







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
gdy u pacjentów nie udaje się odpowiednio kontrolować stężenia lipidów samą statyną
gdy pacjent już otrzymuje statynę i ezetymib.
Homozygotyczna hipercholesterolemia rodzinna
Axocar wskazany jest do stosowania wraz dietą u pacjentów z homozygotyczną hipercholesterolemią rodzinną. Pacjenci mogą także stosować inne, dodatkowe metody leczenia (np. aferezę lipoprotein o małej gęstości [LDL]).
Dawkowanie i sposób podawania Dawkowanie
Przeciwwskazania
Nadwrażliwość na substancje czynne lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Ciąża i laktacja (patrz punkt 4.6).
Czynna choroba wątroby lub utrzymujące się, niewyjaśnione zwiększenie aktywności aminotransferaz w surowicy.
Jednoczesne podawanie silnych inhibitorów CYP3A4 (substancji powodujących co najmniej około pięciokrotne zwiększenie wartości AUC) (np. itrakonazol, ketokonazol, pozakonazol, worykonazol, erytromycyna, klarytromycyna, telitromycyna, inhibitory proteazy wirusa HIV (np. nelfinawir), boceprewir, telaprewir, nefazodon oraz leki zawierające kobicystat) (patrz punkty 4.4 i 4.5).
Jednoczesne podawanie gemfibrozylu, cyklosporyny lub danazolu (patrz punkty 4.4 i 4.5).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
osoby w podeszłym wieku (wiek ≥ 65 lat),
płeć żeńska,
zaburzenia czynności nerek,
nieleczona niedoczynność tarczycy,
indywidualny lub rodzinny wywiad świadczący o dziedzicznych zaburzeniach układu mięśniowego,
wystąpienie w przeszłości uszkadzającego mięśnie działania statyn lub fibratów,
uzależnienie od alkoholu.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Axocar, 10 mg + 10 mg, tabletki Axocar, 10 mg + 20 mg, tabletki Axocar, 10 mg + 40 mg, tabletki Axocar, 10 mg + 80 mg, tabletki
Axocar, 10 mg + 10 mg, tabletki: Każda tabletka zawiera 10 mg ezetymibu oraz 10 mg symwastatyny. Axocar, 10 mg + 20 mg, tabletki: Każda tabletka zawiera 10 mg ezetymibu oraz 20 mg symwastatyny. Axocar, 10 mg + 40 mg, tabletki: Każda tabletka zawiera 10 mg ezetymibu oraz 40 mg symwastatyny. Axocar, 10 mg + 80 mg, tabletki: Każda tabletka zawiera 10 mg ezetymibu oraz 80 mg symwastatyny.
Substancje pomocnicze o znanym działaniu:
Axocar, 10 mg + 10 mg, tabletki: Każda tabletka 10 mg + 10 mg zawiera 51,6 mg laktozy jednowodnej. Axocar, 10 mg + 20 mg, tabletki: Każda tabletka 10 mg + 20 mg zawiera 113,3 mg laktozy jednowodnej. Axocar, 10 mg + 40 mg, tabletki: Każda tabletka 10 mg + 40 mg zawiera 236,5 mg laktozy jednowodnej. Axocar, 10 mg + 80 mg, tabletki: Każda tabletka 10 mg + 80 mg zawiera 483,0 mg laktozy jednowodnej.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka.
Axocar, 10 mg+10 mg, to jasnobrązowe, nakrapiane, okrągłe tabletki o średnicy 6 mm, obustronnie wypukłe, gładkie po jednej stronie, z oznaczeniem „511” po drugiej stronie.
Axocar, 10 mg+20 mg, to jasnobrązowe, nakrapiane, okrągłe tabletki o średnicy 8 mm, obustronnie wypukłe, gładkie po jednej stronie, z oznaczeniem „512” po drugiej stronie.
Axocar, 10 mg+40 mg, to jasnobrązowe, nakrapiane, okrągłe tabletki o średnicy 10 mm, obustronnie wypukłe, gładkie po jednej stronie, z oznaczeniem „513” po drugiej stronie.
Axocar, 10 mg+80 mg, to jasnobrązowe, nakrapiane tabletki w kształcie kapsułki o wymiarach 17,5x7,55 mm, obustronnie wypukłe, gładkie po jednej stronie, z oznaczeniem „515” po drugiej stronie z oznaczeniem „515” po jednej stronie.
Zapobieganie wystąpieniu incydentów sercowo-naczyniowych
Axocar jest wskazany do stosowania, w celu zmniejszenia ryzyka wystąpienia incydentów sercowo- naczyniowych (patrz punkt 5.1) u pacjentów z chorobą wieńcową (ang. CHD, Coronary Heart Disease)
i ostrym zespołem wieńcowym w wywiadzie (ang. ACS, Acute Coronary Syndrome), zarówno u pacjentów leczonych statyną jak i u pacjentów którzy nie byli wcześniej leczeni statyną.
Hipercholesterolemia
Axocar wskazany jest do stosowania wraz z dietą u pacjentów z pierwotną hipercholesterolemią (heterozygotyczną rodzinną lub nierodzinną) lub mieszaną hiperlipidemią, u których wskazane jest zastosowanie produktu leczniczego złożonego:
Hipercholesterolemia
Pacjent powinien stosować odpowiednią dietę zmniejszającą stężenie lipidów we krwi. Należy kontynuować przestrzeganie tej diety w okresie stosowania produktu leczniczego Axocar.
Produkt leczniczy Axocar przyjmuje się doustnie. Zakres dawkowania produktu leczniczego Axocar wynosi od 10 mg + 10 mg na dobę do 10 mg + 80 mg na dobę, wieczorem. W przypadkach klinicznych, w których nie jest możliwe ustalenie optymalnej dawki ezetymibu i symwastatyny stosując produkt Axocar można zastosować inne postaci oraz inną moc ezetymibu i symwastatyny dostępne na rynku. Zwykle podaje się
10 mg + 20 mg na dobę lub 10 mg + 40 mg na dobę, jednorazowo, wieczorem. Dawkę 10 mg + 80 mg zaleca się jedynie u pacjentów z ciężką hipercholesterolemią i zwiększonym ryzykiem wystąpienia powikłań ze strony układu sercowo- naczyniowego, u których nie uzyskano celu terapeutycznego podając lek w niższej dawce, i kiedy spodziewane korzyści przeważają nad potencjalnym ryzykiem (patrz punkty 4.4 i 5.1).
Rozpoczynając leczenie lub dostosowując dawkę dla danego pacjenta, należy uwzględnić takie parametry jak stężenie frakcji cholesterolu LDL (LDL-C), ryzyko rozwoju choroby wieńcowej oraz odpowiedź na dotychczas stosowane leczenie produktami leczniczymi zmniejszającymi stężenie cholesterolu.
Dawkę produktu leczniczego Axocar należy ustalać dla każdego pacjenta indywidualnie, w oparciu o ustaloną skuteczność różnych dawek produktu leczniczego Axocar (patrz punkt 5.1, Tabela 2) i odpowiedź na dotychczas stosowane leczenie produktami leczniczymi zmniejszającymi stężenie cholesterolu. W razie konieczności należy dostosowywać dawkowanie w odstępach nie krótszych niż 4 tygodnie. Axocar może być przyjmowany niezależnie od posiłków. Tabletki nie należy dzielić.
Pacjenci z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie
W badaniu dotyczącym zmniejszenia ryzyka incydentów sercowo-naczyniowych (ang. IMPROVE-IT, IMProved Reduction of Outcomes: Vytorin Efficacy International Trial), początkowa dawka wynosiła 10 mg + 40 mg raz na dobę, podawana wieczorem. Dawkę 10 mg + 80 mg zaleca się jedynie wtedy, kiedy spodziewane korzyści przeważają nad potencjalnym ryzykiem.
Homozygotyczna hipercholesterolemia rodzinna
Zalecana, początkowa dawka produktu leczniczego Axocar u pacjentów z homozygotyczną hipercholesterolemią rodzinną wynosi 10 mg + 40 mg na dobę, wieczorem. Dawkę 10 mg + 80 mg zaleca się jedynie wtedy, kiedy spodziewane korzyści przeważają nad potencjalnym ryzykiem (patrz powyżej; punkty
U pacjentów z homozygotyczną hipercholesterolemią rodzinną podczas jednoczesnego stosowania lomitapidu z produktem leczniczym Axocar w dawkach przekraczających 10 mg + 40 mg na dobę (patrz punkty 4.2, 4.4 i 4.5).
Miopatia i (lub) rabdomioliza
Po wprowadzeniu ezetymibu do obrotu zgłaszano przypadki występowania miopatii i rabdomiolizy. Większość pacjentów, u których wystąpiła rabdomioliza, przyjmowała statyny jednocześnie z ezetymibem. Rabdomioliza występowała jednak bardzo rzadko podczas stosowania ezetymibu w monoterapii oraz bardzo rzadko podczas stosowania ezetymibu z innymi lekami, których podawanie wiąże się ze zwiększeniem ryzyka wystąpienia rabdomiolizy.
Axocar zawiera symwastatynę. Symwastatyna, podobnie jak inne inhibitory reduktazy HMG-CoA, wywołuje niekiedy miopatię objawiającą się bólami mięśni, tkliwością lub osłabieniem z towarzyszącym zwiększeniem aktywności kinazy kreatynowej (CK) do ponad 10 razy powyżej górnej granicy normy (GGN) uznanej za prawidłową (ang. ULN, upper limit of normal). Miopatia czasem przekształca się w rabdomiolizę z lub bez ostrej niewydolności nerek spowodowanej mioglobinurią. Odnotowano bardzo rzadko przypadki zgonów. Duża aktywność inhibitorów reduktazy HMG-CoA w osoczu zwiększa ryzyko wystąpienia miopatii (np. podwyższone stężenia symwastatyny i kwasu symwastatyny w osoczu), którego przyczyną częściowo mogą być wywołujące interakcje produkty lecznicze, które zakłócają metabolizm symwastatyny i (lub) szlaki transporterów (patrz punkt 4.5).
Tak jak w przypadku innych produktów leczniczych z grupy inhibitorów reduktazy HMG-CoA, ryzyko wystąpienia miopatii i (lub) rabdomiolizy zależne jest od wielkości dawki symwastatyny. W badaniu klinicznym, w którym 41 413 pacjentów stosowało symwastatynę, z czego 24 747 (około 60%) osób włączono do badań z okresem obserwacji kontrolnej trwającym średnio przynajmniej 4 lata, częstość występowania miopatii wynosiła około 0,03%, 0,08% i 0,61%, odpowiednio dla dawek 20, 40 i 80 mg na dobę. W badaniach tych pacjenci byli starannie monitorowani, a niektóre produkty lecznicze, mogące wchodzić w interakcje ze stosowanym produktem leczniczym, zostały wykluczone.
W badaniu klinicznym, w którym pacjentów po przebytym zawale mięśnia sercowego leczono symwastatyną w dawce 80 mg na dobę (średni okres obserwacji kontrolnej – 6,7 roku), częstość występowania miopatii wyniosła około 1,0% w porównaniu z 0,02% u pacjentów przyjmujących lek w dawce 20 mg na dobę. Około połowę z tych przypadków miopatii stwierdzono w pierwszym roku leczenia. Częstość występowania miopatii w każdym kolejnym roku leczenia wyniosła około 0,1%. (Patrz punkty 4.8 i 5.1).
Ryzyko wystąpienia miopatii jest większe u pacjentów stosujących ezetymib i symwastatynę w dawce 10 mg
+ 80 mg w porównaniu z innymi rodzajami leczenia opartymi na statynach o podobnej skuteczności
w obniżaniu stężenia cholesterolu LDL. Dlatego produkt leczniczy Axocar w dawce 10 mg + 80 mg należy stosować jedynie u pacjentów z ciężką hipercholesterolemią i z wysokim ryzykiem powikłań sercowo- naczyniowych, u których cel leczenia nie został osiągnięty za pomocą niższych dawek i u których spodziewane korzyści przewyższają potencjalne ryzyko. U pacjentów przyjmujących produkt leczniczy Axocar w dawce 10 mg + 80 mg wymagających podania produktu leczniczego wchodzącego w interakcję należy zastosować niższą dawkę produktu leczniczego Axocar lub alternatywny schemat leczenia oparty na statynie o mniejszym potencjale interakcji z innymi produktami leczniczymi (patrz poniżej Środki zaradcze zmniejszające ryzyko wystąpienia miopatii spowodowanej interakcją produktów leczniczych oraz punkty 4.2, 4.3 i 4.5).
W badaniu IMPROVE-IT 18 144 pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym
w wywiadzie przydzielano w sposób losowy do grupy otrzymującej ezetymib i symawastnynę w dawce 10 mg + 40 mg na dobę (n=9067) lub symwastatynę w dawce 40 mg na dobę (n=9077). Podczas okresu obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania miopatii w grupie otrzymującej ezetymib i symwastatynę oraz w grupie przyjmującej symwastatynę wynosiła odpowiednio 0,2% i 0,1%.
Miopatię zdefiniowano jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością kinazy kreatynowej (CK) w surowicy wynoszącej ≥ 10x GGN lub dwa kolejne epizody zwiększenia aktywności CK wynoszące ≥ 5 i < 10x GGN. Częstość występowania rabdomiolizy w grupie otrzymującej ezetymib
z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,1% i 0,2%. Rabdomiolizę zdefiniowano jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącej ≥ 10x GGN i objawami uszkodzenia nerek, 2 kolejne epizody zwiększenia
aktywności CK wynoszącej ≥ 5 i < 10x GGN z objawami uszkodzenia nerek lub CK ≥ 10 000 j.m./l bez objawów uszkodzenia nerek. (patrz punkt 4.8.).
W badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek randomizowano do grupy leczonej ezetymibem i symwastatyną w dawce 10 mg + 20 mg na dobę (n=4650) lub do grupy przyjmującej placebo (n=4620) (mediana okresu obserwacji 4,9 roku), częstość występowania miopatii wyniosła 0,2% wśród osób przyjmujących ezetymib i symwastatynę oraz 0,1% w grupie placebo. (Patrz punkt 4.8).
W badaniu klinicznym, w którym pacjentom obarczonym wysokim ryzykiem wystąpienia choroby sercowo- naczyniowej podawano symwastatynę w dawce 40 mg/dobę (mediana okresu obserwacji: 3,9 roku), częstość występowania miopatii u pacjentów narodowości innej niż chińska (n = 7367) oraz pacjentów narodowości chińskiej (n = 5468) wynosiła odpowiednio około 0,05% i 0,24%. Mimo, że populacja azjatycka w tym badaniu składała się wyłącznie z pacjentów narodowości chińskiej, należy zachować ostrożność przepisując produkt leczniczy Axocar pacjentom pochodzenia azjatyckiego i stosować u nich najniższą konieczną dawkę.
Zmniejszona aktywność białek transportowych
Zmniejszenie aktywności wątrobowych białek transportowych OATP może wzmagać ekspozycję ogólnoustrojową na kwas symwastatyny i zwiększać ryzyko wystąpienia miopatii i rabdomiolizy. Zmniejszenie aktywności może być skutkiem inhibicji przez leki wchodzące w interakcje (np. cyklosporynę) lub występować u pacjentów będących nosicielami wariantu genotypu SLCO1B1 c.521T> C.
U pacjentów posiadających allel (c.521T>C) genu SLCO1B1, kodujący białko o mniejszej aktywności OATP1B1, występuje wzmożona ekspozycja ogólnoustrojowa na kwas symwastatyny i zwiększone ryzyko miopatii. Bez uwzględnienia testów genetycznych, ryzyko wystąpienia miopatii wywołanej stosowaniem dużych dawek symwastatyny (80 mg) wynosi ok. 1% w populacji ogólnej. Na podstawie wyników badania SEARCH ustalono, iż u homozygotycznych nosicieli allelu C (zwanych CC) leczonych symwastatyną
w dawce 80 mg, ryzyko miopatii wynosi 15% w okresie 1 roku, natomiast u heterozygotycznych nosicieli allelu C (CT) ryzyko to wynosi 1,5%. Ryzyko w przypadku pacjentów z najczęściej występującym genotypem (TT) wynosiło 0,3% (patrz punkt 5.2). Jeżeli to możliwe, należy wziąć pod uwagę wykonanie badań genetycznych na obecność allelu C jako etapu oceny stosunku korzyści do ryzyka przed przepisaniem symwastatyny w dawce 80 mg danemu pacjentowi i unikać przepisywania dużych dawek pacjentom
z genotypem CC. Niemniej jednak, nieobecność wspomnianych alleli podczas genotypowania nie wyklucza wystąpienia miopatii.
Oznaczanie aktywności kinazy kreatynowej
Aktywność kinazy kreatynowej (CK) nie powinna być oznaczana po męczącym wysiłku lub jeśli istnieją jakiekolwiek inne, możliwe przyczyny powodujące zwiększenie aktywności CK, ponieważ może to spowodować trudności w interpretacji oznaczanej wartości. Jeżeli aktywność CK jest znacznie większa od górnej granicy normy (GGN) uznanej za prawidłową (> 5x GGN), należy w celu potwierdzenia wyników powtórzyć badanie po 5. a najpóźniej po 7. dniu.
Przed rozpoczęciem leczenia
Przed rozpoczęciem stosowania produktu leczniczego Axocar lub w przypadku zwiększenia dawki produktu leczniczego Axocar, należy poinformować wszystkich pacjentów o możliwości wystąpienia miopatii i zalecić szybkie zgłoszenie się do lekarza w razie odczuwania trudnych do wyjaśnienia bólów mięśni, ich nadmiernej wrażliwości na dotyk lub osłabienia.
Ostrożnie należy stosować u pacjentów z czynnikami predysponującymi do wystąpienia rabdomiolizy. Aby ustalić referencyjną wartość wyjściową, aktywność CK należy oznaczać przed rozpoczęciem leczenia
w następujących przypadkach:
W powyższych przypadkach należy rozważyć spodziewane korzyści leczenia i związane z tym ryzyko. Zalecane jest monitorowanie stanu zdrowia pacjenta. Jeśli w przeszłości u pacjenta wystąpił szkodliwy wpływ fibratów lub statyn na mięśnie, leczenie jakimkolwiek produktem leczniczym zawierającym statyny (jak produkt leczniczy Axocar) należy rozpocząć ostrożnie. Jeśli aktywność CK jest znacznie powyżej górnej granicy normy (GGN) uznanej za prawidłową (> 5x GGN), nie należy rozpoczynać leczenia.
Podczas leczenia
Jeśli u pacjenta, podczas stosowania produktu leczniczego Axocar, wystąpią bóle mięśni, osłabienie lub tkliwość uciskowa, należy zbadać aktywność CK. Należy przerwać leczenie, jeśli aktywność CK badana przy braku forsownego wysiłku, jest znacznie zwiększona (> 5x GGN). Należy rozważyć przerwanie leczenia, nawet jeśli aktywność CK wynosi < 5x GGN, ale niepożądane objawy ze strony mięśni są nasilone i powodują codzienny dyskomfort. Jeśli z jakichkolwiek powodów podejrzewana jest miopatia, należy przerwać leczenie.
Zgłoszono bardzo rzadkie przypadki wystąpienia immunozależnej miopatii martwiczej (ang. IMNM, Immune-mediated necrotizing myopathy) w trakcie leczenia statynami lub po jego zakończeniu. Cechy kliniczne IMNM to utrzymujące się osłabienie mięśni proksymalnych oraz zwiększona aktywność kinazy kreatynowej w surowicy, utrzymująca się mimo przerwania leczenia statynami (patrz punkt 4.8).
Jeśli objawy ze strony mięśni ustąpią i wartość CK powróci do normy, można rozważyć powtórne podanie produktu leczniczego Axocar lub powtórne podanie innego produktu leczniczego zawierającego statynę w najmniejszej skutecznej dawce, ściśle kontrolując leczenie.
U pacjentów, u których dawkę symwastatyny zwiększono do 80 mg, obserwowano częstsze występowanie miopatii (patrz punkt 5.1). Zaleca się okresowe oznaczanie stężenia CK jako badanie przydatne do identyfikacji subklinicznych przypadków miopatii. Nie ma jednak żadnej pewności, że pozwoli to uniknąć rozwoju miopatii.
Stosowanie produktu leczniczego Axocar należy przerwać na kilka dni przed planowanym, poważnym zabiegiem chirurgicznym lub jeśli konieczne jest podjęcie istotnego leczenia internistycznego lub chirurgicznego.
Środki zaradcze zmniejszające ryzyko wystąpienia miopatii spowodowanej interakcją produktów leczniczych (patrz także punkt 4.5)
Ryzyko miopatii i rabdomiolizy jest znacznie zwiększone podczas jednoczesnego stosowania ezetymibu
i symwastatyny z innymi silnymi inhibitorami CYP3A4 (takimi jak itrakonazol, ketokonazol, pozakonazol, worykonazol, erytromycyna, klarytromycyna, telitromycyna, inhibitory proteazy wirusa HIV
(np. nelfinawir), boceprewir, telaprewir, nefazodon i produkty lecznicze zawierające kobicystat), jak również cyklosporyna, danazol i gemfibrozyl. Stosowanie tych produktów leczniczych jest przeciwwskazane (patrz punkt 4.3).
Ze względu na to, że składnikiem produktu leczniczego Axocar jest symwastatyna, ryzyko miopatii i rabdomiolizy zwiększa się również podczas jednoczesnego stosowania innych fibratów, niacyny w dawkach zmniejszających stężenie lipidów (≥ 1 g na dobę) lub podczas jednoczesnego stosowania
amiodaronu, amlodypiny, werapamilu lub diltiazemu z niektórymi dawkami ezetymibu i symwastatyny (patrz punkty 4.2 i 4.5). Ryzyko miopatii, w tym rabdomiolizy może być zwiększone podczas jednoczesnego podawania kwasu fusydowego z ezetmibem i symwastatyną. U pacjentów z homozygotyczną hipercholesterolemią rodzinną to ryzyko może ulegać zwiększeniu w wyniku jednoczesnego stosowania ezetymibu i symwastatyny z lomitapidem (patrz punkt 4.5).
Przeciwwskazane jest jednoczesne stosowanie ezetymibu i symwastatyny z inhibitorami CYP3A4: itrakonazolem, ketokonazolem, pozakonazolem, worykonazolem, inhibitorami proteazy wirusa HIV (np.
nelfinawirem), boceprewirem, telaprewirem, erytromycyną, klarytromycyną, telitromycyną, nefazodonem i produktami leczniczymi zawierającymi kobicystat (patrz punkty 4.3 i 4.5). Jeśli konieczne jest leczenie silnymi inhibitorami CYP3A4 (substancjami powodującymi co najmniej około pięciokrotne zwiększenie
wartości AUC), należy w tym czasie przerwać stosowanie produktu leczniczego Axocar (i rozważyć podanie innej statyny). Należy ponadto zachować ostrożność podczas podawania produktu leczniczego Axocar
z innymi inhibitorami CYP3A4 o słabszym działaniu: flukonazolem, werapamilem, diltiazemem (patrz punkty 4.2 i 4.5). Należy unikać jednoczesnego przyjmowania produktu leczniczego Axocar i soku grejpfrutowego.
Symwastatyny nie wolno stosować jednocześnie z kwasem fusydowym podawanym ogólnoustrojowo lub w ciągu 7 dni po zakończeniu leczenia kwasem fusydowym. U pacjentów, u których podawanie ogólnoustrojowo kwasu fusydowego uważa się za konieczne, leczenie statynami należy przerwać na cały okres leczenia kwasem fusydowym. Istnieją doniesienia o przypadkach rabdomiolizy (także prowadzących do śmierci) wśród pacjentów leczonych kwasem fusydowym w skojarzeniu ze statynami (patrz punkt 4.5). Pacjentowi trzeba zalecić, aby w razie wystąpienia jakichkolwiek objawów w postaci osłabienia, bólu lub tkliwości mięśni, niezwłocznie zgłosił się do lekarza.
Leczenie statynami można wznowić po upływie 7 dni od daty podania ostatniej dawki kwasu fusydowego. W wyjątkowych okolicznościach, gdy konieczne jest podawanie ogólnoustrojowo kwasu fusydowego,
np. w ramach leczenia ciężkich zakażeń, jednoczesne stosowanie produktu leczniczego Axocar i kwasu fusydowego można rozważyć wyłącznie w przypadkach indywidualnych, pod ścisłym nadzorem lekarza.
Należy unikać podawania ezetymibu i symwastatyny w dawce przekraczającej 10 mg + 20 mg na dobę jednocześnie z niacyną w dawkach obniżających stężenie lipidów (≥1 g/dobę), chyba że istnieje przypuszczenie, że korzyści kliniczne przeważą nad zwiększonym ryzykiem wystąpienia miopatii (patrz punkty 4.2 i 4.5).
Rzadko obserwowano przypadki miopatii i (lub) rabdomiolizy, związane z przyjmowaniem inhibitorów reduktazy HMG-CoA jednocześnie z niacyną (kwasem nikotynowym) w dawkach modyfikujących stężenie lipidów (≥1 g/dobę), każdy lek z obydwu grup stosowany w monoterapii może spowodować rozwój miopatii.
W badaniu klinicznym (mediana okresu obserwacji: 3,9 roku) z udziałem pacjentów obarczonych wysokim ryzykiem wystąpienia choroby sercowo-naczyniowej, z dobrze kontrolowanym poziomem cholesterolu LDL, przyjmujących symwastatynę w dawce 40 mg/dobę w monoterapii lub w skojarzeniu z ezetymibem w dawce 10 mg, nie stwierdzono zwiększenia korzystnego wpływu na układ krążenia po włączeniu niacyny (kwasu nikotynowego) w dawkach modyfikujących stężenie lipidów (≥1 g/dobę). Z tego względu, lekarze rozważający zastosowanie leczenia skojarzonego symwastatyną i niacyną (kwasem nikotynowym) lub lekami zawierającymi niacynę w dawkach modyfikujących stężenie lipidów (≥1 g/dobę) powinni dokładnie ocenić potencjalne korzyści i ryzyko, a także ściśle monitorować pacjentów pod kątem występowania wszelkich objawów podmiotowych i przedmiotowych, w postaci bólu mięśni, tkliwości czy osłabienia siły mięśni, zwłaszcza w pierwszych miesiącach leczenia oraz w przypadku zwiększenia dawki któregokolwiek z leków.
Ponadto, w tym badaniu częstość występowania miopatii u pacjentów narodowości chińskiej przyjmujących symwastatynę w dawce 40 mg lub ezetymib z symwastatyną w dawce 10 mg + 40 mg wynosiła około 0,24% w porównaniu do 1,24% u pacjentów narodowości chińskiej przyjmujących symwastatynę w dawce 40 mg lub ezetymib z symwastatyną w dawce 10 mg + 40 mg w skojarzeniu z kwasem nikotynowym
i laropiprantem w dawce 2000 mg + 40 mg w postaci o przedłużonym uwalnianiu. Mimo, że populacja azjatycka w tym badaniu składała się wyłącznie z pacjentów narodowości chińskiej, z uwagi na fakt, że częstość występowania miopatii jest wyższa u osób pochodzących z Chin niż u pacjentów innych narodowości, nie zaleca się jednoczesnego podawania produktu leczniczego Axocar z niacyną (kwasem nikotynowym) w dawkach modyfikujących stężenie lipidów (≥1 g/dobę) pacjentom pochodzenia azjatyckiego.
Acypimoks pod względem budowy podobny jest do kwasu nikotynowego. Mimo, że acypimoks nie został przebadany, ryzyko działania toksycznego na mięśnie może być podobne do niacyny.
Należy unikać jednoczesnego stosowania ezetymibu i symwastatyny w dawkach większych niż 10 mg +
20 mg na dobę z amiodaronem, amlodypiną, werapamilem lub diltiazemem. U pacjentów z homozygotyczną hipercholesterolemią rodzinną należy unikać jednoczesnego stosowania produktu leczniczego Axocar w dawkach przewyższających 10 mg + 40 mg na dobę z lomitapidem. (patrz punkty 4.2, 4.3 i 4.5).
U pacjentów przyjmujących jednocześnie ezetymib i symwastatynę, zwłaszcza w wyższych dawkach, z innymi lekami uznanymi za umiarkowane inhibitory CYP3A4 w dawkach terapeutycznych, ryzyko wystąpienia miopatii może być większe. W przypadku stosowania produktu leczniczego Axocar jednocześnie z umiarkowanie silnym inhibitorem CYP3A4 (substancją powodującą około 2–5-krotne
zwiększenie wartości AUC) może być konieczne dostosowanie dawki. Jeśli stosowane są pewne substancje mające umiarkowanie hamujący wpływ na inhibitory CYP3A4, np. diltiazem, zaleca się podawanie produktu leczniczego Axocar w dawce wynoszącej maksymalnie 10 mg + 20 mg (patrz punkt 4.2).
Symwastatyna jest substratem transportera pompy lekowej – białka warunkującego oporność w raku piersi (BCRP). Jednoczesne przyjmowanie produktów leczniczych będących inhibitorami BCRP (np. elbaswiru and grazoprewiru) może prowadzić do zwiększenia stężenia symwastatyny w osoczu oraz do zwiększenia ryzyka miopatii; w związku z tym konieczne może być dostosowanie dawki symwastatyny. Nie badano jednoczesnego przyjmowania elbaswiru i grazoprewiru z symwastatyną; jednakże, u pacjentów przyjmujących jednocześnie produkty lecznicze zawierające elbaswir lub grazoprewir, dawka produktu leczniczego Axocar nie powinna przekraczać 10 mg + 20 mg na dobę (patrz punkt 4.5).
Nie przeprowadzono badań dotyczących bezpieczeństwa stosowania i skuteczności ezetymiby i symwastatyny z fibratami. Ryzyko miopatii zwiększa się podczas jednoczesnego stosowania symwastatyny z fibratami (zwłaszcza z gemfibrozylem). Z tego względu, jednoczesne stosowanie produktu leczniczego Axocar i gemfibrozylu jest przeciwwskazane (patrz punkt 4.3) oraz nie zaleca się jednoczesnego stosowania z innymi fibratami (patrz punkt 4.5).
Daptomycyna
Zgłaszano przypadki miopatii i (lub) rabdomiolizy podczas jednoczesnego podawania inhibitorów reduktazy HMG-CoA (np. symwastatyny i ezetymibu z symwastatyną) z daptomycyną. Należy zachować ostrożność przepisując inhibitory reduktazy HMG-CoA z daptomycyną, ponieważ każdy z tych produktów podawany w monoterapii może spowodować wystąpienie miopatii i (lub) rabdomiolizy. Należy rozważyć tymczasowe przerwanie stosowania produktu leczniczego Axocar u pacjentów przyjmujących daptomycynę chyba, że korzyści z jednoczesnego podawania przewyższają ryzyko. Należy zapoznać się z informacją
o daptomycynie w celu uzyskania dalszych informacji na temat wystąpienia ich potencjalnych interakcji z inhibitorami reduktazy HMG-CoA (np. symwastatyny i ezetymibu z symwastatyną) oraz dalszych wskazówek dotyczących monitorowania (patrz punkt 4.5).
Enzymy wątrobowe
Podczas kontrolowanych badań klinicznych, u pacjentów otrzymujących jednocześnie ezetymib
i symwastatynę zaobserwowano zwiększenie aktywności aminotransferaz w surowicy (≥ 3x GGN) (patrz punkt 4.8).
W badaniu IMPROVE-IT 18 144 pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym
w wywiadzie przydzielano w sposób losowy do grupy otrzymującej ezetymib i symwastatynę w dawce 10 mg + 40 mg na dobę (n=9067) lub symwastatynę w dawce 40 mg na dobę (n=9077). Podczas okresu obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania kolejnych wzrostów poziomu transaminaz (≥ 3x GGN) w grupie otrzymującej ezetymib i symwastatynę oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 2,5% i 2,3% (patrz punkt 4.8).
W kontrolowanym badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek randomizowano do grupy leczonej ezetymibem i symwastatyną w dawce 10 mg + 20 mg na dobę (n=4650) lub do grupy przyjmującej placebo (n=4620) (mediana okresu obserwacji 4,9 roku), częstość występowania
następowego zwiększenia aktywności aminotransferaz (> 3x GGN) wyniosła 0,7% wśród osób przyjmujących ezetymib i symwastatynę oraz 0,6% w grupie placebo (patrz punkt 4.8).
Zaleca się wykonywanie testów określających czynność wątroby u wszystkich pacjentów przed rozpoczęciem stosowania produktu leczniczego Axocar a następnie, gdy będzie to wskazane klinicznie.
U pacjentów, u których wymagane jest stosowanie leku w dawce 10 mg + 80 mg należy wykonać dodatkowe badanie przed zmianą dawkowania, 3 miesiące po zmianie dawkowania na 10 mg + 80 mg a następnie okresowo (np. co pół roku) w pierwszym roku leczenia. Należy zwrócić szczególną uwagę na pacjentów,
u których stwierdzono zwiększenie aktywności aminotransferaz. U tych pacjentów należy niezwłocznie powtórzyć badania, a następnie przeprowadzać je częściej. Jeśli następuje dalsze zwiększanie aktywności aminotransferaz, zwłaszcza do wartości trzykrotnie większych od górnej granicy normy (GGN) i utrzymuje się, należy odstawić lek. Należy zwrócić uwagę, że aktywność aminotransferazy alaninowej (ALT) może wywodzić się z tkanki mięśniowej, zatem wzrost aktywności ALT wraz ze wzrostem aktywności CK może wskazywać na miopatię (patrz powyżej „Miopatia i (lub) rabdomioliza”).
W okresie po wprowadzeniu do obrotu zgłaszano rzadkie przypadki niewydolności wątroby bez skutku śmiertelnego i zakończone zgonem, u pacjentów przyjmujących statyny, w tym symwastatynę. Jeżeli podczas leczenia produktem leczniczym Axocar wystąpi ciężkie uszkodzenie wątroby dające objawy kliniczne i (lub) hiperbilirubinemia lub żółtaczka, należy niezwłocznie przerwać leczenie. Jeżeli nie można zidentyfikować innej przyczyny zaburzeń, nie należy wznawiać leczenia produktem leczniczym Axocar.
Axocar należy stosować ostrożnie u osób pijących znaczne ilości alkoholu. Zaburzenia czynności wątroby
Ze względu na to, że nie są znane skutki długotrwałego stosowania ezetymibu u pacjentów
z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby, nie zaleca się stosowania produktu leczniczego Axocar u tych pacjentów (patrz punkt 5.2).
Cukrzyca
Niektóre dane wskazują, że statyny są lekami podnoszącymi stężenie glukozy we krwi; u niektórych pacjentów, u których istnieje wysokie ryzyko wystąpienia cukrzycy w przyszłości, mogą one wywołać hiperglikemię na poziomie wymagającym podania leków przeciwcukrzycowych. To ryzyko jest jednak równoważone przez zdolność statyn do zmniejszenia ryzyka chorób naczyniowych, nie powinno ono zatem być powodem przerwania terapii statynami. U pacjentów z grupy ryzyka (poziom glukozy na czczo 5,6– 6,9 mmol/l, BMI > 30 kg/m2, podwyższony poziom triglicerydów, nadciśnienie) należy prowadzić obserwację kliniczną i monitorować parametry biochemiczne zgodnie z lokalnymi wytycznymi.
Dzieci i młodzież
Bezpieczeństwo stosowania i skuteczność ezetymibu w skojarzeniu z symwastatyną u pacjentów w wieku 10 do 17 lat z heterozygotyczną hipercholesterolemią rodzinną oceniono w badaniu klinicznym z grupą kontrolną z udziałem dorastających chłopców (w fazie II i powyżej w skali Tannera) oraz dziewcząt,
u których pierwsza miesiączka wystąpiła co najmniej 1 rok wcześniej.
W tym ograniczonym badaniu z grupą kontrolną, ogólnie nie stwierdzono wykrywalnego wpływu na wzrost lub dojrzewanie płciowe u dorastających chłopców lub dziewcząt, ani na długość cyklu miesiączkowego
u dziewcząt. Nie przeprowadzono jednak badań dotyczących wpływu stosowania ezetymibu przez okres
> 33 tygodni na wzrost i proces dojrzewania płciowego (patrz punkty 4.2 i 4.8).
Nie przeprowadzono oceny bezpieczeństwa stosowania i skuteczności ezetymibu w skojarzeniu
z symwastatyną w dawkach przekraczających 40 mg na dobę u dzieci i młodzieży w wieku 10 do 17 lat.
Nie przeprowadzono badań dotyczących oceny wpływu ezetymibu u pacjentów poniżej 10 lat, ani u dziewcząt przed pierwszą miesiączką. (patrz punkty 4.2 i 4.8.).
Nie przeprowadzono oceny długotrwałej skuteczności ezetymibu u pacjentów poniżej 17 lat w zakresie zmniejszenia zachorowalności i śmiertelności w wieku dojrzałym.
Fibraty
Nie ustalono bezpieczeństwa stosowania i skuteczności ezetymibu w skojarzeniu z fibratami (patrz powyżej oraz punkty 4.3 i 4.5).
Substancje przeciwzakrzepowe
W przypadku stosowania produktu leczniczego Axocar jednocześnie z warfaryną, inną substancją przeciwzakrzepową z grupy pochodnych kumaryny lub fluindionem należy odpowiednio monitorować wartości INR (międzynarodowy wskaźnik znormalizowany) (patrz punkt 4.5).
Śródmiąższowa choroba płuc
Zgłaszano pojedyncze przypadki rozwoju śródmiąższowej choroby płuc w związku z leczeniem niektórymi statynami (w tym symwastatyną), zwłaszcza leczeniem długotrwałym (patrz punkt 4.8). Choroba może objawiać się dusznością, obecnością suchego kaszlu i pogorszeniem ogólnego stanu zdrowia (zmęczenie, zmniejszenie masy ciała i gorączka). W przypadku podejrzenia śródmiąższowej choroby płuc należy przerwać leczenie produktem leczniczym Axocar.
Substancje pomocnicze
Ten lek zawiera laktozę (w postaci monohydratu). Lek nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy- galaktozy.
Ten produkt zawiera mniej niż 1 mmol sodu (23 mg) w jednej tabletce, to znaczy produkt jest „wolny od sodu”.
Do występowania potencjalnych interakcji z inhibitorami reduktazy HMG-CoA może przyczyniać się wiele mechanizmów. Produkty lecznicze lub produkty ziołowe, które hamują pewne enzymy (np. CYP3A4) i (lub) szlaki transporterów (np. OATP1B), mogą zwiększać stężenie symwastatyny i kwasu symwastatyny
w osoczu oraz mogą prowadzić do zwiększenia ryzyka wystąpienia miopatii i (lub) rabdomiolizy.
Należy zapoznać się z informacją o wszystkich jednocześnie stosowanych produktach leczniczych, w celu uzyskania dalszych informacji na temat wystąpienia ich potencjalnych interakcji
z symwastatyną i (lub) potencjalnych zmian dotyczących enzymów lub transporterów oraz ewentualnego dostosowania dawki i schematów leczenia.
Interakcje farmakodynamiczne
Interakcje z produktami leczniczymi obniżającymi stężenie lipidów, które podawane w monoterapii mogą spowodować miopatię
Ryzyko wystąpienia miopatii, w tym rabdomiolizy jest zwiększone podczas jednoczesnego stosowania symwastatyny i fibratów. Ponadto, farmakokinetyczna interakcja symwastatyny z gemfibrozylem powoduje zwiększenie stężenia symwastatyny w osoczu krwi (patrz poniżej „Interakcje farmakokinetyczne” i punkty
4.3 i 4.4). Rzadko obserwowano przypadki miopatii i (lub) rabdomiolizy związane z przyjmowaniem symwastatyny jednocześnie z niacyną w dawkach modyfikujących stężenie lipidów (≥1 g/dobę) (patrz punkt 4.4).
Fibraty mogą zwiększać wydalanie cholesterolu do żółci, co prowadzi do kamicy żółciowej. W badaniu przedklinicznym na psach, ezetymib zwiększał stężenie cholesterolu w pęcherzyku żółciowym (patrz punkt 5.3). Wprawdzie nie wiadomo, czy wyniki badań przedklinicznych mają znaczenie dla ludzi, jednak nie zaleca się jednoczesnego stosowania produktu leczniczego Axocar i fibratów (patrz punkt 4.4).
Interakcje farmakokinetyczne
Zalecenia odnośnie przepisywania leków wchodzących w interakcje zostały zebrane w tabeli poniżej (dalsze szczegóły zawarte są w tekście, patrz również punkty 4.2, 4.3 i 4.4).
Interakcje lekowe związane ze zwiększonym ryzykiem miopatii i (lub) rabdomiolizy
Leki wchodzące w interakcje | Zalecenia dotyczące przepisywania |
Silne inhibitory CYP3A4, np. Itrakonazol Ketokonazol Pozakonazol Worykonazol Erytromycyna Klarytromycyna Telitromycyna Inhibitory proteazy wirusa HIV (np., nelfinawir) Boceprewir Telaprewir Nefazodon Kobicystat Cyklosporyna Danazol Gemfibrozyl | Przeciwwskazane z produktem Axocar. |
Inne fibraty Kwas fusydowy | Nie zaleca się stosowania z produktem Axocar. |
Niacyna (kwas nikotynowy) (≥ 1 g/dobę) | U pacjentów pochodzenia azjatyckiego nie zaleca się stosowania z Axocar. |
Amiodaron Amlodypina Werapamil Diltiazem Niacyna (≥1 g/dobę) Elbaswir Grazoprewir | Nie stosować dawki większej niż 10 mg + 20 mg produktu Axocar na dobę. |
Lomitapid | U pacjentów z homozygotyczną hipercholesterolemią rodzinną nie stosować dawki większej niż 10 mg + 40 mg produktu Axocar na dobę. |
Daptomycyna | Należy rozważyć tymczasowe przerwanie stosowania produktu leczniczego Axocar u pacjentów przyjmujących daptomycynę chyba, że korzyści z jednoczesnego podawania przewyższają ryzyko (patrz punkt 4.4). |
Tikagrelor | Nie zaleca się stosowania dawek większych niż 10 mg + 40 mg produktu leczniczego Axocar na dobę. |
Sok grejpfrutowy | Unikać soku grejpfrutowego podczas stosowania produktu Axocar. |
Wpływ innych produktów leczniczych na ezetymib z symwastatyną
Ezetymib z symwastatyną Niacyna
W badaniu prowadzonym z udziałem 15 zdrowych osób dorosłych jednoczesne przyjmowanie ezetymibu i symwastatyny (w dawce 10 mg + 20 mg na dobę przez 7 dni) spowodowało niewielki wzrost średniej wartości pola pod krzywą (AUC) niacyny (22%) i kwasu nikotynurenowego (19%) podawanych w postaci
preparatu NIASPAN w tabletkach o przedłużonym uwalnianiu (1000 mg przez 2 dni i 2000 mg przez 5 dni po śniadaniu składającym się z produktów o niskiej zawartości tłuszczu). W tym samym badaniu jednoczesne przyjmowanie preparatu NIASPAN wiązało się z nieznacznym zwiększeniem wartości AUC
ezetymibu (9%), ezetymibu całkowitego (26%), symwastatyny (20%) i kwasu symwastatyny (35%) (patrz
punkty 4.2 i 4.4).
Nie przeprowadzono badań interakcji z symwastatyną podawaną w wyższych dawkach.
Ezetymib
Leki zobojętniające
Jednoczesne podawanie leków zobojętniających zmniejsza szybkość wchłaniania ezetymibu, ale nie ma wpływu na jego biodostępność. Zmniejszenie szybkości wchłaniania nie jest uważane za znamienne klinicznie.
Kolestyramina
Jednoczesne podawanie kolestyraminy zmniejsza średnią wartość pola pod krzywą (AUC) ezetymibu całkowitego (ezetymib + glukuronid ezetymibu) o około 55%. Efekt zwiększonej redukcji stężenia cholesterolu LDL po włączeniu ezetymibu i symwastatyny do leczenia kolestyraminą może ulec osłabieniu w wyniku tej interakcji (patrz punkt 4.2).
Cyklosporyna
W badaniu przeprowadzonym z udziałem ośmiu pacjentów po przebytej operacji przeszczepienia nerki, u których klirens kreatyniny wynosił > 50 ml/min, stosujących cyklosporynę w ustalonej dawce, podanie ezetymibu w dawce jednorazowej 10 mg spowodowało 3,4-krotne zwiększenie (zakres 2,3- do 7,9-raza) średniej wartości AUC całkowitego ezetymibu, w porównaniu z grupą kontrolną w postaci populacji zdrowych osób otrzymujących ezetymib w monoterapii w innym badaniu (n=17). W innym badaniu,
u pacjenta po przeszczepieniu nerki, z ciężkimi zaburzeniami czynności nerek, który przyjmował cyklosporynę i wiele innych leków, wykazano 12- krotne zwiększenie stężenia całkowitego ezetymibu, w porównaniu z osobami z grupy kontrolnej otrzymującymi ezetymib w monoterapii. W badaniu skrzyżowanym złożonym z 2-óch okresów, w którym uczestniczyło dwunastu zdrowych ochotników,
stosowanie 20 mg ezetymibu na dobę przez 8 dni oraz podanie pojedynczej dawki cyklosporyny wynoszącej 100 mg w 7. dniu spowodowało średnie zwiększenie AUC cyklosporyny o 15% (zakres 10% zmniejszenia do 51% zwiększenia) w porównaniu z podaniem wyłącznie pojedynczej dawki cyklosporyny wynoszącej 100 mg. Nie przeprowadzono badania kontrolowanego dotyczącego wpływu jednoczesnego stosowania ezetymibu na narażenie na cyklosporynę u pacjentów po przeszczepieniu nerki. Jednoczesne stosowanie produktu leczniczego Axocar i cyklosporyny jest przeciwwskazane (patrz punkt 4.3).
Fibraty
Podczas jednoczesnego przyjmowania fenofibratu lub gemfibrozylu następuje zwiększenie stężeń całkowitego ezetymibu o odpowiednio 1,5 i 1,7 raza. Mimo, że zwiększenie to nie jest uważane za istotne klinicznie, jednoczesne podawanie produktu leczniczego Axocar z gemfibrozylem jest przeciwwskazane, a z innymi fibratami nie jest zalecane (patrz punkty 4.3 i 4.4).
Symwastatyna
Symwastatyna jest substratem cytochromu P450 3A4. Podczas leczenia symwastatyną, silne inhibitory cytochromu P450 3A4 zwiększają ryzyko miopatii i rabdomiolizy, na skutek zwiększenia stężenia inhibitorów reduktazy HMG-CoA w osoczu. Do takich inhibitorów należą: itrakonazol, ketokonazol, pozakonazol, worykonazol, erytromycyna, klarytromycyna, telitromycyna, inhibitory proteazy wirusa HIV (np. nelfinawir), boceprewir, telaprewir, nefazodon i produkty lecznicze zawierające kobicystat. Jednoczesne podanie itrakonazolu spowodowało ponad 10-krotne zwiększenie narażenia na kwas symwastatyny (aktywny metabolit beta-hydroksykwas). Telitromycyna spowodowała 11-krotnie większe narażenie na kwas symwastatyny.
Przeciwwskazane jest jednoczesne podawanie symwastatyny z itrakonazolem, ketokonazolem, pozakonazolem, worykonazolem, inhibitorami proteazy wirusa HIV (np. nelfinawirem), boceprewirem, telaprewirem, erytromycyną, klarytromycyną, telitromycyną, nefazodonem i produktami leczniczymi zawierającymi kobicystat, jak również z gemfibrozylem, cyklosporyną i danazolem (patrz punkt 4.3). Jeśli konieczne jest leczenie silnymi inhibitorami CYP3A4 (substancjami powodującymi co najmniej około pięciokrotne zwiększenie wartości AUC), należy podczas tego leczenia zaprzestać stosowania produktu
leczniczego Axocar (i rozważyć podanie innej statyny). Należy ostrożnie stosować produkt leczniczy Axocar jednocześnie z innymi słabszymi inhibitorami CYP3A4: flukonazolem, werapamilem lub diltiazemem (patrz punkty 4.2 i 4.4).
Tikagrelor
Jednoczesne stosowanie symwastatyny z tikagrelorem powodowało zwiększenie Cmax symwastatyny o 81% i AUC o 56% oraz zwiększenie Cmax kwasu symwastatyny o 64% i jego AUC o 52% z pojedynczymi przypadkami zwiększenia 2- lub 3-krotnego. Jednoczesne stosowanie tikagrelolu i symwastatyny w dawce większej niż 40 mg na dobę mogłoby spowodować wystąpienie działań niepożądanych symwastatyny
i dlatego należy je uwzględnić w ocenie potencjalnych korzyści tego skojarzenia. Nie stwierdzono wpływu symwastatyny na stężenie tikagreloru w osoczu. Nie zaleca się jednoczesnego stosowania tikagreloru
z symwastatyną w dawkach większych niż 40 mg.
Flukonazol
Zgłaszano rzadkie przypadki rabdomiolizy związane z jednoczesnym stosowaniem symwastatyny i flukonazolu (patrz punkt 4.4).
Cyklosporyna
Ryzyko miopatii i (lub) rabdomiolizy zwiększone jest podczas jednoczesnego stosowania cyklosporyny z ezetymibem i symwastatynaą; z tego względu, jednoczesne stosowanie z cyklosporyną jest przeciwwskazane (patrz punkty 4.3 i 4.4). Mimo, że mechanizm tej interakcji nie jest w pełni wyjaśniony wykazano, iż cyklosporyna zwiększa AUC inhibitorów reduktazy HMG-CoA. Zwiększenie AUC kwasu symwastatyny przypuszczalnie wynika z hamowania CYP3A4 i (lub) OATP1B1.
Danazol
Jednoczesne stosowanie danazolu i ezetymibu z symwastatyną zwiększa ryzyko miopatii i rabdomiolizy; z tego względu jednoczesne stosowanie z danazolem jest przeciwwskazane (patrz punkty 4.3 i 4.4).
Gemfibrozyl
Gemfibrozyl zwiększa 1,9-krotnie wartość AUC kwasu symwastatyny, prawdopodobnie w wyniku zahamowania szlaku glukuronidowego i (lub) OATP1B1 (patrz punkty 4.3 i 4.4).
Jednoczesne stosowanie z gemfibrozylem jest przeciwwskazane.
Kwas fusydowy
Ryzyko miopatii, w tym rabdomiolizy może być zwiększone podczas jednoczesnego stosowania kwasu fusydowego ze statynami. Mechanizm leżący u podłoża tej interakcji (niezależnie od tego, czy ma ona charakter farmakodynamiczny, czy farmakokinetyczny, czy zarówno farmakodynamiczny, jak
i farmakokinetyczny) jest jeszcze nieznany. Istnieją doniesienia o przypadkach rabdomiolizy (także prowadzących do śmierci) wśród pacjentów leczonych takim skojarzeniem. W wyniku zastosowania takiego skojarzenia może dojść do wzrostu stężenia obu leków w osoczu.
U pacjentów, u których ogólnoustrojowe podawanie kwasu fusydowego uważa się za konieczne, leczenie produktem leczniczym Axocar należy przerwać na cały okres leczenia kwasem fusydowym. Patrz także punkt 4.4.
Amiodaron
Ryzyko miopatii i rabdomiolizy zwiększone jest podczas jednoczesnego stosowania amiodaronu
z symwastatyną (patrz punkt 4.4). W badaniu klinicznym odnotowano wystąpienie miopatii u 6% pacjentów otrzymujących symwastatynę w dawce 80 mg i amiodaron. Z tego względu, u pacjentów otrzymujących jednocześnie amiodaron, dawka produktu leczniczego Axocar nie powinna być większa niż 10 mg + 20 mg na dobę.
Leki blokujące kanał wapniowy
Werapamil: Ryzyko miopatii i rabdomiolizy zwiększone jest podczas jednoczesnego stosowania werapamilu z symwastatyną w dawce 40 mg lub 80 mg (patrz punkt 4.4). W badaniach farmakokinetycznych jednoczesne podawanie symwastatyny z werapamilem powoduje 2,3-krotne
zwiększenie narażenia na kwas symwastatyny, przypuszczalnie na skutek hamowania CYP3A4.
Z tego względu, u pacjentów otrzymujących jednocześnie werapamil, dawka produktu leczniczego Axocar nie powinna być większa niż 10 mg + 20 mg na dobę.
Diltiazem: Ryzyko miopatii i rabdomiolizy zwiększone jest podczas jednoczesnego stosowania diltiazemu z symwastatyną w dawce 80 mg (patrz punkt 4.4). W badaniach farmakokinetycznych jednoczesne podawanie symwastatyny z diltiazemem powoduje 2,7-krotne zwiększenie narażenia na kwas symwastatyny, przypuszczalnie na skutek hamowania CYP3A4. Z tego względu, dawka produktu leczniczego Axocar u pacjentów otrzymujących jednocześnie diltiazem nie powinna być większa niż 10 mg + 20 mg na dobę.
Amlodypina: U pacjentów przyjmujących amlodypinę jednocześnie z symwastatyną ryzyko rozwoju miopatii było większe. W badaniu farmakokinetycznym jednoczesne podawanie amlodypiny powoduje 1,6-krotne zwiększenie narażenia na kwas symwastatyny. Z tego względu, u pacjentów otrzymujących jednocześnie amlodypinę, dawka produktu leczniczego Axocar nie powinna być większa niż 10 mg + 20 mg na dobę.
Lomitapid
Ryzyko rozwoju miopatii i rabdomiolizy może być zwiększone w wyniku jednoczesnego podawania lomitapidu z symwastatyną (patrz punkty 4.3 i 4.4), dlatego w przypadku pacjentów z homozygotyczną hipercholesterolemią rodzinną dawka produktu leczniczego Axocar nie może przekraczać 10 mg + 40 mg na dobę u pacjentów przyjmujących jednocześnie lomitapid.
Umiarkowane inhibitory CYP3A4
U pacjentów stosujących jednocześnie ezetymib z symwastatyną, zwłaszcza w wyższych dawkach, z innymi lekami uznanymi za mające umiarkowany wpływ na układ enzymatyczny CYP3A4, ryzyko wystąpienia miopatii może być większe (patrz punkt 4.4).
Inhibitory białka transportowego OATP1B1
Kwas symwastatyny jest substratem dla białka transportowego OATP1B1. Jednoczesne podawanie produktów leczniczych będących inhibitorami białka transportowego OATP1B1 może prowadzić do podwyższonego stężenia kwasu symwastatyny w osoczu oraz podwyższonego ryzyka wystąpienia miopatii (patrz punkty 4.3 i 4.4).
Inhibitory białka warunkującego oporność w raku piersi (BCRP)
Jednoczesne przyjmowanie produktów leczniczych będących inhibitorami BCRP, w tym produktów leczniczych zawierających elbaswir lub grazoprewir, może prowadzić do zwiększenia stężenia symwastatyny w osoczu oraz do zwiększenia ryzyka miopatii (patrz punkty 4.2 i 4.4).
Sok grejpfrutowy
Sok grejpfrutowy hamuje cytochrom P450 3A4. Jednoczesne spożywanie dużych ilości soku grejpfrutowego (ponad 1 l na dobę) i symwastatyny powoduje 7-krotne zwiększenie narażenia na kwas symwastatyny.
Wypicie 240 ml soku grejpfrutowego rano i przyjęcie wieczorem symwastatyny powoduje także 1,9-krotne zwiększenie narażenia. Z tego względu, należy unikać picia soku grejpfrutowego podczas stosowania produktu leczniczego Axocar.
Kolchicyna
U pacjentów z zaburzeniami czynności nerek zgłaszano przypadki miopatii i rabdomiolizy podczas jednoczesnego stosowania kolchicyny i symwastatyny. Zalecane jest uważne monitorowanie pacjentów przyjmujących jednocześnie oba te leki.
Ryfampicyna
Ponieważ ryfampicyna jest silnym induktorem CYP3A4, u pacjentów długotrwale leczonych ryfampicyną (np. leczenie gruźlicy) może nastąpić utrata skuteczności działania symwastatyny. W badaniu farmakokinetycznym u zdrowych ochotników wielkość pola pod krzywą (AUC) wartości stężenia osoczowego dla kwasu symwastatytny zmniejszyła się o 93% podczas jednoczesnego stosowania
ryfampicyny.
Niacyna
Podczas podawania symwastatyny jednocześnie z niacyną w dawkach modyfikujących poziom lipidów (≥1 g/dobę) obserwowano przypadki miopatii i (lub) rabdomiolizy (patrz punkt 4.4).
Daptomycyna
Ryzyko miopatii i (lub) rabdomiolizy może być zwiększone podczas jednoczesnego podawania inhibitorów reduktazy HMG-CoA (np. symwastatyny i ezetymibu z symwastatyną) i daptomycyny (patrz punkt 4.4).
Wpływ ezetymibu z symwastatyną na farmakokinetykę innych produktów leczniczych
Ezetymib
W badaniach przedklinicznych wykazano, że ezetymib nie indukuje metabolizujących leki enzymów z grupy cytochromu P450. Nie obserwowano istotnych klinicznie interakcji farmakokinetycznych pomiędzy ezetymibem a lekami, o których wiadomo, że metabolizowane są przez cytochromy P450 1A2, 2D6, 2C8, 2C9 i 3A4 lub N-acetylotransferazę.
Substancje przeciwzakrzepowe
Jednoczesne stosowanie ezetymibu (10 mg raz na dobę) nie miało istotnego wpływu na biodostępność warfaryny i czas protrombinowy w badaniu przeprowadzonym z udziałem dwunastu zdrowych dorosłych mężczyzn. Jednakże, istnieją doniesienia z okresu po wprowadzeniu leku do obrotu dotyczące zwiększenia wartości INR u pacjentów stosujących warfarynę lub fluindion, u których dołączono leczenie ezetymibem. W przypadku stosowania produktu leczniczego Axocar jednocześnie z warfaryną, inną substancją przeciwzakrzepową z grupy pochodnych kumaryny lub fluindionem należy odpowiednio monitorować wartości INR (patrz punkt 4.4).
Symwastatyna
Symwastatyna nie wpływa hamująco na cytochrom P450 3A4. Z tego względu, symwastatyna nie wpływa na stężenie w osoczu substancji metabolizowanych przez cytochrom P450 3A4.
Doustne leki przeciwzakrzepowe
W 2 badaniach klinicznych, jednym obejmującym zdrowych ochotników i drugim obejmującym pacjentów z hipercholesterolemią, symwastatyna w dawce 20-40 mg na dobę umiarkowanie nasilała działanie substancji przeciwzakrzepowych z grupy pochodnych kumaryny: czas protrombinowy, podawany jako wskaźnik INR zwiększył się z wartości sprzed podania produktu leczniczego 1,7 do 1,8 oraz z 2,6 do 3,4 odpowiednio u zdrowych ochotników i osób chorych. Odnotowano bardzo rzadkie przypadki zwiększenia wartości INR. U pacjentów przyjmujących substancje przeciwzakrzepowe z grupy pochodnych kumaryny, czas protrombinowy należy ocenić przed rozpoczęciem stosowania produktu leczniczego Axocar, jak
i odpowiednio często w początkowym okresie trwania leczenia, aby upewnić się, że nie występują istotne zmiany w czasie protrombinowym. Po stwierdzeniu stabilności czasu protrombinowego, czasy protrombinowe należy monitorować z częstością zalecaną podczas leczenia pacjentów substancjami przeciwzakrzepowymi z grupy pochodnych kumaryny. Procedurę należy powtórzyć w przypadku zmiany dawki produktu leczniczego Axocar lub przerwania stosowania. U pacjentów, którzy nie przyjmują produktów przeciwzakrzepowych, leczenie symwastatyną nie było związane z występowaniem krwawień lub zmian czasu protrombinowego.
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono wyłącznie u dorosłych.
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Zaburzenia snu, w tym koszmary senne
Zaburzenia funkcji seksualnych
Cukrzyca: Częstość zależy od obecności lub braku czynników ryzyka (poziom glukozy na czczo
Przedawkowanie
Ciąża
Miażdżyca jest chorobą przewlekłą a przerwanie podczas ciąży stosowania leków zmniejszających stężenie lipidów, powinno mieć niewielki wpływ na długotrwałe ryzyko związane z pierwotną hipercholesterolemią.
Axocar
Axocar jest przeciwwskazany do stosowania w ciąży. Brak jest danych klinicznych dotyczących stosowania ezetymibu z symwastatyną w czasie ciąży. Badania na zwierzętach dotyczące leczenia skojarzonego wykazały toksyczny wpływ na reprodukcję. (patrz punkt 5.3).
Symwastatyna
Nie ustalono bezpieczeństwa stosowania symwastatyny u kobiet w ciąży. Nie przeprowadzono kontrolowanych badań klinicznych dotyczących stosowania symwastatyny u kobiet w ciąży. Obserwowano rzadkie przypadki występowania wad wrodzonych po wewnątrzmacicznym narażeniu na inhibitory reduktazy HMG-CoA. Jednak, w analizie danych dotyczących około 200 ciężarnych, które były prospektywnie obserwowane a w pierwszym trymestrze ciąży przyjmowały symwastatynę lub inny zbliżony inhibitor reduktazy HMG-CoA o podobnej strukturze chemicznej, częstość występowania wad wrodzonych nie była większa od tej, jaka występuje w ogólnej populacji. Liczba odnotowanych przypadków ciąży była statystycznie wystarczająca, aby wykluczyć 2,5-krotne lub większe zwiększenie występowania wad wrodzonych w stosunku do ogólnej częstości występowania.
Chociaż nie ma dowodów świadczących, że przypadki wad wrodzonych u potomstwa pacjentów przyjmujących symwastatynę lub inny zbliżony inhibitor reduktazy HMG-CoA różnią się od tych obserwowanych ogólnie w populacji, leczenie symwastatyną w okresie ciąży może zmniejszyć u płodu stężenie mewalonianu, który jest prekursorem biosyntezy cholesterolu. Z tego względu, nie wolno stosować produktu leczniczego Axocar u kobiet w ciąży, starających się zajść w ciążę lub tych, u których podejrzewana jest ciąża. Leczenie produktem leczniczym Axocar musi być przerwane w okresie ciąży lub do czasu ustalenia czy pacjentka nie jest w ciąży (patrz punkt 4.3).
Ezetymib
Brak jest danych klinicznych dotyczących stosowania ezetymibu w ciąży.
Karmienie piersią
Axocar jest przeciwwskazany do stosowania w okresie laktacji. Badania na szczurach wykazały, że ezetymib jest wydzielany z mlekiem. Nie wiadomo, czy aktywne składniki produktu leczniczego Axocar są wydzielane z mlekiem kobiecym (patrz punkt 4.3).
Płodność
Ezetymib
Nie ma dostępnych danych z badań klinicznych dotyczących wpływu ezetymibu na płodność u człowieka. Nie stwierdzono wpływu ezetymibu na płodność u samców i samic szczura (patrz punkt 5.3).
Symwastatyna
Brak dostępnych danych z badań klinicznych dotyczących wpływu symwastatyny na płodność u ludzi. Symwastatyna nie miała wpływu na płodność samców i samic szczurów (patrz punkt 5.3).
Nie przeprowadzono badań dotyczących wpływu na zdolność prowadzenia pojazdów i obsługiwania maszyn. Jednakże, podczas prowadzenia pojazdów lub obsługiwania maszyn, należy wziąć pod uwagę, że zgłaszano występowanie zawrotów głowy.
W badaniach klinicznych obejmujących około 12 000 pacjentów oceniono bezpieczeństwo stosowania ezetymibu i symwastatyny.
W badaniach klinicznych obserwowano następujące działania niepożądane u pacjentów leczonych ezetymibem z symwastatyną (n=2404), które pojawiły się z większą częstością niż u pacjentów przyjmujących placebo (n=1340), u pacjentów leczonych ezetymibem z symwastatyną (n=9595), które pojawiły się z większą częstością niż u pacjentów przyjmujących wyłącznie statyny (n=8883) w badaniach klinicznych ezetymibu
lub symwastatyny i (lub) zgłoszone po wprowadzeniu ezetymibu z symwastatyną lub ezetymibu lub symwastatyny do obrotu. Działania te przedstawiono według klasyfikacji układów i narządów oraz częstości występowania w Tabeli 1.
Częstość występowania działań niepożądanych uszeregowano zgodnie z następującą klasyfikacją: bardzo często (≥ 1/10), często (≥ 1/100 do <1/10), niezbyt często (≥ 1/1000 do < 1/100), rzadko (≥ 1/10 000 < do 1/1000), bardzo rzadko (< 1/10 000) w tym pojedyncze przypadki oraz częstość nieznana (częstość nie może być określona na podstawie dostępnych danych)
Tabela 1. Działania niepożądane
Klasyfikacja układów i narządów Częstość występowania | Działanie niepożądane |
Zaburzenia krwi i układu chłonnego | |
Częstość nieznana | trombocytopenia; niedokrwistość |
Zaburzenia układu immunologicznego | |
Bardzo rzadko | anafilaksja |
Częstość nieznana | reakcje nadwrażliwości |
Zaburzenia metabolizmu i odżywiania | |
Częstość nieznana | zmniejszenie łaknienia |
Zaburzenia psychiczne | |
Niezbyt często | zaburzenia snu, bezsenność |
Częstość nieznana | depresja |
Zaburzenia układu nerwowego | |
Niezbyt często | zawroty głowy; bóle głowy; parestezje |
Częstość nieznana | neuropatia obwodowa; zaburzenia pamięci |
Zaburzenia oka | |
Rzadko | niewyraźne widzenie; zaburzenia widzenia |
Zaburzenia naczyniowe | |
Częstość nieznana | uderzenia gorąca; nadciśnienie tętnicze |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Częstość nieznana | kaszel; duszność; śródmiąższowa choroba płuc (patrz punkt 4.4) |
Zaburzenia żołądka i jelit | |
Niezbyt często | ból brzucha; dyskomfort w jamie brzusznej; bóle w nadbrzuszu; niestrawność; wzdęcia; nudności; wymioty; wzdęcie brzucha; biegunka; suchość jamy ustnej; choroba refluksowa przełyku i żołądka |
Częstość nieznana | zaparcia; zapalenie trzustki; zapalenie żołądka |
Zaburzenia wątroby i dróg żółciowych | |
Częstość nieznana | zapalenie wątroby/żółtaczka; niewydolność wątroby bez skutku śmiertelnego i zakończona zgonem; kamica żółciowa; zapalenie pęcherzyka żółciowego |
Zaburzenia skóry i tkanki podskórnej | |
Niezbyt często | świąd; wysypka; pokrzywka |
Bardzo rzadko | polekowe zmiany liszajowate |
Częstość nieznana | łysienie; rumień wielopostaciowy; obrzęk naczynioruchowy |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często | bóle mięśni |
Niezbyt często | bóle stawów; kurcze mięśni; osłabienie siły mięśni; dolegliwości układu mięśniowo- szkieletowego; bóle szyi; bóle kończyn; bóle pleców; bóle mięśniowo-szkieletowe |
Bardzo rzadko | zerwanie mięśni |
Częstość nieznana | kurcze mięśni; miopatia* (w tym zapalenie mięśni); rabdomioliza z ostrą niewydolnością nerek lub bez (patrz punkt 4.4); tendinopatia, czasami powikłana zerwaniem ścięgien; immunozależna miopatia martwicza (IMNM)** |
Zaburzenia układu rozrodczego i piersi | |
Bardzo rzadko | ginekomastia |
Częstość nieznana | zaburzenie erekcji |
Zaburzenia ogólne i stany w miejscu podania | |
Niezbyt często | astenia; ból w klatce piersiowej; zmęczenie; złe samopoczucie; obrzęki obwodowe |
Częstość nieznana | ból |
Badania diagnostyczne | |
Często | zwiększenie aktywności AlAT i (lub) AspAT; zwiększenie aktywności kinazy kreatynowej (CK) we krwi |
Niezbyt często | zwiększenie stężenia bilirubiny we krwi; zwiększenie stężenia kwasu moczowego we krwi; zwiększenie aktywności gamma- glutamylotransferazy (GGTP), zwiększenie wartości międzynarodowego wskaźnika znormalizowanego (INR); obecność białka w moczu; zmniejszenie masy ciała |
Częstość nieznana | zwiększenie aktywności fosfatazy zasadowej; nieprawidłowe wyniki testów czynnościowych wątroby |
* W badaniu klinicznym u pacjentów leczonych symwastatyną w dawce 80 mg na dobę miopatia występowała częściej niż u osób przyjmujących 20 mg na dobę (odpowiednio 1,0% w porównaniu z 0,02%) (patrz punkty 4.4 i 4.5).
** Zgłoszono bardzo rzadkie przypadki immunozależnej miopatii martwiczej (ang. IMNM, immune- mediated necrotising myopathy), miopatii autoimmunologicznej, w trakcie leczenia statynami lub po jego zakończeniu. Cechy kliniczne IMNM to: utrzymujące się osłabienie mięśni proksymalnych oraz zwiększona aktywność kinazy kreatynowej w surowicy, utrzymująca się mimo przerwania leczenia statynami; biopsja mięśni wykazuje martwiczą miopatię bez istotnego zapalenia; poprawa następuje w wyniku stosowania leków immunosupresyjnych (patrz punkt 4.4).
Populacja pediatryczna
W badaniu z udziałem młodzieży (w wieku 10 do 17 lat) z heterozygotyczną hipercholesterolemią rodzinną (n=248), u 3% pacjentów (4 pacjentów) leczonych ezetymibem z symwastatyną obserwowano podwyższenie aktywności AlAT i (lub) AspAT (≥ 3x GGN, nieprzerwanie) w porównaniu z 2% osób (2 pacjentami)
z grupy stosującej symwastatynę w monoterapii; podwyższenie stężenia CPK (≥ 10x GGN) stwierdzono odpowiednio u 2% (2 pacjentów) i 0% uczestników badania. Nie zaobserwowano przypadków miopatii.
To badanie nie było dostosowane do oceny porównawczej rzadko występujących działań niepożądanych na lek.
Pacjenci z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie
W badaniu IMPROVE-IT (patrz punkt 5.1) z udziałem 18 144 pacjentów leczonych ezetymibem i symwastatyną w dawce 10 mg + 40 mg (n=9067, przy czym u 6% pacjentów dawkę ezetymibu
i symwastatyny zwiększono do 10 mg + 80 mg) lub symwastatyną w dawce 40 mg (n=9077, przy czym u 27% pacjentów dawkę symwastatyny zwiększono do 80 mg) w okresie obserwacji, którego mediana
wynosiła 6,0 lat, obserwowano zbliżone profile bezpieczeństwa. Wskaźnik przerwania leczenia z powodu działań niepożądanych wynosił 10,6% u pacjentów leczonych ezetymibem i symwastatyną oraz 10,1%
u pacjentów leczonych symwastatyną. Częstość występowania miopatii w grupie otrzymującej ezetymib i symwastatynę oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,2% i 0,1%. Miopatię zdefiniowano, jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącą ≥ 10x GGN lub 2 kolejne epizody zwiększenia aktywności CK wynoszące ≥ 5 i < 10x GGN. Częstość występowania rabdomiolizy w grupie otrzymującej ezetymib i symwastatynę oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,1% i 0,2%. Rabdomiolizę zdefiniowano jako
niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącą ≥ 10x GGN i objawami uszkodzenia nerek, dwa kolejne epizody zwiększenia aktywności CK wynoszącej ≥ 5x GGN
i < 10x GGN z objawami uszkodzenia nerek lub CK ≥ 10 000 j.m./l bez objawów uszkodzenia nerek. Częstość występowania kolejnych wzrostów poziomu transaminaz (≥ 3x GGN) w grupie otrzymującej ezetymib i symwastatynę oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 2,5% i 2,3% (patrz punkt 4.4.). Działania niepożądane ze strony pęcherzyka żółciowego zgłoszono u 3,1% pacjentów z grupy otrzymującej ezetymib i symwastatynę oraz u 3,5% pacjentów z grupy leczonej symwastatyną.
Częstość hospitalizacji z powodu cholecystektomii w obu leczonych grupach wynosiła 1,5%. Nowotwór (zdefiniowany jako dowolny nowy nowotwór) zdiagnozowano podczas badania u odpowiednio 9,4% i 9,5% pacjentów.
Pacjenci z przewlekłą chorobą nerek
W badaniu Study of Heart and Renal Protection (SHARP) (patrz punkt 5.1), z udziałem ponad 9000 pacjentów przyjmujących ezetymib i symwastatynę w dawce wynoszącej 10 mg + 20 mg na dobę (n=4650) lub placebo (n=4620) profile bezpieczeństwa były porównywalne w okresie obserwacji wynoszącym średnio 4,9 roku. W tym badaniu rejestrowano tylko ciężkie działania niepożądane i przypadki rezygnacji z powodu wystąpienia jakichkolwiek działań niepożądanych. Częstość przerywania leczenia z powodu wystąpienia działań niepożądanych była porównywalna (10,4% wśród pacjentów leczonych ezetymibem i symwastatyną i 9,8% wśród pacjentów przyjmujących placebo). Częstość występowania miopatii i (lub) rabdomiolizy wyniosła 0,2% w przypadku pacjentów leczonych ezetymibem i symwastatyną i 0,1% wśród pacjentów przyjmujących placebo. Przypadki zwiększenia aktywności aminotransferaz (> 3x GGN) stwierdzono
u 0,7% pacjentów leczonych ezetymibem i symwastatyną w porównaniu z 0,6% pacjentów przyjmujących placebo (patrz punkt 4.4). W tym badaniu nie odnotowano statystycznie istotnego wzrostu częstości występowania określonych wcześniej działań niepożądanych, w tym nowotworów (9,4% w grupie leczonej ezetymibem i symwastatyną, 9,5% w grupie przyjmującej placebo), zapalenia wątroby, operacji usunięcia pęcherzyka żółciowego lub powikłań kamicy żółciowej, lub zapalenia trzustki.
Badania diagnostyczne
W badaniach dotyczących leczenia skojarzonego częstość występowania istotnego klinicznie zwiększenia aktywności aminotransferaz w surowicy krwi (aktywność AlAT i (lub) AspAT ≥ 3x GGN) u pacjentów leczonych ezetymibem i symwastatyną wynosiła 1,7%. Zwiększenie aktywności aminotransferaz przebiegało zwykle bezobjawowo, nie towarzyszyła mu cholestaza, a wartości enzymów powracały do poziomu wyjściowego po przerwaniu leczenia lub w przypadku kontynuowania leczenia (patrz punkt 4.4).
Istotne klinicznie zwiększenie aktywności CK (≥10x GGN) zaobserwowano u 0,2% pacjentów leczonych ezetymibem i symwastatyną .
Działania niepożądane obserwowane po wprowadzeniu do obrotu
Rzadko zgłaszano występowanie zespołu nadwrażliwości, obejmującego niektóre z następujących objawów: obrzęk naczynioruchowy, zespół toczniopodobny, zespół bólu wielomięśniowego, zapalenie skórno- mięśniowe, zapalenie naczyń, trombocytopenia, eozynofilia, zwiększenie szybkości sedymentacji krwinek czerwonych, zapalenie stawów i ból stawów, pokrzywka, reakcja nadwrażliwości na światło, gorączka, uderzenia gorąca, duszność i złe samopoczucie.
U pacjentów przyjmujących statyny, w tym symwastatynę, zgłaszano przypadki wzrostu stężenia HbA1c i glukozy w surowicy na czczo.
W okresie po wprowadzeniu do obrotu zgłaszano rzadkie przypadki zaburzeń funkcji poznawczych (np. utrata pamięci, słaba pamięć, amnezja, zaburzenia pamięci, splątanie) u pacjentów przyjmujących statyny, w tym symwastatynę. Zgłaszane problemy były na ogół łagodne i ustępowały po przerwaniu leczenia statyną. Obserwowano zmienny czas do wystąpienia (od 1 dnia do kilku lat) i ustąpienia (mediana
3 tygodnie) objawów.
Zgłaszano występowanie następujących działań niepożądanych u pacjentów przyjmujących niektóre statyny:
≥ 5,6 mmol/l, BMI > 30 kg/m2, podwyższony poziom triglicerydów, nadciśnienie w wywiadzie).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych
i Produktów Biobójczych, Al. Jerozolimskie 181C, 02-222 Warszawa, tel.: + 48 22 49-21-301,
fax: + 48 22 49-21-309, strona internetowa: https://smz.ezdrowie.gov.pl
Dzięki zgłaszaniu działań niepożądanych będzie można zgromadzić więcej informacji na temat bezpieczeństwa stosowania leku.
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu lub przedstawicielowi podmiotu odpowiedzialnego w Polsce.
Ezetymib z symwastatyną
W przypadku przedawkowania należy zastosować leczenie objawowe i podtrzymujące. W badaniach ostrej toksyczności, po podaniu doustnym u myszy i szczurów, jednoczesne stosowanie ezetymibu (1000 mg/kg) i symwastatyny (1000 mg/kg) było dobrze tolerowane. Nie zaobserwowano żadnych klinicznych objawów toksyczności u tych zwierząt. Szacowana doustna wartość LD50 dla obu gatunków wynosiła dla ezetymibu
≥ 1000 mg/kg i dla symwastatyny ≥ 1000 mg/kg.
Ezetymib
W badaniach klinicznych, w których dawkę 50 mg na dobę podawano 15 zdrowym ochotnikom do 14 dni lub dawkę 40 mg na dobę 18 pacjentom z hipercholesterolemią pierwotną przez okres do 56 dni, ezetymib był dobrze tolerowany. Odnotowano kilka przypadków przedawkowania; większość nie była związana
z występowaniem działań niepożądanych. Odnotowane działania niepożądane nie były poważne. U zwierząt nie obserwowano działania toksycznego po przyjęciu doustnie pojedynczych dawek wynoszących
5000 mg/kg ezetymibu u szczurów i myszy oraz 3000 mg/kg u psów.
Symwastatyna
Odnotowano kilka przypadków przedawkowania; maksymalna przyjęta dawka wynosiła 3,6 g. Wszyscy pacjenci powrócili do zdrowia bez następstw.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Grupa farmakoterapeutyczna: inhibitory reduktazy HMG-CoA w skojarzeniu z innymi lekami wpływającymi na lipidy, kod ATC: C10BA02.
Ezetymib z symwastatyną jest produktem zmniejszającym stężenie lipidów, który wybiórczo hamuje wchłanianie cholesterolu i pochodnych steroli roślinnych w jelitach oraz hamuje syntezę endogennego cholesterolu.
Mechanizm działania
Ezetymib z symwastatyną
Cholesterol obecny w osoczu wchłaniany jest z przewodu pokarmowego oraz syntetyzowany w organizmie. Axocar zawiera ezetymib i symwastatynę, 2 składniki zmniejszające stężenie lipidów, których mechanizmy działania wzajemnie się uzupełniają. Ezetymib z symwastatyną zmniejsza stężenie podwyższonego cholesterolu całkowitego (total-C), cholesterolu LDL, apolipoproteiny B (Apo B), triglicerydów (TG)
i frakcji nie-HDL (non-HDL-C) oraz zwiększenie stężenia cholesterolu HDL (HDL-C) poprzez zahamowanie zarówno wchłaniania, jak i syntezy cholesterolu.
Ezetymib
Ezetymib hamuje wchłanianie cholesterolu z przewodu pokarmowego. Ezetymib działa po podaniu doustnym, a jego mechanizm działania różni się od mechanizmu działania związków zmniejszających stężenie cholesterolu, należących do innych klas (np. statyny, sekwestranty kwasów żółciowych [żywice], pochodne kwasu fibrynowego i stanole roślinne). Celem działania ezetymibu na poziomie molekularnym jest przenośnik sterolu - białko Niemann-Pick C1-Like 1 (NPC1L1), które odpowiada za wychwyt cholesterolu
i fitosteroli w przewodzie pokarmowym.
Ezetymib wiąże się z rąbkiem szczoteczkowym jelita cienkiego i hamuje wchłanianie cholesterolu. Prowadzi to do zmniejszenia ilości cholesterolu transportowanego do wątroby. Statyny zmniejszają syntezę cholesterolu w wątrobie. Ze względu na różne mechanizmy działania obu leków możliwe jest uzupełniające się zmniejszenie stężenia cholesterolu. W trwającym 2 tygodnie badaniu z udziałem 18 pacjentów
z hipercholesterolemią, ezetymib hamował wchłanianie cholesterolu z przewodu pokarmowego o 54% w porównaniu z placebo.
Przeprowadzono szereg badań przedklinicznych, aby określić czy działanie ezetymibu hamujące absorpcję cholesterolu jest wybiórcze. Ezetymib hamował wchłanianie cholesterolu znakowanego izotopem węgla 14C, bez wpływu na wchłanianie triglicerydów, kwasów tłuszczowych, kwasów żółciowych, progesteronu, etynyloestradiolu lub witamin rozpuszczalnych w tłuszczach, takich jak witaminy A i D.
Symwastatyna
Po podaniu doustnym, symwastatyna, będąca nieaktywnym laktonem, jest hydrolizowana w wątrobie do odpowiedniego, aktywnego beta-hydroksykwasu, który jest inhibitorem reduktazy HMG-CoA (reduktazy 3- hydroksy-3-metyloglutarylo-koenzymu A) o dużej aktywności. Enzym ten katalizuje przemianę HMG-CoA w mewalonian, wczesny etap przemian prowadzących do powstawania cholesterolu. Ograniczenie tego procesu ma wpływ na szybkość syntezy cholesterolu.
Wykazano, że symwastatyna powoduje zmniejszenie zarówno prawidłowego, jak i zwiększonego stężenia cholesterolu LDL. LDL składa się z protein o bardzo małej gęstości (VLDL) i katabolizowany jest poprzez receptor LDL o bardzo wysokim powinowactwie. Mechanizm zmniejszania stężenia cholesterolu LDL przez symwastatynę może być wynikiem zarówno zmniejszenia stężenia cholesterolu VLDL (VLDL-C), jak
i indukcji receptora LDL, co prowadzi do zmniejszenia wytwarzania oraz zwiększonego katabolizmu cholesterolu LDL. Podczas leczenia symwastatyną znacznie zmniejsza się stężenie apolipoproteiny B. Ponadto, symwastatyna powoduje niewielkie zwiększenie stężenia cholesterolu HDL oraz zmniejszenie stężenia TG w osoczu. W wyniku tych działań stosunek całkowitego cholesterolu do cholesterolu HDL oraz
cholesterolu LDL do cholesterolu HDL jest zmniejszony. Skuteczność kliniczna i bezpieczeństwo stosowania
W kontrolowanych badaniach klinicznych, stosowanie ezetymibu i symwastatyny wiązało się z istotnym zmniejszeniem stężenia cholesterolu całkowitego, cholesterolu LDL, Apo B, TG, i frakcji non-HDL-C oraz zwiększeniem stężenia cholesterolu HDL u pacjentów z hipercholesterolemią.
Zapobieganie wystąpieniu incydentów sercowo-naczyniowych
Wykazano, że ezetymib i symwastatyna zmniejsza ryzyko wystąpienia poważnych incydentów sercowo- naczyniowych u pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie.
Badanie IMPROVE-IT było wieloośrodkowym, randomizowanym badaniem klinicznym, z podwójnie ślepą próbą i grupą kontrolną przyjmującą aktywny lek, przeprowadzonym z udziałem 18 144 pacjentów włączonych do badania w ciągu 10 dni od hospitalizacji z powodu wystąpienia ostrego zespołu wieńcowego (OZW, tj. ostrego zawału mięśnia sercowego lub niestabilnej dławicy piersiowej). W czasie przyjęcia do szpitala z powodu OZW u pacjentów, którzy nie przyjmowali leczenia obniżającego poziom lipidów, stężenie cholesterolu LDL-C wynosiło ≤ 125 mg/dl (≤ 3,2 mmol/l), a u pacjentów, którzy przyjmowali leczenie obniżające poziom lipidów, wynosiło ≤ 100 mg/dl (≤ 2,6 mmol/l). Wszystkich pacjentów przydzielano w sposób losowy w stosunku 1:1 do grupy otrzymującej ezetymib z symwastatyną w dawce 10 mg+ 40 mg (n=9067) lub symwastatynę w dawce 40 mg (n=9077). Mediana okresu obserwacji wynosiła 6,0 lat.
Średnia wieku pacjentów wynosiła 63,6 lat. 76% pacjentów stanowili mężczyźni, 84% pacjentów było rasy kaukaskiej, a 27% pacjentów chorowało na cukrzycę. Średnie stężenie cholesterolu LDL-C w czasie wystąpienia incydentu kwalifikującego do badania u pacjentów przyjmujących leczenie obniżające poziom lipidów (n=6390) i u tych, którzy nie przyjmowali leczenia obniżającego poziom lipidów (n=11 594) wynosiło odpowiednio 80 mg/dl (2,1 mmol/l) i 101 mg/dl (2,6 mmol/l). Przed hospitalizacją z powodu wystąpienia OZW kwalifikującego do badania 34% pacjentów przyjmowało statynę. Po upływie 1 roku średnie stężenie cholesterolu LDL-C u pacjentów kontynuujących leczenie wynosiło 53,2 mg/dl (1,4 mmol/l) w grupie otrzymującej ezetymib i symwastatynę i 69,9 mg/dl (1,8 mmol/l) w grupie przyjmującej symwastatynę w monoterapii. Poziom lipidów badano na ogół u pacjentów, którzy w dalszym ciągu przyjmowali badane leczenie.
Na pierwszorzędowy punkt końcowy składał się zgon z przyczyn naczyniowo-sercowych, poważne incydenty wieńcowe (zdefiniowane jako zawał mięśnia sercowego niezakończony zgonem, udokumentowana niestabilna dławica piersiowa wymagająca hospitalizacji lub jakakolwiek procedura rewaskularyzacji wieńcowej przeprowadzona przynajmniej 30 dni po losowym przydzieleniu leczenia) oraz udar mózgu niezakończony zgonem. Badanie wykazało, że leczenie ezetymibem i symwastatyną pozwalało uzyskać dodatkowe korzyści w zmniejszeniu pierwszorzędowego złożonego punktu końcowego, tj. zgonu z przyczyn sercowo-naczyniowych, poważnego incydentu wieńcowego i udaru mózgu niezakończonego zgonem, w porównaniu z samą symwastatyną (zmniejszenie ryzyka względnego o 6,4%, p=0,016).
Pierwszorzędowy punkt końcowy wystąpił u 2572 z 9067 pacjentów (7-letni wskaźnik Kaplana-Meiera [KM] wynosił 32,72%) w grupie przyjmującej ezetymib i symwastatynę oraz u 2742 z 9077 pacjentów (7- letni wskaźnik KM wynosił 34,67%) w grupie otrzymującej samą symwastatynę (patrz Rycina 1 i Tabela 2.). Całkowita liczba zgonów w tej grupie podwyższonego ryzyka nie uległa zmianie (patrz Tabela 2).
Stwierdzono ogólną korzyść w przypadku wszystkich udarów mózgu; jakkolwiek odnotowano niewielkie, nieistotne zwiększenie liczby udarów krwotocznych w grupie ezetymib z symwastatyną w porównaniu do grupy pacjentów otrzymujących samą symwastatynę (patrz Tabela 2). Ryzyko udaru krwotocznego
w przypadku jednoczesnego podawania ezetymibu w skojarzeniu ze statyną o silniejszym działaniu nie zostało ocenione w długoterminowych badaniach.
Efekt leczenia ezetymibem z symwastatyną był na ogół spójny z efektami uzyskiwanymi w szeregu podgrup, tj. zależnie od płci, wieku, rasy, cukrzycy w wywiadzie, wyjściowego poziomu lipidów, wcześniejszego leczenia statyną, przebytego udaru mózgu i nadciśnienia.
Rycina 1: Wpływ leczenia ezetymibem i symwastatyną na pierwszorzędowy złożony punkt końcowy, tj. zgon z przyczyn sercowo-naczyniowych, poważny incydent wieńcowy lub udar mózgu niezakończony zgonem
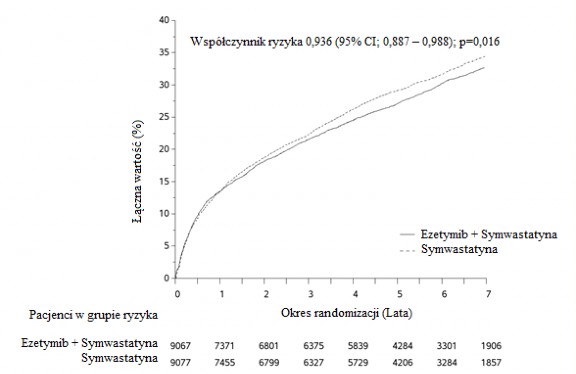
Tabela 2: Poważne incydenty sercowo-naczyniowe u wszystkich przydzielanych w sposób losowy pacjentów w badaniu IMPROVE-IT z podziałem na grupy leczenia.
Wynik | Ezetymib+Symwastatyna 10 mg+40 mga (N=9067) | Symwastatyna 40 mgb (N=9077) | Współczynnik ryzyka (95% CI) | Wartość p | ||
n | K-M(%)c | n | K- M(%) c | |||
Pierwszorzędowy złożony punkt końcowy skuteczności | ||||||
(zgon z przyczyn sercowo- | 2572 | 32,72 | 2742 | 34,67 | 0,936 | 0,016 |
naczyniowych, poważny | (0,887-0,988) | |||||
incydent wieńcowy i udar | ||||||
mózgu niezakończony | ||||||
zgonem) | ||||||
Drugorzędowe złożone punkty końcowe skuteczności | ||||||
Zgon związany z chorobą | 1322 | 17,52 | 1448 | 18,88 | 0,912 | 0,016 |
wieńcową, zawał mięśnia | (0,847-0,983) | |||||
sercowego niezakończony | ||||||
zgonem, rewaskularyzacja | ||||||
wieńcowa w trybie pilnym po | ||||||
30 dniach | ||||||
Poważny incydent wieńcowy, | 3089 | 38,65 | 3246 | 40,25 | 0,948 | 0,035 |
udar mózgu niezakończony | (0,903-0,996) | |||||
zgonem, zgon (z dowolnej | ||||||
przyczyny) | ||||||
Zgon z przyczyn sercowo- naczyniowych, zawał mięśnia sercowego niezakończony zgonem, niestabilna dławica piersiowa wymagająca hospitalizacji, jakakolwiek procedura rewaskularyzacji, udar mózgu niezakończony zgonem | 2716 | 34,49 | 2869 | 36,20 | 0,945 (0,897-0,996) | 0,035 |
Składowe pierwszorzędowego złożonego punktu końcowego skuteczności i wybrane punkty końcowe skuteczności (pierwsze wystąpienie określonego zdarzenia w dowolnym momencie) | ||||||
Zgon z przyczyn sercowo- naczyniowych | 537 | 6,89 | 538 | 6,84 | 1,000 (0,887-1,127) | 0,997 |
Poważny incydent wieńcowy: | ||||||
Zawał mięśnia sercowego niezakończony zgonem | 945 | 12,77 | 1083 | 14,41 | 0,871 (0,798- 0,950) | 0,002 |
Niestabilna dławica piersiowa wymagająca hospitalizacji | 156 | 2,06 | 148 | 1,92 | 1,059 (0,846- 1,326) | 0,618 |
Rewaskularyzacja wieńcowa po 30 dniach | 1690 | 21,84 | 1793 | 23,36 | 0,947 (0,886-1,012) | 0,107 |
Udar mózgu niezakończony zgonem | 245 | 3,49 | 305 | 4,24 | 0,802 (0,678-0,949) | 0,010 |
Wszystkie przypadki zawału mięśnia sercowego (zakończone i niezakończone zgonem) | 977 | 13,13 | 1118 | 14,82 | 0,872 (0,800-0,950) | 0,002 |
Wszystkie przypadki udaru (zakończone i niezakończone zgonem) | 296 | 4,16 | 345 | 4,77 | 0,857 (0,734-1,001) | 0,052 |
Udar niekrwotocznyd | 242 | 3,48 | 305 | 4,23 | 0,793 (0,670-0,939) | 0,007 |
Udar krwotoczny | 59 | 0,77 | 43 | 0,59 | 1,377 (0,930-2,040) | 0,110 |
Zgon z dowolnej przyczyny | 1215 | 15,36 | 1231 | 15,28 | 0,989 (0,914-1,070) | 0,782 |
a
u 6% pacjentów dawkę ezetymibu + symwastatyny zwiększono do 10 mg + 80 mg.
b
u 27% pacjentów dawkę symwastatyny zwiększono do 80 mg.
c
estymator Kaplana-Meiera po 7 latach.
d
w tym udar niedokrwienny mózgu lub udar niekreślonego rodzaju.
Pierwotna hipercholesterolemia
W trwającym 8 tygodni badaniu z podwójnie ślepą próbą, kontrolowanym placebo, 240 pacjentów
z hipercholesterolemią stosujących symwastatynę w monoterapii, u których nie uzyskano docelowego stężenia cholesterolu LDL-C (2,6 do 4,1 mmol/l [100 do 160 mg/dl], w zależności od stężenia początkowego) według amerykańskiego National Cholesterol Education Program (NCEP), randomizowano do grupy otrzymującej ezetymib w dawce 10 mg jednocześnie z symwastatyną lub do grupy otrzymującej placebo, w połączeniu z aktualnie stosowaną symwastatyną. W przypadku pacjentów leczonych symwastatyną, u których w punkcie wyjścia nie uzyskano docelowego stężenia cholesterolu LDL-C (~80%), w punkcie końcowym badania stwierdzono docelowe stężenie cholesterolu LDL-C u istotnie większej liczby pacjentów stosujących ezetymib jednocześnie z symwastatyną, niż u pacjentów przyjmujących placebo jednocześnie z symwastatyną, odpowiednio u 76% i 21,5%.
Zmniejszenie stężenia cholesterolu LDL-C u pacjentów stosujących ezetymib lub placebo jednocześnie z symwastatyną także różniło się istotnie w obydwu grupach (odpowiednio 27% lub 3%). Ponadto, jednoczesne podawanie ezetymibu i symwastatyny wiązało się z istotnym zmniejszeniem stężenia cholesterolu całkowitego-C, Apo B oraz TG w porównaniu ze stosowaniem placebo jednocześnie
z symwastatyną.
W wieloośrodkowym, trwającym 24 tygodnie badaniu z podwójnie ślepą próbą, 214 pacjentów z cukrzycą typu 2 leczonych produktem należącym do grupy tiazolidynodionów (rozyglitazon lub pioglitazon) przez co najmniej 3 miesiące i symwastatyną w dawce 20 mg przez co najmniej 6 tygodni, u których średnie stężenie cholesterolu LDL wynosiło 2,4 mmol/l (93 mg/dl), randomizowano do grupy otrzymującej symwastatynę
w dawce 40 mg lub składniki aktywne równoważne ezetymib i symwastatynę w dawce 10 mg + 20 mg.
Ezetymib i symwastatyna w dawce 10 mg + 20 mg działały znacznie skuteczniej niż symwastatyna
w podwójnej dawce wynoszącej 40 mg pod względem dalszego zmniejszenia stężenia cholesterolu LDL-C (odpowiednio -21% i 0%), cholesterolu całkowitego-C (odpowiednio -14% i -1%), Apo B (odpowiednio - 14% i -2%) oraz frakcji nie-HDL-C (odpowiednio -20% i -2%) w porównaniu ze zmniejszeniem zaobserwowanym w przypadku stosowania symwastatyny w dawce 20 mg. Różnice w stężeniach cholesterolu HDL-C i TG pomiędzy 2 leczonymi grupami nie były istotne. Na wyniki nie miał wpływu rodzaj stosowanego produktu z grupy tiazolidynodionów.
Skuteczność działania różnych dawek ezetymibu i symwastatyny (10 mg + 10 mg do 10 mg + 80 mg na dobę) wykazano w wieloośrodkowym, trwającym 12 tygodni badaniu z podwójnie ślepą próbą, kontrolowanym placebo, w którym oceniano wszystkie dostępne dawki ezetymibu i symwastatyny oraz wszystkie istotne dawki symwastatyny. Porównując wyniki uzyskane w grupie pacjentów stosujących wszystkie dawki ezetymibu i symwastatyny z wynikami uzyskanymi w grupie pacjentów stosujących symwastatynę we wszystkich dawkach stwierdzono, że stosowanie ezetymibu i symwastatyny wiązało się z istotnym zmniejszeniem stężenia cholesterolu całkowitego-C, cholesterolu LDL-C i TG (patrz Tabela 3), jak również Apo B (odpowiednio -42% i -29%), frakcji nie-HDL-C (odpowiednio -49% i 34%) i białka C- reaktywnego (odpowiednio -33% i -9%). Wpływ stosowania ezetymibu i symwastatyny na stężenie cholesterolu HDL-C był zbliżony do wpływu symwastatyny. Dalsza analiza wykazała, że stosowanie
ezetymibu i symwastatyny wiązało się z istotnym zwiększeniem stężenia cholesterolu HDL-C w porównaniu z placebo.
Tabela 3: Reakcja na zastosowanie ezetymibu i symwastatyny u pacjentów z pierwotną hipercholesterolemią (Średnia a % zmiana w stosunku do wartości wyjściowychb)
Leczenie | |||||
(dawka dobowa) | N | Total-C | LDL-C | HDL-C | TGa |
Dane łączne (wszystkie dawki ezetymibu i symwastatyny)c | 353 | -38 | -53 | +8 | -28 |
Dane łączne (wszystkie dawki symwastatyny)c | 349 | -26 | -38 | +8 | -15 |
Ezetymib 10 mg | 92 | -14 | -20 | +7 | -13 |
Placebo | 93 | +2 | +3 | +2 | -2 |
Ezetymib i symwastatyna w dawkach | |||||
10 mg + 10 mg | 87 | -32 | -46 | +9 | -21 |
10 mg + 20 mg | 86 | -37 | -51 | +8 | -31 |
10 mg + 40 mg | 89 | -39 | -55 | +9 | -32 |
10 mg + 80 mg | 91 | -43 | -61 | +6 | -28 |
Symwastatyna w dawkach | |||||
10 mg | 81 | -21 | -31 | +5 | -4 |
20 mg | 90 | -24 | -35 | +6 | -14 |
40 mg | 91 | -29 | -42 | +8 | -19 |
80 mg | 87 | -32 | -46 | +11 | -26 |
a Dla triglicerydów, średnia % zmiana w stosunku do wartości wyjściowych.
b Wartości wyjściowe – w przypadku pacjentów niestosujących leków zmniejszających stężenie lipidów.
c Ezetymib i symwastatyna we wszystkich dawkach (10 mg + 10 mg – 10 mg + 80 mg) spowodował istotne zmniejszenie stężenia cholesterolu całkowitego-C, cholesterolu LDL-C i TG w porównaniu
z symwastatyną, a także istotne zwiększenie stężenia cholesterolu HDL-C w porównaniu z placebo.
W podobnie zaprojektowanym badaniu, wyniki dotyczące wszystkich parametrów związanych ze stężeniem lipidów były na ogół zgodne. Łączna analiza danych pochodzących z 2 wyżej wymienionych badań wykazała, że reakcja na leczenie ezetymibem i symwastatyną była zbliżona w przypadku pacjentów,
u których stężenie triglicerydów (TG) było wyższe lub niższe niż 200 mg/dl.
W wieloośrodkowym badaniu klinicznym z podwójnie ślepą próbą i grupą kontrolną (ENHANCE) 720 pacjentów chorych na heterozygotyczną hipercholesterolemię rodzinną randomizowano do grupy leczonej przez 2 lata ezetymibem w dawce 10 mg w skojarzeniu z symwastatyną w dawce 80 mg (n = 357) lub symwastatyną w monoterapii w dawce 80 mg (n = 363). Celem pierwszoplanowym tego badania była ocena wpływu stosowania terapii skojarzonej ezetymibem i symwastatyną na grubość błony środkowo- wewnętrznej (ang. IMT, intima-media thickness) tętnicy szyjnej w porównaniu z symwastatyną stosowaną w monoterapii. Dotychczas nie wykazano wpływu tego markera zastępczego na wskaźniki zachorowalności i śmiertelności z powodu chorób układu sercowo-naczyniowego.
Na podstawie wyników pomiarów wykonywanych podczas badań ultradźwiękowych (USG) w projekcji B nie wykazano istotnych różnic między 2 grupami leczonymi (p = 0,29) w zakresie pierwszoplanowego punktu końcowego – zmiany średniej wartości IMT we wszystkich 6 ocenianych odcinkach tętnicy szyjnej.
W okresie 2 lat badania grubość błony środkowo-wewnętrznej (IMT) tętnicy szyjnej zwiększyła się
o 0,0111 mm i 0,0058 mm odpowiednio w grupie leczonej ezetymibem w dawce 10 mg w skojarzeniu z symwastatyną w dawce 80 mg i w grupie leczonej symwastatyną w monoterapii w dawce wynoszącej 80 mg (średnia wyjściowa wartość IMT tętnicy szyjnej wynosiła odpowiednio 0,68 mm i 0,69 mm).
Leczenie ezetymibem w dawce 10 mg w skojarzeniu z symwastatyną w dawce 80 mg wiązało się z istotnie większym obniżeniem stężeń frakcji cholesterolu LDL-C, cholesterolu całkowitego-C, Apo B oraz TG
w porównaniu z monoterapią symwastatyną w dawce 80 mg. Procentowy wzrost stężenia frakcji cholesterolu HDL-C był zbliżony w 2 grupach leczonych. Działania niepożądane zgłaszane w grupie leczonej ezetymibem w dawce 10 mg w skojarzeniu z symwastatyną w dawce 80 mg odpowiadały ustalonemu profilowi bezpieczeństwa tej terapii skojarzonej.
W skład produktu leczniczego Axocar wchodzi symwastatyna. Wpływ leczenia symwastatyną u pacjentów należących do grupy wysokiego ryzyka wystąpienia incydentów wieńcowych z powodu stwierdzonej choroby niedokrwiennej serca, cukrzycy, choroby naczyń obwodowych oraz udaru lub innej choroby naczyń mózgowych oceniano w 2 dużych badaniach klinicznych kontrolowanych placebo: Scandinavian Simvastatin Survival Study (20-40 mg; N=4444 pacjentów) oraz Heart Protection Study (40 mg; N=20 536 pacjentów). W badaniach tych dowiedziono, iż stosowanie symwastatyny wiąże się ze zmniejszeniem: ryzyka całkowitej śmiertelności wskutek zmniejszenia liczby zgonów w wyniku choroby wieńcowej; ryzyka wystąpienia zawału i udaru bez skutku śmiertelnego; oraz konieczności wykonania procedur rewaskularyzacji naczyń wieńcowych i innych naczyń krwionośnych.
W badaniu Study of Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) oceniano wpływ leczenia symwastatyną w dawce 80 mg vs leczenie dawką 20 mg (mediana czasu obserwacji kontrolnej – 6,7 lat) na występowanie poważnych zdarzeń naczyniowych (MVE, zdefiniowanych jako choroba wieńcowa zakończona zgonem, zawał mięśnia sercowego bez skutku śmiertelnego, procedura rewaskularyzacji naczyń wieńcowych, udar mózgu ze skutkiem śmiertelnym lub bez lub procedura rewaskularyzacji naczyń obwodowych) u 12 064 pacjentów po przebytym zawale mięśnia sercowego. Nie stwierdzono istotnej różnicy częstości występowania MVE między 2 grupami leczonymi; symwastatyną
w dawce 20 mg (n = 1553; 25,7%) w porównaniu z symwastatyną w dawce 80 mg (n = 1477; 24,5%); RR 0,94, 95% CI: 0,88 do 1,01. Bezwzględna różnica stężenia frakcji cholesterolu LDL-C odnotowana między 2 grupami leczonymi w przebiegu badania wyniosła 0,35 ± 0,01 mmol/l. Profil bezpieczeństwa był zbliżony
w 2 grupach, poza częstością występowania miopatii wynoszącą około 1,0% w grupie leczonej
symwastatyną w dawce 80 mg w porównaniu z 0,02% w grupie leczonej symwastatyną w dawce 20 mg. Około połowę z tych przypadków miopatii stwierdzono w pierwszym roku leczenia. Częstość występowania miopatii w każdym kolejnym roku leczenia wyniosła około 0,1%.
Populacja pediatryczna
W wieloośrodkowym badaniu z podwójnie ślepą próbą i grupą kontrolną, 142 chłopców (w fazie II i powyżej w skali Tannera) oraz 106 dziewcząt, u których wystąpiła już pierwsza miesiączka, w wieku 10 do 17 lat (średnia wieku 14,2 roku) z heterozygotyczną hipercholesterolemią rodzinną (HeFH) i stężeniem wyjściowym cholesterolu LDL-C pomiędzy 4,1 a 10,4 mmol/l przydzielono losowo albo do grupy przyjmującej ezetymib w dawce 10 mg w skojarzeniu z symwastatyną (10, 20 lub 40 mg), albo do grupy otrzymującej wyłącznie symwastatynę (10, 20 lub 40 mg) przez okres 6 tygodni, ezetymib podawany
w skojarzeniu z symwastatyną w dawce 40 mg lub symwastatynę w monoterapii w dawce 40 mg przez okres kolejnych 27 tygodni, a następnie metodą terapii otwartej ezetymibem w skojarzeniu z symwastatyną (10, 20 lub 40 mg) przez kolejne 20 tygodni.
W 6. tygodniu wykazano, że ezetymib stosowany w skojarzeniu z symwastatyną (we wszystkich dawkach) znacząco obniżał stężenie cholesterolu całkowitego-C (38% w porównaniu z 26%), cholesterolu LDL-C (49% w porównaniu z 34%), Apo B (39% w porównaniu z 27%) i nie-HDL-C (47% w porównaniu z 33%) w porównaniu z symwastatyną w monoterapii (we wszystkich dawkach). W odniesieniu do stężenia triglicerydów (TG) i cholesterolu HDL-C wyniki otrzymane w 2 grupach leczonych były zbliżone (odpowiednio -17% w porównaniu z -12% oraz +7% w porównaniu z +6%). W tygodniu 33. uzyskane wyniki były zgodne z otrzymanymi w tygodniu 6. i znacząco większa liczba pacjentów otrzymujących ezetymib w skojarzeniu z symwastatyną w dawce 40 mg (62%), osiągnęła idealną docelową wartość stężenia cholesterolu LDL-C według kryteriów NCEP AAP (< 2,8 mmol/l [110 mg/dl]) w porównaniu z pacjentami przyjmującymi wyłącznie symwastatynę w dawce 40 mg (25%). W tygodniu 53., pod koniec fazy przedłużenia badania prowadzonego metodą terapii otwartej, utrzymywały się wyniki leczenia w zakresie parametrów lipidowych.
Nie przeprowadzono oceny bezpieczeństwa stosowania i skuteczności leczenia skojarzonego ezetymibem i symwastatyną w dawkach przekraczających 40 mg na dobę u dzieci w wieku 10 do 17 lat. Nie przeprowadzono oceny długoterminowej skuteczności leczenia ezetymibem u pacjentów poniżej 17 lat
w zakresie zmniejszenia zachorowalności i śmiertelności w wieku dojrzałym.
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań skojarzenia ezetymibu
i symwastatyny we wszystkich podgrupach populacji pediatrycznej w hipercholesterolemii (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Homozygotyczna hipercholesterolemia rodzinna
Przeprowadzono trwające 12 tygodni, randomizowane badanie kliniczne z podwójnie ślepą próbą,
z udziałem pacjentów z homozygotyczną hipercholesterolemią rodzinną rozpoznaną na podstawie objawów klinicznych i (lub) badań genotypu. Analiza obejmowała dane uzyskane w podgrupie pacjentów (n=14) otrzymujących w punkcie wyjścia symwastatynę w dawce 40 mg. Zwiększenie dawki symwastatyny z 40 do 80 mg (n=5) spowodowało zmniejszenie stężenia cholesterolu LDL-C o 13% w stosunku do wartości wyjściowych, gdy symwastatyna podawana była w dawce 40 mg. Jednoczesne podawanie ezetymibu
i symwastatyny, równoważne stosowaniu ezetymibu i symwastatyny (10 mg + 40 mg i 10 mg + 80 mg łącznie, n=9), wiązało się ze zmniejszeniem stężenia cholesterolu LDL-C o 23% względem wartości wyjściowych, gdy symwastatyna podawana była w dawce 40 mg. U tych pacjentów otrzymujących jednocześnie ezetymib i symwastatynę, równoważnych leczeniu ezetymibem z symwastatyną (10 mg +
80 mg, n=5) uzyskano zmniejszenie stężenia cholesterolu LDL-C o 29% względem wartości wyjściowych, gdy symwastatyna podawana była w dawce 40 mg.
Zapobieganie poważnym zdarzeniom naczyniowym w przewlekłej chorobie nerek (ang. CKD, chronic kidney disease)
Badanie Study of Heart and Renal Protection (SHARP) było wielonarodowym badaniem z randomizacją, grupą kontrolną placebo i podwójnie ślepą próbą przeprowadzonym z udziałem 9438 pacjentów z przewlekłą chorobą nerek, z których 1/3 stosowała dializoterapię w punkcie wyjścia. W sumie 4650 pacjentów
przydzielono do grupy ezetymib i symwastatynę w dawce 10 mg + 20 mg, a 4620 do grupy placebo i prowadzono obserwację średnio przez 4,9 roku. Średnia wieku pacjentów wyniosła 62 lata, 63% uczestników stanowili mężczyźni, 72% osób było rasy kaukaskiej, u 23% stwierdzono cukrzycę,
a w przypadku pacjentów niedializowanych średni szacunkowy wskaźnik filtracji kłębuszkowej (ang. eGFR, estimated glomerular filtration rate) wynosił 26,5 ml/min/1,73 m2. W kryteriach przystąpienia do badania nie uwzględniono parametrów lipidowych. Średnie wyjściowe stężenie frakcji cholesterolu LDL-C wynosiło 108 mg/dl. Od oznaczenia wykonanego w 1. roku stężenie cholesterolu LDL-C zmniejszyło się względem placebo o 26% w grupie leczonej symwastatyną w monoterapii w dawce wynoszącej 20 mg i o 38%
w grupie stosującej ezetymib i symwastatynę w dawce 10 mg + 20 mg.
Podstawowe porównanie określone w protokole badania SHARP stanowiła analiza ITT (zgodna
z zaplanowanym leczeniem) „poważnych zdarzeń naczyniowych” (ang. MVE, major vascular events; zdefiniowanych jako zawał mięśnia sercowego bez skutku śmiertelnego lub zgon z przyczyn sercowych, udar mózgu lub jakakolwiek procedura rewaskularyzacji) wyłącznie u pacjentów randomizowanych na początku do grupy leczonej ezetymibem i symwastatyną (n=4193) lub do grupy placebo (n=4191).
W analizach dodatkowych uwzględniono tę samą kombinację analizowaną w całej kohorcie osób randomizowanych (w punkcie wyjścia lub po upływie 1 roku) do grupy leczonej ezetymibem
i symwastatyną (n=4650) lub do grupy placebo (n=4620), a także poszczególne elementy tej kombinacji.
W analizie pierwszorzędowego punktu końcowego wykazano, że stosowanie ezetymibu i symwastatyny wiązało się z istotnym zmniejszeniem ryzyka wystąpienia poważnych zdarzeń naczyniowych (749 pacjentów ze stwierdzonymi zdarzeniami w grupie placebo w porównaniu z 639 przypadkami w grupie leczonej ezetymibem i symwastatyną), przy czym ryzyko względne zmniejszyło się o 16% (p=0,001).
W planie tego badania nie uwzględniono jednak odrębnie udziału jednoskładnikowego ezetymibu w skuteczności w istotnym zmniejszaniu ryzyka występowania poważnych zdarzeń naczyniowych u pacjentów z przewlekłą chorobą nerek (ang. CKD).
W Tabeli 4 przedstawiono poszczególne elementy parametru poważnych zdarzeń naczyniowych (ang. MVE) stwierdzone u wszystkich randomizowanych pacjentów. Stosowanie ezetymibu i symwastatyny wiązało się z istotnym zmniejszeniem ryzyka wystąpienia udaru mózgu oraz wykonania jakiejkolwiek procedury rewaskularyzacji, przy nieistotnych różnicach liczbowych na korzyść ezetymibu i symwastatyny
w odniesieniu do zdarzeń w postaci zawału mięśnia sercowego bez skutku śmiertelnego i zgonu z przyczyn sercowych.
Tabela 4: Poważne zdarzenia naczyniowe w zależności do grupy leczonej stwierdzone u wszystkich pacjentów randomizowanych w badaniu SHARPa
Rezultat | Ezetymib i symwastatyna, 10 mg + 20 mg (N=4650) | Placebo (N=4620) | Współczynnik ryzyka (95% CI) | Wartość p |
Poważne zdarzenia naczyniowe | 701 (15,1%) | 814 (17,6%) | 0,85 (0,77-0,94) | 0,001 |
Zawał mięśnia sercowego bez skutku śmiertelnego | 134 (2,9%) | 159 (3,4%) | 0,84 (0,66-1,05) | 0,12 |
Zgon z przyczyn sercowych | 253 (5,4%) | 272 (5,9%) | 0,93 (0,78-1,10) | 0,38 |
Jakikolwiek udar mózgu | 171 (3,7%) | 210 (4,5%) | 0,81 (0,66-0,99) | 0,038 |
Niekrwotoczny udar mózgu | 131 (2,8%) | 174 (3,8%) | 0,75 (0,60-0,94) | 0,011 |
Krwotoczny udar mózgu | 45 (1,0%) | 37 (0,8%) | 1,21 (0,78-1,86) | 0,40 |
Jakakolwiek procedura rewaskularyzacji | 284 (6,1%) | 352 (7,6%) | 0,79 (0,68-0,93) | 0,004 |
Poważne zdarzenia miażdżycowe (ang. MAE, Major Atherosclerotic Events)b | 526 (11,3%) | 619 (13,4%) | 0,83 (0,74-0,94) | 0,002 |
aAnaliza ITT z uwzględnieniem wszystkich uczestników badania SHARP randomizowanych w punkcie
wyjścia lub po upływie 1 roku do grupy leczonej ezetymibem i symwastatyną lub do grupy placebo.
b
MAE; zdefiniowane jako kombinacja zawału mięśnia sercowego bez skutku śmiertelnego, zgonu
z przyczyn wieńcowych, niekrwotocznego udaru mózgu lub jakiejkolwiek procedury rewaskularyzacji.
W przypadku pacjentów, u których stężenie cholesterolu LDL-C było mniejsze (< 2,5 mmol/l) w punkcie wyjścia, oraz pacjentów poddawanych dializie w punkcie wyjścia całkowite zmniejszenie stężenia frakcji LDL cholesterolu uzyskane za pomocą ezetymibu i symwastatyny było mniejsze niż w przypadku pozostałych pacjentów, a zmniejszenie ryzyka w 2 tych grupach było analogicznie osłabione.
Zwężenie aorty
Badanie Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis (SEAS) było trwającym średnio 4,4 roku wieloośrodkowym badaniem z podwójnie ślepą próbą i grupą kontrolną placebo, które przeprowadzono z udziałem 1873 pacjentów z bezobjawowym zwężeniem aorty (ang. AS, aortic stenosis), udokumentowanym na podstawie mierzonej metodą Dopplera szczytowej prędkości przepływu w aorcie mieszczącej się w zakresie od 2,5 do 4,0 m/s. Do badania włączono wyłącznie pacjentów, w przypadku których stwierdzono brak konieczności leczenia statynami w celu zmniejszenia ryzyka rozwoju miażdżycy układu sercowo-naczyniowego. Pacjentów randomizowano w stosunku liczbowym 1:1 do grupy przyjmującej placebo lub do grupy leczonej ezetymibem w dawce 10 mg w skojarzeniu z symwastatyną
w dawce wynoszącej 40 mg na dobę.
Pierwszorzędowym punktem końcowym badania był punkt złożony w postaci poważnych zdarzeń sercowo- naczyniowych (ang. MCE, major cardiovascular events), takich jak zgon z przyczyn sercowo-naczyniowych, operacja wymiany zastawki aortalnej (ang. AVR, aortic valve replacement), zastoinowa niewydolność serca (ang. CHF, congestive heart failure) w wyniku postępującego zwężenia aorty, zawał mięśnia sercowego bez skutku śmiertelnego, operacja pomostowania aortalno wieńcowego (ang. CABG, coronary artery bypass grafting), przezskórna interwencja wieńcowa (ang. PCI, percutaneous coronary intervention), hospitalizacja z powodu niestabilnej dusznicy bolesnej oraz udar mózgu inny niż krwotoczny. Najważniejsze drugorzędowe punkty końcowe stanowiły połączone w podgrupach kategorie zdarzeń będących elementami pierwszorzędowego punktu końcowego.
W porównaniu z placebo leczenie skojarzeniem ezetymibu z symwastatyną w dawce 10 mg + 40 mg nie wiązało się z istotnym zmniejszeniem ryzyka MCE.
Pierwszorzędowy punkt końcowy stwierdzono u 333 pacjentów (35,3%) z grupy leczonej skojarzeniem ezetymib i symwastatyna i u 355 pacjentów (38,2%) z grupy placebo (współczynnik ryzyka (HR) w grupie leczonej skojarzeniem ezetymib i symwastatyna, 0,96; 95% CI, 0,83 - 1,12; p = 0,59). Operację wymiany zastawki aortalnej przeprowadzono u 267 pacjentów (28,3%) z grupy leczonej skojarzeniem ezetymib
i symwastatyna i u 278 pacjentów (29,9%) z grupy placebo (HR 1,00; 95% CI, 0,84 - 1,18; p = 0,97). Niedokrwienne zdarzenia sercowo naczyniowe wystąpiły u mniejszej liczby pacjentów z grupy leczonej skojarzeniem ezetymib i symwastatyna (n=148) w porównaniu z grupą pacjentów przyjmujących placebo (n=187) (HR 0,78; 95% CI, 0,63 - 0,97; p = 0,02), głównie ze względu na mniejszą liczbę pacjentów,
u których wykonano operację pomostowania aortalno-wieńcowego.
W grupie leczonej skojarzeniem ezetymib i symwastatyna stwierdzono częstsze występowanie zmian nowotworowych (105 vs 70, p = 0,01). Istotność kliniczna tej obserwacji jest niepewna, ponieważ w większym badaniu SHARP całkowita liczba pacjentów, u których wystąpiły nowotwory (438 w grupie leczonej skojarzeniem ezetymib i symwastatyna vs 439 w grupie otrzymującej placebo) nie różniła się. Ponadto w badaniu IMPROVE-IT całkowita liczba pacjentów z nowo zdiagnozowanym nowotworem złośliwym (853 w grupie ezetymib i symwastatyna vs. 863 w grupie otrzymującej tylko symwastatynę) nie różniła się znacząco, a zatem wyniki uzyskane w badaniu SEAS nie zostały potwierdzone przez badanie SHARP lub IMPROVE-IT.
Nie stwierdzono istotnych klinicznie interakcji farmakokinetycznych podczas jednoczesnego stosowania ezetymibu i symwastatyny.
Wchłanianie
Ezetymib z symwastatyną
Stosowanie skojarzenia ezetymibu z symwastanyną jest biorównoważne z jednoczesnym stosowaniem ezetymibu i symwastatyny.
Ezetymib
Po podaniu doustnym ezetymib jest szybko wchłaniany i w znacznym stopniu sprzęgany do postaci czynnego farmakologicznie glukuronidu fenolowego (glukuronid ezetymibu). Średnie maksymalne stężenia (Cmax) występują w ciągu 1 do 2 godzin w przypadku glukuronidu ezetymibu i w ciągu 4 do 12 godzin w przypadku ezetymibu. Nie można określić bezwzględnej biodostępności ezetymibu, ponieważ substancja ta jest prawie całkowicie nierozpuszczalna w roztworach wodnych, które mogą być stosowane do wstrzykiwań.
Jednoczesne przyjmowanie pokarmów (z dużą lub małą zawartością tłuszczu) nie ma wpływu na biodostępność ezetymibu podczas stosowania w postaci tabletek 10 mg.
Symwastatyna
Po podaniu doustnym symwastatyny, wchłanianie beta-hydroksykwasu do krążenia ogólnego było mniejsze niż 5% dawki, wynika to ze znacznego jej wychwytu w wątrobie. Głównymi metabolitami symwastatyny obecnymi w ludzkim osoczu są beta-hydroksykwas i 4 dodatkowe aktywne metabolity.
W porównaniu do stanu na czczo, profil osoczowy aktywnych i całkowitych inhibitorów nie zależał od tego czy symwastatyna była podawana bezpośrednio przed posiłkiem testowym.
Dystrybucja
Ezetymib
Ezetymib i glukuronid ezetymibu wiążą się z białkami ludzkiego osocza odpowiednio w 99,7% i 88 do 92%.
Symwastatyna
Symwastatyna i beta-hydroksykwas są wiązane z białkiem ludzkiego osocza (95%).
Farmakokinetyka po podaniu pojedynczych i wielokrotnych dawek symwastatyny wskazuje na brak kumulacji po wielokrotnym podaniu. We wszystkich wyżej wymienionych badaniach farmakokinetycznych, maksymalne stężenie inhibitorów w osoczu wystąpiło między 1,3 a 2,4 godziną po podaniu dawki.
Metabolizm
Ezetymib
Ezetymib jest metabolizowany głównie w jelicie cienkim i w wątrobie poprzez sprzęganie z kwasem glukuronowym (reakcja II fazy), a następnie wydalany z żółcią. U wszystkich badanych gatunków obserwowano minimalny metabolizm oksydacyjny (reakcja I fazy). Ezetymib i glukuronid ezetymibu są głównymi pochodnymi leku wykrywanymi w osoczu, stanowiąc odpowiednio około 10 do 20% i 80 do 90% całkowitego stężenia leku w osoczu. Zarówno ezetymib, jak i glukuronid ezetymibu są powoli usuwane
z osocza, w wyniku znacznego krążenia jelitowo-wątrobowego. Okres półtrwania ezetymibu i glukuronidu ezetymibu wynosi około 22 godziny.
Symwastatyna
Symwastatyna jest nieaktywnym laktonem, który in vivo łatwo ulega hydrolizie do odpowiedniego beta- hydroksykwasu, silnego inhibitora reduktazy HMG-CoA. Proces hydrolizy odbywa się głównie w wątrobie; w osoczu ludzkim przebiega bardzo powoli.
Symwastatyna jest dobrze wchłaniana i znaczny jej wychwyt następuje w wątrobie. Wychwyt w wątrobie zależy od przepływu krwi w wątrobie. Symwastatyna działa przede wszystkim w wątrobie, a następnie metabolity wydzielane są z żółcią. Wskutek tego, dostępność aktywnego leku w układzie krążenia jest bardzo mała.
Po dożylnym podaniu metabolitu beta-hydroksykwasu, okres półtrwania wynosił przeciętnie 1,9 godziny.
Eliminacja
Ezetymib
Po doustnym podaniu ludziom ezetymibu znakowanego [14C] (20 mg), całkowity ezetymib w osoczu krwi stanowił około 93% całkowitej aktywności promieniotwórczej. W stolcu i w moczu wykrywano odpowiednio około 78% i 11% całkowitej dawki izotopu promieniotwórczego w okresie 10-dniowej zbiórki.
Po 48 godzinach od podania nie stwierdzono wykrywalnego poziomu aktywności promieniotwórczej w osoczu.
Symwastatyna
Kwas symwastatyny jest aktywnie wprowadzany do hepatocytów przez białko transportujące OATP1B1.
Symwastatyna jest substratem transportera pompy lekowej – białka warunkującego oporność w raku piersi (BCRP).
Po doustnym podaniu ludziom znakowanej symwastatyny, w ciągu 96 godzin 13% dawki wydzielane jest z moczem a 60% z kałem. Zawartość wykryta w kale odpowiadała ilości wchłoniętych metabolitów, które zostały wydzielone z żółcią oraz lekowi niewchłoniętemu. Po podaniu dożylnym metabolitu beta- hydroksykwasu, przeciętnie tylko 0,3% dawki podanej dożylnie było wydalane z moczem w postaci inhibitorów.
Szczególne grupy pacjentów
Populacja pediatryczna
Wchłanianie i metabolizm ezetymibu są zbliżone u dzieci i młodzieży (10 do 18 lat) oraz u dorosłych. Nie stwierdzono znamiennych różnic pod względem właściwości farmakokinetycznych pomiędzy młodzieżą a dorosłymi w przeliczeniu na całkowity ezetymib. Brak danych dotyczących właściwości farmakokinetycznych w populacji dzieci w wieku < 10 lat. Doświadczenie kliniczne u dzieci i młodzieży obejmuje pacjentów z homozygotyczną hipercholesterolemią rodzinną, heterozygotyczną hipercholesterolemią rodzinną lub sitosterolemią (patrz punkt 4.2).
Pacjenci w podeszłym wieku
Stężenie ezetymibu całkowitego w osoczu krwi u osób w wieku podeszłym (≥65 lat) jest około 2 razy większe niż u osób młodych (18 do 45 lat). Zmniejszenie stężenia cholesterolu LDL-C oraz profil bezpieczeństwa są porównywalne podczas stosowania ezetymibu u osób w podeszłym wieku i u osób młodszych (patrz punkt 4.2).
Zaburzenia czynności wątroby
Po podaniu ezetymibu w pojedynczej dawce 10 mg, średnia wartość AUC całkowitego ezetymibu była zwiększona o około 1,7-krotnie u pacjentów z łagodnymi zaburzeniami czynności wątroby (5-6 punktów wg skali Child-Pugh) w porównaniu z osobami zdrowymi. W 14-dniowym badaniu, w którym podawano dawki wielokrotne leku (10 mg na dobę) pacjentom z umiarkowanymi zaburzeniami czynności wątroby (7-
9 punktów wg skali Child-Pugh), stwierdzono około 4-krotne zwiększenie średniego AUC całkowitego ezetymibu pomiędzy dobą 1. i 14. w porównaniu z osobami zdrowymi. Nie jest konieczne dostosowanie dawkowania u pacjentów z łagodnymi zaburzeniami czynności wątroby. Ze względu na to, że nie jest znany wpływ zwiększonego narażenia na ezetymib u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby (> 9 punktów wg skali Child-Pugh) nie zaleca się stosowania ezetymibu u tych pacjentów (patrz punkty 4.2 i 4.4).
Zaburzenia czynności nerek Ezetymib
Po podaniu ezetymibu w pojedynczej dawce 10 mg pacjentom z ciężką chorobą nerek (n=8, średni
CrCl ≤ 30 ml/min), średnie AUC całkowitego ezetymibu zwiększyło się około 1,5- krotnie w porównaniu z osobami zdrowymi (n=9) (patrz punkt 4.2).
U dodatkowego pacjenta uczestniczącego w tym badaniu (stan po przeszczepieniu nerki, otrzymującego wiele leków, w tym cyklosporynę) stężenie ezetymibu całkowitego zwiększyło się 12-krotnie.
Symwastatyna
W badaniu, w którym uczestniczyli pacjenci z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny
< 30 ml/min) stężenia całkowite inhibitorów w osoczu po podaniu pojedynczej dawki inhibitora reduktazy HMG-CoA były około 2-krotnie większe niż u zdrowych ochotników.
Płeć
U kobiet stwierdzono nieco większe (około < 20%) stężenia całkowite ezetymibu w osoczu niż u mężczyzn.
Nie stwierdzono różnic pod względem zmniejszenia stężenia cholesterolu LDL i profilu bezpieczeństwa u mężczyzn i kobiet leczonych ezetymibem.
Polimorfizm genu SLCO1B1
Osoby posiadające allel c.521T>C genu SLCO1B1 mają mniejszą aktywność białka transportującego OATP1B1. Średnia ekspozycja (AUC) na główny aktywny metabolit, kwas symwastatyny, ma wartość 120% u heterozygotycznych nosicieli allelu C (CT) oraz 221% u nosicieli homozygotycznych (CC)
w stosunku do pacjentów o najbardziej powszechnym genotypie (TT). W populacji europejskiej allel C występuje z częstotliwością 18%. U pacjentów z polimorfizmem genu SLCO1B1 występuje ryzyko zwiększonej ekspozycji na kwas symwastatyny, co prowadzić może do zwiększenia ryzyka wystąpienia rabdomiolizy (patrz punkt 4.4).
Ezetymib i symwastatyna
W badaniach dotyczących jednoczesnego stosowania ezetymibu i symwastatyny zaobserwowane działania toksyczne były w zasadzie takie, jak wywoływane zazwyczaj przez statyny. Niektóre z działań toksycznych były bardziej nasilone niż te występujące podczas stosowania statyn w monoterapii. Przypisuje się to wystąpieniu interakcji farmakokinetycznych i (lub) farmakodynamicznych w przypadku leczenia skojarzonego. W badaniach klinicznych takie interakcje nie występowały. U szczurów miopatie występowały jedynie po narażeniu na dawki kilkakrotnie przewyższające dawkę terapeutyczną stosowaną u ludzi (dawki większe około 20 x od wartości AUC dla symwastatyny i dawki większe 1800 x od wartości
AUC dla aktywnego metabolitu). Nie ma dowodów na to, że równoczesne stosowanie ezetymibu wpływa na możliwość wywoływania działań toksycznych przez symwastatynę.
Podczas jednoczesnego stosowania ezetymibu i statyn u psów (≤ 1 x AUC określonego dla ludzi), obserwowano wpływ małych dawek na wątrobę. Odnotowano wyraźne zwiększenie aktywności enzymów wątrobowych (AlAT, AspAT) bez towarzyszącej martwicy tkanki wątrobowej. U psów, którym podawano jednocześnie ezetymib i symwastatynę obserwowano zmiany histopatologiczne wątroby (hiperplazję nabłonka dróg żółciowych, akumulację barwnika, nacieki komórek jednojądrzastych i obecność małych hepatocytów). Zmiany te nie postępowały w miarę wydłużania czasu podawania tych preparatów do 14 miesięcy. Po przerwaniu stosowania preparatów obserwowano zazwyczaj cofanie się zmian wątrobowych. Zmiany te odpowiadały tym opisywanym podczas stosowania inhibitorów reduktazy HMG-CoA lub wiązały się z uzyskanym u badanych psów bardzo niskim stężeniem cholesterolu.
Jednoczesne podawanie ezetymibu i symwastatyny nie wykazywało teratogennego wpływu na szczury. Zaobserwowano niewielką liczbę przypadków deformacji układu kostnego u płodów królików (zrośnięcie kręgów ogonowych, zmniejszenie liczby kręgów ogonowych).
W cyklu prób przeprowadzonych in vivo i in vitro stosowanie ezetymibu w monoterapii lub jednocześnie z symwastatyną nie wiązało się z możliwością wystąpienia działań genotoksycznych.
Ezetymib
Badania na zwierzętach dotyczące przewlekłego działania toksycznego nie wykazały istnienia narządów szczególnie zagrożonych takim działaniem. U psów, którym podawano ezetymib przez okres 4 tygodni
(≥ 0,03 mg/kg na dobę) stwierdzono zwiększenie stężenia cholesterolu w żółci znajdującej się w pęcherzyku żółciowym o 2,5 do 3,5 razy. Niemniej jednak w badaniu, w którym psom podawano lek w dawkach do
300 mg/kg na dobę przez okres 1 roku, nie stwierdzono zwiększenia zapadalności na kamicę żółciową, ani innego oddziaływania na wątrobę i drogi żółciowe. Nie wiadomo, czy wyniki tych badań mają odniesienie
do ludzi. Nie można wykluczyć ryzyka powstawania kamieni żółciowych w przypadku stosowania ezetymibu w dawkach terapeutycznych.
Wyniki długotrwałych badań dotyczących działania rakotwórczego ezetymibu były negatywne.
Ezetymib nie miał wpływu na płodność samic i samców szczurów, nie wykryto działania teratogennego u szczurów i królików ani wpływu na rozwój przedurodzeniowy i pourodzeniowy. Ezetymib stosowany w dawkach wielokrotnych wynoszących 1000 mg/kg na dobę przenikał przez barierę łożyskową
u ciężarnych samic szczurów i królików.
Symwastatyna
W oparciu o konwencjonalne badania na zwierzętach, dotyczące farmakodynamiki, toksyczności po podaniu wielokrotnym, genotoksyczności i rakotwórczości stwierdzono, że dla pacjenta nie występuje inne zagrożenie poza tym, jakie może wyniknąć z samego mechanizmu działania. Po podaniu maksymalnej tolerowanej dawki symwastatyny, zarówno u szczurów jak i królików, nie stwierdzono zniekształceń płodów i wpływu na płodność, funkcje rozrodcze lub rozwój noworodków.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
Rdzeń tabletki Laktoza jednowodna
Hypromeloza (typ 2910) Kroskarmeloza sodowa Celuloza mikrokrystaliczna Kwas askorbowy
Kwas cytrynowy Butylohydroksyanizol (E320) Propylu galusan
Magnezu stearynian
Mieszanina barwników (Pigment blend PB-220001 Yellow): Laktoza jednowodna
Żelaza tlenek żółty (E 172) Żelaza tlenek czerwony (E 172) Żelaza tlenek czarny (E 172)
Nie dotyczy.
2 lata.
Nie przechowywać w temperaturze powyżej 25°C.
Blistry OPA/Aluminium/PVC/Aluminium. Wielkości opakowań:
Axocar, 10 mg + 10 mg: 10, 30, 50, 100 i 300 tabletek.
Axocar, 10 mg + 20 mg, 10 mg + 40 mg, 10 mg + 80 mg: 30, 50, 100 i 300 tabletek. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Brak szczególnych wymagań.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
Zentiva, k.s., U kabelovny 130, Dolní Měcholupy, 102 37 Praga 10, Republika Czeska
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
Axocar, 10 mg+10 mg: 25533
Axocar, 10 mg+20 mg: 25534
Axocar, 10 mg+40 mg: 25535
Axocar, 10 mg+80 mg: 25536
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 5 września 2019 r.
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
09/2021








