, z wrodzonym wydłużeniem odstępu QT, pacjenci przyjmujący produkty lecznicze przeciwarytmiczne lub inne produkty lecznicze, które powodują wydłużenie odstępu QT, a także osoby leczone dużą dawką skumulowaną antracykliny. Przed podaniem dazatynibu należy uzupełnić niedobór potasu oraz magnezu.
Działania niepożądane związane z sercem
Dazatynib badano w randomizownym badaniu klinicznym u 519 pacjentów z nowo rozpoznaną CML w fazie przewlekłej, obejmującym pacjentów z chorobami serca w wywiadzie. U pacjentów otrzymujących dazatynib stwierdzono działania niepożądane związane z sercem, takie jak zastoinowa niewydolność serca/zaburzenia czynności serca, wysięk osierdziowy, zaburzenia rytmu serca, kołatanie serca, wydłużenie odstępu QT i zawał serca (w tym zakończony zgonem).
Działania niepożądane związane z sercem występowały dużo częściej u pacjentów z czynnikami ryzyka lub chorobą serca w wywiadzie. Pacjentów z czynnikami ryzyka (np. nadciśnienie, hiperlipidemia, cukrzyca) lub z chorobą serca w wywiadzie (np. wcześniejsza przezskórna interwencja wieńcowa, udokumentowana choroba naczyń wieńcowych) należy dokładnie monitorować w kierunku objawów podmiotowych i przedmiotowych związanych z niewydolnością serca, takich jak ból w piersiach, skrócenie oddechu i obfite pocenie się.
Jeśli wystąpią ww. objawy podmiotowe i przedmiotowe, zaleca się, aby lekarz przerwał podawanie dazatynibu i rozważył konieczność zastosowania alternatywnego, swoistego leczenia CML. Po powrocie do zdrowia, przed wznowieniem podawania dazatynibu, należy wykonać ocenę czynnościową. Dazatynib można podać w niezmienionej dawce w przypadku działań niepożądanych łagodnych do umiarkowanych (≤ stopnia 2.), a w przypadku ciężkich działań niepożądanych (≥ stopnia 3.) w zmniejszonej dawce (patrz punkt 4.2). Pacjentów kontynuujących leczenie należy okresowo badać.
Pacjenci z niewyrównaną lub poważną chorobą układu sercowo-naczyniowego nie byli włączani do badań klinicznych.
Mikroangiopatia zakrzepowa (ang. thrombotic microangiopathy, TMA)
Inhibitory kinazy tyrozynowej BCR-ABL są kojarzone z mikroangiopatią zakrzepową (TMA), w tym zgłoszenia pojedynczych przypadków dotyczących dazatynibu (patrz punkt 4.8). Jeśli u pacjenta otrzymującego dazatynib wyniki badań laboratoryjnych lub klinicznych wskazują na wystąpienie TMA, należy przerwać leczenie dazatynibem i przeprowadzić dokładną ocenę TMA, w tym oznaczenie aktywności ADAMTS13 (ang. a disintegrin and metalloproteinase with thrombospondin motif) i przeciwciał przeciwko ADAMTS13. Nie należy wznawiać leczenia dazatynibem w przypadku zwiększonego miana przeciwciał przeciwko ADAMTS13 w połączeniu z niską aktywnością ADAMTS13.
Reaktywacja wirusowego zapalenia wątroby typu B
U pacjentów będących przewlekłymi nosicielami wirusa zapalenia wątroby typu B dochodziło do reaktywacji zapalenia wątroby po otrzymaniu przez nich inhibitorów kinazy tyrozynowej BCR- ABL. Niektóre przypadki prowadziły do ostrej niewydolności wątroby lub piorunującego zapalenia wątroby, a w konsekwencji do przeszczepienia wątroby lub zgonu pacjenta.
U pacjentów należy wykonać badania pod kątem zakażenia wirusem HBV przed rozpoczęciem leczenia dazatynibem. Przed rozpoczęciem leczenia u pacjentów z dodatnim wynikiem badania serologicznego w kierunku wirusowego zapalenia wątroby typu B (w tym u pacjentów z aktywną chorobą) i w przypadku pacjentów z dodatnim wynikiem badania w kierunku zakażenia wirusem HBV w trakcie leczenia należy skonsultować się ze specjalistami w zakresie chorób wątroby i leczenia wirusowego zapalenia wątroby typu B. Nosiciele wirusa HBV, którzy wymagają leczenia dazatynibem, powinni być poddawani ścisłej obserwacji pod kątem objawów podmiotowych i przedmiotowych aktywnego zakażenia wirusem HBV w trakcie całego okresu leczenia i przez kilka miesięcy po jego zakończeniu (patrz punkt 4.8).
Wpływ na wzrost i rozwój dzieci i młodzieży
W badaniach z udziałem dzieci i młodzieży oceniających stosowanie dazatynibu u pacjentów z Ph+ CML CP z opornością lub nietolerancją imatynibu oraz u dzieci i młodzieży z Ph+ CML CP dotychczas nieleczonych, po co najmniej 2 latach leczenia zdarzenia niepożądane związane z leczeniem, dotyczące wzrostu kości i rozwoju, zgłoszono u 6 (4,6%) pacjentów, przy czym jedno z nich miało nasilenie ciężkie (opóźnienie wzrostu stopnia 3). Te 6 przypadków obejmowało opóźnienie zrastania się nasad kości, osteopenię, opóźnienie wzrostu i ginekomastię (patrz punkt 5.1). Wyniki te są trudne do zinterpretowania w kontekście chorób przewlekłych, takich jak CML i wymagają długotrwałej obserwacji.
W badaniach z udziałem dzieci i młodzieży, w których oceniano stosowanie dazatynibu w skojarzeniu z chemioterapią u dzieci i młodzieży z nowo rozpoznaną Ph+ ALL, po maksymalnie 2 latach leczenia zdarzenia niepożądane związane z leczeniem, dotyczące wzrostu kości i rozwoju, zgłoszono u 1 (0,6%) pacjenta. Była to osteopenia stopnia 1.
Substancje pomocnicze
Laktoza
Ten produkt leczniczy zawiera laktozę jednowodną. Produkt leczniczy nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, niedoborem laktazy całkowitej lub zespołem złego wchłaniania glukozy-galaktozy.
Sód
Ten produkt leczniczy zawiera sód (w postaci kroskarmelozy).
Produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) w każdej tabletce, co oznacza, że jest w zasadzie „wolny” od sodu.
Substancje czynne, które mogą zwiększać stężenie dazatynibu w osoczu
Z badań przeprowadzonych in vitro wynika, że dazatynib jest substratem CYP3A4. Jednoczesne stosowanie dazatynibu i produktów leczniczych lub substancji, które silnie hamują CYP3A4 (np. ketokonazolu, itrakonazolu, erytromycyny, klarytromycyny, rytonawiru, telitromycyny, soku grejpfrutowego) może zwiększać ekspozycję na dazatynib. Dlatego, nie zaleca się ogólnoustrojowego podawania silnych inhibitorów CYP3A4 u pacjentów przyjmujących dazatynib (patrz punkt 4.2).
W oparciu o badania in vitro stwierdzono, że dazatynib, w stężeniach istotnych klinicznie, wiąże się z białkami osocza w około 96%. Nie wykonano badań oceniających interakcje dazatynibu z innymi produktami leczniczymi wiążącymi się z białkami. Potencjał wypierania i jego kliniczne znaczenie nie są znane.
Substancje czynne, które mogą zmniejszać stężenie dazatynibu w osoczu
W przypadku, gdy dazatynib był podany po uprzednim 8-dniowym podawaniu wieczorem 600 mg ryfampicyny, leku silnie indukującego CYP3A4, pole pod krzywą (AUC) dazatynibu zmniejszyło się o 82%. Inne produkty lecznicze zwiększające aktywność CYP3A4 (np. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital lub produkty roślinne zawierające ziele dziurawca [Hypericum perforatum]) mogą także zwiększać metabolizm i zmniejszać stężenie dazatynibu w osoczu. Dlatego, nie zaleca się stosowania silnych induktorów CYP3A4 z dazatynibem. W przypadku pacjentów, u których wskazane jest stosowanie ryfampicyny lub innych induktorów CYP3A4, należy stosować alternatywne produkty lecznicze o słabszym działaniu indukującym enzymy. Dozwolone jest jednoczesne stosowanie deksametazonu, słabego induktora CYP3A4, z dazatynibem; przewiduje się, że w przypadku jednoczesnego stosowania deksametazonu AUC dla dazatynibu zmniejszy się o około 25%, co prawdopodobnie nie będzie miało znaczenia klinicznego.
Antagoniści receptora histaminowego typu 2 oraz inhibitory pompy protonowej
Długotrwałe hamowanie wydzielania żołądkowego przez antagonistów H2 lub inhibitory pompy protonowej (np. famotydyny i omeprazolu) może prawdopodobnie zmniejszać ekspozycję na dazatynib. Badanie, w którym podawano pojedynczą dawkę leku zdrowym ochotnikom wykazało,
że podanie famotydyny 10 godzin przed podaniem jednorazowej dawki dazatynibu zmniejszało ekspozycję na dazatynib o 61%. W badaniu na 14 zdrowych ochotnikach, którym podano pojedynczą dawkę 100 mg dazatynibu 22 godziny po 4-dniowym podawaniu 40 mg omeprazolu w stanie stacjonarnym, stwierdzono zmniejszenie wartości AUC dazatynibu o 43% i wartości Cmax dazatynibu o 42%. U pacjentów leczonych dazatynibem, zamiast stosowania antagonistów receptora H2 lub inhibitorów pompy protonowej należy rozważyć leki zobojętniające sok żołądkowy (patrz punkt 4.4).
Leki zobojętniające sok żołądkowy
Z danych nieklinicznych wynika, że rozpuszczalność dazatynibu zależy od pH. Równoczesne podanie wodorotlenku glinu/wodorotlenku magnezu z dazatynibem u zdrowych ochotników zmniejszało AUC pojedynczej dawki dazatynibu o 55%, a Cmax o 58%. Jednakże, w przypadku podawania leków zobojętniających kwas żołądkowy 2 godziny przed jednorazową dawką dazatynibu nie obserwowano żadnych istotnych zmian w stężeniu lub w ekspozycji na dazatynib. W związku z tym, leki zobojętniające mogą być podawane do 2 godzin przed lub w 2 godziny po podaniu dazatynibu (patrz punkt 4.4).
Substancje czynne, których stężenie w osoczu może ulec zmianie pod wpływem dazatynibu Jednoczesne stosowanie dazatynibu i substratów CYP3A4 może zwiększyć ekspozycję na substrat CYP3A4. W badaniu przeprowadzonym na zdrowych ochotnikach, podanie dazatynibu w dawce 100 mg zwiększało ekspozycję na symwastatynę, która jest znanym substratem CYP3A4, przez zwiększenie AUC o 20% oraz Cmax o 37%. Nie można wykluczyć, że działanie to jest silniejsze po wielokrotnym podaniu dazatynibu. Dlatego należy zachować ostrożność stosując substraty CYP3A4 o wąskim zakresie dawek terapeutycznych (jak np. astemizol, terfenadyna, cyzapryd, pimozyd, chinidyna, beprydil lub alkaloidy sporyszu [ergotamina, dihydroergotamina]) u pacjentów otrzymujących dazatynib (patrz punkt 4.4).
Z danych in vitro wynika możliwe ryzyko interakcji z substratami CYP2C8, takimi jak glitazony.
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono tylko u dorosłych.
Kobiety w wieku rozrodczym/antykoncepcja u mężczyzn i kobiet
Zarówno mężczyźni aktywni seksualnie, jak i kobiety w wieku rozrodczym, w trakcie leczenia powinni stosować skuteczne metody antykoncepcji.
Ciąża
Na podstawie danych dotyczących stosowania u ludzi istnieje podejrzenie, że dazatynib wywołuje wady wrodzone, w tym wady cewy nerwowej i wykazuje szkodliwe działanie farmakologiczne na płód, gdy jest podawany podczas ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3).
Dazatynibu nie wolno stosować w okresie ciąży, chyba że stan kliniczny kobiety wymaga leczenia dazatynibem. W przypadku stosowania dazatynibu w czasie ciąży, pacjentka musi być poinformowana o potencjalnym ryzyku dla płodu.
Karmienie piersią
Dane dotyczące wydzielania dazatynibu w mleku kobiecym lub mleku zwierząt są niewystarczające/ograniczone. Dane fizyko-chemiczne oraz dostępne dane farmakodynamiczne/toksykologiczne dotyczące dazatynibu wskazują na wydzielanie z mlekiem kobiecym, w związku z czym nie można wykluczyć ryzyka dla dziecka karmionego piersią.
Podczas leczenia dazatynibem należy przerwać karmienie piersią.
Płodność
W badaniach na zwierzętach leczenie dazatynibem nie miało wpływu na płodność samców i samic szczurów (patrz punkt 5.3). Lekarze i inni pracownicy ochrony zdrowia powinni udzielić porad pacjentom płci męskiej w odpowiednim wieku na temat możliwego wpływu dazatynibu na płodność, przy czym porady te mogą uwzględniać możliwość oddania nasienia do banku.
Dazatynib wywiera niewielki wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Pacjentów należy poinformować o możliwości wystąpienia w trakcie leczenia dazatynibem takich działań niepożądanych jak zawroty głowy lub zaburzenia widzenia. Dlatego należy zachować ostrożność podczas prowadzenia pojazdów mechanicznych lub obsługiwania urządzeń mechanicznych w ruchu.
Podsumowanie profilu bezpieczeństwa
Dane opisane poniżej dotyczą ekspozycji na dazatynib stosowany w monoterapii, we wszystkich dawkach ocenianych w badaniach klinicznych (N=2900), w tym u 324 dorosłych pacjentów z nowo rozpoznaną CML w fazie przewlekłej, 2388 dorosłych pacjentów z opornością lub nietolerancją imatynibu z CML w fazie przewlekłej lub zaawansowanej, lub z Ph+ ALL oraz 188 dzieci i młodzieży.
U 2712 dorosłych pacjentów z CML w fazie przewlekłej, CML w fazie zaawansowanej lub z Ph+ ALL, mediana czasu leczenia wynosiła 19,2 miesiąca (zakres 0 do 93,2 miesiąca).
W randomizowanym badaniu obejmującym pacjentów z nowo rozpoznaną CML w fazie przewlekłej mediana czasu leczenia wynosiła około 60 miesięcy. U 1618 dorosłych pacjentów z CML w fazie przewlekłej mediana czasu leczenia wynosiła 29 miesięcy (zakres 0 do 92,9 miesiąca). U 1094 dorosłych pacjentów z CML w fazie zaawansowanej lub Ph+ ALL mediana czasu leczenia wynosiła 6,2 miesiąca (zakres 0 do 93,2 miesiąca). Spośród 188 pacjentów w badaniach z udziałem dzieci i młodzieży mediana czasu leczenia wynosiła 26,3 miesiąca (zakres 0 do 99,6 miesiąca). W podgrupie obejmującej 130 dzieci i młodzieży z CML w fazie przewlekłej leczonych dazatynibem mediana czasu leczenia wynosiła 42,3 miesiąca (zakres 0,1 do 99,6 miesiąca).
U większości pacjentów leczonych dazatynibem w pewnym okresie leczenia występowały działania niepożądane. W ogólnej populacji 2712 pacjentów leczonych dazatynibem, u 520 (19%) pacjentów wystąpiły działania niepożądane prowadzące do przerwania leczenia.
Ogólny profil bezpieczeństwa stosowania dazatynibu u dzieci i młodzieży z Ph+ CML CP był podobny do profilu bezpieczeństwa w populacji dorosłych, bez względu na postać farmaceutyczną, z wyjątkiem braku opisanych przypadków wysięku osierdziowego, wysięku opłucnowego, obrzęku płuc czy nadciśnienia płucnego u dzieci i młodzieży. Ze 130 dzieci i młodzieży z CML CP leczonych dazatynibem u 2 (1,5%) wystąpiły działania niepożądane prowadzące do przerwania leczenia.
Tabelaryczne zestawienie działań niepożądanych
U pacjentów leczonych dazatynibem w monoterapii w badaniach klinicznych oraz po wprowadzeniu produktu do obrotu wystąpiły następujące działania niepożądane, z wyjątkiem nieprawidłowości w wynikach badań laboratoryjnych (Tabela 5.). Działania niepożądane przedstawiono według klasyfikacji układów narządowych oraz częstości występowania. Częstości występowania określono następująco: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); niezbyt często (≥ 1/1 000 do < 1/100); rzadko (≥ 1/10 000 do < 1/1 000); częstość nieznana (nie może być określona na podstawie dostępnych danych po wprowadzeniu do obrotu).
W obrębie każdej grupy o określonej częstości występowania działania niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 5.: Tabelaryczne podsumowanie działań niepożądanych
Zakażenia i zarażenia pasożytnicze | |
Bardzo często | zakażenia (w tym bakteryjne, wirusowe, grzybicze, nieokreślone) |
Często | zapalenie płuc (w tym bakteryjne, wirusowe oraz grzybicze), zakażenia/zapalenie górnych dróg oddechowych, zakażenia wirusem Herpes (w tym cytomegalowirusem CMV), zakażenia przewodu pokarmowego, posocznica (w tym niezbyt częste przypadki zakończone zgonem) |
Częstość nieznana | reaktywacja wirusowego zapalenia wątroby typu B |
Zaburzenia krwi i układu chłonnego | |
Bardzo często | mielosupresja (w tym niedokrwistość, neutropenia, małopłytkowość) |
Często | neutropenią z gorączką |
Niezbyt często | powiększenie węzłów chłonnych, limfopenia |
Rzadko | aplazja układu czerwonokrwinkowego |
Zaburzenia układu immunologicznego | |
Niezbyt często | nadwrażliwość (w tym rumień guzowaty) |
Rzadko | wstrząs anafilaktyczny |
Zaburzenia endokrynologiczne | |
Niezbyt często | niedoczynność tarczycy |
Rzadko | nadczynność tarczycy, zapalenie tarczycy |
Zaburzenia metabolizmu i odżywiania | |
Często | zaburzenia łaknieniaa, hiperurykemia |
Niezbyt często | zespół rozpadu guza, odwodnienie, hipoalbuminemia, hipercholesterolemia |
Rzadko | cukrzyca |
Zaburzenia psychiczne | |
Często | depresja, bezsenność |
Niezbyt często | lęk, stan splątania, niestabilność emocjonalna, zmniejszenie libido |
Zaburzenia układu nerwowego | |
Bardzo często | bóle głowy |
Często | neuropatia (w tym neuropatia obwodowa), zawroty głowy, zaburzenia smaku, senność |
Niezbyt często | krwawienie w OUN*b, omdlenie, drżenia, niepamięć, zaburzenia równowagi |
Rzadko | udar mózgu, przemijające napady niedokrwienne, drgawki, zapalenie nerwu wzrokowego, porażenie nerwu VII, otępienie, ataksja |
Zaburzenia oka | |
Często | zaburzenia widzenia (w tym zaburzenia widzenia, zamglone widzenie i zmniejszenie ostrości wzroku), suchość oczu |
Niezbyt często | upośledzenie widzenia, zapalenia spojówek, światłowstręt, nasilone łzawienie |
Zaburzenia ucha i błędnika | |
Często | szumy uszne |
Niezbyt często | utrata słuchu, zawroty głowy |
Zaburzenia serca | |
Często | zastoinowa niewydolność serca/zaburzenia czynności serca*c, wysięk osierdziowy*, zaburzenia rytmu serca (w tym tachykardia), kołatanie serca |
Niezbyt często | zawał serca (czasem zakończony zgonem)*, wydłużenie odstępu QT*, zapalenie osierdzia, arytmia komorowa (w tym częstoskurcz komorowy), dławica piersiowa, powiększenie serca, nieprawidłowości załamka T w EKG, zwiększenie aktywności troponiny |
Rzadko | cor pulmonale, zapalenie mięśnia serca, ostry zespół wieńcowy, wydłużenie odstępu PR w elektrokardiogramie, choroba wieńcowa, zapalenie opłucnej i osierdzia |
Częstość nieznana | migotanie przedsionków/trzepotanie przedsionków |
Zaburzenia naczyniowe | |
Bardzo często | krwotok*d |
Często | nadciśnienie tętnicze, zaczerwienienia twarzy |
Niezbyt często | niedociśnienie, zakrzepowe zapalenie żył, zakrzepica |
Rzadko | zakrzepica żył głębokich, zatorowość, sinica marmurkowata |
Częstość nieznana | mikroangiopatia zakrzepowa |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Bardzo często | wysięk w jamie opłucnej*, duszność |
Często | obrzęk płuc*, nadciśnienie płucne*, nacieki w płucach, zapalenie płuc, kaszel |
Niezbyt często | nadciśnienie tętnicze płucne, skurcz oskrzeli, astma |
Rzadko | zakrzepica płucna, ostry zespół zaburzeń oddychania |
Częstość nieznana | śródmiąższowa choroba płuc |
Zaburzenia żołądka i jelit | |
Bardzo często | biegunka, wymioty, nudności, ból brzucha |
Często | krwawienie z przewodu pokarmowego*, zapalenie okrężnicy (w tym agranulocytowe zapalenie okrężnicy), zapalenie żołądka, zapalenie błony śluzowej (w tym zapalenie śluzówki/zapaleniejamy ustnej), niestrawność, wzdęcia, zaparcia, zaburzenia tkanek miękkich jamy ustnej |
Niezbyt często | zapalenie trzustki (w tym ostre zapalenie trzustki), owrzodzenie górnego odcinka przewodu pokarmowego, zapalenie przełyku, wodobrzusze*, szczelina odbytu, dysfagia, choroba refluksowa przełyku |
Rzadko | gastroenteropatia związana z utratą białka, niedrożność jelita, przetoka odbytu |
Częstość nieznana | śmiertelne krwawienie z przewodu pokarmowego* |
Zaburzenia wątroby i dróg żółciowych | |
Niezbyt często | zapalenie wątroby, zapalenie pęcherzyka żółciowego, cholestaza |
Zaburzenia skóry i tkanki podskórnej | |
Bardzo często | wysypka skórnae |
Często | łysienie, zapalenie skóry (w tym wyprysk), świąd, trądzik, suchość skóry, pokrzywka, nadmierne pocenie się |
Niezbyt często | agranulocytowe zapalenie skóry, nadwrażliwość na światło, zaburzenia pigmentacji, zapalenie tkanki podskórnej, owrzodzenia skóry, zmiany pęcherzowe, zmiany dotyczące paznokci, zespół erytrodysestezji dłoniowo- podeszwowej, zaburzenia dotyczące włosów |
Rzadko | leukoklastyczne zapalenie naczyń, zwłóknienie skóry |
Częstość nieznana | zespół Stevensa-Johnsonaf |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Bardzo często | bóle kostno-mięśnioweg |
Często | bóle stawów, bóle mięśni, osłabienie mięśni, sztywność układu mięśniowo- szkieletowego, kurcz mięśni |
Niezbyt często | rabdomioliza, martwica kości, zapalenie mięśni, zapalenie ścięgien, zapalenie |
Rzadko | opóźnienie zrastania się nasad kościh, opóźnienie wzrostuh |
Zaburzenia nerek i dróg moczowych | |
Niezbyt często | zaburzenie czynności nerek (w tym niewydolność nerek), częste oddawanie moczu, białkomocz |
Częstość nieznana | zespół nerczycowy |
Ciąża, połóg i okres okołoporodowy | |
Rzadko | poronienie |
Zaburzenia układu rozrodczego i piersi | |
Niezbyt często | ginekomastia, zaburzenia miesiączkowania |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często | obrzęk obwodowyi, zmęczenie, gorączka, obrzęk twarzyj |
Często | osłabienie, ból, ból w klatce piersiowej, obrzęk uogólniony*k, dreszcze |
Niezbyt często | złe samopoczucie, inne obrzęki powierzchniowel |
Rzadko | zaburzenia chodu |
Badania diagnostyczne | |
Często | zmniejszenie masy ciała, zwiększenie masy ciała |
Niezbyt często | zwiększona aktywność fosfokinazy kreatynowej w krwi, zwiększona aktywność gamma-glutamylotransferazy |
Urazy, zatrucia i powikłania po zabiegach | |
Często | stłuczenia |
a W tym zmniejszenie apetytu, wczesne uczucie sytości, zwiększenie apetytu.
b W tym krwotok w obrębie ośrodkowego układu nerwowego, krwiak mózgowy, krwotok mózgowy, krwiak nadtwardówkowy, krwotok śródczaszkowy, udar krwotoczny, krwotok podpajęczynówkowy, krwiak podtwardówkowy i krwotok podtwardówkowy.
c W tym zwiększone stężenie mózgowego peptydu natriuretycznego, zaburzenia czynności komór, zaburzenie czynności lewej komory, zaburzenie czynności prawej komory, niewydolność serca, ostra niewydolność serca, przewlekła niewydolność serca, zastoinowa niewydolność serca, kardiomiopatia, kardiomiopatia zastoinowa, niedomoga rozkurczowa, zmniejszenie frakcji wyrzutowej i niewydolność komorowa, niewydolność lewej komory, niewydolność prawej komory, zmniejszona ruchliwość komór.
d Z wyłączeniem krwawień z przewodu pokarmowego i krwawień w OUN, te działania niepożądane wymieniono odpowiednio w
„Zaburzeniach żołądka i jelit" i „Zaburzeniach układu nerwowego".
e W tym wysypka polekowa, rumień, rumień wielopostaciowy, zaczerwienienie skóry (erythrosis), wysypka złuszczająca, rumień uogólniony, wysypka narządów płciowych, potówka czerwona, prosaki, potówki, łuszczyca krostkowa, wysypka, wysypka rumieniowa, wysypka pęcherzykowa, uogólniona wysypka, wysypka plamista, wysypka grudkowo-plamista, wysypka grudkowa, wysypka swędząca, wysypka krostkowa, wysypka pęcherzykowa, złuszczanie się skóry, podrażnienie skóry, toksyczne wykwity skórne, pokrzywka pęcherzykowa i wysypka związana z zapaleniem naczyń.
f Po wprowadzeniu do obrotu, odnotowano pojedyncze przypadki wystąpienia zespołu Stevensa-Johnsona. Nie można było ustalić, czy te śluzówkowo-skórne działania niepożądane były bezpośrednio związane z dazatynibem czy z produktami leczniczymi stosowanymi jednocześnie.
8 W trakcie leczenia lub po jego przerwaniu odnotowano ból mięśniowo-szkieletowy
h W badaniach z udziałem dzieci i młodzieży częstość występowania odnotowana jako często.
i Obrzęk opadowy, obrzęk miejscowy, obrzęk obwodowy.
j Obrzęk spojówek, obrzęk oczu, opuchnięcie oczu, obrzęk powiek, obrzęk twarzy, obrzęk warg, obrzęk plamki, obrzęk ust, obrzęk oczodołu, obrzęk okołooczodołowy, opuchnięcie twarzy.
k Przeciążenie płynami, zatrzymanie płynów, obrzęki w przewodzie pokarmowym, obrzęk uogólniony, obrzęki obwodowe, obrzęk, obrzęk z powodu choroby serca, wysięk okołonerkowy, obrzęk pozabiegowy, obrzęk trzewny.
1 Obrzmienie narządów płciowych, obrzęk w miejscu nacięcia, obrzęk narządów płciowych, obrzęk prącia, opuchnięcie prącia, obrzęk moszny, opuchnięcie skóry, opuchnięcie jądra, opuchnięcie sromu i pochwy.
* Dodatkowe informacje - patrz punkt „Opis wybranych działań niepożądanych"
Opis wybranych działań niepożądanych
Mielosupresja
Leczenie dazatynibem jest związane z występowaniem niedokrwistości, neutropenii i małopłytkowości. Występują one wcześniej i częściej u pacjentów z CML w fazie zaawansowanej lub z Ph+ ALL niż u pacjentów z CML w fazie przewlekłej (patrz punkt 4.4).
Krwawienia
U pacjentów przyjmujących dazatynib zgłaszano polekowe działania niepożądane związane z krwawieniem, począwszy od wybroczyn i krwawienia z nosa po krwotoki z przewodu pokarmowego oraz krwawienie w ośrodkowym układzie nerwowym stopnia 3. lub 4. (patrz punkt 4.4).
Retencja płynów
Różne działania niepożądane, takie jak wysięk opłucnowy, wodobrzusze, obrzęk płuc oraz wysięk osierdziowy z obrzękiem powierzchownym lub bez, można określić łącznie jako „zatrzymanie płynów". W badaniu dotyczącym nowo rozpoznanej CML w fazie przewlekłej, po co najmniej 60 miesiącach obserwacji, działania niepożądane związane z zatrzymaniem płynów po leczeniu dazatynibem obejmowały wysięk opłucnowy (28%), obrzęki powierzchowne (14%), nadciśnienie płucne (5%), obrzęk uogólniony (4%) i wysięk osierdziowy (4%). Zastoinową niewydolność serca/zaburzenia czynności serca i obrzęk płuc odnotowano u < 2% pacjentów. Skumulowana w czasie częstość występowania wysięku opłucnowego (dowolnego stopnia) u pacjentów leczonych dazatynibem wynosiła 10% po 12 miesiącach, 14% po 24 miesiącach, 19% po 36 miesiącach, 24% po 48 miesiącach i 28% po 60 miesiącach. Nawrotowy wysięk opłucnowy wystąpił łącznie u 46 pacjentów leczonych dazatynibem. U siedemnastu pacjentów wystąpiły 2 osobne działania niepożądane, u 6 wystąpiły 3 działania niepożądane, u 18 od 4 do 8 działań niepożądanych, a u 5 > 8 epizodów wysięku opłucnowego.
Mediana czasu do wystąpienia pierwszego związanego z dazatynibem wysięku opłucnowego stopnia 1. lub 2. wynosiła 114 tygodni (zakres 4 do 299 tygodni). Wysięk opłucnowy związany z dazatynibem oceniono jako ciężki (stopnia 3. lub 4.) u mniej niż 10% pacjentów z wysiękiem opłucnowym. Mediana czasu do wystąpienia pierwszego związanego z dazatynibem wysięku opłucnowego stopnia ≥ 3 wynosiła 175 tygodni (zakres 114 do 274 tygodni). Mediana czasu trwania związanego z dazatynibem wysięku opłucnowego (dowolnego stopnia) wynosiła 283 dni (około 40 tygodni).
Wysięk opłucnowy był zazwyczaj odwracalny i ustępował po odstawieniu dazatynibu
i zastosowaniu leków moczopędnych lub innych odpowiednich metod leczenia wspomagającego (patrz punkty 4.2 i 4.4). Wśród pacjentów leczonych dazatynibem, u których wystąpił związany z lekiem wysięk opłucnowy (n=73), u 45 (62%) przerwano podawanie leku, a u 30 (41%) zmniejszono jego dawkę. Dodatkowo 34 (47%) pacjentów otrzymało leki moczopędne, 23 (32%) otrzymało kortykosteroidy, a 20 (27%) otrzymało zarówno kortykosteroidy, jak i leki moczopędne. U dziewięciu (12%) pacjentów wykonano terapeutyczny drenaż jamy opłucnej. Sześć procent pacjentów leczonych dazatynibem przerwało leczenie z powodu związanego z lekiem wysięku opłucnowego.
Wysięk opłucnowy nie miał niekorzystnego wpływu na zdolność pacjentów do uzyskania odpowiedzi terapeutycznej. Wśród pacjentów leczonych dazatynibem z wysiękiem opłucnowym u 96% uzyskano odpowiedź cCCyR, u 82% - MMR, a u 50% osiągnięto odpowiedź MR4.5 pomimo przerwania leczenia lub zmodyfikowania dawki.
Dodatkowe informacje dotyczące pacjentów z CML w fazie przewlekłej oraz z CML w fazie zaawansowanej lub Ph+ ALL przedstawiono w punkcie 4.4.
Tętnicze nadciśnienie płucne (TNP)
TNP (przedwłośniczkowe tętnicze nadciśnienie płucne potwierdzone poprzez cewnikowanie prawej komory i przedsionka serca) zgłaszano w związku z leczeniem dazatynibem. W tych przypadkach TNP było zgłaszane po rozpoczęciu leczenia dazatynibem, w tym po ponad roku leczenia. Pacjenci, u których zgłaszano występowanie TNP podczas leczenia dazatynibem, zwykle przyjmowali inne produkty lecznicze lub występowały u nich choroby współistniejące z podstawową chorobą nowotworową. U pacjentów z TNP obserwowano poprawę parametrów hemodynamicznych i klinicznych po zaprzestaniu leczenia dazatynibem.
Wydłużenie odstępu QT
W badaniu III fazy u pacjentów z nowo rozpoznaną CML w fazie przewlekłej, u 1 pacjenta (<1%) leczonego dazatynibem stwerdzono QTcF > 500 ms po co najmniej 12 miesiącach obserwacji (patrz punkt 4.4). Nie zgłoszono żadnych dodatkowych przypadków z QTcF > 500 ms po co najmniej 60 miesiącach obserwacji.
W 5 badaniach klinicznych II fazy u pacjentów z opornością lub nietolerancją na wcześniejsze leczenie imatynibem, kilkakrotne zapisy EKG wykonywane przed przystąpieniem do leczenia i we wcześniej wyznaczonych odstępach czasowych, wykonano u 865 pacjentów otrzymujących dazatynib w dawce 70 mg dwa razy na dobę. Wyniki badań EKG odczytywano centralnie.
Obliczając długość odstępu QT stosowano poprawkę uwzględniającą częstość rytmu serca, zgodnie ze wzorem Fridericia. We wszystkich punktach czasowych w 8. dniu od rozpoczęcia podawania leku, średnia zmiana odstępu QTcF w porównaniu z wartościami wyjściowymi wynosiła 4 - 6 ms, przy górnym 95% przedziale ufności < 7 ms. W badaniach klinicznych u 15 (1%) z 2182 pacjentów z opornością lub nietolerancją na wcześniejsze leczenie imatynibem, którzy otrzymywali dazatynib zgłoszono wydłużenie QTc jako działanie niepożądane. U dwudziestu jeden pacjentów (1%) obserwowano wydłużenie QTcF > 500 ms (patrz punkt 4.4).
Działania niepożądane związane z sercem
Pacjentów z czynnikami ryzyka lub z chorobą serca w wywiadzie należy dokładnie monitorować w kierunku objawów podmiotowych i przedmiotowych związanych z niewydolnością serca oraz należy w odpowiedni sposób oceniać i leczyć (patrz punkt 4.4).
Reaktywacja wirusowego zapalenia wątroby typu B
Opisywano reaktywację wirusowego zapalenia wątroby typu B związaną ze stosowaniem inhibitorów kinazy tyrozynowej BCR-ABL. Niektóre przypadki prowadziły do ostrej niewydolności wątroby lub piorunującego zapalenia wątroby, a w konsekwencji do przeszczepienia wątroby lub zgonu pacjenta (patrz punkt 4.4).
W badaniu III fazy dotyczącym optymalizacji dawkowania, u pacjentów w fazie przewlekłej CML z opornością lub nietolerancją na wcześniejsze leczenie imatynibem (mediana czasu leczenia 30 miesiące), częstość występowania wysięku w jamie opłucnej i zastoinowej niewydolność serca/zaburzenia czynności serca była mniejsza u pacjentów leczonych dazatynibem w dawce 100 mg raz na dobę niż u pacjentów leczonych dazatynibem w dawce 70 mg podawanej dwa razy na dobę. Również mielosupresję obserwowano rzadziej w leczonych grupach po podaniu 100 mg raz na dobę (patrz poniżej Badania diagnostyczne). Mediana czasu leczenia w grupie z zastosowaniem dawki 100 mg raz na dobę wynosiła 37 miesięcy (zakres 1-91 miesięcy). Skumulowana częstość występowania wybranych działań niepożądanych, które zaobserwowano po zastosowaniu zalecanej dawki początkowej wynoszącej 100 mg raz na dobę, przedstawiono w Tabeli 6a.
Tabela 6a: Wybrane działania niepożądane obserwowane w badaniu III fazy dotyczącym optymalizacji dawkowania (nietolerancja lub oporność na imatynib w fazie przewlekłej CML)a
Co najmniej 2-letni okres obserwacji | Co najmniej 5-letni okres obserwacji | Co najmniej 7-letni okres obserwacji |
Wszystkie stopnie | Stopień 3/4 | Wszystkie stopnie | Stopień 3/4 | Wszystkie stopnie | Stopień 3/4 | |
Zalecane określenie | Odsetek pacjentów (%) | |||||
Biegunka | 27 | 2 | 28 | 2 | 28 | 2 |
Retencja płynów | 34 | 4 | 42 | 6 | 48 | 7 |
Obrzęk powierzchowny | 18 | 0 | 21 | 0 | 22 | 0 |
Wysięk opłucnowy | 18 | 2 | 24 | 4 | 28 | 5 |
Obrzęk uogólniony | 3 | 0 | 4 | 0 | 4 | 0 |
Wysięk osierdziowy | 2 | 1 | 2 | 1 | 3 | 1 |
Nadciśnienie płucne | 0 | 0 | 0 | 0 | 2 | 1 |
Krwotok | 11 | 1 | 11 | 1 | 12 | 1 |
Krwawienie z przewodu pokarmowego | 2 | 1 | 2 | 1 | 2 | 1 |
a Wyniki badania III fazy dotyczącego optymalizacji dawkowania w populacji z zastosowaniem zalecanej dawki początkowej 100 mg raz na dobę (n=165)
W badaniu III fazy dotyczącym optymalizacji dawkowania, u pacjentów z CML w fazie zaawansowanej i Ph+ ALL, mediana leczenia wynosiła 14 miesięcy dla fazy akceleracji w CML, 3 miesiące dla mieloblastycznej postaci przełomu blastycznego w CML, 4 miesiące dla limfoblastycznej postaci przełomu blastycznego w CML i 3 miesiące dla Ph+ ALL. Wybrane działania niepożądane, które zaobserwowano po zastosowaniu zalecanej dawki początkowej
140 mg raz na dobę przedstawiono w Tabeli 6b. Oceniano również schemat dawkowania 70 mg dwa razy na dobę. Schemat dawkowania 140 mg raz na dobę miał profil skuteczności porównywalny ze schematem dawkowania 70 mg dwa razy na dobę, lecz z korzystniejszym profilem bezpieczeństwa.
Tabela 6b: Wybrane działania niepożądane obserwowane w badaniu III fazy dotyczącym optymalizacji dawkowania: CML w fazie zaawansowanej i Ph+ ALLa
140 mg raz na dobę
n=304 Wszystkie stopnie Stopień3/4
Odsetek pacjentów (%)
Zalecane określenie
Biegunka | 28 | 3 |
Retencja płynów | 33 | 7 |
Obrzęk powierzchowny | 15 | < 1 |
Wysięk e opłucnej | 20 | 6 |
Obrzęk uogólniony | 2 | 0 |
Zastoinowa niewydolność serca/ zaburzenia czynności sercab | 1 | 0 |
Wysięk w osierdziu | 2 | 1 |
Obrzęk płuc | 1 | 1 |
Krwawienie | 23 | 8 |
Krwawienie z przewodu pokarmowego | 8 | 6 |
a Wyniki badania III fazy dotyczącego optymalizacji dawkowania w populacji z zastosowaniem zalecanej dawki początkowej 140 mg raz na dobę (n=304) w 2-letnim okresie końcowej obserwacji.
b W tym zaburzenia czynności komór, niewydolność serca, zastoinowa niewydolność serca, kardiomiopatia, kardiomiopatia
zastoinowa, niedomoga rozkurczowa, zmniejszenie frakcji wyrzutowej i niewydolność komorowa.
Ponadto, przeprowadzono dwa badania w grupie 161 u dzieci i młodzieży z Ph+ ALL, którym podawano dazatynib w skojarzeniu z chemioterapią. W badaniu głównym wzięło udział 106 dzieci i młodzieży, którzy otrzymywali dazatynib w skojarzeniu z chemioterapią w sposób ciągły. W badaniu wspomagającym wzięło udział 55 dzieci i młodzieży, z czego 35 pacjentów otrzymywało dazatynib
w skojarzeniu z chemioterapią w schemacie leczenia przerywanego (dwa tygodnie leczenia, a następnie jeden do dwóch tygodni bez leczenia), a 20 pacjentów otrzymywało dazatynib w skojarzeniu z chemioterapią w sposób ciągły. W grupie 126 dzieci i młodzieży z Ph+ ALL leczonych dazatynibem w sposób ciągły, mediana czasu leczenia wynosiła 23,6 miesiąca (zakres od 1,4 do 33 miesięcy).
U 2 (1,6%) ze 126 dzieci i młodzieży z Ph+ ALL leczonych w sposób ciągły wystąpiły działania niepożądane prowadzące do przerwania leczenia. W Tabeli 7. wymieniono działania niepożądane występujące w tych dwóch badaniach z udziałem dzieci i młodzieży z częstością ≥10% u pacjentów leczonych w sposób ciągły. Warto zauważyć, że wysięk opłucnowy odnotowano u 7 (5,6%) pacjentów w tej grupie i dlatego też nie został ujęty w tabeli.
Tabela 7.: Działania niepożądane odnotowane u ≥10% dzieci i młodzieży z Ph+ ALL leczonych dazatynibem w sposób ciągły w skojarzeniu z chemioterapią (N=126)a
Odsetek pacjentów (%)
Działanie niepożądane | Wszystkie stopnie | Stopień 3/4 |
Neutropenia z gorączką | 27,0 | 26,2 |
Nudności | 20,6 | 5,6 |
Wymioty | 20,6 | 4,8 |
Ból brzucha | 14,3 | 3,2 |
Biegunka | 12,7 | 4,8 |
Gorączka | 12,7 | 5,6 |
Ból głowy | 11,1 | 4,8 |
Osłabienie łaknienia | 10,3 | 4,8 |
Zmęczenie | 10,3 | 0 |
a W badaniu głównym łącznie 24 ze 106 pacjentów stosowało proszek do sporządzania zawiesiny doustnej przynajmniej raz, z czego 8 pacjentów stosowało wyłącznie proszek do sporządzania zawiesiny doustnej.
Nieprawidłowości w wynikach badań laboratoryjnych Hematologia
W badaniu dotyczącym nowo rozpoznanej CML w fazie przewlekłej, u pacjentów otrzymujących dazatynib, po co najmniej 12 miesiącach obserwacji stwierdzono następujące nieprawidłowości w wynikach badań laboratoryjnych stopnia 3. lub 4.: neutropenia (21%), małopłytkowość (19%) i niedokrwistość (10%). Po co najmniej 60 miesiącach obserwacji łączne wskaźniki występowania neutropenii, małopłytkowości i niedokrwistości wynosiły, odpowiednio 29%, 22% i 13%.
U pacjentów z nowo rozpoznaną CML w fazie przewlekłej leczonych dazatynibem, u których stwierdzono mielosupresję stopnia 3. lub 4., powrót do stanu wyjściowego następował zwykle po krótkim zaprzestaniu podawania i (lub) zmniejszeniu dawki, natomiast u 1,6% pacjentów, po co najmniej 12 miesiącach obserwacji, zaprzestano leczenia na stałe. Po co najmniej 60 miesiącach obserwacji łączny wskaźnik zaprzestania leczenia na stałe z powodu mielosupresji stopnia 3. lub 4. wynosił 2,3%.
U pacjentów z CML z opornością lub nietolerancją na wcześniejsze leczenie imatynibem stwierdzano zgodnie niedobór krwinek (małopłytkowość, neutropenię oraz niedokrwistość). Jednakże, występowanie niedoboru krwinek było oczywiście zależne od stadium choroby. Częstość występowania zaburzeń hematologicznych stopnia 3. lub 4. przedstawiono w Tabeli 8.
Tabela 8.: Zaburzenia hematologiczne stopnia 3./4. wg CTC w badaniach klinicznych u pacjentów z opornością lub nietolerancją na wcześniejsze leczenie imatynibema
Mieloblastyczna | Limfoblastyczna | |||
postać | postać przełomu | |||
Faza | przełomu | blastycznego | ||
przewlekła | Faza akceleracji blastycznego | i Ph+ ALL | ||
(n= 165)b | (n= 157)c | (n= 74)c | (n= 168)c | |
Odsetek pacjentów (%) | ||||
Wskaźniki hematologiczne | ||||
Neutropenia | 36 | 58 | 77 | 76 |
Małopłytkowość | 23 | 63 | 78 | 74 |
Niedokrwistość | 13 | 47 | 74 | 44 |
a Wyniki badania III fazy dotyczącego optymalizacji dawkowania odnotowane w 2 -letnim okresie obserwacji.
b Wyniki badania CA180-034 w zalecanej dawce początkowej wynoszącej 100 mg raz na dobę.
c Wyniki badania CA180-035 w zalecanej dawce początkowej wynoszącej 140 mg raz na dobę.
Stopnie wg CTC: neutropenia (stopień 3. ≥ 0,5 - < 1,0 x 109/l, stopień 4. < 0,5 x 109/l); małopłytkowość (stopień 3. ≥ 25 -
< 50 x 109/l, stopień 4. < 25 x 109/l); niedokrwistość (hemoglobina stopień 3. ≥ 65 - < 80 g/l, stopień 4. < 65 g/l).
Łączne wskaźniki dotyczące cytopenii stopnia 3. lub 4. wśród pacjentów leczonych dawką 100 mg raz na dobę były podobne po 2 i 5 latach, w tym dla: neutropenii (35% vs. 36%), małopłytkowości (23% vs. 24%) i niedokrwistości (13% vs. 13%)..
U pacjentów, u których wystąpiło zahamowanie czynności szpiku kostnego stopnia 3. lub 4., powrót wartości morfotycznych krwi do normy następował w większości przypadków po krótkim okresie wstrzymania leku i (lub) zmniejszenia dawki. W przypadku 5% pacjentów konieczne było całkowite zakończenie podawania preparatu. W przeważającej części pacjenci kontynuowali leczenie bez nawrotu objawów supresji szpiku kostnego.
Biochemia
W badaniu dotyczącym nowo rozpoznanej CML w fazie przewlekłej, hipofosfatemię stopnia 3. lub
4. stwierdzono u 4% pacjentów otrzymujących dazatynib, a zwiększenie aktywności transaminaz oraz stężenia kreatyniny i bilirubiny stopnia 3. lub 4. stwierdzono u ≤ 1% pacjentów po co najmniej 12 miesiącach obserwacji. Po co najmniej 60 miesiącach obserwacji łączny wskaźnik występowania hipofosfatemii stopnia 3. lub 4. wynosił 7%, zwiększonego stężenia kreatyniny i bilirubiny stopnia 3. lub 4. wynosił 1%, a zwiększenia aktywności transaminaz stopnia 3. lub 4. pozostał na poziomie 1%. Nie było przypadków zaprzestania leczenia dazatynibem z powodu zaburzeń opisanych biochemicznych wskaźników laboratoryjnych.
2-letni okres obserwacji
Podwyższoną aktywność aminotransferaz lub stężenie bilirubiny stopnia 3. lub 4. obserwowano u 1% pacjentów z CML (opornych na lub nietolerujących imatynibu) w fazie przewlekłej, natomiast u 1 do 7% pacjentów z zaawansowanymi postaciami CML oraz z Ph+ ALL. Poziomy te wyrównywały się zazwyczaj po zmniejszeniu dawki lub przerwaniu leczenia. W badaniu III fazy dotyczącym optymalizacji dawkowania w fazie przewlekłej CML podwyższoną aktywność aminotransferaz lub stężenie bilirubiny stopnia 3. lub 4. obserwowano u ≤ 1% pacjentów z podobnie małą częstością w czterech leczonych grupach. W badaniu III fazy dotyczącym optymalizacji dawkowania w fazie zaawansowanej CML i Ph+ ALL, podwyższoną aktywność aminotransferaz lub stężenie bilirubiny stopnia 3. lub 4. obserwowano u od 1% do 5% pacjentów w grupach badanych.
U około 5% pacjentów leczonych dazatynibem, u których wyjściowe stężenie wapnia w surowicy krwi było prawidłowe, w którymś momencie leczenia występowała przemijająca hipokalcemia stopnia 3. lub 4. W przeważającej większości przypadków nie obserwowano związku pomiędzy obniżeniem stężenia wapnia a objawami klinicznymi. U pacjentów z hipokalcemią stopnia 3. lub
Doświadczenie dotyczące przedawkowania dazatynibu w badaniach klinicznych jest ograniczone do pojedynczych przypadków. Największe przedawkowanie po podaniu 280 mg na dobę przez jeden tydzień opisano u dwóch pacjentów i u obu stwierdzono istotne zmniejszenie ilości płytek.
Ponieważ podawanie dazatynibu jest związane z mielosupresją stopnia 3. lub 4. (patrz punkt 4.4), pacjenci, którzy przyjmują dawkę większą niż zalecana powinni być ściśle monitorowani w kierunku mielosupresji i należy wdrożyć odpowiednie leczenie podtrzymujące.
Grupa farmakoterapeutyczna: leki przeciwnowotworowe, inhibitory kinazy proteinowej, kod ATC: L01XE06
Farmakodynamika
Dazatynib hamuje aktywność kinazy BCR-ABL oraz rodziny kinaz SRC równocześnie hamuje też inne, liczne, wybrane kinazy onkogenne, w tym c-KIT, kinazy receptora efryny (EPH) oraz receptora PDGF (3. Dazatynib jest silnym, subnanomolarnym inhibitorem kinazy BCR-ABL, działającym w zakresie stężeń 0,6-0,8 nM. Łączy się on zarówno z nieaktywną jak i aktywną postacią enzymu BCR-ABL.
Mechanizm działania
In vitro dazatynib wykazuje aktywność w liniach komórek białaczkowych reprezentujących odmiany zarówno białaczki wrażliwej jak i opornej na imatynib. Badania przedkliniczne wskazują, że dazatynib może przełamać oporność wynikającą ze zwiększonej ekspresji BCR-ABL, z mutacji domeny kinazy BCR-ABL, aktywacji alternatywnych dróg sygnalizacyjnych obejmujących kinazy rodziny SRC (LYN, HCK) oraz zwiększoną ekspresję genu determinującego oporność ielolekową. Dodatkowo, dazatynib hamuje kinazy rodziny SRC w stężeniach subnanomolarnych.
In vivo, w kilku oddzielnych eksperymentach z zastosowaniem mysiego modelu CML, dazatynib zapobiegał progresji fazy przewlekłej CML do fazy blastycznej i przedłużał czas przeżycia myszy
z komórkami CML przeniesionymi od pacjentów i umiejscowionymi w różnych miejscach, w tym także w ośrodkowym układzie nerwowym.
Skuteczność kliniczna i bezpieczeństwo stosowania
W badaniach I fazy uzyskano odpowiedź hematologiczną i cytogenetyczną we wszystkich fazach CML i w Ph+ ALL u pierwszych 84 pacjentów, którzy byli leczeni i obserwowani aż do 27 miesięcy. Odpowiedź była trwała w przypadku wszystkich faz CML i Ph+ ALL.
Przeprowadzono cztery otwarte badania kliniczne II fazy, z jednym ramieniem, bez grupy kontrolnej, w celu określenia bezpieczeństwa stosowania i skuteczności dazatynibu u pacjentów z CML w fazie przewlekłej, w fazie akceleracji lub w mieloblastycznej postaci przełomu blastycznego, u których stwierdzono oporność lub nietolerancję imatynibu. Przeprowadzono także jedno nieporównawcze badanie z randomizacją pacjentów w fazie przewlekłej, u których nie uzyskano odpowiedzi po wstępnym leczeniu imatynibem w dawce 400 lub 600 mg. Początkowa dawka wynosiła 70 mg dazatynibu dwa razy na dobę. Dopuszczalne były zmiany dawki w celu poprawy skuteczności działania lub opanowania toksyczności (patrz punkt 4.2).
Przeprowadzono dwa randomizowane, otwarte badania III fazy oceniające skuteczność dazatynibu podawanego raz na dobę w porównaniu do dazatynibu podawanego dwa razy na dobę. Ponadto przeprowadzono jedno otwarte, randomizowane, porównawcze badanie III fazy u dorosłych pacjentów z nowo rozpoznaną CML w fazie przewlekłej.
Skuteczność dazatynibu oceniana jest na podstawie odpowiedzi hematologicznych
i cytogenetycznych.Trwałość odpowiedzi i oszacowany wskaźnik przeżycia dostarcza dodatkowych dowodów klinicznej skuteczności dazatynibu.
Badaniami klinicznymi objęto 2712 pacjentów, 23% pośród nich miało ≥ 65 lat, a 5% miało ≥ 75 lat.
Faza przewlekła CML - Pacjenci nowo zdiagnozowani
Przeprowadzono międzynarodowe, otwarte, wieloośrodkowe, randomizowane, porównawcze badanie III fazy u dorosłych pacjentów z nowo rozpoznaną CML w fazie przewlekłej. Pacjentów randomizowano do grupy otrzymującej dazatynib w dawce 100 mg raz na dobę lub imatynib w dawce 400 mg raz na dobę. Pierwszorzędowym punktem końcowym był procent potwierdzonej całkowitej odpowiedzi cytogenetycznej (ang. complete cytogenetic response, cCCyR) w ciągu 12 miesięcy.
Drugorzędowe punkty końcowe obejmowały czas trwania cCCyR (miara trwałości odpowiedzi), czas do wystąpienia cCCyR, odsetek chorych z większą odpowiedzią molekularną (ang. major molecular response, MMR), czas do wystąpienia MMR, czas przeżycia wolny od progresji (ang. progression free survival, PFS) i całkowite przeżycie (ang. overall survival, OS). Inne istotne wyniki skuteczności obejmowały odsetek chorych z CCyR i z całkowitą odpowiedzią molekularną (CMR). Badanie jest w toku.
Do badanych grup zrandomizowano ogółem 519 pacjentów: 259 do grupy leczonej dazatynibrm i 260 do grupy otrzymującej imatynib. Wyjściowa charakterystyka pacjentów była dobrze zrównoważona między obu badanymi grupami w odniesieniu do wieku (mediana wieku wynosiła 46 lat w grupie otrzymującej dazatynib i 49 lat w grupie otrzymującej imatynib; odpowiednio, 10% i 11% stanowili pacjenci w wieku 65 lat i więcej), płci (kobiety odpowiednio 44% i 37%) i rasy (odpowiednio, 51% i 55% rasa kaukaska; 42% i 37% rasa azjatycka). Na początku badania rozkład wg skali Hasforda był podobny w grupach otrzymujących dazatynib i imatynib (odpowiednio, niskie ryzyko: 33% i 34%; pośrednie ryzyko 48% i 47%; wysokie ryzyko: 19% i 19%,).
W minimalnym okresie obserwacji wynoszącym 12 miesięcy, 85% pacjentów randomizowanych do grupy otrzymującej dazatynib i 81% pacjentów randomizowanych do grupy otrzymującej imatynib wciąż otrzymywało leczenie pierwszej linii. Zaprzestanie leczenia w ciągu 12 miesięcy z powodu progresji wystąpiło u 3% pacjentów leczonych dazatynibem i 5% pacjentów leczonych imatynibem.
W co najmniej 60-miesięcznym okresie obserwacji, 60% pacjentów randomizowanych do grupy otrzymującej dazatynib i 63% pacjentów randomizowanych do grupy otrzymującej imatynib wciąż otrzymywało leczenie pierwszej linii. Zaprzestanie leczenia w ciągu 60 miesięcy z powodu progresji wystąpiło u 11% pacjentów leczonych dazatynibem i 14% pacjentów leczonych imatynibem.
Wyniki skuteczności przedstawiono w Tabeli 9. W ciągu pierwszych 12 miesięcy leczenia cCCyR osiągnięto u statystycznie większego odsetka pacjentów w grupie otrzymującej dazatynib w porównaniu do grupy otrzymującej imatynib. Skuteczność dazatynibu konsekwentnie wykazano w różnych podgrupach, w tym dotyczących wieku, płci i stopnia ryzyka wg skali Hasforda przed rozpoczęciem leczenia.
Tabela 9.: Wyniki skuteczności uzyskane w badaniu III fazy u pacjentów z nowo rozpoznaną CML w fazie przewlekłej
dasatynib n= 259 | imatynib n= 260 | wartość p | |
Odsetek odpowiedzi (95% CI) | |||
Odpowiedź cytogenetyczna w ciągu 12 miesięcy | |||
cCCyRa | 76,8% (71,2–81,8) | 66,2% (60,1–71,9) | p< 0,007* |
CCyRb | 85,3% (80,4-89,4) | 73,5% (67,7-78,7) | ⎯ |
w ciągu 24 miesięcy | |||
cCCyRa | 80,.3% | 74,2% | ⎯ |
CCyRb | 87,3% | 82,3% | ⎯ |
w ciągu 36 miesięcy | |||
cCCyRa | 82,.6% | 77,3% | ⎯ |
CCyRb | 88,0% | 83,5% | ⎯ |
w ciągu 48 miesięcy | |||
cCCyRa | 82,6% | 78,5% | ⎯ |
CCyRb | 87,6% | 83,8% | ⎯ |
w ciągu 60 miesięcy | |||
cCCyRa | 83,0% | 78,.5% | ⎯ |
CCyRb | 88,0% | 83,8% | ⎯ |
Większa odpowiedź molekularnac | |||
12 miesięcy | 52,1% (45,9–58,3) | 33,8% (28,1–39,.9) | p< 0,00003* |
24 miesięcy | 64,5% (58,3-70,3) | 50% (43,8-56,2) | ⎯ |
36 miesięcy | 69,1% (63,1-74,7) | 56,2% (49,9-62,3) | ⎯ |
48 miesięcy | 75,7% (70,0-80,8) | 62,7% (56,5-68,6) | ⎯ |
60 miesięcy | 76,4% (70,8-81,5) | 64,2% (58,1-70,1) | p=0,0021 |
Współczynnik ryzyka (HR) w ciągu 12 miesięcy (99,99% CI) | |||
Czas do cCCyR | 1,55 (1,0-2,3) | p< 0,0001* | |
Czas do MMR | 2,01 (1,2-3,4) | p< 0,0001* | |
Trwałość cCCyR | 0,7(0,4-1,4) | p< 0,035 | |
w ciągu 24 miesięcy (95% CI) | |||
Czas do cCCyR | 1,49 (1,22-1,82) | — | |
Czas do MMR | 1,69(1,34-2,12) | — | |
Trwałość cCCyR | 0,77(0,55-1,10) | — | |
w ciągu 36 miesięcy (95% CI) | |||
Czas do cCCyR | 1,48 (1,22-1,80) | — | |
Czas do MMR | 1,59 (1,28-1,99) | — |
Trwałość cCCyR | 0,77(0,53-1,11) | — |
w ciągu 48 miesięcy (95% CI) | ||
Czas do cCCyR | 1,45 (1,20-1,77) | — |
Czas do MMR | 1,55 (1,26-1,91) | — |
Trwałość cCCyR | 0,81 (0,56-1,17) | — |
w ciągu 60 miesięcy (95% CI) | ||
Czas do cCCyR | 1,46 (1,20-1,77) | p=0,0001 |
Czas do MMR | 1,54 (1,25-1,89) | p<0,0001 |
Trwałość cCCyR | 0,79(0,55-1,13) | p=0,1983 |
a Potwierdzona pełna odpowiedź cytogenetyczna (cCCyR) jest definiowana jako odpowiedź stwierdzona w dwóch kolejnych oznaczeniach (w co najmniej 28 dniowym odstępie czasowym).
b Pełna odpowiedź cytogenetyczna (CCyR) oceniana jest na podstawie jednorazowej oceny cytogenetycznej szpiku kostnego.
c Większa odpowiedź molekularna (w jakimkolwiek czasie) była definiowana wg współczynnika BCR-ABL ≤ 0,1% w oznaczeniu metodą RQ-PCR w próbkach krwi obwodowej ze standaryzacją zgodnie ze skalą międzynarodową. Są to łączne wskaźniki reprezentujące minimalny okres obserwacji w ciągu określonego czasu.
*Dostosowany do skali Hasforda i wskazujący istotność statystyczną w określonym na początku nominalnym poziomie istotności.
CI = przedział ufności
U pacjentów z potwierdzoną CCyR mediana czasu do wystąpienia cCCyR po 60-miesięcznym okresie obserwacji wynosiła 3,1 miesiąca w grupie otrzymującej dazatynib i 5,8 miesiąca w grupie otrzymującej imatynib. U pacjentów z MMR mediana czasu do uzyskania MMR po 60- miesięcznym okresie obserwacji wynosiła 9,3 miesiąca w grupie otrzymującej dazatynib i 15,0 miesięcy w grupie otrzymującej imatynib. Wyniki te są zgodne z wartościami uzyskanymi po 12, 24 i 36 miesiącach.
Czas do uzyskania odpowiedzi MMR przedstawiono graficznie na wWykresie 1. Czas do uzyskania MMR był stale krótszy u pacjentów leczonych dazatynibem w porównaniu z pacjentami otrzymującymi imatynib.
Wykres 1.: Kaplana-Meiera estymator czasu do większej odpowiedzi molekularnej (MMR)
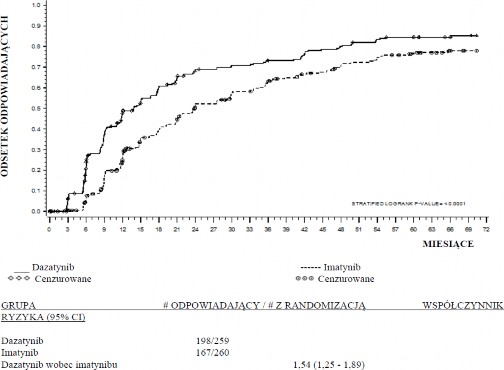
Wskaźniki cCCyR, odpowiednio, w grupach leczonych dazatynibem i imatynibem w ciągu 3 miesięcy (54% i 30%), 6 miesięcy (70% i 56%), 9 miesięcy (75% i 63%), 24 miesięcy (80% i 74%),
36 miesięcy (83% i 77%), 48 miesięcy (83% i 79%) i 60 miesięcy (83% i 79%) były zgodne z
pierwszorzędowym punktem końcowym. Wskaźniki MMR, odpowiednio, w grupach leczonych dazatynibem i imatynibem w ciągu 3 miesięcy (8% i 0,4%), 6 miesięcy (27% i 8%), 9 miesięcy
(39% i 18%), 12 miesięcy (46% i 28%), 24 miesięcy (64% i 46%), 36 miesięcy (67% i 55 %), 48
miesięcy (73% i 60%) i 60 miesięcy (76% i 64%) były także zgodne z pierwszorzędowym punktem końcowym.
Wskaźniki MMR w poszczególnych punktach czasowych przedstawiono graficznie na wykresie 2. Wskaźniki MMR były stale wyższe wśród pacjentów leczonych dazatynibem w porównaniu z pacjentami leczonymi imatynibem.
Wykres 2.: Wskaźniki MMR w czasie - wszyscy zrandomizowani pacjenci w badaniu III fazy z udziałem pacjentów z nowo rozpoznaną CML w fazie przewlekłej
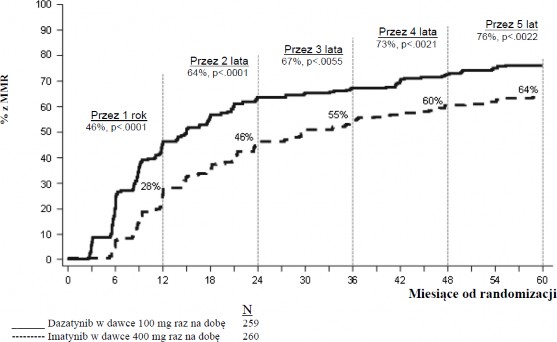
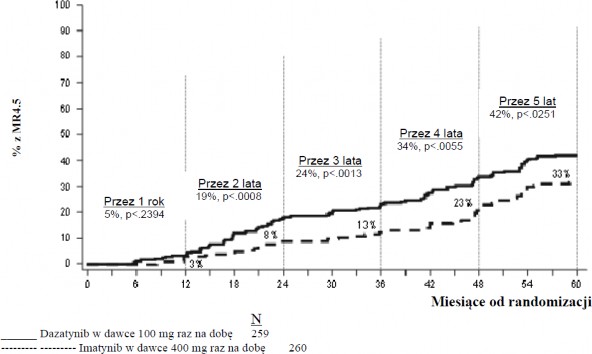
Odsetek pacjentów osiągających współczynnik BCR-ABL o wartości ≤ 0,01% (zmniejszenie 4-log) w każdym czasie był większy w grupie leczonych dazatynibem w porównaniu z grupą leczonych imatynibem (54,1% w stosunku do 45%). Odsetek pacjentów osiągających współczynnik BCR-ABL o wartości ≤0,0032% (zmniejszenie 4,5-log) w każdym czasie był większy w grupie leczonych dazatynibem w porównaniu z grupą leczonych imatynibem (44% w stosunku do 34%).
Wskaźniki MR4.5 w czasie przedstawiono graficznie na Wykresie 3. Odsetki MR4.5 w czasie były stale wyższe wśród pacjentów leczonych dazatynibem w porównaniu z pacjentami leczonymi imatynibem.
Wykres 3.: Wskaźniki MR4.5 w czasie - wszyscy zrandomizowani pacjenci w badaniu III fazy z udziałem pacjentów z nowo rozpoznaną CML w fazie przewlekłej
Wskaźnik MMR w każdym czasie, w każdej z grup ryzyka określonych przez wskaźnik Hasforda był większy w grupie leczonych dazatynibem w porównaniu z grupą leczonych imatynibem (niskiego ryzyka: 90% i 69%; pośredniego ryzyka: 71% i 65%; wysokiego ryzyka: 67% i 54%, odpowiednio).
W dodatkowej analizie, więcej pacjentów leczonych dazatynibem (84%) uzyskało wczesną odpowiedź molekularną (definiowaną jako poziom BCR-ABL ≤ 10% po 3 miesiącach) w porównaniu do pacjentów leczonych imatynibem (64%). Pacjenci uzyskujący wczesną odpowiedź molekularną mieli mniejsze ryzyko transformacji, wyższy wskaźnik czasu przeżycia wolnego od progresji (PFS) oraz wyższy wskaźnik całkowitego przeżycia (OS), jak przedstawiono w Tabeli 10.
Tabela 10.: Pacjenci leczeni dazatynibem z BCR-ABL ≤ 10% i > 10% po 3 miesiącach
Dasatinib N = 235 | Pacjenci z BCR-ABL ≤ 10% po 3 miesiącach | Pacjenci z BCR-ABL > 10% po 3 miesiącach |
Liczba pacjentów (%) | 198 (84,3) | 37 (15,7) |
Transformacja po 60 miesiącach, n/N (%) | 6/198 (3,0) | 5/37 (13,5) |
Wskaźnik PFS po 60 miesiącach (95% CI) | 92,0% (89,6, 95,2) | 73,8% (52,0, 86,8) |
Wskaźnik OS po 60 miesiącach (95% CI) | 93,8% (89,3, 96,4) | 80,6% (63,5, 90,2) |
Wartość OS w poszczególnych punktach czasowych przedstawiono graficznie na wykresie 4. Wartość OS była stale wyższa wśród pacjentów leczonych dazatynibem, u których osiągnięto poziom BCR-ABL ≤10% po 3 miesiącach, niż wśród pacjentów, u których nie osiągnięto takiego poziomu.
Wykres 4.: Orientacyjny wykres całkowitego czasu przeżycia dla dazatynibu w zależności od poziomu BCR-ABL (≤10% lub > 10%) po 3 miesiącach w badaniu III fazy z udziałem pacjentów z nowo rozpoznaną CML w fazie przewlekłej
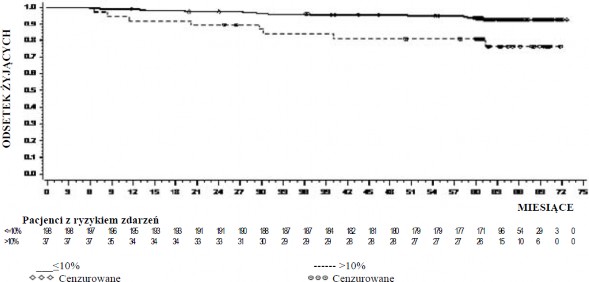
GRUPA | # ZGONY / # Land Patient | MEDIANA (95% CI) | WSPÓŁCZTNNIK RYZYKA (95% CI) |
≤10% | 14/198 | .(. - .) | |
>10% | 8/37 | .(. - .) | |
0,29 (0,12 – 0,69) |
Progresję choroby definiowano jako zwiększenie liczby białych krwinek pomimo odpowiedniego leczenia, utratę CHR, częściowej CyR lub CCyR, przejście do fazy akceleracji lub przełomu blastycznego lub zgon. Szacowany 60-miesięczny odsetek przeżyć wolnych od progresji wynosił 88,9% (CI: 84% - 92,4%) zarówno w grupie leczonej dazatynibem jak i imatynibem. Przejście w ciągu 60 miesięcy do fazy akceleracji lub przełomu blastycznego występowało u mniejszej liczby pacjentów leczonych dazatynibem (n=8; 3%) w porównaniu z pacjentami leczonymi imatynibem (n=15; 5,8%). Szacowany 60-miesięczny odsetek przeżyć pacjentów leczonych dazatynibem i imatynibem wynosił odpowiednio 90,9% (CI: 86,6% - 93,8%) i 89,6% (CI: 85,2% - 92,8%). Nie było różnic w całkowitym czasie przeżycia (OS) (HR 1,01; 95% CI: 0,58 - 1,73; p=0,9800) i czasie przeżycia bez progresji choroby (PFS) (HR 1,00; 95% CI: 0,58 - 1,72; p=0,9998) między dazatynibem a imatynibem.
W przypadku pacjentów, którzy zgłosili progresję choroby lub przerwali leczenie dazatynibem lub imatynibem, przeprowadzono sekwencjonowanie BCR-ABL na próbkach krwi, o ile były dostępne. W obu grupach leczenia zaobserwowano podobne wskaźniki mutacji. Mutacje wykryte wśród pacjentów leczonych dazatynibem obejmowały T315I, F317I/L i V299L. W grupie leczonych imatynibem stwierdzono odmienny zakres mutacji. W oparciu o dane in vitro wydaje się, że dazatynib nie jest aktywny przeciw mutacji T315I.
Faza przewlekła CML - Oporność lub nietolerancja na wcześniejsze leczenie imatynibem
U pacjentów z opornością lub nietolerancją imatynibu przeprowadzono dwa badania kliniczne. Pierwszorzędnym punktem końcowym dotyczącym skuteczności w tych badaniach była duża odpowiedź cytogenetyczna (ang. major cytogenetic response, MCyR):
Badanie 1.
U pacjentów, u których wstępne leczenie imatynibem w dawce 400 lub 600 mg było nieskuteczne przeprowadzono badanie otwarte, randomizowane, nieporównawcze wieloośrodkowe. Pacjenci byli losowo przydzielani (2:1) do grupy otrzymującej dazatynib (70 mg dwa razy na dobę) lub imatynib (400 mg dwa razy na dobę). Przeniesienie do alternatywnego ramienia badania było dopuszczalne,
jeśli u pacjentów stwierdzano progresję choroby lub nietolerancję leczenia mimo zmiany dawkowania
Pierwszorzędowym punktem końcowym była MCyR w 12 tygodniu. Otrzymane wyniki obejmują grupę 150 pacjentów: 101 było randomizowanych do grupy otrzymującej dazatynib, a 49 do grupy otrzymującej imatynib (wszyscy byli oporni na imatynib). Mediana czasu od momentu rozpoznania do randomizacji wynosiła 64 miesiące dla pacjentów otrzymujących dazatynib i 52 miesiące dla pacjentów otrzymujących imatynib. Wszyscy pacjenci byli wcześniej intensywnie leczeni. Po wcześniejszym leczeniu imatynibem całkowitą odpowiedź hematologiczną (ang. complete haematologic response, CHR) po leczeniu uzyskano u 93% spośród wszystkich pacjentów.
Wcześniejszą dużą odpowiedź cytogenetyczną (MCyR) po leczeniu imatynibem osiągnęło 28% pacjentów w ramieniu dazatynibu i 29% pacjentów w ramieniu imatynibu.
Mediana czasu leczenia wyniosła 23 miesiące dla dazatynibu (przy 44% pacjentów leczonych > 24 do chwili obecnej) oraz 3 miesiące dla imatynibu (przy 10% pacjentów leczonych > 24 do chwili obecnej). Dziewięćdziesiąt trzy procent pacjentów w ramieniu dazatynibu i 82% pacjentów w ramieniu imatynibu osiągnęło całkowitą odpowiedź hematologiczną (CHR) przed przeniesieniem do innego ramienia leczenia.
W 3 miesiącu w ramieniu dazatynibu obserwowano częściej dużą odpowiedź cytogenetyczną (MCyR) (36%) niż w ramieniu imatynibu (29%). Warto zauważyć, że w ramieniu dazatynibu 22% pacjentów osiągnęło całkowitą odpowiedź cytogenetyczną (ang. complete cytogenetic response, CCyR), podczas gdy w ramieniu imatynibu tylko 8% pacjentów uzyskało taką odpowiedź. Po dłuższym czasie leczenia i okresie obserwacji (mediana 24 miesięcy) MCyR uzyskało 53% pacjentów leczonych dazatynibem (CCyR u 44%) i 33% pacjentów leczonych imatynibem (CCyR u 18%) przed przeniesieniem do innego ramienia leczenia. Spośród pacjentów, którzy przed włączeniem do badania otrzymywali imatynib w dawce 400 mg, MCyR uzyskało 61% pacjentów otrzymujących dazatynib i 50% pacjentów otrzymujących imatynib.
Na podstawie oceny wg Kaplana-Meiera, procent pacjentów, u których MCyR utrzymała się przez 1 rok wynosił 92% (95% CI: [85%-100%]) dla dazatynibu (CCyR 97%, 95% CI: [92%-100%]) i
74% (95% CI: [49%-100%]) dla imatynibu (CCyR 100%). Procent pacjentów, u których MCyR
utrzymał się przez 18 miesięcy wynosił 90% (95% CI: [82%-98%]) dla dazatynibu (CCyR 94%,
95% CI: [87%-100%]) i 74% (95% CI: [49%-100%]) dla imatynibu (CCyR 100%).
Na podstawie oceny wg Kaplana-Meiera, odsetek pacjentów z rocznym okresem przeżycia bez postępu choroby (ang. progression-free survival, PFS) wynosił 91% (95% CI: [85%-97%]) dla dazatynibu i 73% (95% CI: [54%-91%]) dla imatynibu. Odsetek pacjentów z dwuletnim PFS wynosił 86% (95% CI: [78%-93%]) dla dazatynibu i 65% (95% CI: [43%-87%]) dla imatynibu.
Niepowodzenie leczenia, zdefiniowane jako progresja choroby lub przeniesienie do innego programu leczenia (brak odpowiedzi, nietolerancja badanego produktu leczniczego, itp), obserwowano u 43% pacjentów z ramienia dazatynibu oraz u 82% pacjentów z ramienia imatynibu.
Procent większej odpowiedzi molekularnej (definiowanej jako BCR-ABL/kontrolowane transkrypty
≤ 0,1% przez RQ-PCR w próbkach krwi obwodowej) przed przeniesieniem do innego ramienia leczenia wynosił 29% dla dazatynibu i 12% dla imatynibu.
Badanie 2.
U pacjentów wykazujących oporność lub nietolerancję na imatynib przeprowadzono wieloośrodkowe, otwarte badanie z jednym ramieniem (tj. pacjentów, u których w czasie leczenia imatynibem wystąpiły istotne objawy toksyczności, uniemożliwiające dalsze leczenie). Ogółem 387 pacjentów otrzymywało dazatynib w dawce 70 mg dwa razy na dobę (288 z opornością oraz 99 z brakiem tolerancji). Mediana czasu od momentu rozpoznania do momentu rozpoczęcia leczenia wyniosła 61 miesiące. Większość pacjentów (53%) otrzymywała wcześniej imatynib przez ponad 3 lata. Większość pacjentów z opornością (72%) otrzymywała > 600 mg imatynibu. Dodatkowo do leczenia imatynibem, 35% pacjentów otrzymywała wcześniejszą chemioterapię, 65% otrzymywało wcześniej interferon, a u 10% wykonywano uprzednio przeszczepienie komórek macierzystych szpiku. U trzydziestu ośmiu procent pacjentów stwierdzono obecność podstawowych mutacji, o których wiadomo, że są odpowiedzialne za oporność na imatynib. Mediana czasu leczenia dazatynibem wyniosła 24 miesiąca, przy 51% pacjentów leczonych > 24 do chwili obecnej.
Wyniki skuteczności przedstawiono w Tabeli 11. MCyR osiągnięto u 55% pacjentów opornych na
imatynib i u 82% pacjentów, którzy nie tolerowali imatynibu. Po minimalnym czasie obserwacji wynoszącym 24 miesięcy, u 21 spośród 240 pacjentów, którzy osiągnęli dużą odpowiedź cytogenetyczną (MCyR) stwierdzono progresję choroby i u tych pacjentów nie osiągnięta została mediana czasu trwania dużej odpowiedzi cytogenetycznej (MCyR).
Na podstawie oceny wg Kaplana-Meiera, 95% (95% CI: [92%-98%]) pacjentów utrzymała MCyR przez 1 rok, a 88% (95% CI: [83%-93%]) utrzymała MCyR przez 2 lata. Odsetek pacjentów, u
których CCyR utrzymała się przez 1 rok wynosił 97% (95% CI: [94%-99%]), a przez 2 lata 90% (95% CI: [86%-95%]). Czterdzieści dwa procent pacjentów opornych na imatynib, którzy nie uzyskali wcześniej MCyR po zastosowaniu imatynibu (n= 188) uzyskało MCyR po podaniu dazatynibu. Stwierdzono 45 różnych typów mutacji BCR-ABL u 38% pacjentów włączonych do tego badania. Całkowitą odpowiedź hematologiczną lub MCyR uzyskano u pacjentów z licznymi typami mutacji BCR-ABL związanymi z opornością na imatynib z wyjątkiem T315I. Procent MCyR w 2 roku był podobny, bez względu na yo, czy na początku leczenia u pacjentów występowały mutacje BCR-ABL, P-loop mutacje czy nie mieli mutacji (odpowiednio 63%, 61% i 62%).
Dla pacjentów opornych na imatynib, przewidywany PFS wynosił 88% (95% CI: [84%-92%]) dla 1 roku i 75% (95% CI: [69%-81%]) dla 2 lat. Dla pacjentów, którzy nie tolerują imatynibu
przewidywany PFS wynosił 98% (95% CI: [95%-100%]) dla 1 roku i 94% (95% CI: [88%-99%])
dla 2 lat.
Większa odpowiedź molekularna po 24 miesiącach wynosiła 45% (35% dla pacjentów opornych na imatynib i 74% dla pacjentów, którzy nie tolerują imatynibu).
Faza akceleracji CML
U pacjentów wykazujących oporność lub nietolerancję imatynibu przeprowadzono wieloośrodkowe, otwarte badanie z jednym ramieniem. Ogółem 174 pacjentów otrzymywało dazatynib w dawce 70 mg dwa razy na dobę (161 z opornością oraz 13 z nietolerancją imatynibu). Mediana czasu od momentu rozpoznania do momentu rozpoczęcia leczenia wynosiła 82 miesiące. Mediana czasu leczenia dazatynibem wyniosła 14 miesięcy przy 31% pacjentów leczonych > 24 do chwili obecnej. Współczynnik większej odpowiedzi molekularnej po 24 miesiącach wynosił 46% (oceniony u 41 pacjentów z CCyR). Inne wyniki skuteczności przedstawiono w Tabeli 11.
Mieloblastyczna postać przełomu blastycznego CML
U pacjentów wykazujących oporność lub nietolerancję imatynibu przeprowadzono wieloośrodkowe, otwarte badanie z jednym ramieniem. Ogółem 109 pacjentów otrzymywało dazatynib w dawce 70 mg dwa razy na dobę (99 z opornością oraz 10 z nietolerancją imatynibu). Mediana czasu od momentu rozpoznania do momentu rozpoczęcia leczenia wynosiła 48 miesięcy. Mediana czasu leczenia dazatynibem wyniosła 3,5 miesiąca przy 12% pacjentów leczonych > 24 do chwili obecnej. Współczynnik większej odpowiedzi molekularnej po 24 miesiącach wynosił 68% (oceniony u 19 pacjentów z CCyR). Inne wyniki skuteczności przedstawiono w Tabeli 11.
Limfoblastyczna postać przełomu blastycznego CML i Ph+ ALL
U pacjentów z limfoblastyczną postacią przełomu blastycznego CML oraz z Ph+ ALL wykazujących oporność lub nietolerancję na wcześniejsze leczenie imatynibem przeprowadzono wieloośrodkowe, otwarte badanie z jednym ramieniem. Ogółem 48 pacjentów z limfoblastyczną postacią przełomu blastycznego CML otrzymywało dazatynib w dawce 70 mg dwa razy na dobę (42 z opornością oraz 6 z nietolerancją imatynibu). Mediana czasu od momentu rozpoznania do momentu rozpoczęcia leczenia wynosiła 28 miesięcy. Mediana czasu leczenia dazatynibem wyniosła 3 miesiące przy 2% pacjentów leczonych > 24 miesięcy do chwili obecnej.
Współczynnik większej odpowiedzi molekularnej po 24 miesiącach wynosił 50% (oceniony u wszystkich 22 leczonych pacjentów z CCyR). Dodatkowo 46 pacjentów z Ph+ ALL otrzymywało dazatynib w dawce 70 mg dwa razy na dobę (44 z opornością oraz 2 z brakiem tolerancji imatynibu). Mediana czasu od momentu rozpoznania do momentu rozpoczęcia leczenia wynosiła 18 miesięcy. Mediana czasu leczenia dazatynibem wyniosła 3 miesiące przy 7% pacjentów leczonych > 24 miesięcy do chwili obecnej. Współczynnik większej odpowiedzi molekularnej po 24 miesiącach wynosił 52% (oceniony u wszystkich 25 leczonych pacjentów z CCyR). Inne wyniki skuteczności przedstawiono w Tabeli 11.Warto zauważyć, że szybko uzyskano dużą odpowiedź hematologiczną (MaHR) (w większości przypadków w ciągu 35 dni od pierwszej dawki dazatynibu
dla pacjentów z limfoblastyczną postacią przełomu blastycznego CML oraz w ciągu 55 dni dla pacjentów z Ph+ ALL).
Tabela 11.: Skuteczność w badaniach II fazy z jednym ramieniema
Limfoblasty | |||||
Mieloblasty | -czna postać | ||||
-czna postać | przełomu | ||||
Faza | Faza | przełomu | blastyczne- | ||
przewlekła | akceleracji | blastyczne- | go | Ph+ ALL | |
(n= 387) | (n=174) | go (n= 109) | (n=48) | (n=46) | |
Odpowiedź hematologicznab (%) | |||||
MaHR (95% CI) | n/a | 64% (57-72) | 33% (24-43) | 35% (22-51) | 41% (27-57) |
CHR (95% CI) | 91% (88-94) | 50% (42-58) | 26% (18-35) | 29% (17-44) | 35% (21-50) |
NEL (95% CI) | n/a | 14% (10-21) | 7% (3-14) | 6% (1-17) | 7% (1-18) |
Czas trwania MaHR (%; ocena wg Kaplana | |||||
1 rok | n/a | 79% (71-87) | 71% (55-87) | 29% (3-56) | 32% (8-56) |
2 lata | n/a | 60% (50-70) | 41% (21-60) | 10% (0-28) | 24% (2-47) |
Odpowiedź cytogenetycznac (%) | |||||
MCyR (95% CI) | 62% (57-67) | 40% (33-48) | 34% (25-44) | 52% (37-67) | 57% (41-71) |
CCyR (95% CI) | 54% (48-59) | 33% (26-41) | 27% (19-36) | 46% (31-61) | 54% (39-69) |
Przeżycie (%; ocena wg Kaplana-Meiera) | |||||
Bez postępu | |||||
1 rok | 91% (88-94) | 64% (57-72) | 35% (25-45) | 14% (3-25) | 21% (9-34) |
2 lata | 80% (75-84) | 46% (38-54) | 2% ( 2- 23) | 5% (0-13) | 12% (2-23) |
Ogółem | |||||
1 rok | 97% (95-99) | 83% (77-89) | 48% (38-59) | 30% (14-47) | 35% (20-51) |
2 lata | 94% (91-97) | 72% (64-79) | 38% (27-50) | 26% (10-42) | 31% (16-47) |
Dane zebrane w tej tabeli pochodzą z badań, w których dawką początkową było 70 mg podawane dwa razy na dobę. Zalecana dawka początkowa patrz punkt 4.2.
a Wyniki zebrane dla pierwszorzędowych punktów końcowych zostały wytłuszczone.
b Kryteria odpowiedzi hematologicznej (wszystkie potwierdzone po 4 tygodniach): Duża odpowiedź hematologiczna: (MaHR - Major hematologic response) = całkowita odpowiedź hematologiczna (CHR - complete haematologic response)
+ brak objawów białaczki (NEL - no evidence of leukemia).
CHR (faza przewlekła CML): Krwinki białe (WBC) ≤ GGN laboratoryjnej w danej instytucji, płytki krwi
< 450000/mm3, brak blastów lub promielocytów w krwi obwodowej, < 5% mielocytów oraz metamielocytów w krwi obwodowej, < 20% granulocytów zasadochłonnych w krwi obwodowej oraz brak ognisk białaczkowych poza szpikiem.
CHR (zaawansowana CML/Ph+ ALL): WBC ≤ GGN laboratoryjnej w danej instytucji, ANC ≥ 1000/mm3,
płytki ≥ 100000/mm3, brak blastów lub promielocytów w krwi obwodowej, liczba blastów w szpiku kostnym < 5%, < 5% mielocytów oraz metamielocytów w krwi obwodowej, < 20% bazofili w krwi obwodowej oraz brak ognisk białaczkowych poza szpikiem. NEL: te same kryteria jak w przypadku CHR, ale ANC ≥ 500/mm3 i < 1000/mm3, lub płytki krwi ≥ 20000/mm3 i ≤ 100000/mm3.
c Kryteria odpowiedzi cytogenetycznej: całkowita (0% metafaz Ph+) lub częściowa (> 0%-35%). MCyR (0%-35%) obejmuje zarówno odpowiedź całkowitą jak i częściową, n/a = nie dotyczy;
CI = przedział ufności; GGN = górna granica normy.
Wyniki u pacjentów z przeszczepem szpiku po leczeniu dazatynibem nie zostały w pełni ocenione.
Badania kliniczne III fazy u pacjentów z CML w fazie przewlekłej, w fazie akceleracji lub w fazie przełomu blastycznego i Ph+ ALL, którzy byli oporni lub nie tolerowali imatynibu
Wykonano dwa randomizowane badania otwarte oceniające skuteczność dazatynibu podawanego raz na dobę w porównaniu do dazatynibu podawanego dwa razy na dobę. Wyniki opisane pPoniżej pochodzą z co najmniej 2-letniej i 7-letniej obserwacji od rozpoczęcia leczenia dazatynibem.
Badanie 1.
W badaniu fazy przewlekłej CML u pacjentów opornych na imatynib głównym punktem końcowym była MCyR. Drugorzędowym punktem końcowym była wartość MCyR podzielona przez dawkę dobową u pacjentów opornych na imatynib. Do innych punktów drugorzędowych należały czas trwania MCyR, PFS i całkowity czas przeżycia. Całkowitą liczbę 670 pacjentów, z których 497 było opornych na imatynib, randomizowano do grup otrzymujących dazatynib w dawce 100 mg raz na dobę, 140 mg raz na dobę, 50 mg dwa razy na dobę lub 70 mg dwa razy na dobę. Mediana czasu leczenia dla wszystkich pacjentów nadal leczonych z co najmniej 5-letnim
okresem obserwacji (n=205) wynosiła 59 miesięcy (zakres 28 66 miesięcy). Mediana czasu leczenia dla wszystkich pacjentów po 7-letniej obserwacji wynosiła 29,8 miesięcy (zakres < 1-92,9 miesięcy).
Skuteczność osiągnięto we wszystkich grupach leczonych dazatynibem podawanym jeden raz na dobę. Dla pierwszorzędowego punktu końcowego (różnica w MCyR 1,9%; z 95% przedziałem ufności [-6,8% - 10,6%]) wykazano porównywalną skuteczność (nie stwierdzono mniejszej) z podawaniem 2 razy na dobę; jednakże przy zastosowaniu schematu dawkowania 100 mg raz na dobę wykazano większe bezpieczeństwo i tolerancję. Wyniki skuteczności przedstawiono w Tabelach 12. i 13.
Tabela 12.: Skuteczność produktu Dasatinib Alvogen w badaniu III fazy dotyczącym optymalizacji dawkowania: oporność lub nietolerancja na imatynib w fazie przewlekłej CML (wyniki po 2 latach)a
Wszyscy pacjenci | n=167 |
Pacjenci oporni na imatynib | n=124 |
Odsetek odpowiedzi hematologicznejb | (%) (95% CI) |
CHR | 92% (86-95) |
Odpowiedź cytogenetycznac (%) (95% CI) | |
MCyR | |
Wszyscy pacjenci | 63% (56-71) |
Pacjenci oporni na imatynib | 59% (50-68) |
CCyR | |
Wszyscy pacjenci | 50% (42-58) |
Pacjenci oporni na imatynib | 44% (35-53) |
Większa odpowiedź molekularna u pacjentów osiągających CCyRd (%) (95% CI) | |
Wszyscy pacjenci | 69% (58-79) |
Pacjenci oporni na imatynib | 72% (58-83) |
a Wyniki odnotowane po zastosowaniu zalecanej dawki początkowej wynoszącej 100 mg raz na dobę.
b Kryteria odpowiedzi hematologicznej (wszystkie potwierdzone po 4 tygodniach): całkowita odpowiedź hematologiczna (CHR) (faza przewlekła CML): Krwinki białe (WBC) ≤ GGN laboratoryjnej w danej instytucji, płytki krwi < 450000/mm3, brak blastów lub promielocytów w krwi obwodowej, < 5% mielocytów oraz metamielocytów w krwi obwodowej, liczba granulocytów zasadochłonnych w krwi obwodowej < 20% oraz brak ognisk białaczkowych poza szpikiem.
c Kryteria odpowiedzi cytogenetycznej: całkowita (0% metafaz Ph+) lub częściowa (> 0%—35%). Duża odpowiedź cytogenetyczna (MCyR) (0%-35%) obejmuje zarówno odpowiedź całkowitą jak i częściową.
d Kryteria większej odpowiedzi molekularnej: określonajako współczynnik BCR-ABL/gen kontrolny ≤ 0,1% oznaczony metodą RQ- PCR w próbkach krwi obwodowej.
Tabela 13.: Długoterminowa skuteczność dazatynibu w badaniu fazy III dotyczącym optymalizacji dawkowania: pacjenci z CML w fazie przewlekłej z opornością lub nietolerancją na imatyniba
Minimalny okres obserwacji
1rok | 2 lata | 5 lat | 7 lat | ||
Większa odpowiedź molekularna | |||||
Wszyscy pacjenci | NA | 37% (57/154) | 44% | (71/160) | 46% (73/160) |
Pacjenci oporni na | NA | 35% (41/117) | 42% | (50/120) | 43% (51/120) |
imatynib | |||||
Pacjenci z nietolerancją | NA | 43% (16/37) | 53% | (21/40) | 55% (22/40) |
imatynibu | |||||
Czas przeżycia bez progresji chorobyb | |||||
Wszyscy pacjenci | 90% (86, 95) | 80% (73, 87) | 51% | (41, 60) | 42% (33, 51) |
Pacjenci oporni na | 88% (82, 94) | 77% (68, 85) | 49% | (39, 59) | 39% (29, 49) |
imatynib | |||||
Pacjenci z nietolerancją | 97% (92, 100) | 87% (76, 99) | 56% | (37, 76) | 51% (32, 67) |
imatynibu | |||||
Całkowity czas przeżycia | |||||
Wszyscy pacjenci | 96% (93, 99) | 91% (86, 96) | 78% | (72, 85) | 65% (56, 72) |
Pacjenci oporni na | 94% (90, 98) | 89% (84, 95) | 77% | (69, 85) | 63% (53, 71) |
imatynib | |||||
Pacjenci z nietolerancją | 100% (100, 100) | 95% (88, 100) | 82% | (70, 94) | 70% (52, 82) |
imatynibu |
a Wyniki odnotowane po zastosowaniu zalecanej dawki początkowej wynoszącej 100 mg raz na dobę
b Progresję definiowano jako zwiększenie liczby białych krwinek (WBC), utratę CHR lub MCyR ≥30% zwiększenie liczby metafaz Ph+, potwierdzoną chorobę AP/BP lub zgon. PFS oceniano według zasad zgodnych z zaplanowanym leczeniem, a pacjenci byli obserwowani pod względem zdarzeń łącznie z kolejną terapią.
Na podstawie oceny wg Kaplana-Meiera, odsetek pacjentów leczonych dazatynibem w dawce 100 mg raz na dobę, u których MCyR utrzymywała się przez 18 miesięcy, wynosił 93% (95% CI: [88%-98%]).
Skuteczność oceniano również u pacjentów, którzy nie tolerowali imatynibu. W tej populacji pacjentów, którzy otrzymywali 100 mg raz na dobę, MCyR uzyskano u 77% a CCyR u 67%.
Badanie 2.
W badaniu fazy zaawansowanej CML i Ph+ ALL głównym punktem końcowym była MaHR. Całkowitą liczbę 611 pacjentów randomizowano albo do grupy otrzymującej dazatynib w dawce 140 mg raz na dobę lub 70 mg dwa razy na dobę. Mediana czasu leczenia wynosiła około 6 miesięcy (zakres 0,03-31 miesięcy).
Dla głównego punktu końcowego określającego skuteczność podawanie raz na dobę miało porównywalną skuteczność (nie stwierdzono mniejszej) do podawania dwa razy na dobę (różnica MaHR 0,8%; z 95% przedziałem ufności [-7,1% - 8,7%]); jednakże przy zastosowaniu schematu dawkowania 140 mg raz na dobę wykazano zwiększone bezpieczeństwo i tolerancję. Wskaźniki odpowiedzi przedstawiono w Tabeli 14.
Tabela 14.: Skuteczność dazatynibu w badaniach III fazy dotyczących optymalizacji dawkowania: faza zaawansowana CML i Ph+ ALL (wyniki po 2 latach) a
Mieloblastyczna | Limfoblastyczna | |||
postać przełomu | postać przełomu | |||
Faza akceleracji | blastycznego | blastycznego | Ph+ ALL | |
(n= 158) | (n= 75) | (n= 33) | (n= 40) | |
MaHRb | 66% | 28% | 42% | 38% |
(95% CI) | (59-74) | (18-40) | (26-61) | (23-54) |
CHRb | 47% | 17% | 21% | 33% |
(95% CI) | (40-56) | (10-28) | (9-39) | (19-49) |
NELb | 19% | 11% | 21% | 5% |
(95% CI) | (13-26) | (5-20) | (9-39) | (1-17) |
MCyRc | 39% | 28% | 52% | 70% |
(95% CI) | (31-47) | (18-40) | (34-69) | (54-83) |
CCyR | 32% | 17% | 39% | 50% |
(95% CI) | (25-40) | (10-28) | (23-58) | (34-66) |
a Wyniki odnotowane po zastosowaniu zalecanej dawki początkowej wynoszącej 140 mg raz na dobę (patrz punkt 4.2).
b Kryteria odpowiedzi hematologicznej (wszystkie potwierdzone po 4 tygodniach): Duża odpowiedź hematologiczna: (MaHR - Major hematologic response) = całkowita odpowiedź hematologiczna (CHR - complete haematologic response) + brak objawów białaczki (NEL - no evidence of leukemia).
CHR: Krwinki białe (WBC) ≤ GGN laboratoryjnej w danej instytucji, ANC ≥ 1000/mm3 płytki krwi ≥ 100 000/mm3, brak blastów lub promielocytów w krwi obwodowej, liczba blastów w szpiku kostnym ≤ 5%, < 5% mielocytów oraz metamielocytów w krwi obwodowej, < 20% granulocytów zasadochłonnych w krwi obwodowej oraz brak ognisk białaczkowych poza szpikiem.
NEL: te same kryteria jak w przypadku CHR, ale ANC ≥ 500/mm3 i < 1000/mm3, lub płytki krwi ≥ 20000/mm3 i ≤ 100000/mm3.
c MCyR obejmuje zarówno odpowiedź całkowitą (0% Ph+ metaphases) jak i częściową (> 0%-35%). CI = przedział ufności; GGN = górna granica normy.
U pacjentów z CML w fazie akceleracji leczonych schematem 140 mg raz na dobę nie osiągnięto mediany MaHR i mediany całkowitego przeżycia, a mediana PFS wynosiła 25 miesięcy.
U pacjentów z mieloblastyczną postacią przełomu blastycznego leczonych schematem 140 mg raz na dobę mediana MaHR wynosiła 8 miesięcy, mediana PFS wynosiła 4 miesiące, a mediana całkowitego przeżycia 8 miesięcy. U pacjentów z limfoblastyczną postacią przełomu blastycznego
leczonych zgodnie ze schematem 140 mg raz na dobę mediana MaHR wynosiła 5 miesięcy, mediana PFS wynosiła 5 miesięcy, a mediana całkowitego przeżycia wynosiła 11 miesięcy.
U pacjentów z Ph+ ALL leczonych schematem 140 mg raz na dobę mediana MaHR wynosiła 5 miesięcy, mediana PFS wynosiła 4 miesiące, a mediana całkowitego przeżycia 7 miesięcy.
Dzieci i młodzież
Dzieci i młodzież z CML
Spośród 130 pacjentów z CML w fazie przewlekłej (CML CP) leczonych w dwóch badaniach z udziałem dzieci i młodzieży: w badaniu I fazy prowadzonym metodą otwartej próby bez randomizacji, mającym na celu ustalenie właściwej dawki leku, oraz w badaniu II fazy prowadzonym metodą otwartej próby bez randomizacji, 84 pacjentów (wyłącznie z badania II fazy) to pacjenci z nowo rozpoznaną CML CP, a 46 pacjentów (17 z badania I fazy i 29 z badania II fazy) z nietolerancją lub opornością na wcześniejsze leczenie imatynibem. Dziewięćdziesięcioro siedmioro ze 130 dzieci i młodzieży z CML CP leczono dazatynibem w tabletkach w dawce 60 mg/m2 pc. raz na dobę (dawka maksymalna 100 mg raz na dobę u pacjentów z dużą powierzchnią ciała). Pacjenci byli leczeni do chwili wystąpienia progresji choroby lub nieakceptowalnej toksyczności.
Kluczowymi punktami końcowymi dotyczącymi skuteczności były: całkowita odpowiedź cytogenetyczna (CCyR), większa odpowiedź cytogenetyczna (MCyR) i większa odpowiedź molekularna (MMR). Wyniki przedstawiono w Tabeli 15.
Tabela 15.: Wyniki skuteczności produktu Dasatinib Alvogen u dzieci i młodzieży z CML-CP Łączna odpowiedź w czasie uwzględniając minimalny okres obserwacji
3 miesiące | 6 miesięcy | 12 miesięcy | 24 miesiące | |
CCyR | ||||
(95% CI) | ||||
Nowo zdiagnozowani | 43,1% | 66,7% | 96,1% | 96,1% |
(N = 51)a | (29,3; 57,8) | (52,1; 79,2) | (86,5; 99,5) | (86,5; 99,5) |
Leczeni wcześniej imatynibem | 45,7% | 71,7% | 78,3% | 82,6% |
(N = 46)b | (30,9; 61,0) | (56,5; 84,0) | (63,6; 89,1) | (68,6; 92,2) |
MCyR | ||||
(95% CI) | ||||
Nowo zdiagnozowani | 60,8% | 90,2% | 98,0% | 98,0% |
(N = 51)a | (46,1; 74,2) | (78,6; 96,7) | (89,6; 100) | (89,6; 100) |
Leczeni wcześniej imatynibem | 60,9% | 82,6% | 89,1% | 89,1% |
(N = 46)b | (45,4; 74,9) | (68,6; 92,2) | (76,4; 96,4) | (76,4; 96,4) |
MMR | ||||
(95% CI) | ||||
Nowo zdiagnozowani | 7,8% | 31,4% | 56,9% | 74,5% |
(N = 51)a | (2,2; 18,9) | (19,1; 45,9) | (42,2; 70,7) | (60,4; 85,7) |
Leczeni wcześniej imatynibem | 15,2% | 26,1% | 39,1% | 52,2% |
(N = 46)b | (6,3; 28,9) | (14,3; 41,1) | (25,1; 54,6) | (36,9; 67,1) |
a Pacjenci z badania II fazy z udziałem dzieci i młodzieży, dotyczącego nowo rozpoznanej CML CP otrzymujący produkt leczniczy w postaci tabletek doustnych
b Pacjenci z badań I fazy i II fazy z udziałem dzieci i młodzieży, dotyczących CML CP, z opornością lub nietolerancją imatynibu, otrzymujący produkt leczniczy w postaci tabletek doustnych
W badaniu z udziałem dzieci i młodzieży I fazy po co najmniej 7 latach obserwacji, u 17 pacjentów z CML CP z opornością lub nietolerancją imatynibu, mediana czasu trwania PFS wynosiła 53,6 miesiąca, a wskaźnik OS wynosił 82,4%.
W badaniu z udziałem dzieci i młodzieży II fazy, u pacjentów otrzymujących produkt leczniczy w postaci tabletek, oszacowany wskaźnik 24-miesięcznych PFS wśród 51 pacjentów z nowo rozpoznaną CML CP wynosił 94,0% (82,6; 98,0), a wśród 29 pacjentów z CML CP z opornością lub nietolerancją imatynibu wynosił 81,7% (61,4; 92,0). Po 24 miesiącach obserwacji wskaźnik OS u pacjentów z nowo rozpoznaną chorobą wynosił 100%, a u pacjentów z opornością lub nietolerancją imatynibu wynosił 96,6%.
W badaniu z udziałem dzieci i młodzieży II fazy u 1 pacjenta z nowo rozpoznaną chorobą i u
2 pacjentów z opornością lub nietolerancją imatynibu nastąpiła progresja choroby do CML w fazie blastycznej.
U 33 dzieci i młodzieży z nowo rozpoznaną CML CP stosowano dazatynib w postaci proszku do sporządzania zawiesiny doustnej w dawce 72 mg/m2 pc. Ta dawka odpowiada ekspozycji mniejszej o 30% w porównaniu z zalecaną dawką (patrz punkt 5.2 Charakterystyki Produktu Leczniczego w postaci proszku do sporządzania zawiesiny doustnej). U tych pacjentów wskaźnik CCyR wynosił 87,9% [95% CI: (71,8-96,6)], a wskaźnik MMR 45,5% [95% CI: (28,1-63,6)] po 12 miesiącach.
Wśród dzieci i młodzieży z CML CP leczonych dazatynibem, którzy wcześniej otrzymywali imatynib, mutacjami wykrywanymi pod koniec leczenia były: T315A, E255K i F317L. Jednak przed leczeniem mutacje E255K i F317L również były wykrywane. U pacjentów z nowo rozpoznaną CML CP nie wykryto żadnych mutacji pod koniec leczenia.
Dzieci i młodzież z ALL
Skuteczność dazatynibu w skojarzeniu z chemioterapią oceniano w badaniu głównym z udziałem dzieci powyżej jednego roku życia i młodzieży z nowo rozpoznaną Ph+ ALL.
W tym wieloośrodkowym badaniu II fazy, z historyczną grupą kontrolną oceniano dazatynib podawany jako uzupełnienie chemioterapii standardowej; 106 dzieciom i młodzieży z nowo rozpoznaną Ph+ ALL, z czego 104 pacjentów miało potwierdzoną Ph+ ALL; podawano dazatynib w dawce dobowej 60 mg/m2 w sposób ciągły przez maksymalnie 24 miesiące, w skojarzeniu z chemioterapią. Osiemdziesięciu dwóch pacjentów otrzymywało wyłącznie dazatynib w postaci tabletek, a 24 pacjentów przynajmniej raz przyjęło dazatynib w postaci proszku do sporządzania zawiesiny doustnej, z czego 8 pacjentów stosowało wyłącznie dazatynib w postaci proszku do sporządzania zawiesiny doustnej. Schemat chemioterapii podstawowej był taki sam jak w badaniu AIEOP-BFM ALL 2000 (protokół standardowej chemioterapii wielolekowej). Głównym punktem końcowym do oceny skuteczności był odsetek 3-letnich przeżyć wolnych od zdarzeń (ang. event- free survival, EFS), który wynosił 65,5% (55,5; 73,7).
Odsetek ujemnych wyników badania w kierunku minimalnej choroby resztkowej (ang, minimal residual disease, MRD) oceniany na podstawie rearanżacji genów Ig/TCR wyniósł 71,7% do końca konsolidacji u wszystkich leczonych pacjentów. Kiedy odsetek ten określono u 85 pacjentów z możliwymi do oceny wynikami badania Ig/TCR, jego szacunkowa wartość wyniosła 89,4%.
Odsetek ujemnych wyników badania w kierunku minimalnej choroby resztkowej na koniec indukcji i konsolidacji mierzony metodą cytometrii przepływowej wyniósł odpowiednio 66,0% i 84,0%.
Właściwości farmakokinetyczne dazatynibu oceniano w grupie 229 zdrowych, dorosłych ochotników oraz w grupie 84 pacjentów.
Wchłanianie
Dazatynib jest szybko wchłaniany po podaniu doustnym u pacjentów; stężenie maksymalne osiąga po 0,5-3 godzinach. Po podaniu doustnym, zwiększenie średniej ekspozycji (AUCX) jest w przybliżeniu proporcjonalny do zwiększenia dawki, w zakresie dawek od 25 mg do 120 mg dwa razy na dobę. Średni końcowy okres półtrwania dazatynibu u pacjentów wynosił w przybliżeniu 5 - 6 godzin.
Dane uzyskane u zdrowych ochotników po podaniu jednorazowej dawki 100 mg dazantynibu 30 minut po posiłku bogatotłuszczowym wykazały 14% wzrost średniej wartości AUC dazatynibu. Ubogotłuszczowy posiłek podany 30 minut przed dawką dazatynibu powodował 21% wzrost średniej wartości AUC dazatynibu. Obserwowany wpływ pożywienia nie zmienia ekspozycji w sposób klinicznie istotny.
Dystrybucja
Objętość dystrybucji dazatynibu u pacjentów jest duża (2505 l, współczynnik zmienności (CV% 93%) co wskazuje, że lek ten przenika w znacznym stopniu do przestrzeni pozanaczyniowej. Z badań in vitro wynika, że dazantynib w klinicznie istotnych stężeniach wiąże się z białkami osocza w około 96%.
Metabolizm
U ludzi dazatynib jest w znacznym stopniu metabolizowany, z udziałem licznych enzymów. Po podaniu zdrowym ochotnikom 100 mg dazatynibu znakowanego węglem [14C], dazatynib w postaci niezmienionej stanowił 29% z całości radioaktywnej krążącej w osoczu. Stężenia metabolitów w osoczu oraz ich aktywność oznaczana in vitro wskazują, że metabolity nie mogą odgrywać istotnej roli w obserwowanych właściwościach farmakologicznych dazatynibu. Głównym enzymem odpowiedzialnym za metabolizm dazatynibu jest CYP3A4.
Eliminacja
Średni końcowy okres półtrwania dazatynibu wynosi 3 godziny do 5 godzin. Średni pozorny klirens po podaniu doustnym wynosi 363,8 l/godz. (CV% 81,3%).
Dazatynib wydalany jest w przeważającej części z kałem, głównie w postaci metabolitów. Po doustnym podaniu pojedynczej dawki dazatynibu znakowanego węglem [14C] około 89% tej dawki zostało wydalone w ciągu 10 dni, przy 4% radioaktywności stwierdzanej w moczu i 85% stwierdzanej w kale. Dazatynib w postaci niezmienionej stanowił 0,1% całości wydalanej z moczem i 19% całości wydalanej w kale, przy czym pozostałą część stanowiły metabolity.
Zaburzenia czynności wątroby i nerek
Wpływ zaburzeń czynności wątroby na farmakokinetykę dazatynibu po podaniu jednorazowym oceniono u 8 osób z umiarkowanymi zaburzeniami czynności wątroby, którym podano dawkę 50 mg oraz u 5 osób z ciężkimi zaburzeniami czynności wątroby, którym podawano dawkę 20 mg i porównywano z odpowiednio dobranymi zdrowymi osobami, którym podano dawkę 70 mg dazatynibu. U pacjentów z umiarkowanymi zaburzeniami czynności wątroby w porównaniu do osób z prawidłową czynnością wątroby średnie wartości Cmax i AUC dazatynibu, dostosowane do dawki 70 mg, były zmniejszone odpowiednio o 47% i 8%. U osób z ciężkimi zaburzeniami czynności wątroby w porównaniu do osób z prawidłową czynnością wątroby średnie wartości Cmax i AUC dazatynibu, dostosowane do dawki 70 mg, były zmniejszone odpowiednio o 43% i 28% (patrz punkty 4.2 i 4.4).
Dazatynib i jego metabolity są w znikomym stopniu wydalane przez nerki. Dzieci i młodzież
Farmakokinetykę dazatynibu oceniano u 104 dzieci i młodzieży z białaczką lub guzami litymi (72 z nich otrzymywało produkt w postaci tabletek, a 32 w postaci proszku do sporządzania zawiesiny doustnej).
W badaniu farmakokinetyki z udziałem dzieci i młodzieży ekspozycja na dazatynib z normalizacją dawki (Cavg, Cmin i Cmax) wydawała się być zbliżona u 21 pacjentów z przewlekłą białaczką szpikową w fazie przewlekłej (CP CML) oraz 16 pacjentów z Ph+ ALL.
Farmakokinetykę dazatynibu w postaci tabletek oceniano u 72 dzieci i młodzieży z nawrotową lub oporną na leczenie białaczką lub z guzami litymi. Lek podawano w dawkach doustnych od 60 do 120 mg/m2 pc. raz na dobę i od 50 do 110 mg/m2 pc. dwa razy na dobę. Połączono dane z dwóch badań, które wykazały, że dazatynib był szybko wchłaniany. Średnią wartość Tmax obserwowano po upływie od 0,5 do 6 godzin, a średni czas półtrwania wynosił od 2 do 5 godzin w przypadku wszystkich poziomów dawkowania i wszystkich grup wiekowych. Farmakokinetyka dazatynibu była proporcjonalna do dawki, stwierdzono zależne od dawki zwiększenie ekspozycji u dzieci i młodzieży. Nie stwierdzono istotnych różnic dotyczących farmakokinetyki dazatynibu pomiędzy
dziećmi a młodzieżą. Średnia geometryczna wartości Cmax, AUC (0-T) i AUC (INF) znormalizowanych względem dawki dazatynibu okazała się podobna u dzieci i młodzieży po zastosowaniu różnych poziomów dawkowania. Na podstawie symulacji opartej na modelu farmakokinetyki populacyjnej przewidziano, że zalecane dawkowanie zależne od przedziałów masy ciała, opisane dla tabletek w punkcie 4.2, powinno zapewnić podobną ekspozycję, jak dawka tabletek wynosząca 60 mg/m2 pc. Dane te należy wziąć pod uwagę, jeśli u pacjentów planuje się zmianę tabletek na proszek do sporządzania zawiesiny doustnej lub odwrotnie.
Profil bezpieczeństwa stosowania dazatynibu oceniano w nieklinicznych badaniach in vitro oraz in vivo przeprowadzonych na myszach, szczurach, małpach oraz królikach.
Toksyczność pierwotna wystąpiła w układzie pokarmowym, krwiotwórczym i limfatycznym. U szczurów i małp czynnikiem ograniczającym dawkę była toksyczność dotycząca układu pokarmowego, ponieważ u obu tych gatunków jelito było organem docelowym. U szczurów zmiany parametrów krwinek czerwonych o nasileniu nieznacznym lub umiarkowanymtowarzyszyły zmianom w obrębie szpiku kostnego; podobne zmiany, lecz z mniejszą częstością, obserwowano u małp. Toksyczność dotycząca układu limfatycznego obejmowała u szczurów zmniejszenie liczby limfocytów w węzłach chłonnych, śledzionie oraz grasicy i zmniejszeniu się masy organów limfatycznych. Zmiany dotyczące układu pokarmowego, krwiotwórczego i limfatycznego były odwracalne i ustępowały po zakończeniu leczenia.
Zmiany w nerkach obserwowane u małp leczonych aż do 9 miesięcy ograniczały się do zwiększenia śródmiąższowej mineralizacji nerek. W badaniu przeprowadzonym na małpach, w którym oceniano ostrą toksyczność, po doustnym podaniu pojedynczej dawki leku wystąpił krwotok skórny. Podobnych objawów nie obserwowano ani w badaniach przeprowadzonych na małpach ani na szczurach, w których podawano wielokrotne dawki leku. U szczurów dazatynib hamował agregację płytek krwi in vitro i wydłużał czas krwawienia in vivo, lecz nie powodował samoistnych krwotoków.
Działanie dazatynibu na kanał potasowy hERG oraz na włókna. Purkinje'go stwierdzone w badaniach in vitro wskazuje na możliwość wydłużania czasu repolaryzacji komór (odstęp QT). Jednakże, w badaniu przeprowadzonym in vivo po podaniu jednorazowej dawki dazatynibu małpom, z zachowanym stanem świadomości, u których uzyskiwano zapis EKG metodą telemetryczną, nie stwierdzono zmian odstępu QT ani innych zmian w zapisie EKG.
Nie wykazano mutagenności dazatynibu w badaniach przeprowadzonych in vitro na komórkach bakteryjnych (test Amesa). Nie wykazano także genotoksyczności dazatynibu w teście mikrojądrowym przeprowadzonym in vivo u szczurów. Dazatynib wywoływał in vitro efekt klastogenny na dzielące się komórki jajnika chomika chińskiego (ang. Chinese Hamster Ovary, CHO).
Dazatynib nie wpływał na płodność samców i samic w standardowych badaniach płodności i rozwoju wczesnopłodowego u szczurów, ale w dawkach podobnych do stosowanych u ludzi, indukował obumieranie płodów. W badaniach dotyczących rozwoju embrionalno-płodowego, dazatynib powodował również obumieranie zarodków oraz związaną z tym mniejszą liczebność miotów u szczurów, jak też zmiany kośćca płodu u szczurów i u królików. Zmiany te występowały po dawkach, które nie powodowały toksyczności u matek wskazując, że dazatynib wykazuje selektywną toksyczność w okresie od implantacji do zakończenia organogenezy. Nieznany jest wpływ dazatynibu na płodność kobiet i mężczyzn.
U myszy dazatynib powodował immunosupresję, która była zależna od dawki i którą można było skutecznie kontrolować zmniejszając dawkę i (lub) zmieniając schemat dawkowania. Dazatynib wykazywał działanie fototoksyczne w badaniach przeprowadzonych in vitro na mysich fibroblastach, w których oceniano fototoksyczność w oparciu o wychwyt neutralnego promieniowania czerwonego. Stwierdzono, że dazatynib nie ma działania fototoksycznego in vivo po jednorazowym podaniu samicom bezwłosych myszy dawek powodujących narażenie do 3 razy większe niż narażenie występujące u ludzi po podaniu zalecanych dawek leczniczych (na podstawie AUC).
W dwuletnim badaniu oceniającym rakotwórczość dazatynib podawano doustnie szczurom w dawkach 0,3; 1 i 3 mg/kg/dobę. Zastosowanie największej dawki było związane z poziomem ekspozycji w osoczu (AUC) zasadniczo równoważnym ekspozycji u ludzi podczas stosowania dawki początkowej z zalecanego zakresu, wynoszącego od 100 mg do 140 mg na dobę.
Zaobserwowano statystycznie istotny wzrost łącznej częstości występowania raka płaskonabłonkowego oraz brodawczaków w macicy i szyjce macicy u samic otrzymujących duże dawki, a także gruczolaka prostaty u samców otrzymujących małe dawki. Znaczenie wyników badań rakotwórczości prowadzonych na szczurach dla ludzi nie jest znane.
Rdzeń tabletki
Celuloza mikrokrystaliczna (typ 101) Celuloza mikrokrystaliczna (typ 102) Laktoza jednowodna Hydroksypropyloceluloza Kroskarmeloza sodowa
Magnezu stearynian
Otoczka tabletki Hypromeloza
Tytanu dwutlenek (E171) Trietylu cytrynian
Nie dotyczy.
30 miesięcy
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Dasatinib Alvogen 20 mg, Dasatinib Alvogen 50 mg i Dasatinib Alvogen 70 mg, tabletki powlekane
Blistry OPA/Aluminium/PVC/Aluminium zawierające 30, 56 lub 60 tabletek powlekanych.
Butelki HDPE zawierająca środek pochłaniający wilgoć (żel krzemionkowy), z nakrętką z polipropylenu z zabezpieczeniem przed dostępem dzieci, zawierające 60 tabletek powlekanych.
Dasatinib Alvogen 80 mg, Dasatinib Alvogen 100 mg i Dasatinib Alvogen 140 mg, tabletki powlekane
Blistry OPA/Aluminium/PVC/Aluminium zawierające 30 lub 60 tabletek powlekanych.
Butelki HDPE zawierająca środek pochłaniający wilgoć (żel krzemionkowy), z nakrętką z polipropylenu z zabezpieczeniem przed dostępem dzieci, zawierające 30 tabletek powlekanych.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Tabletki powlekane złożone są z rdzenia pokrytego otoczką, która zapobiega narażeniu personelu medycznego na substancje czynną. Zaleca się stosowanie rękawiczek lateksowych lub nitrylowych do prawidłowego usunięcia produktu leczniczego w przypadku zetknięcia się z tabletkami, które zostały przypadkowo rozkruszone lub połamane, w celu zmniejszenia ryzyka narażenia na kontakt ze skórą.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Alvogen Pharma Trading Europe EOOD 86 Bulgaria blvd.,
1680 Sofia, Bułgaria
Dasatinib Alvogen, 20 mg, tabletki powlekane:
Dasatinib Alvogen, 50 mg, tabletki powlekane:
Dasatinib Alvogen, 70 mg, tabletki powlekane:
Dasatinib Alvogen, 80 mg, tabletki powlekane:
Dasatinib Alvogen, 100 mg, tabletki powlekane:
Dasatinib Alvogen, 140 mg, tabletki powlekane:
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:








