







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi lekami i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Szczególne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Erlotinib Vipharm, 25 mg, tabletki powlekane Erlotinib Vipharm, 100 mg, tabletki powlekane Erlotinib Vipharm, 150 mg, tabletki powlekane
Erlotinib Vipharm 25 mg tabletki powlekane: Każda tabletka powlekana zawiera 25 mg erlotynibu (w postaci erlotynibu chlorowodorku).
Substancje pomocnicze o znanym działaniu:
Każda tabletka powlekana zawiera 17,7 mg laktozy jednowodnej. Każda tabletka powlekana zawiera mniej niż 1 mmol sodu (23 mg).
Erlotinib Vipharm 100 mg tabletki powlekane: Każda tabletka powlekana zawiera 100 mg erlotynibu (w postaci erlotynibu chlorowodorku).
Substancje pomocnicze o znanym działaniu
Każda tabletka powlekana zawiera 70,7 mg laktozy jednowodnej. Każda tabletka powlekana zawiera mniej niż 1 mmol sodu (23 mg).
Erlotinib Vipharm 150 mg tabletki powlekane: Każda tabletka powlekana zawiera 150 mg erlotynibu (w postaci erlotynibu chlorowodorku).
Substancje pomocnicze o znanym działaniu
Każda tabletka powlekana zawiera 106 mg laktozy jednowodnej. Każda tabletka powlekana zawiera mniej niż 1 mmol sodu (23 mg).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana
Erlotinib Vipharm 25 mg tabletki powlekane:
białe, okrągłe, obustronnie wypukłe tabletki powlekane o średnicy około 6 mm z wytłoczonym oznakowaniem „E9OB” po jednej stronie i „25” po drugiej stronie.
Erlotinib Vipharm 100 mg tabletki powlekane:
białe, okrągłe, obustronnie wypukłe tabletki powlekane o średnicy około 10 mm z linią podziału po obu stronach z wytłoczonym oznakowaniem „E9OB” powyżej linii podziału i „100” poniżej linii podziału. Tabletkę można podzielić na równe dawki .
Erlotinib Vipharm 150 mg tabletki powlekane:
białe, okrągłe, obustronnie wypukłe tabletki powlekane o średnicy około 10,4 mm z wytłoczonym oznakowaniem „E9OB” po jednej stronie napisem i ”150” po drugiej stronie.
Niedrobnokomórkowy rak płuca (NDRP)
Erlotinib Vipharm jest wskazany w leczeniu pierwszego rzutu u pacjentów z niedrobnokomórkowym rakiem płuca (NDRP) miejscowo zaawansowanym lub z przerzutami, z aktywującymi mutacjami EGFR.
Erlotinib Vipharm jest także wskazany w terapii podtrzymującej u pacjentów z miejscowo zaawansowanym NDRP lub z NDRP z przerzutami, z aktywującymi mutacjami EGFR, u których nastąpiła stabilizacja choroby po chemioterapii pierwszego rzutu.
Erlotinib Vipharm jest także wskazany w leczeniu pacjentów z miejscowo zaawansowanym NDRP lub NDRP z przerzutami, u których doszło do niepowodzenia leczenia po uprzednim zastosowaniu co najmniej jednego schematu chemioterapii.
Zastosowanie produktu leczniczego Erlotinib Vipharm u pacjentów z nowotworami bez mutacji aktywujących EGFR jest wskazane tylko wtedy, gdy inne opcje leczenia uznane są za nieodpowiednie.
Przy przepisywaniu produktu leczniczego Erlotinib Vipharm należy wziąć pod uwagę czynniki związane z wydłużeniem przeżycia.
Nie wykazano korzyści, co do czasu przeżycia ani innych istotnych klinicznie skutków leczenia u pacjentów z nowotworami, w których w badaniu immunohistochemicznym (IHC)
nie stwierdzano ekspresji receptora dla naskórkowego czynnika wzrostu (EGFR) (patrz punkt 5.1).
Rak trzustki
Erlotinib Vipharm w skojarzeniu z gemcytabiną jest wskazany w leczeniu pacjentów z rakiem trzustki z przerzutami.
Przy przepisywaniu produktu leczniczego Erlotinib Vipharm należy wziąć pod uwagę czynniki związane z wydłużeniem przeżycia (patrz punkty 4.2 i 5.1).
Nie wykazano korzyści co do czasu przeżycia u pacjentów z chorobą miejscowo zaawansowaną.
Leczenie produktem leczniczym Erlotinib Vipharm powinno być nadzorowane przez lekarza mającego doświadczenie w stosowaniu leków przeciwnowotworowych.
Pacjenci z niedrobnokomórkowym rakiem płuca
Należy wykonać badanie na obecność mutacji EGFR zgodnie z zatwierdzonymi wskazaniami (patrz punkt 4.1).
Zalecana dawka dobowa produktu leczniczego Erlotinib Vipharm wynosi 150 mg i powinna być przyjmowana co najmniej jedną godzinę przed posiłkiem lub co najmniej dwie godziny po posiłku.
Pacjenci z rakiem trzustki
Zalecana dawka dobowa produktu leczniczego Erlotinib Vipharm wynosi 100 mg i powinna być przyjmowana co najmniej jedną godzinę przed posiłkiem lub co najmniej dwie godziny po posiłku, w skojarzeniu z gemcytabiną (patrz: Charakterystyka Produktu Leczniczego dla gemcytabiny w raku trzustki).
W przypadku pacjentów, u których nie wystąpiła wysypka w ciągu pierwszych 4-8 tygodni terapii, zasadność kontynuacji leczenia erlotynibem powinna być powtórnie oceniona (patrz punkt 5.1).
Jeżeli konieczna jest modyfikacja dawki, należy ją zmniejszać stopniowo po 50 mg (patrz punkt 4.4). Erlotynib Vipharm jest dostępny w dawkach 25 mg, 100 mg i 150 mg.
W przypadku jednoczesnego stosowania substratów i leków wpływających na CYP3A4 może być konieczna modyfikacja dawki (patrz punkt 4.5).
Zaburzenia czynności wątroby
Erlotynib jest metabolizowany w wątrobie i wydalany z żółcią. Pomimo że ekspozycja na erlotynib była podobna w grupie pacjentów z umiarkowanie zaburzoną czynnością wątroby (punktacja Child- Pugh 7-9) jak u pacjentów z prawidłową czynnością wątroby, należy zachować ostrożność podając Erlotinib Vipharm pacjentom z niewydolnością wątroby. W przypadku wystąpienia ciężkich działań niepożądanych należy zredukować dawkę lub przerwać podawanie produktu leczniczego. Nie badano bezpieczeństwa i skuteczności działania erlotynibu u pacjentów z ciężkimi zaburzeniami czynności wątroby (aktywność AspAT i AIAT pięć razy większa niż wartości górnej granicy normy). Nie należy stosować produktu leczniczego Erlotinib Vipharm u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkt 5.2).
Zaburzenia czynności nerek
Nie badano bezpieczeństwa i skuteczności stosowania erlotynibu u pacjentów z zaburzeniami czynności nerek (stężenie kreatyniny w surowicy > 1,5 razy większe niż górna granica wartości prawidłowych). Na podstawie danych farmakokinetycznych nie wydaje się, aby zmiana dawkowania była konieczna u pacjentów z łagodnymi lub umiarkowanymi zaburzeniami czynności nerek (patrz punkt 5.2). Nie zaleca się stosowania produktu leczniczego Erlotinib Vipharm u pacjentów z ciężkimi zaburzeniami czynności nerek.
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania i skuteczności erlotynibu stosowanego w zarejestrowanych wskazaniach u pacjentów w wieku poniżej 18 lat. Nie zaleca się stosowania produktu leczniczego Erlotinib Vipharm u dzieci i młodzieży.
Stosowanie u palaczy tytoniu
Wykazano, że palenie tytoniu zmniejsza ekspozycję na erlotynib o 50-60%. Maksymalna tolerowana dawka erlotynibu u pacjentów z niedrobnokomórkowym rakiem płuca, którzy aktualnie palą papierosy wynosiła 300 mg. Skuteczność dawki 300 mg w leczeniu drugiego rzutu po niepowodzeniu chemioterapii w porównaniu z zalecaną dawką 150 mg nie była większa u pacjentów, którzy nadal palą papierosy. Dane dotyczące bezpieczeństwa stosowania były porównywalne pomiędzy dawkami
300 mg i 150 mg; stwierdzono jednak liczbowe zwiększenie częstości występowania wysypki, choroby śródmiąższowej płuc i biegunki u pacjentów otrzymujących większą dawkę erlotynibu. Osobom aktualnie palącym należy zalecić zaprzestanie palenia (patrz punkty 4.4, 4.5, 5.1 i 5.2).
Nadwrażliwość na erlotynib lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Ocena statusu mutacji EGFR
Rozważając zastosowanie produktu Erlotinib Vipharm w leczeniu pierwszego rzutu lub leczeniu podtrzymującym z powodu miejscowo zaawansowanego NDRP lub NDRP z przerzutami ważne jest określenie statusu mutacji EGFR u pacjenta.
Należy przeprowadzić zwalidowany, wiarygodny, rzetelny i czuły test z określonym progiem czułości i o udowodnionej przydatności do określania statusu mutacji EGFR, z wykorzystaniem DNA guza pochodzącego z próbki tkanki lub wolnego DNA znajdującego się w krwiobiegu (cfDNA) pochodzącego z próbki krwi (osocza), zgodnie z lokalnie przyjętą praktyką medyczną.
W przypadku wykorzystania testu cfDNA z osocza i otrzymania wyniku ujemnego dla mutacji aktywujących, należy w miarę możliwości wykonać test tkankowy z uwagi na możliwość otrzymania wyników fałszywie ujemnych w badaniu osocza.
Stosowanie u palaczy tytoniu
Osobom palącym tytoń należy doradzić, aby zaprzestały palenia, ponieważ stężenie erlotynibu w osoczu osób palących jest mniejsze niż u osób niepalących. Prawdopodobnie stopień zmniejszenia stężenia ma znaczenie kliniczne (patrz punkty 4.2, 4.5, 5.1 i 5.2).
Śródmiąższowa choroba płuc
U pacjentów otrzymujących erlotynib z powodu niedrobnokomórkowego raka płuca (NDRP), raka trzustki lub z powodu innych zaawansowanych nowotworów litych, donoszono o niezbyt częstych przypadkach śródmiąższowej choroby płuc (ang. interstitial lung disease - ILD) lub stanach
o podobnym obrazie chorobowym, w tym również przypadkach śmiertelnych. W badaniu głównym BR.21 pacjentów z NDRP, częstość występowania ILD (0,8%) była taka sama w grupie otrzymującej placebo jak w grupie otrzymującej erlotynib.. W metaanalizie randomizowanych, kontrolowanych badań klinicznych pacjentów z NDRP (wyłączając badanie fazy I i jednoramienne badanie fazy II
z powodu braku grupy kontrolnej) częstość występowania stanów o podobnym obrazie chorobowym do ILD wynosiła 0,9 % u otrzymujących erlotynib w porównaniu do 0,4 % w ramionach kontrolnych. W badaniu dotyczącym stosowania leku u pacjentów z rakiem trzustki w skojarzeniu z gemcytabiną, częstość występowania przypadków podobnych do ILD wynosiła 2,5% w grupie otrzymującej erlotynib w skojarzeniu z gemcytabiną w porównaniu do 0,4% w grupie otrzymującej placebo
w skojarzeniu z gemcytabiną. U pacjentów, u których podejrzewano przypadki podobne do ILD, zgłaszano następujące rozpoznania: zapalenie płuc, popromienne zapalenie płuc, zapalenie płuc z nadwrażliwości, śródmiąższowe zapalenie płuc, śródmiąższową chorobę płuc, obliteracyjne zapalenie oskrzelików, zwłóknienie płuc, zespół ostrej niewydolności oddechowej (ang. Acute Respiratory Distress Syndrome - ARDS), zapalenie pęcherzyków płucnych i nacieki w płucach.
Początek objawów występował po kilku dniach do kilku miesięcy po rozpoczęciu leczenia erlotynibem. Często towarzyszyły im czynniki zakłócające lub współdziałające, takie jak jednocześnie lub uprzednio stosowana chemioterapia, uprzednia radioterapia, występujące uprzednio zmiany miąższowe płuc, przerzuty do płuc lub zakażenia płuc. Większą częstość występowania ILD (około 5%, ze wskaźnikiem śmiertelności wynoszącym 1,5%) zaobserwowano wśród pacjentów w badaniach prowadzonych w Japonii.
U pacjentów, u których nagle wystąpią nowe i (lub) narastające, niewyjaśnione objawy ze strony płuc, takie jak duszność, kaszel i gorączka, należy przerwać stosowanie erlotynibu do czasu przeprowadzenia oceny diagnostycznej. Pacjentów leczonych erlotynibem równocześnie
z gemcytabiną należy uważnie monitorować w kierunku możliwości wystąpienia śródmiąższowej choroby płuc. W przypadku rozpoznania ILD, erlotynib należy odstawić i w razie konieczności wdrożyć odpowiednie leczenie (patrz punkt 4.8).
Biegunka, odwodnienie, zaburzenia elektrolitów i zaburzenia czynności nerek
Biegunka (w tym bardzo rzadkie przypadki zakończone zgonem) występowała u około 50% pacjentów przyjmujących erlotynib. W przypadku biegunki o umiarkowanym lub ciężkim nasileniu, należy wdrożyć leczenie, np. loperamidem. W niektórych przypadkach może zachodzić konieczność zmniejszenia dawki. W badaniach klinicznych dawki zmniejszano stopniowo o 50 mg.
Nie przeprowadzano badań dotyczących zmniejszania dawki o 25 mg. W przypadku ciężkiej lub uporczywej biegunki, nudności, jadłowstrętu lub wymiotów z towarzyszącym odwodnieniem, należy przerwać stosowanie erlotynibu i wdrożyć postępowanie mające na celu przeciwdziałanie odwodnieniu (patrz punkt 4.8). Rzadko donoszono o przypadkach hipokaliemii i niewydolności nerek (w tym
z przypadkami zgonów). Niektóre z nich były spowodowane ciężkim odwodnieniem wywołanym biegunką, wymiotami i (lub) jadłowstrętem, podczas gdy inne były następstwem równocześnie otrzymywanej chemioterapii. W przypadku ciężkiej lub uporczywej biegunki lub w przypadkach prowadzących do odwodnienia, szczególnie w grupach pacjentów obciążonych czynnikami ryzyka (w szczególności równoczesne przyjmowanie chemioterapii i innych leków, predysponujące objawy, choroby lub inne czynniki, w tym podeszły wiek), leczenie erlotynibem należy przerwać i podjąć
odpowiednie działania w celu intensywnego nawodnienia pacjentów drogą dożylną. Ponadto, u pacjentów zagrożonych odwodnieniem należy monitorować czynność nerek oraz stężenia elektrolitów w surowicy krwi, w tym stężenie potasu.
Hepatotoksyczność
Podczas przyjmowania erlotynibu zgłaszano poważne przypadki polekowego uszkodzenia wątroby (DILI), w tym zapalenie wątroby, ostre zapalenie wątroby i niewydolność wątroby (w tym przypadki śmiertelne). Czynniki ryzyka mogą obejmować istniejącą wcześniej chorobę wątroby czy jednoczesne przyjmowanie leków działających toksycznie na wątrobę. Zaleca się okresowe badanie czynności wątroby podczas leczenia erlotynibem. Należy zwiększyć częstotliwość monitorowania czynności wątroby u pacjentów z istniejącymi wcześniej zaburzeniami funkcji wątroby lub niedrożnością dróg żółciowych. U pacjentów zgłaszających objawy mogące świadczyć o uszkodzeniu wątroby należy przeprowadzić szybką ocenę kliniczną i testy czynnościowe wątroby. Należy przerwać stosowanie erlotynibu, jeśli występują ciężkie zmiany parametrów czynności wątroby (patrz punkt 4.8). Erlotynib nie jest zalecany u pacjentów z ciężkimi zaburzeniami czynności wątroby.
Perforacja przewodu pokarmowego
U pacjentów otrzymujących erlotynib istnieje zwiększone ryzyko wystąpienia perforacji przewodu pokarmowego, obserwowanych niezbyt często (łącznie z przypadkami zakończonymi zgonem pacjenta). Zwiększone ryzyko dotyczy pacjentów przyjmujących jednocześnie inhibitory angiogenezy, kortykosteroidy, NLPZ i (lub) chemioterapię z zastosowaniem taksanów, a także pacjentów z wrzodem trawiennym lub chorobą uchyłkową jelit w wywiadzie. U pacjentów, u których dojdzie do perforacji przewodu pokarmowego, należy całkowicie zaprzestać leczenia erlotynibem (patrz punkt 4.8).
Pęcherzowe oraz złuszczające zmiany skórne
Zgłaszano występowanie pęcherzowych i złuszczających zmian skórnych oraz zmian o charakterze pryszczy, w tym bardzo rzadkich przypadków przypominających zespół Stevensa-Johnsona/martwicę toksyczno-rozpływną naskórka, w niektórych przypadkach prowadzących do zgonu (patrz punkt 4.8). Jeśli u pacjenta wystąpią nasilone zmiany skórne pęcherzowe, złuszczające lub o charakterze pryszczy, należy czasowo przerwać lub całkowicie zaprzestać leczenia erlotynibem. Pacjenci z pęcherzowymi oraz złuszczającymi zmianami skórnymi powinni być badani pod kątem zakażenia skóry i leczeni zgodnie z obowiązującymi wytycznymi.
Zaburzenia oka
Pacjenci z objawami podmiotowymi i przedmiotowymi sugerującymi zapalenie rogówki, takimi jak ostre lub pogarszające się: zapalenie oka, łzawienie, nadwrażliwość na światło, nieostre widzenie, ból oka i (lub) zaczerwienienie oka powinni zostać niezwłocznie skonsultowani przez okulistę.
W przypadku potwierdzenia rozpoznania wrzodziejącego zapalenia rogówki, leczenie erlotynibem należy przerwać lub zakończyć. W przypadku zdiagnozowania zapalenia rogówki, należy ocenić stosunek korzyści do ryzyka dla dalszego stosowania erlotynibu. Szczególną ostrożność należy zachować w przypadku stosowania erlotynibu u osób chorujących wcześniej na zapalenie rogówki, wrzodziejące zapalenie rogówki lub ciężką postać suchości oka. Stosowanie soczewek kontaktowych jest również czynnikiem ryzyka zapalenia rogówki oraz owrzodzenia. Podczas stosowania erlotynibu zgłaszano występowanie bardzo rzadkich przypadków perforacji lub owrzodzenia rogówki (patrz punkt 4.8).
Interakcje z innymi produktami leczniczymi
Substancje silnie indukujące CYP3A4 mogą zmniejszać skuteczność erlotynibu, natomiast silne inhibitory CYP3A4 mogą prowadzić do nasilenia toksyczności. Należy unikać jednoczesnego stosowania leków tego typu z erlotynibem (patrz punkt 4.5).
Inne rodzaje interakcji
Erlotynib charakteryzuje się zmniejszeniem rozpuszczalności przy pH powyżej 5. Produkty lecznicze, które zmieniają pH górnego odcinka przewodu pokarmowego, jak: inhibitory pompy protonowej, antagoniści receptora H2 czy leki zobojętniające kwas solny w żołądku mogą upośledzać rozpuszczalność erlotynibu oraz ograniczać jego biodostępność. Zwiększenie dawki erlotynibu
podawanego razem ze wspomnianymi lekami, nie jest w stanie zrekompensować utraty ekspozycji na lek. Należy unikać jednoczesnego leczenia erlotynibem i inhibitorami pompy protonowej. Nie badano wpływu leków zobojętniających kwas solny w żołądku oraz antagonistów receptora H2 na wchłanianie erlotynibu, lecz leki te mogą upośledzać biodostępność erlotynibu. Dlatego należy unikać jednoczesnego stosowania erlotynibu z tymi lekami (patrz punkt 4.5). Jeżeli podczas leczenia erlotynibem konieczne jest podanie leków zobojętniających kwas solny w żołądku, należy zastosować je co najmniej 4 godziny przed lub 2 godziny po dobowej dawce erlotynibu.
Pozostałe składniki
Tabletki produktu leczniczego Erlotinib Vipharm zawierają laktozę.
Produkt leczniczy nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Każda tabletka zawiera mniej niż 1 mmol (23 mg), sodu to znaczy produkt leczniczy uznaje się za
„wolny od sodu”.
Badania dotyczące interakcji prowadzono wyłącznie u dorosłych pacjentów. Erlotynib i inne substraty CYP
Erlotynib jest silnie działającym inhibitorem CYP1A1 i umiarkowanym inhibitorem CYP3A4 i CYP2C8, a w warunkach in vitro jest on również silnym inhibitorem reakcji sprzęgania
z kwasem glukuronowym, która przebiega za pośrednictwem UGT1A1.
Fizjologiczne znaczenie silnych właściwości hamujących wobec CYP1A1 nie jest znane, z uwagi na znacznie ograniczoną ekspresję CYP1A1 w tkankach ludzkich.
Gdy erlotynib podawano z cyprofloksacyną, umiarkowanym inhibitorem CYP1A2 ekspozycja na erlotynib [AUC] znacząco wzrosła o 39%, bez znamiennej statystycznie zmiany stężenia maksymalnego Cmax. Podobnie, ekspozycja na jego aktywny metabolit wzrosła o około 60% i o 48% odpowiednio dla AUC i Cmax. Nie wiadomo, jakie jest kliniczne znaczenie tego faktu. Należy zachować ostrożność, stosując jednocześnie cyprofloksacyną lub silny inhibitor CYP1A2 (np. fluwoksaminę) w skojarzeniu z erlotynibem. Jeżeli wystąpią działania niepożądane zależne od erlotynibu, można zmniejszyć jego dawkę.
Uprzednie leczenie lub jednoczesne stosowanie erlotynibu z prototypowymi substratami CYP3A4, jak midazolam i erytromycyna nie wpływało na ich klirens. Powodowało jednak zmniejszenie biodostępności podawanego doustnie midazolamu do 24%. W innym badaniu klinicznym wykazano, że erlotynib nie wpływa na farmakokinetykę stosowanego jednocześnie substratu CYP3A4/2C8, paklitakselu. Dlatego wystąpienie istotnych interakcji wpływających na klirens innych substratów CYP3A4 jest mało prawdopodobne.
Hamowanie reakcji sprzęgania z kwasem glukuronowym może wywoływać interakcje z lekami, które są substratami UGT1A1 i są wydalane wyłącznie na tej drodze. U pacjentów, u których stopień ekspresji UGT1A1 jest niski lub u pacjentów z genetycznie uwarunkowanymi zaburzeniami reakcji sprzęgania z kwasem glukuronowym (np. choroba Gilberta) mogą być zwiększone stężenia bilirubiny w surowicy. Pacjenci ci muszą być leczeni z ostrożnością.
U ludzi, erlotynib jest metabolizowany przez układ cytochromów (CYP) w wątrobie, głównie przy udziale CYP3A4 i w mniejszym stopniu CYP1A2. Metabolizm pozawątrobowy przebiegający
z udziałem CYP3A4 w jelicie, CYP1A1 w płucach i CYP1B1 w tkankach nowotworowych może odgrywać rolę w klirensie metabolicznym erlotynibu. Mogą występować potencjalne interakcje
z lekami, które są metabolizowane przez te enzymy lub są ich inhibitorami bądź induktorami.
Silne inhibitory CYP3A4 powodują zmniejszenie metabolizmu erlotynibu i zwiększenie jego stężenia w osoczu. W badaniu klinicznym jednoczesne stosowanie erlotynibu i ketokonazolu (200 mg doustnie dwa razy na dobę przez 5 dni), który jest silnym inhibitorem CYP3A4, zwiększało ekspozycję na erlotynib (AUC o 86% i Cmax o 69%). Dlatego należy zachować ostrożność w przypadku jednoczesnego leczenia erlotynibem i silnym inhibitorem CYP3A4, np. z azolowymi lekami przeciwgrzybiczymi (np. ketokonazolem, itrakonazolem, worykonazolem), inhibitorami proteazy, erytromycyną lub klarytromycyną. W razie konieczności należy zmniejszyć dawkę erlotynibu, zwłaszcza w przypadku zaobserwowania objawów toksyczności.
Silne induktory aktywności CYP3A4 powodują zwiększenie metabolizmu erlotynibu i istotne zmniejszenie jego stężenia w osoczu. W badaniu klinicznym jednoczesne stosowanie erlotynibu i ryfampicyny (600 mg doustnie raz na dobę przez 7 dni), która jest silnym induktorem CYP3A4,
spowodowało zmniejszenie o 69% mediany AUC erlotynibu. Równoczesne podawanie ryfampicyny z pojedynczą dawką 450 mg erlotynibu spowodowało średnie obniżenie AUC erlotynibu do 57,5% wartości występującej po podaniu pojedynczej dawki 150 mg erlotynibu bez równoczesnego
stosowania ryfampicyny. Z tego powodu należy unikać jednoczesnego podawania erlotynibu z silnymi induktorami CYP3A4. Pacjentom wymagającym jednoczesnego leczenia erlotynibem oraz silnym induktorem CYP3A4 np. ryfampicyną należy rozważyć zwiększenie dawki erlotynibu do 300 mg.
Jednocześnie należy ściśle kontrolować u nich bezpieczeństwo leczenia (w tym czynność nerek
i wątroby oraz stężenie elektrolitów w surowicy krwi). Jeżeli takie leczenie jest dobrze tolerowane przez okres dłuższy niż 2 tygodnie można rozważyć kolejne zwiększenie dawki do 450 mg uważnie kontrolując bezpieczeństwo leczenia. Zmniejszenie ekspozycji na erlotynib może również występować pod wpływem innych induktorów CYP3A4, np. fenytoiny, karbamazepiny, barbituranów lub ziela dziurawca (Hypericum perforatum). W przypadku jednoczesnego stosowania tych leków
z erlotynibem należy zachować ostrożność. W miarę możliwości należy rozważyć zastosowanie innych leków, które nie są induktorami CYP3A4.
Erlotynib i leki przeciwzakrzepowe z grupy pochodnych kumaryny
U pacjentów przyjmujących erlotynib zgłaszano interakcję z lekami przeciwzakrzepowymi z grupy pochodnych kumaryny, w tym z warfaryną, która prowadziła do zwiększenia wartości znormalizowanego współczynnika międzynarodowego (ang. International Normalised Ratio - INR)
i incydentów krwawień, niekiedy zkończonych zgonem. Pacjenci przyjmujący leki przeciwzakrzepowe z grupy pochodnych kumaryny powinni być regularnie monitorowani pod kątem jakichkolwiek zmian parametrów czasu protrombinowego lub INR.
Erlotynib i statyny
Jednoczesne stosowanie erlotynibu i leków z grupy statyn może zwiększać ryzyko miopatii wywołanej statynami, łącznie z rzadko występującą rabdomiolizą.
Erlotynib i palacze tytoniu
Wyniki farmakokinetycznego badania interakcji z udziałem palących i niepalących zdrowych ochotników wykazały, że w wyniku palenia tytoniu po 24 godzinach po podaniu erlotynibu znacznie zmniejsza się AUCinf, Cmax oraz stężenie leku w osoczu, odpowiednio 2,8; 1,5 i 9 razy. Dlatego pacjentów, którzy nadal palą tytoń należy zachęcać do jak najszybszego zaprzestania palenia przed rozpoczęciem leczenia erlotynibem, ponieważ palenie papierosów może zmniejszać stężenie leku
w osoczu. Na podstawie danych z badania CURRENTS nie uzyskano dowodów na występowanie jakichkolwiek korzyści ze stosowania większej dawki erlotynibu wynoszącej 300 mg w porównaniu z zalecaną dawką 150 mg u czynnych palaczy tytoniu. Dane dotyczące bezpieczeństwa stosowania były porównywalne pomiędzy dawkami 300 mg a 150 mg; stwierdzono jednak liczbowe zwiększenie
częstości występowania wysypki, choroby śródmiąższowej płuc i biegunki u pacjentów otrzymujących większą dawkę erlotynibu (patrz punkty 4.2, 4.4, 5.1 i 5.2).
Erlotynib i inhibitory glikoproteiny-P
Erlotynib jest substratem dla glikoproteiny-P transportującej substancję czynną. Jednoczesne stosowanie inhibitorów glikoproteiny-P, np. cyklosporyny lub werapamilu może prowadzić do zmiany dystrybucji i(lub) zmiany eliminacji erlotynibu. Następstwa tych interakcji z punktu widzenia np. toksyczności dla OUN nie są ustalone. W takich sytuacjach należy zachować ostrożność.
Erlotynib i produkty lecznicze zmieniające pH
Erlotynib charakteryzuje się zmniejszeniem rozpuszczalności przy pH powyżej 5. Produkty lecznicze, które zmieniają pH w górnym odcinku przewodu pokarmowego, mogą zmieniać rozpuszczalność erlotynibu, a zatem także jego biodostępność. Podawanie jednoczesne erlotynibu z omeprazolem, inhibitorem pompy protonowej, spowodowało zmniejszenie ekspozycji na erlotynib [AUC] oraz jego stężenia maksymalnego [Cmax] odpowiednio o 46% i 61%. Nie zaobserwowano zmian czasu stężenia maksymalnego Tmax czy okresu półtrwania. Jednoczesne podawanie erlotynibu z antagonistą receptora H2 ranitydyną w dawce 300 mg zmniejsza ekspozycję na erlotynib [AUC] oraz maksymalne stężenie [Cmax] o odpowiednio 33% i 54%. Zwiększenie dawki erlotynibu podawanego razem ze wspomnianymi lekami nie jest w stanie zrekompensować utraty ekspozycji na lek. Jednakże, kiedy podawano erlotynib naprzemiennie z ranitydyną, tzn. 2 godziny przed podaniem lub 10 godzin po podaniu ranitydyny stosowanej w dawce 150 mg 2 razy na dobę, ekspozycja na erlotynib [AUC] oraz stężenie maksymalne [Cmax] zmniejszały się odpowiednio tylko o 15% i 17%. Nie badano wpływu leków zobojętniających kwas solny w żołądku oraz antagonistów receptora H2 na wchłanianie erlotynibu, lecz leki te mogą upośledzać jego wchłanianie prowadząc do zmniejszenia stężenia leku
w osoczu. Podsumowując, należy unikać jednoczesnego leczenia erlotynibem i inhibitorami pompy protonowej. Jeżeli podczas leczenia erlotynibem konieczne jest podanie leków zobojętniających kwas solny w żołądku, należy zastosować je co najmniej 4 godziny przed podaniem lub 2 godziny
po podaniu dobowej dawki erlotynibu. W przypadku, kiedy rozważane jest podawanie ranitydyny, oba leki powinny być podawane naprzemiennie, tzn. erlotynib musi być przyjęty 2 godziny przed podaniem lub 10 godzin po podaniu ranitydyny.
Erlotynib i gemcytabina
W badaniu fazy Ib nie obserwowano istotnego wpływu gemcytabiny na farmakokinetykę erlotynibu ani istotnego wpływu erlotynibu na farmakokinetykę gemcytabiny.
Erlotynib i karboplatyna/paklitaksel
Erlotynib zwiększa stężenie platyny w surowicy krwi. W badaniu klinicznym równoczesne stosowanie erlotynibu z karboplatyną i paklitakselem prowadziło do zwiększenia całkowitej wartości AUC 0-48 platyny o 10,6%. Pomimo znamienności statystycznej, kliniczne znaczenie tej zmiany nie jest istotne. W praktyce klinicznej mogą występować inne czynniki prowadzące do zwiększonej ekspozycji na karboplatynę, jak niewydolność nerek. Nie zanotowano znaczącego wpływu karboplatyny lub pakalitakselu na właściwości farmakokinetyczne erlotynibu.
Erlotynib i kapecytabina
Kapecytabina może zwiększyć stężenie erlotynibu w surowicy krwi. Gdy erlotynib podawano jednocześnie z kapecytabiną odnotowano statystycznie istotne zwiększenie AUC erlotynibu
i graniczne zwiększenie Cmax w porównaniu do tych wartości w innym badaniu, w którym erlotynib podawano jako pojedynczy lek. Nie zaobserwowano istotnego wpływu erlotynibu na farmakokinetykę kapecytabiny.
Erlotynib i inhibitory proteasomów
Ze względu na mechanizm działania, inhibitory proteasomów, w tym bortezomib, mogą wpływać na działanie inhibitorów EGFR, w tym erlotynibu. Istnieją nieliczne dane z badań klinicznych
i przedklinicznych ukazujące wpływ proteasomów na degradację EGFR.
Ciąża
Brak odpowiednich danych dotyczących stosowania erlotynibu u kobiet w okresie ciąży. Badania na zwierzętach nie wykazały działania teratogennego, ani nieprawidłowego porodu. Jednakże nie można wykluczyć niepożądanego działania leku na ciążę, ponieważ badania na szczurach i królikach wykazały zwiększenie śmiertelności zarodka i płodu (patrz punkt 5.3). Potencjalne ryzyko stosowania leku u ludzi nie jest znane.
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym muszą wiedzieć, że należy unikać zachodzenia w ciążę podczas stosowania erlotynibu. W trakcie leczenia i co najmniej przez 2 tygodnie po jego zakończeniu należy stosować odpowiednie skuteczne metody antykoncepcji. U kobiet ciężarnych leczenie można kontynuować tylko wtedy, gdy potencjalne korzyści dla matki przewyższają ryzyko dla płodu.
Karmienie piersią
Nie wiadomo, czy erlotynib przenika do mleka matki. Nie przeprowadzono badań w celu oceny wpływu erlotynibu na wytwarzanie mleka lub obecności produktu leczniczego w mleku matki. Ponieważ potencjalny szkodliwy wpływ na karmione piersią niemowlę jest nieznany, należy odradzać karmienie piersią podczas przyjmowania erlotynibu oraz przez co najmniej 2 tygodnie po przyjęciu ostatniej dawki.
Płodność
Badania na zwierzętach nie wykazały zaburzenia płodności. Nie można jednak wykluczyć niepożądanego działania na płodność, ponieważ w badaniach na zwierzętach wykazano wpływ leku na parametry związane z rozrodczością (patrz punkt 5.3). Potencjalne ryzyko stosowania leku u ludzi nie jest znane.
Nie przeprowadzono badań nad wpływem produktu na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu; wiadomo jednak, że erlotynib nie powoduje upośledzenia sprawności umysłowej.
Ocenę bezpieczeństwa stosowania erlotynibu przeprowadzono na podstawie danych dotyczących ponad 1500 pacjentów leczonych co najmniej jedną dawką 150 mg erlotynibu w monoterapii i ponad 300 pacjentów, którzy otrzymali erlotynib w dawce 100 mg lub 150 mg, w skojarzeniu z gemcytabiną.
Częstość występowania działań niepożądanych (ang. adverse drug reactions, ADR) w badaniach klinicznych dotyczących erlotynibu w monoterapii lub w skojarzeniu z chemioterapią podsumowano w skali National Cancer Institute-Common Toxicity Criteria (NCI-CTC) w Tabeli 1. Wymienione ADR zgłaszano u co najmniej 10% pacjentów (w grupie otrzymującej erlotynib), przy czym występowały one częściej (≥3%) u pacjentów leczonych erlotynibem niż produktem porównawczym. Inne ADR, w tym obserwowane w innych badaniach, podsumowano w Tabeli 2.
Niepożądane działania polekowe obserwowane w badaniach klinicznych (Tabela 1) są wymienione według klasyfikacji układów i narządów MedDRA. Kategorie częstości występowania poszczególnych niepożądanych reakcji polekowych oparto na następującej konwencji: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), niezbyt często (od ≥ 1/1000 do < 1/100), rzadko (od ≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000).
W każdej grupie o określonej częstości występowania, działania niepożądane wymieniono w kolejności według malejącego nasilenia.
Niedrobnokomórkowy rak płuca (erlotynib stosowany w monoterapii)
Leczenie pierwszego rzutu u pacjentów z mutacjami EGFR
W otwartym randoizowanymbadaniu fazy III, ML20650, przeprowadzonym z udziałem 154 pacjentów, bezpieczeństwo stosowania erlotynibu w leczeniu pierwszego rzutu u pacjentów z NDRP z mutacjami aktywującymi EGFR oceniano u 75 osób; nie zaobserwowano nowych sygnałów dotyczących bezpieczeństwa u tych pacjentów.
Najczęstszymi ADR obserwowanymi u pacjentów leczonych erlotynibem w badaniu ML20650 były wysypka i biegunka (dowolnego stopnia odpowiednio 80% i 57%), przy czym większość tych reakcji miała nasilenie stopnia 1/2 i ustępowała bez interwencji. Wysypka i biegunka stopnia 3 wystąpiły
u odpowiednio 9% i 4% pacjentów. Nie zaobserwowano wysypki ani biegunki stopnia 4. Zarówno wysypka jak i biegunka doprowadziły do odstawienia erlotynibu u 1% pacjentów. Modyfikacja dawki (odstawienie lub zmniejszenie) z powodu wysypki i biegunki była konieczna u odpowiednio 11% i 7% pacjentów.
Leczenie podtrzymujące
W dwóch innych randomizowanych badaniach klinicznych fazy III prowadzonych metodą podwójnie ślepej próby z kontrolą placebo, BO18192 (SATURN) i BO25460 (IUNO), erlotynib był podawany jako leczenie podtrzymujące po chemioterapii pierwszego rzutu. Badania te przeprowadzono
z udziałem łącznie 1532 pacjentów z zaawansowanym, nawracającym lub z przerzutami NDRP po standardowej chemioterapii pierwszego rzutu, zawierającej związki platyny. Nie zidentyfikowano nowych sygnałów dotyczących bezpieczeństwa.
Najczęstszymi ADR obserwowanymi u pacjentów leczonych erlotynib w badaniach BO18192
i BO25460 były wysypka (BO18192: wszystkie stopnie 49,2%; stopień 3: 6,0%; BO25460: wszystkie stopnie 39,4%; stopień 3: 5,0%) i biegunka (BO18192: wszystkie stopnie 20,3%; stopień 3: 1,8%; BO25460: wszystkie stopnie 24,2%; stopień 3: 2,5%). W żadnym z badań nie zaobserwowano wysypki ani biegunki stopnia 4. Wysypka i biegunka doprowadziły do odstawienia erlotynibu u odpowiednio 1% i < 1% pacjentów w badaniu BO18192, podczas gdy w badaniu BO25460 żaden z pacjentów nie przerwał leczenia z powodu wysypki lub biegunki. Modyfikacja dawki (odstawienie lub zmniejszenie) z powodu wysypki i biegunki była konieczna u odpowiednio 8,3% i 3% pacjentów w badaniu BO18192 oraz odpowiednio 5,6% i 2,8% pacjentów w badaniu BO25460.
Leczenie drugiego rzutu i późniejsze linie leczenia
W randomizowanym, podwójnie zaślepionym badaniu (BR.21; erlotynib stosowany w leczeniu drugiego rzutu, do najczęściej zgłaszanych niepożądanych reakcji na lek należały wysypka (75%)
i biegunka (54%). W większości przypadków były to reakcje 1 lub 2 stopnia i dawały się opanować bez konieczności leczenia. Wysypka i biegunka o stopniu nasilenia 3 lub 4 występowały z częstością odpowiednio 9% i 6% u pacjentów leczonych erlotynibem; każda z tych reakcji była przyczyną przerwania udziału w badaniu przez 1% pacjentów. Zmniejszenie dawki z powodu wysypki i biegunki było konieczne odpowiednio u 6% i 1% pacjentów. W badaniu BR.21, mediana czasu do wystąpienia wysypki wynosiła 8 dni, a mediana czasu do wystąpienia biegunki 12 dni.
Wysypka najczęściej objawia się jako łagodne lub umiarkowanie nasilone zmiany rumieniowe lub grudkowo-krostkowe, mogące pojawić się lub nasilać na skórze wystawionej na działanie promieni słonecznych. Pacjentom, którzy wystawieni są na działanie promieni słonecznych, można zalecić stosowanie odzieży ochronnej i(lub) kremów chroniących przed słońcem (np. zawierających minerały).
Rak trzustki (erlotynib podawany jednocześnie z gemcytabiną)
Najczęstszymi działaniami niepożądanymi w kluczowym badaniu PA.3 u pacjentów z rakiem trzustki leczonych erlotynibem w dawce 100 mg w skojarzeniu z gemcytabiną były: zmęczenie, wysypka
i biegunka. W grupie otrzymującej erlotynib w skojarzeniu z gemcytabiną wysypkę i biegunkę stopnia 3/4 zgłoszono u 5% pacjentów. Mediana czasu do wystąpienia wysypki i biegunki wyniosła odpowiednio10 dni i 15 dni. Wysypka i biegunka doprowadziły do zmniejszenia dawki u 2% pacjentów oraz do przerwania udziału w badaniu u nawet 1% pacjentów otrzymujących erlotynib
w skojarzeniu z gemcytabiną.
Tabela 1: ADR występujące u ≥ 10% pacjentów w badaniach BR.21 (leczonych erlotynibem) i PA.3 (leczonych erlotynibem w skojarzeniu z gemcytabiną) oraz ADR występujące częściej (≥ 3%) niż
w grupie otrzymującej placebo w badaniu BR.21 (u pacjentów erlotynibem) i badaniu PA.3 (u pacjentów leczonych erlotynibem w skojarzeniu z gemcytabiną)
Erlotynib N = 485 | Erlotynib N = 259 | Najczęściej obserwowana kategoria częstości występowania | |||||
Stopień wg NCI-CTC | Każdy stopień | 3 | 4 | Każdy stopień | 3 | 4 | |
Terminologia MedDRA | % | % | % | % | % | % | |
Zakażenia i infestacje | 24 | 4 | 0 | 31 | 3 | <1 | bardzo często |
Zakażenia* | |||||||
Zaburzenia metabolizmu i odżywiania | |||||||
Jadłowstręt | 52 | 8 | 1 | - | - | - | bardzo często |
Zmniejszenie masy ciała | - | - | - | 39 | 2 | 0 | bardzo często |
Zaburzenia oka | |||||||
Suche zapalenie rogówki i spojówek | 12 | 0 | 0 | - | - | - | bardzo często |
Zapalenie spojówek | 12 | <1 | 0 | - | - | - | bardzo często |
Zaburzenia psychiczne | - | - | - | 19 | 2 | 0 | bardzo często |
Depresja | |||||||
Zaburzenia układu nerwowego | |||||||
Neuropatia | - | - | - | 13 | 1 | < 1 | bardzo często |
Ból głowy | - | - | - | 15 | < 1 | 0 | bardzo często |
Zaburzenia oddechowe, klatki piersiowej i śródpiersia | |||||||
Duszność | 41 | 17 | 11 | - | - | - | bardzo często |
Kaszel | 33 | 4 | 0 | 16 | 0 | 0 | bardzo często |
Zaburzenia żołądka i jelit | |||||||
Biegunka** | 54 | 6 | <1 | 48 | 5 | < 1 | bardzo często |
Nudności | 33 | 3 | 0 | - | - | - | bardzo często |
Wymioty | 23 | 2 | <1 | - | - | - | bardzo często |
Zapalenie jamy ustnej | 17 | <1 | 0 | 22 | < 1 | 0 | bardzo często |
Ból brzucha | 11 | 2 | <1 | - | - | - | bardzo często |
Niestrawność | - | - | - | 17 | < 1 | 0 | bardzo często |
Wzdęcia | - | - | - | 13 | 0 | 0 | bardzo często |
Zaburzenia skóry i tkanki podskórnej | |||||||
Wysypka*** | 75 | 8 | <1 | 69 | 5 | 0 | bardzo często |
Świąd | 13 | <1 | 0 | - | - | - | bardzo często |
Suchość skóry | 12 | 0 | 0 | - | - | - | bardzo często |
Łysienie | - | - | - | 14 | 0 | 0 | bardzo często |
Zaburzenia ogólne i stany w miejscu podania | |||||||
Zmęczenie | 52 | 14 | 4 | 73 | 14 | 2 | bardzo często |
Gorączka | - | - | - | 36 | 3 | 0 | bardzo często |
Dreszcze | - | - | - | 12 | 0 | 0 | bardzo często |
* Ciężkie zakażenia, z neutropenią lub bez neutropenii, obejmowały zapalenie płuc, posocznicę i zapalenie tkanki łącznej.
** Może prowadzić do odwodnienia, hipokaliemii i niewydolności nerek.
*** Wysypka w tym wysypka trądzikopodobna.
- odpowiada odsetkowi poniżej wartości granicznej
Tabela 2: Podsumowanie reakcji niepożądanych pod względem częstości występowania
Grupa | Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko | Nieznana |
układowonarząd | (≥1/10) | (≥1/100 do <1/10) | (≥1/1 000 do | (≥1/10 000 do | (<1/10 000) | (częstość nie |
owa | <1/100) | <1/1 000) | może być | |||
określona na | ||||||
podstawie | ||||||
dostępnych | ||||||
danych) | ||||||
Zaburzenia oka | Zapalenie rogówki Zapalenie spojówek1 | Zmiany dotyczące rzęs2 | Perforacje rogówki Owrzodzenia rogówki Zapalenie błony | |||
naczyniowej oka | ||||||
Zaburzenia oddechowe, klatki piersiowej | Krwawienie z nosa | Śródmiąższowa choroba płuc (ILD)3 | ||||
i śródpiersia | ||||||
Zaburzenia żołądka i jelit | Biegunka7 | Krwawienie z przewodu pokarmowego4,7 | Perforacje przewodu pokarmowego7 | Śródścienna odma jelitowa | ||
Zaburzenia | Nieprawidłowe | Niewydolność | Ostre zapalenie | |||
wątroby i dróg | wyniki badań | wątroby6 | wątroby | |||
żółciowych | czynności | Zapalenie wątroby | ||||
wątroby5 |
Zaburzenia skóry i tkanki podskórnej | Wysypka | Łysienie Suchość skóry1 Zanokcica Zapalenie mieszków włosowych Trądzik/ Trądzikowate zapalenie skóry Pęknięcia skóry | Nadmierne owłosienie Zmiany dotyczące brwi oraz kruche i wiotkie paznokcie Łagodne reakcje skórne, takie jak przebarwienia | Zespół erytrodyzestezji dłoniowopodeszw owej | Zespół Stevensa- Johnsona/ Martwica toksycznorozpływ na naskórka7 | |
Zaburzenia nerek i dróg moczowych | Niewydolność nerek1 | Zapalenie nerek1 Białkomocz1 |
Objawy
Tolerowane były pojedyncze dawki doustne erlotynibu do 1000 mg erlotynibu u zdrowych osób i do 1600 mg u chorych na nowotwory. Dawki 200 mg podawane dwa razy na dobę zdrowym osobom były źle tolerowane po zaledwie kilku dniach stosowania. Jak wynika z danych uzyskanych na podstawie
tych badań, ciężkie działania niepożądane, takie jak biegunka, wysypka i ewentualnie zwiększona aktywność aminotransferaz, mogą występować po podaniu dawki większej niż zalecana.
Postępowanie
W przypadku, gdy podejrzewane jest przedawkowanie, należy zaprzestać leczenia erlotynibem i wdrożyć leczenie objawowe.
Grupa farmakoterapeutyczna: leki przeciwnowotworowe inhibitory kinazy białkowej, kod ATC: L01EB02.
Mechanizm działania
Erlotynib jest inhibitorem kinazy tyrozynowej receptora dla naskórkowego czynnika wzrostu/receptora typu 1 dla ludzkiego naskórkowego czynnika wzrostu (EGFR znanego także jako HER1). Erlotynib silnie hamuje wewnątrzkomórkową fosforylację EGFR. EGFR ulega ekspresji na powierzchni komórek prawidłowych i nowotworowych. W modelach nie-klinicznych hamowanie fosfotyrozyny EGFR prowadzi do zatrzymania podziałów komórki i (lub) jej śmierci.
Mutacje EGFR mogą prowadzić do istotnej aktywacji antyapoptotycznych oraz proliferacyjnych szlaków sygnałowych. Znacząca skuteczność erlotynibu w blokowaniu przekaźnictwa sygnału poprzez ścieżkę związaną z EGFR w komórkach guzów nowotworowych wykazujących mutację EGFR jest przypisywana ścisłemu wiązaniu się erlotynibu z miejscem wiążącym ATP w zmutowanej domenie kinazowej receptora EGFR. W wyniku blokowania przekaźnictwa zstępującego zostaje zatrzymana proliferacja komórek oraz indukowana śmierć komórki poprzez wewnętrzną ścieżkę apoptozy.
W modelach mysich z wymuszoną ekspresją receptora EGFR wykazującego aktywującą mutację, obserwuje się regresję guza.
Skuteczność kliniczna
Leczenie pierwszego rzutu pacjentów z niedrobnokomórkowym rakiem płuca (NDRP) z aktywującymi mutacjami EGFR (erlotynib podawany w monoterapii):
Skuteczność erlotynibu w leczeniu pierwszego rzutu pacjentów z NDRP z mutacjami aktywującymi EGFR, wykazano w otwartym, randomizowanym badaniu III fazy (ML20650, EURTAC). Badanie to przeprowadzono z udziałem pacjentów rasy kaukaskiej z przerzutowym lub miejscowo zaawansowanym NDRP (stadium IIIB i IV), którzy nie otrzymywali dotychczas chemioterapii ani żadnego układowego leczenia przeciwnowotworowego w związku z zaawansowanym nowotworem, i u których występują mutacje w domenie kinazy tyrozynowej EGFR (delecja
w egzonie 19. lub mutacja w egzonie 21.). Pacjenci zostali losowo przydzieleni w stosunku 1:1 do grup mających otrzymywać erlotynib w dawce 150 mg na dobę lub do 4 cykli dwulekowego schematu chemioterapii opartego na pochodnych platyny.
Pierwszorzędowym punktem końcowym badania był oceniany przez badaczy czas przeżycia bez progresji choroby (PFS).
Wyniki dotyczące skuteczności są podsumowane w Tabeli 3.
Wykres 1: Krzywa Kaplana-Meiera dla ocenianego przez badaczy PFS w badaniu ML20650 (EURTAC) (data zakończenia zbierania danych: kwiecień 2012)
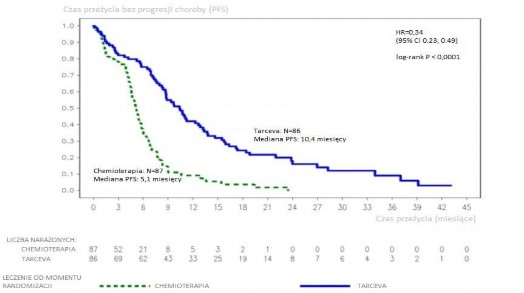
Tabela 3: Wyniki dotyczące skuteczności erlotynibu względem chemioterapii w badaniu ML20650 (EURTAC)
Erlotynib | Chemioterapia | Hazard względny (95% CI) | Wartość p | ||
n=77 | n=76 | ||||
Wcześniej | Pierwszorzędowy punkt końcowy: Przeżycie bez progresji choroby (PFS, mediana w miesiącach)* Ocena badacza ** Niezależna ocena** | ||||
zaplanowana analiza | |||||
pośrednia (35% | 9,4 | 5,2 | 0,42 | p<0,0001 | |
dojrzałość danych | [0,27-0,64] | ||||
dotyczących przeżycia | p=0,003 | ||||
całkowitego) (n=153) | 10,4 | 5,4 | 0,47 [0,27-0,78] | ||
data zakończenia zbierania | Całkowity odsetek | ||||
danych: sierpień 20100 | najlepszych odpowiedzi | 54,5% | 10,5% | p<0,0001 | |
(CR/PR) | |||||
Przeżycie całkowite (OS) | 22,9 | 18,8 | 0,80 | p=0,4170 | |
(miesiące) | [0,47-1,37] | ||||
Analiza | n=86 | n=87 | |||
eksploracyjna (40% dojrzałość danych dotyczących przeżycia | |||||
PFS (mediana w miesiącach), Ocena badacza | 9,7 | 5,2 | 0,37 [0,27-0,54] | p<0,0001 | |
całkowitego) | Całkowity odsetek | ||||
(n=173) | najlepszych odpowiedzi | 58,1% | 14,9% | p<0,0001 | |
(CR/PR) | |||||
data zakończenia zbierania danych: styczeń 2011 | Przeżycie całkowite (OS) (miesiące) | 19,3 | 19,5 | 1,04 [0,65-1,68] | p=0,8702 |
Analiza | n=86 | n=87 | |||
zaktualizowana | |||||
10,4 | 5,1 | p<0,0001 | |||
(62% dojrzałość | PFS (mediana w | 0,34 | |||
danych dotyczących | miesiącach) | [0,23-0,49] |
przeżycia całkowitego) (n=173) data zakończenia zbierania danych: kwiecień 2012 | OS*** (miesiące) | 22,9 | 20,8 | 0,93 [0,64-1,36] | p=0,7149 |
CR (ang. complete response) - odpowiedź całkowita; PR (ang. partial response) - odpowiedź częściowa
* Odnotowano zmniejszenie ryzyka progresji choroby lub zgonu o 58%
** Ogólny współczynnik zgodności pomiędzy oceną badaczy i niezależnej komisji wyniósł 70%
*** W wyniku nasilonego zjawiska “crossover”, 82% pacjentów otrzymało późniejsze leczenie z zastosowaniem inhibitora kinazy tyrozynowej EGFR; wszyscy pacjenci z wyjątkiem 2, otrzymali erlotynib.
Leczenie podtrzymujące pacjentów NDRP po chemioterapii pierwszego rzutu (erlotynib podawany w monoterapii):
Skuteczność i bezpieczeństwo erlotynibu w leczeniu podtrzymującym po chemioterapii pierwszego rzutu pacjentów z NDRP, badano w randomizowanym badaniu z podwójnie ślepą próbą, kontrolowanym placebo (BO18192, SATURN). Badanie to przeprowadzono u 889 pacjentów
z miejscowo zaawansowanym NDRP lub NDRP z przerzutami, u których nie doszło do progresji choroby po otrzymaniu czterech cykli chemioterapii składających się z dwóch leków opartych na pochodnych platyny. Pacjenci byli randomizowani w stosunku 1:1 do grupy otrzymującej doustnie erlotynib w dawce 150 mg lub placebo raz na dobę, dopóki nie wystąpiła progresja choroby.
Pierwszorzędowy punkt końcowy badania uwzględniał czas przeżycia bez progresji choroby (ang. progression-free survival) w całej grupie pacjentów. Cechy demograficzne oraz cechy opisujące chorobę nowotworową były równomiernie rozłożone w obu grupach pacjentów. Pacjenci, u których stwierdzano stan sprawności >1 stopnia wg ECOG oraz pacjenci z istotnymi współistniejącymi chorobami wątroby lub nerek nie byli włączani do badania.
W badaniu wykazano korzyść pod względem pierwszorzędowego punktu końcowego, jakim był PFS (HR=0,71 p< 0,0001), oraz drugorzędowego punktu końcowego, jakim był OS (HR=0,81 p=0,0088) w całej populacji pacjentów. Jednakże największą korzyść obserwowano, na podstawie zaplanowanej wcześniej analizy eksploracyjnej, u pacjentów z aktywującymi mutacjami EGFR (n=49), u których wykazano znaczącą korzyść pod względem PFS (HR=0,10, 95% CI, 0,04 do 0,25; p<0,0001), a HR dotyczący czasu przeżycia całkowitego wyniósł 0,83 (95% CI, 0,34 do 2,02). Sześćdziesiąt siedem procent pacjentów przyjmujących placebo w podgrupie z aktywująca mutacją EGFR otrzymało leczenie drugiego lub dalszego rzutu inhibitorami kinazy tyrozynowej EGFR (IKT EGFR).
Badanie BO25460 (IUNO) przeprowadzono z udziałem 643 pacjentów z zaawansowanym NDRP,
u których w guzach nowotworowych nie stwierdzono obecności aktywującej mutacji EGFR (delecja w eksonie 19 lub mutacja L858R w eksonie 21) oraz u których nie doszło do progresji choroby po czterech cyklach chemioterapii opartej na pochodnych platyny.
Celem badania było porównanie czasu przeżycia całkowitego w leczeniu podtrzymującym z udziałem erlotynibu do czasu przeżycia całkowitego w przypadku erlotynibu podawanego w momencie progresji choroby. Badanie nie osiągnęło pierwszorzędowego punktu końcowego. Czas przeżycia całkowitego
u pacjentów przyjmujących erlotynib w leczeniu podtrzymującym nie był lepszy niż u pacjentów przyjmujących erlotynib w drugim rzucie, u których w guzach nowotworowych nie stwierdzono obecności aktywującej mutacji EGFR (HR= 1,02, 95% CI, 0,85 do 1,22, p=0,82). Drugorzędowy punkt końcowy jakim był PFS nie wykazał żadnej różnicy pomiędzy erlotynibem i placebo w leczeniu podtrzymującym (HR=0,94, 95% CI, 0,80 do 1,11; p=0,48).
Na podstawie wyników badania BO25460 (IUNO), erlotynib nie jest zalecany w leczeniu podtrzymującym u pacjentów bez aktywującej mutacji EGFR.
Leczenie pacjentów z NDRP po niepowodzeniu przynajmniej jednej wcześniejszej chemioterapii (erlotynib podawany w monoterapii):
Skuteczność i bezpieczeństwo stosowania erlotynibu w leczeniu drugiego i trzeciego rzutu wykazano w randomizowanym badaniu z podwójnie ślepą próbą, kontrolowanym placebo (BR.21), przeprowadzonym u 731 pacjentów z miejscowo zaawansowanym niedrobnokomórkowym rakiem płuca lub niedrobnokomórkowym rakiem płuca z przerzutami, u których doszło do niepowodzenia po uprzednim zastosowaniu co najmniej jednego schematu chemioterapii. Pacjenci byli losowo przydzielani w stosunku 2:1 do grupy otrzymującej doustnie erlotynib w dawce 150 mg lub placebo raz na dobę. Punkty końcowe badania obejmowały czas przeżycia całkowitego, czas przeżycia bez progresji choroby (PFS), odsetek odpowiedzi, czas trwania odpowiedzi, czas do wystąpienia nasilenia objawów raka płuca (kaszel, duszność i ból) i tolerancję. Pierwszoplanowym punktem końcowym był czas przeżycia.
Cechy demograficzne były równomiernie rozłożone w obu grupach pacjentów. Około dwie trzecie pacjentów stanowili mężczyźni i u około jednej trzeciej wyjściowy stan sprawności (PS) wg ECOG wynosił 2, a u 9% pacjentów stwierdzano stan sprawności 3 stopnia wg ECOG. Dziewięćdziesiąt trzy procent i 92% wszystkich pacjentów, odpowiednio, z grupy otrzymującej erlotynib i placebo, uprzednio poddawano leczeniu z zastosowaniem schematów zawierających pochodne platyny,
a u odpowiednio 36% i 37% wszystkich pacjentów zastosowano uprzednio taksany.
Skorygowany stosunek ryzyka HR zgonu w grupie pacjentów otrzymujących erlotynib w stosunku do grupy otrzymującej placebo wynosił 0,73 (95% przedział ufności - CI, 0,60 do 0,87)
(p = 0,001). Odsetek pacjentów pozostających przy życiu po 12 miesiącach wynosił 31,2% oraz 21,5%, odpowiednio dla grupy leczonej erlotynibem i grupy otrzymującej placebo. Mediana czasu przeżycia całkowitego wynosiła 6,7 miesięcy w grupie otrzymującej erlotynib (95% przedział ufności - CI; 5,5 do 7,8 miesięcy) w porównaniu do 4,7 miesięcy w grupie otrzymującej placebo (95% CI; 4,1 do 6,3 miesięcy).
Badano wpływ na czas przeżycia całkowitego w różnych podgrupach pacjentów. Wpływ leczenia erlotynibem na czas przeżycia całkowitego był podobny u pacjentów z wyjściowym stopniem sprawności PS 2-3 wg ECOG (HR = 0,77; 95% CI 0,6-1,0) i PS 0-1 (HR = 0,73; 95% CI 0,6-0,9),
u mężczyzn (HR = 0,76; 95% CI 0,6-0,9) i u kobiet (HR = 0,80; 95% CI 0,6-1,1), u pacjentów w wieku < 65 lat (HR = 0,75; 95% CI 0,6-0,9) i u starszych pacjentów (HR = 0,79; 95% CI 0,6-1,0), u pacjentów, u których wcześniej zastosowano jeden schemat chemioterapii (HR = 0,76; 95% CI 0,61,0) lub więcej niż jeden schemat chemioterapii (HR = 0,75; 95% CI 0,6-1,0), u pacjentów rasy
kaukaskiej (HR = 0,79; 95% CI 0,6-1,0) i u pacjentów rasy azjatyckiej (HR = 0,61; 95% CI 0,4-1,0), u pacjentów z rakiem gruczołowym (HR = 0,71; 95% CI 0,6-0,9) i u pacjentów z rakiem płaskonabłonkowym (HR = 0,67; 95% CI 0,5-0,9), lecz nie u pacjentów z innymi typami histologicznymi (HR = 1,04; 95% CI 0,7-1,5), u pacjentów w IV stopniu klinicznego zaawansowania przy rozpoznaniu choroby (HR = 0,92; 95% CI 0,7-1,2) i u pacjentów ze stopniem klinicznego zaawansowania < IV przy rozpoznaniu choroby (HR = 0,65; 95% CI 0,5-0,8). Pacjenci, którzy nigdy nie palili tytoniu odnieśli dużo większą korzyść z leczenia erlotynibem (HR dla przeżycia = 0,42; 95% CI 0,28-0,64) w porównaniu do pacjentów, którzy obecnie lub w przeszłości palili tytoń (HR = 0,87; 95% CI 0,71-1,05).
U 45% pacjentów, u których oznaczono ekspresję receptora EGFR, współczynnik ryzyka wynosił 0,68 (95% CI 0,49-0,94) dla pacjentów, u których wykazano ekspresję receptora EGFR, i 0,93 (95% CI 0,63-1,36) dla pacjentów, u których wyniki oznaczenia ekspresji receptora EGFR były negatywne (oznaczane w badaniu immunohistochemicznym przy użyciu zestawu EGFR pharmDx i definiujące status EGFR-ujemny jako mniej niż 10% wybarwionych komórek nowotworowych). U pozostałych 55% pacjentów, u których stopień ekspresji receptora EGFR był nieznany, iloraz ryzyka wynosił 0,77 (95% CI 0,61-0,98)
Mediana PFS wynosiła 9,7 tygodni w grupie leczonej erlotynibem (95% CI, 8,4 do 12,4 tygodni) w porównaniu do 8 tygodni w grupie otrzymującej placebo (95% CI, 7,9 do 8,1 tygodni).
Odsetek obiektywnych odpowiedzi oceniany wg skali RECIST w grupie pacjentów leczonych erlotynibem wynosił 8,9% (95% CI, 6,4 do 12,0%). Pierwszych 330 pacjentów oceniano centralnie (odsetek odpowiedzi 6,2%); 401 pacjentów oceniali badacze (odsetek odpowiedzi 11,2%).
Mediana czasu trwania odpowiedzi wynosiła 34,3 tygodni i wahała się od 9,7 do 57,6+ tygodni. Odsetek pacjentów, u których uzyskano całkowitą odpowiedź, częściową odpowiedź lub stabilizację choroby wynosił 44,0% i 27,5% odpowiednio w grupie leczonej erlotynibem i w grupie otrzymującej placebo (p = 0,004).
Korzyść z leczenia erlotynibem pod względem przeżycia obserwowano także u pacjentów, u których nie uzyskano obiektywnej odpowiedzi ze strony nowotworu (wg skali RECIST). Dowodzi tego wartość stosunku ryzyka zgonu wynosząca 0,82 (95% CI, 0,68 do 0,99) u pacjentów, u których najlepszą uzyskaną odpowiedzią była stabilizacja lub progresja choroby.
Leczenie erlotynibem przyniosło korzyści dotyczące objawów choroby poprzez znaczące wydłużenie czasu do wystąpienia nasilenia kaszlu, duszności i bólu w porównaniu do placebo.
W randomizowanym, prowadzonym metodą podwójnie ślepej próby badaniu III fazy (MO22162, CURRENTS), w którym porównywano dwie dawki erlotynibu (300 mg i 150 mg) u pacjentów aktualnie palących tytoń (średnio 38 paczkolat) z miejscowo zaawansowanym lub z przerzutami NDRP w leczeniu drugiego rzutu po niepowodzeniu chemioterapii, nie wykazano przewagi dawki 300 mg erlotynibu pod względem korzystnego wpływu na PFS w porównaniu do dawki zalecanej (odpowiednio 7,00 i 6,86 tygodnia).
Wszystkie drugorzędowe punkty końcowe oceny skuteczności były spójne z pierwszorzędowym punktem końcowym i nie stwierdzono różnicy w OS pomiędzy pacjentami leczonymi erlotynibem
w dawce 300 mg a 150 mg na dobę (HR 1,03, 95% CI 0,80 do 1,32). Dane dotyczące bezpieczeństwa stosowania były porównywalne pomiędzy dawkami 300 mg a 150 mg; odnotowano jednak liczbowe zwiększenie częstości występowania wysypki, choroby śródmiąższowej płuc i biegunki u pacjentów otrzymujących większą dawkę erlotynibu. Na podstawie danych z badania CURRENTS nie uzyskano dowodów na występowanie jakiejkolwiek korzyści z zastosowania większej dawki erlotynibu wynoszącej 300 mg w porównaniu z zalecaną dawką 150 mg u czynnych palaczy tytoniu.
Pacjenci w tym badaniu nie byli dobierani na podstawie statusu mutacji EGF. Patrz punkty 4.2, 4.4, 4.5 i 5.2.
Rak trzustki (erlotynib podawany równocześnie z gemcytabiną w badaniu PA.3):
Skuteczność i bezpieczeństwo stosowania erlotynibu w skojarzeniu z gemcytabiną w leczeniu pierwszego rzutu oceniano w randomizowanym badaniu klinicznym z podwójnie ślepą próbą, kontrolowanym placebo, przeprowadzonym z udziałem pacjentów z miejscowo zaawansowanym, nieoperacyjnym rakiem trzustki lub rakiem trzustki z przerzutami. Pacjenci byli losowo przydzielani do grupy otrzymującej erlotynib lub placebo raz na dobę w sposób ciągły oraz gemcytabinę dożylnie (1000 mg/m2; cykl 1. – dni 1, 8, 15, 22, 29, 36 i 43 w cyklu trwającym 8 tygodni; cykl 2. i następne – dni 1, 8 i 15 w cyklu trwającym 4 tygodnie [dawka i sposób podawania zatwierdzone dla raka trzustki
– patrz charakterystyka produktu leczniczego dla gemcytabiny]). Erlotynib lub placebo przyjmowane były doustnie raz na dobę do czasu wystąpienia progresji choroby lub niemożliwych do zaakceptowania objawów toksyczności. Pierwszorzędowym punktem końcowym był czas przeżycia całkowitego.
Cechy demograficzne i charakterystyka choroby były wyjściowo podobne w obu grupach pacjentów – grupie otrzymującej erlotynib w dawce 100 mg wraz z gemcytabiną oraz w grupie otrzymującej placebo wraz z gemcytabiną – z wyjątkiem nieznacznie większego odsetka kobiet w grupie erlotynib/gemcytabina w porównaniu z grupą placebo/gemcytabina:
Dane wyjściowe | Erlotynib | Placebo |
Kobiety | 51% | 44% |
Wyjściowy stan sprawności wg ECOG (PS) = 0 | 31% | 32% |
Wyjściowy stan sprawności wg ECOG (PS) = 1 | 51% | 51% |
Wyjściowy stan sprawności wg ECOG (PS) = 2 | 17% | 17% |
Choroba rozsiana wyjściowo | 77% | 76% |
Przeżycie oceniano w populacji zgodnej z zaplanowanym leczeniem (ang. Intent-to-treat – ITT) na podstawie danych dotyczących przeżycia zebranych w okresie obserwacji. Wyniki przedstawiono w poniższej tabeli (wyniki dla grup pacjentów z chorobą rozsianą i miejscowo zaawansowaną pochodzą z analizy podgrup).
Parametr | Erlotynib (miesiące) | Placebo (miesiące) | ∆ (miesiące) | CI dla ∆ | HR | CI dla HR | Wartość p |
Cała populacja | |||||||
Mediana czasu przeżycia całkowitego | 6,4 | 6,0 | 0,41 | -0,54 - 1.64 | 0,82 | 0,69-0,98 | 0,028 |
Średni czas | 8,8 | 7,6 | 1,16 | -0,05-2,34 | |||
przeżycia całkowitego | |||||||
Populacja z chorobą rozsianą | |||||||
Mediana czasu przeżycia całkowitego | 5,9 | 5,1 | 0,87 | -0,26-1,56 | 0,80 | 0,66-0,98 | 0,029 |
Średni czas przeżycia całkowitego | 8,1 | 6,7 | 1,43 | 0,17-2,66 | |||
Populacja z chorobą miejscowo zaawansowaną | |||||||
Mediana czasu przeżycia całkowitego | 8,5 | 8,2 | 0,36 | -2,43-2,96 | 0,93 | 0,65-1,35 | 0,713 |
Średni czas przeżycia całkowitego | 10,7 | 10,5 | 0,19 | -2,43-2,69 | |||
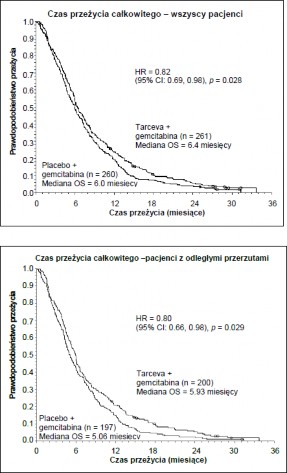
W analizie retrospektywnej („post-hoc”) stwierdzono, że pacjenci z korzystnym wyjściowym stanem klinicznym (ból o małym nasileniu, dobra jakość życia, dobry stan sprawności) mogą uzyskać większą korzyść ze stosowania erlotynibu. Korzyść ta jest szczególnie silnie skorelowana z małym nasileniem bólu.
W analizie retrospektywnej („post-hoc”) wykazano, że pacjenci przyjmujący erlotynib, u których pojawiła się wysypka, osiągali dłuższy czas przeżycia całkowitego w porównaniu z pacjentami, u których wysypka nie wystąpiła (mediana OS = 7,2 mies. vs 5 mies., HR: 0,61).
U 90% pacjentów leczonych erlotynibem wysypka wystąpiła w ciągu pierwszych 44 dni. Mediana czasu do wystąpienia wysypki wynosiła 10 dni.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań erlotynibu we wszystkich podgrupach populacji dzieci i młodzieży we wskazaniu niedrobnokomórkowy rak płuca i rak trzustki (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Wchłanianie
Maksymalne stężenia erlotynibu w osoczu uzyskuje się w ciągu około 4 godzin po podaniu doustnym. Badanie przeprowadzone u zdrowych ochotników pozwoliło uzyskać szacunkową wartość biodostępności całkowitej równą 59%. Ekspozycja po podaniu doustnym może ulegać zwiększeniu pod wpływem pożywienia.
Dystrybucja
Średnia rzeczywista objętość dystrybucji erlotynibu wynosi 232 l. Erlotynib jest dystrybuowany do tkanki nowotworowej u ludzi. W badaniu przeprowadzonym z udziałem 4 pacjentów (3
z niedrobnokomórkowym rakiem płuc - NDRP, i 1 z rakiem krtani) otrzymujących erlotynib doustnie w dawce 150 mg na dobę, stężenie erlotynibu w próbkach guza po wycięciu chirurgicznym w 9 dniu leczenia wynosiły średnio 1,185 ng/g tkanki. Odpowiada to ogólnej średniej wartości sięgającej 63% (zakres 5-161%) maksymalnych stężeń w osoczu w stanie stacjonarnym. Główne czynne metabolity były obecne w guzie nowotworowym w stężeniach wynoszących średnio 160 ng/g tkanki, co odpowiada ogólnej średniej wartości 113% (zakres 88-130%) maksymalnych stężeń w osoczu w stanie stacjonarnym. Erlotynib wiąże się z białkami osocza w około 95%. Erlotynib wiąże się z albuminami surowicy i z kwaśną α1-glikoproteiną (AAG).
Metabolizm
U ludzi erlotynib jest metabolizowany w wątrobie przez układ enzymatyczny cytochromu (CYP), głównie przez CYP3A4 i w mniejszym stopniu CYP1A2. Metabolizm pozawątrobowy przebiegający z udziałem CYP3A4 w jelicie, CYP1A1 w płucach i CYP1B1 w tkankach nowotworowych, może być elementem klirensu metabolicznego erlotynibu.
Zidentyfikowano trzy główne szlaki metaboliczne: 1) O-demetylację po jednej lub obu stronach łańcucha, z późniejszym utlenianiem do kwasów karboksylowych; 2) utlenianie grupy acetylenowej, a następnie hydroliza do kwasu arylokarboksylowego; 3) hydroksylację pierścienia aromatycznego grupy fenyloacetylenowej. Główne metabolity erlotynibu, OSI-420 i OSI-413, powstałe w wyniku O-
demetylacji po jednej stronie łańcucha, wykazywały siłę działania porównywalną do erlotynibu w nie- klinicznych testach in vitro oraz w modelach nowotworów in vivo. Występują one w osoczu
w stężeniach <10% stężenia erlotynibu i wykazują podobną farmakokinetykę jak erlotynib.
Wydalanie
Erlotynib jest wydalany przede wszystkim w postaci metabolitów z kałem (>90%), a wydalanie nerkowe stanowi tylko niewielką część (około 9%) dawki doustnej. Mniej niż 2% dawki doustnej wydala się w postaci macierzystego leku. Farmakokinetyczna analiza populacyjna u 591 pacjentów przyjmujących erlotynib jako jedyny lek wykazała, że średni całkowity klirens wynosi 4,47 l/h,
a mediana okresu półtrwania wynosi 36,2 godzin. Można więc oczekiwać, że czas potrzebny do uzyskania stanu stacjonarnego stężenia w osoczu będzie wynosił około 7-8 dni.
Farmakokinetyka w szczególnych populacjach
Na podstawie farmakokinetycznej analizy populacyjnej nie stwierdzono klinicznie istotnej zależności pomiędzy przewidywaną wartością całkowitego klirensu a wiekiem, masą ciała, płcią i grupą etniczną pacjenta. Czynniki zależne od pacjenta, które korelowały z farmakokinetyką erlotynibu, obejmowały stężenie bilirubiny całkowitej w surowicy, AAG i aktualne palenie papierosów. Zwiększone stężenia bilirubiny całkowitej i AAG były związane z wolniejszym wydalaniem erlotynibu. Kliniczne znaczenie tych różnic nie jest jasne. Jednakże, u palaczy szybkość wydalania erlotynibu była większa.
Potwierdzono to w badaniu farmakokinetycznym osób zdrowych niepalących i aktualnie palących papierosy, które otrzymały pojedynczą dawkę doustną 150 mg erlotynibu. Średnia geometryczna Cmax wynosiła 1056 ng/ml u osób niepalących i 689 ng/ml u osób palących, przy średnim stosunku osób palących do niepalących wynoszącym 65,2% (95% CI: 44,3 do 95,9, p = 0,031). Średnia geometryczna AUC0-inf wynosiła 18726 ng•h/ml wśród osób niepalących i 6718 ng•h/ml wśród osób palących, przy średnim stosunku wynoszącym 35,9% (95% CI: 23,7 do 54,3, p < 0,0001). Średnia geometryczna C24h wynosiła 288 ng/ml wśród osób niepalących i 34,8 ng/ml wśród osób palących, przy średnim stosunku wynoszącym 12,1% (95% CI: 4,82 do 30,2, p = 0,0001). W badaniu rejestracyjnym III fazy
u pacjentów z niedrobnokomórkowym rakiem płuca, u osób aktualnie palących papierosy, średnie wartości najmniejszych stężeń leku w surowicy krwi wynosiły 0,65 µg/ml (n=16) i były one około dwukrotnie mniejsze niż u pacjentów uprzednio palących papierosy lub nigdy niepalących (1,28 µg/ml, n=108). Efektowi temu towarzyszyło zwiększenie klirensu erlotynibu o 24%. W badaniu I fazy z eskalacją dawki erlotynibu u pacjentów z niedrobnokomórkowym rakiem płuca, którzy byli aktualnymi palaczami papierosów, na podstawie analiz farmakokinetycznych wykazano proporcjonalny wzrost średnich stężeń erlotynibu wraz ze zwiększeniem dawki erlotynibu ze 150 mg do maksymalnej dawki tolerowanej 300 mg. Po podaniu dawki 300 mg średnie wartości najmniejszych
stężeń leku w surowicy krwi u pacjentów aktualnie palących papierosy wynosiły 1,22 µg/ml (n=17). Patrz punkty 4.2, 4.4, 4.5 i 5.1.
Na podstawie wyników badań dotyczących farmakokinetyki, osobom palącym papierosy należy doradzić zaprzestanie palenia w trakcie przyjmowania erlotynibu, ponieważ palenie tytoniu może zmniejszać stężenie leku w osoczu.
Na podstawie wyników populacyjnej analizy dotyczącej farmakokinetyki, obecność opioidów wydaje się zwiększać ekspozycję o około 11%.
Inna farmakokinetyczna analiza populacyjna została przeprowadzona na podstawie danych dotyczących erlotynibu zebranych u 204 pacjentów z rakiem trzustki, którzy otrzymywali erlotynib wraz z gemcytabiną. Analiza ta wykazała, że czynniki wpływające na klirens erlotynibu u pacjentów w badaniu dotyczącym raka trzustki były bardzo podobne do czynników obserwowanych we wcześniejszej analizie farmakokinetycznej gdzie stosowano lek w monoterapii. Nie zidentyfikowano nowych działań zależnych od zmiennych towarzyszących. Równoczesne podawanie gemcytabiny nie wpływało na klirens osoczowy erlotynibu.
Dzieci i młodzież
Nie prowadzono specjalnych badań u dzieci i młodzieży.
Pacjenci w podeszłym wieku
Nie prowadzono specjalnych badań u pacjentów w podeszłym wieku.
Zaburzenia czynności wątroby
Erlotynib jest wydalany przede wszystkim przy udziale wątroby. W grupie pacjentów z litym guzami i umiarkowaną niewydolnością wątroby (punktacja w skali Child-Pugh 7-9), średnie geometryczna AUC0-t i Cmax erlotynibu wynosiły odpowiednio: 27000 ng•h/ml i 805 ng/ml, w porównaniu do 29300 ng•h/ml i 1090 ng/ml w grupie pacjentów z prawidłową czynnością wątroby, w tym pacjentów
z pierwotnym rakiem wątroby lub przerzutami do wątroby. Pomimo, że Cmax było znamiennie mniejsze w grupie pacjentów z umiarkowaną niewydolnością wątroby, różnica ta nie jest klinicznie istotna. Brak danych dotyczących wpływu ciężkich zaburzeń czynności wątroby na farmakokinetykę erlotynibu. W farmakokinetycznej analizie populacyjnej zwiększone stężenia całkowitej bilirubiny w surowicy wiązały się z wolniejszym wydalaniem erlotynibu.
Zaburzenia czynności nerek
Erlotynib i jego metabolity nie są w znaczącym stopniu wydalane przez nerki, ponieważ mniej niż 9% pojedynczej dawki jest wydalane z moczem. W farmakokinetycznej analizie populacyjnej nie stwierdzono klinicznie istotnej zależności między wydalaniem erlotynibu a klirensem kreatyniny, ale nie ma danych dotyczących pacjentów, u których klirens kreatyniny wynosi <15 ml/min.
Działania toksyczne po podaniu wielokrotnym, obserwowane u co najmniej jednego gatunku zwierząt lub w co najmniej jednym badaniu, obejmowały zmiany w rogówce (zanik, owrzodzenie), skórze (zwyrodnienie i zapalenie mieszków włosowych, zaczerwienienie, łysienie), jajnikach (zanik), wątrobie (martwica wątroby), nerkach (martwica brodawek nerkowych i poszerzenie cewek)
i przewodzie pokarmowym (opóźnione opróżnianie żołądka i biegunka). Parametry czerwonokrwinkowe były obniżone, natomiast liczba krwinek białych, głównie granulocytów obojętnochłonnych, ulegała zwiększeniu. Stwierdzano związane z leczeniem zwiększenie aktywności AlAT, AspAT i stężenia bilirubiny we krwi. Wyniki te obserwowano przy ekspozycji na lek znacznie poniżej wartości istotnych klinicznie.
Na podstawie mechanizmu działania erlotynibu można wnioskować, że może on mieć właściwości teratogenne. Dane pochodzące z badań działania toksycznego na reprodukcję u szczurów i królików
w dawkach zbliżonych do maksymalnej dawki tolerowanej i(lub) dawek toksycznych dla matki wykazały toksyczny wpływ na reprodukcję (embriotoksyczność u szczurów, resorpcję zarodka i działanie toksyczne na płód u królików) i rozwój (zmniejszenie wzrastania i przeżywalności
potomstwa u szczurów), ale nie wykazały działania teratogennego i szkodliwego wpływu na płodność. Obserwowano to przy ekspozycji na lek na poziomie wartości istotnych klinicznie.
Erlotynib dawał negatywne wyniki w konwencjonalnych badaniach genotoksyczności. Dwuletnie badania dotyczące działania rakotwórczego z zastosowaniem erlotynibu u szczurów i myszy
w dawkach nieprzekraczających dawek terapeutycznych stosowanych u ludzi, dały negatywne wyniki (do dawek dwukrotnie i dziesięciokrotnie większych, odpowiednio, na podstawie Cmax i(lub) AUC).
Skórną reakcję fototoksyczną o niewielkim nasileniu obserwowano u szczurów po naświetlaniu promieniowaniem UV.
Rdzeń tabletki:
Laktoza jednowodna
Celuloza mikrokrystaliczna i wapnia wodorofosforan Karboksymetyloskrobia sodowa (typ A)
Krzemionka koloidalna bezwodna Celuloza mikrokrystaliczna
Sodu laurylosiarczan Magnezu stearynian
Otoczka tabletki:
Hypromeloza 2910, 6 mPas Hydroksypropyloceluloza Tytanu dwutlenek (E 171) Makrogol
Nie dotyczy.
3 lata
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Pudełko z blistrami OPA/Aluminum/PVC/Aluminum zawierającymi 30 tabletek.
Bez specjalnych wymagań dotyczących usuwania.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Vipharm S.A.
ul. A. i F. Radziwiłłów 9
05-850 Ożarów Mazowiecki
tel.: (+4822) 679 5135
fax: (+4822) 678 92 87
e-mail: vipharm@vipharm.com.pl
Erlotinib Vipharm, 25 mg Pozwolenie nr 25537 Erlotinib Vipharm, 100 mg Pozwolenie nr 25538 Erlotinib Vipharm, 150 mg Pozwolenie nr 25539
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 05-09-2019
18/11/2022








