







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODULKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na substancję czynną, orzeszki ziemne, soję lub którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Ciąża (patrz punkt 4.6).
Kobiety w wieku rozrodczym, które nie stosują skutecznych metod antykoncepcji (patrz punkty 4.4 i 4.6).
Karmienie piersią (patrz punkt 4.6).
Ciężkie zaburzenia czynności wątroby (z marskością wątroby lub bez) (patrz punkt 4.2).
Wyjściowe wartości aminotransferaz wątrobowych (aminotransferazy asparaginianowej (AspAT) i (lub) aminotransferazy alaninowej (AlAT)) >3xGGN (patrz punkty 4.2 i 4.4).
Idiopatyczne zwłóknienie płuc, z wtórnym nadciśnieniem płucnym lub bez (patrz punkt 5.1).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
Ambrisentan Zentiva, 5 mg, tabletki powlekane Ambrisentan Zentiva, 10 mg, tabletki powlekane
Ambrisentan Zentiva, 5 mg, tabletki powlekane
Każda tabletka zawiera 5 mg ambrisentanu.
Ambrisentan Zentiva, 10 mg, tabletki powlekane
Każda tabletka zawiera 10 mg ambrisentanu. Substancje pomocnicze o znanym działaniu:
Ambrisentan Zentiva, 5 mg, tabletki powlekane:
każda tabletka zawiera około 47,5 mg laktozy jednowodnej, 0,14 mg lecytyny sojowej i 0,022 mg barwnika
czerwień Allura AC (E 129).
Ambrisentan Zentiva, 10 mg, tabletki powlekane
każda tabletka zawiera około 95 mg laktozy jednowodnej, 0,21 mg lecytyny sojowej i 0,405 mg barwnika czerwień Allura AC (E 129).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana.
Ambrisentan Zentiva, 5 mg, tabletki powlekane
Jasnoróżowe, kwadratowe, wypukłe tabletki powlekane z wytłoczonym oznakowaniem „5” po jednej stronie, gładkie po drugiej, o długości nominalnej/średnicy około 5,9 mm.
Ambrisentan Zentiva, 10 mg, tabletki powlekane
Różowe, owalne, obustronnie wypukłe tabletki powlekane z wytłoczonym oznakowaniem „10” po jednej stronie, gładkie po drugiej, o długości nominalnej około 11,1 mm i średnicy nominalnej około 5,6 mm.
Produkt leczniczy Ambrisentan Zentiva jest wskazany w leczeniu tętniczego nadciśnienia płucnego (ang. pulmonary arterial hypertension, PAH) u dorosłych pacjentów sklasyfikowanych według WHO do klasy czynnościowej II i III, w tym w leczeniu skojarzonym (patrz punkt 5.1). Wykazano jego skuteczność w idiopatycznym PAH (ang. IPAH), jak również w PAH związanym z chorobami tkanki łącznej.
Produkt leczniczy Ambrisentan Zentiva jest wskazany w PAH u młodzieży i dzieci (w wieku od 8 lat do ukończenia 18. roku życia) sklasyfikowanych według WHO do klasy czynnościowej II i III, w tym wleczeniu skojarzonym. Wykazano jego skuteczność w IPAH, jak również w PAH o podłożu rodzinnym, PAH związanym ze skorygowaną wadą wrodzoną oraz w PAH związanym z chorobami tkanki łącznej (patrz punkt 5.1).
Leczenie powinien rozpoczynać wyłącznie lekarz mający doświadczenie w leczeniu PAH.
Dawkowanie
Dorośli
Monoterapia ambrisentanem
Produkt leczniczy Ambrisentan Zentiva należy przyjmować doustnie, rozpoczynając od dawki 5 mg raz na dobę i w zależności od zaobserwowanej odpowiedzi i tolerancji, dawka ta może zostać zwiększona do 10 mg raz na dobę.
Ambrisentan w leczeniu skojarzonym z tadalafilem
W leczeniu skojarzonym z tadalafilem dawkę produktu Ambrisentan Zentiva należy ustalić na poziomie 10 mg raz na dobę.
W badaniu AMBITION, pacjentom podawano ambrisentan w dawce 5 mg raz na dobę przez pierwszych 8 tygodni, po czym, w zależności od tolerancji, zwiększano dawkę do 10 mg (patrz punkt 5.1). W leczeniu skojarzonym z tadalafilem pacjentów początkowo leczono dawką 5 mg ambrisentanu i 20 mg tadalafilu. W zależności od tolerancji, dawka tadalafilu była zwiększana do 40 mg po 4 tygodniach leczenia, a dawka ambrisentanu była zwiększana do 10 mg po 8 tygodniach leczenia.
Osiągnięto to u ponad 90% pacjentów. W zależności od tolerancji możliwe było również zmniejszanie dawek.
Z ograniczonej liczby danych wynika, że gwałtowne przerwanie przyjmowania ambrisentanu nie wiąże się z pogorszeniem przebiegu PAH.
Ambrisentan w leczeniu skojarzonym z cyklosporyną A
U osób dorosłych, podczas jednoczesnego stosowania z cyklosporyną A, dawkę ambrisentanu należy ograniczyć do 5 mg raz na dobę i uważnie monitorować stan pacjenta (patrz punkty 4.5 i 5.2).
Dzieci i młodzież w wieku od 8 lat do ukończenia 18. roku życia
Monoterapia ambrisentanem lub terapia ambrisentanem w skojarzeniu z innymi terapiami PAH Produkt leczniczy Ambrisentan Zentiva należy przyjmować doustnie, zgodnie ze schematem dawkowania opisanym poniżej:
Masa ciała (kg) | Początkowa dawka przyjmowana raz na dobę (mg) | Stopniowe zwiększanie dawki przyjmowanej raz na dobę (mg)a |
≥50 | 5 | 10 |
≥35 to <50 | 5 | 7,5 |
≥20 to <35 | 2,5 | 5 |
a = w zależności od zaobserwowanej odpowiedzi klinicznej i tolerancji (patrz punkt 5.1) | ||
Ambrisentan w leczeniu skojarzonym z cyklosporyną A
U dzieci i młodzieży, podczas jednoczesnego stosowania z cyklosporyną A, dawkę ambrisentanu u pacjentów
≥ 50 kg należy ograniczyć do 5 mg raz na dobę, a u pacjentów ≥20 kg do <50 kg należy ograniczyć do 2,5 mg
raz na dobę. Należy uważnie monitorować stan pacjenta (patrz punkty 4.5 i 5.2). Szczególne grupy pacjentów
Pacjenci w podeszłym wieku
Nie jest konieczne dostosowanie dawek u pacjentów w wieku powyżej 65 lat (patrz punkt 5.2).
Pacjenci z zaburzeniami czynności nerek
Nie jest konieczne dostosowanie dawek u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2). Doświadczenie w stosowaniu ambrisentanu u pacjentów z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny <30 ml/min) jest ograniczone; w tej podgrupie leczenie należy rozpoczynać ostrożnie i zwrócić szczególną uwagę podczas zwiększania dawki ambrisentanu do 10 mg.
Pacjenci z zaburzeniami czynności wątroby
Nie badano ambrisentanu u pacjentów z zaburzeniami czynności wątroby (z marskością wątroby lub bez). Ponieważ główne szlaki metabolizmu ambrisentanu to glukuronidacja i utlenianie z następczym wydalaniem z żółcią, można oczekiwać, że zaburzenie czynności wątroby zwiększy ekspozycję (Cmax i AUC) na ambrisentan. Dlatego nie wolno rozpoczynać leczenia ambrisentanem u pacjentów z ciężkimi zaburzeniami czynności wątroby bądź z istotnym klinicznie zwiększeniem aktywności aminotransferaz wątrobowych [ponad 3 krotnie przekraczającym górną granicę normy (>3 x GGN); patrz punkty 4.3 i 4.4].
Dzieci i młodzież
Nie oceniano bezpieczeństwa i skuteczności stosowania ambrisentanu u dzieci w wieku poniżej 8 lat. Nie ma dostępnych danych z badań klinicznych (dane z badań przeprowadzonych na młodych zwierzętach, patrz punkt 5.3).
Sposób podawania
Produkt Ambrisentan Zentiva jest przeznaczony do przyjmowania doustnie. Zaleca się połykanie tabletki w całości oraz przyjmowanie podczas posiłków lub niezależnie od posiłków. Zaleca się, aby tabletka nie była dzielona, kruszona ani żuta.
Ambrisentanu nie badano u wystarczającej ilości pacjentów, aby ustalić stosunek korzyści do ryzyka w klasie czynnościowej I PAH według WHO.
Nie określono skuteczności stosowania ambrisentanu w monoterapii u pacjentów w klasie czynnościowej IV PAH według WHO. Jeżeli stan kliniczny ulegnie pogorszeniu, wówczas należy rozważyć leczenie zalecane w ciężkich stadiach zaawansowania choroby (np. epoprostenol).
Czynność wątroby
Zaburzenia czynności wątroby bywają związane z PAH. Po zastosowaniu ambrisentanu obserwowano przypadki zbliżone do autoimmunologicznego zapalenia wątroby, w tym możliwe zaostrzenie uprzednio występującego autoimmunologicznego zapalenia wątroby, uszkodzenie wątroby oraz zwiększenie aktywności enzymów wątrobowych potencjalnie związane z leczeniem (patrz punkty 4.8 i 5.1). Dlatego przed rozpoczęciem leczenia ambrisentanem należy skontrolować aktywność aminotransferaz wątrobowych (AlAT i AspAT) i nie należy rozpoczynać leczenia ambrisentanem u pacjentów z wyjściowymi wartościami AlAT i (lub) AspAT >3 x GGN (patrz punkt 4.3).
Należy monitorować pacjentów w celu wykluczenia objawów uszkodzenia wątroby i zaleca się comiesięczne monitorowanie aktywności AlAT i AspAT. Jeżeli u pacjentów wystąpi trwałe, niewyjaśnione, klinicznie
istotne zwiększenie aktywności AlAT i (lub) AspAT, bądź jeżeli zwiększeniu aktywności AlAT i (lub) AspAT towarzyszą objawy przedmiotowe lub podmiotowe uszkodzenia wątroby (np. żółtaczka), leczenie ambrisentanem należy przerwać.
U pacjentów bez objawów klinicznych uszkodzenia wątroby lub żółtaczki, można rozważyć ponowne rozpoczęcie leczenia ambrisentanem, gdy aktywność enzymów wątrobowych powróci do normy.
Zaleca się konsultację hepatologa.
Stężenie hemoglobiny
Zmniejszenie stężenia hemoglobiny i hematokrytu były związane ze stosowaniem leków z grupy antagonistów receptora endoteliny (ERA), w tym ambrisentanu. Większość tego typu przypadków stwierdzano w pierwszych 4 tygodniach leczenia, później stężenie hemoglobiny zazwyczaj ulegało stabilizacji. W długoterminowym badaniu, prowadzonym na zasadzie otwartej próby (będącym rozszerzeniem głównego badania klinicznego fazy 3) średnie zmniejszenie stężenia hemoglobiny względem wartości wyjściowych (w przedziale od 0,9 do 1,2 g/dl) utrzymywało się podczas leczenia ambrisentanem do 4 lat. Po wprowadzeniu do obrotu obserwowano przypadki niedokrwistości wymagające przetoczenia komórek krwi (patrz punkt 4.8).
Nie zaleca się rozpoczynania leczenia ambrisentanem u pacjentów z klinicznie istotną niedokrwistością. Zaleca się monitorowanie stężenia hemoglobiny i (lub) hematokrytu podczas leczenia ambrisentanem, na przykład po 1 miesiącu, 3 miesiącach, a następnie okresowo, zgodnie z praktyką kliniczną. W razie zaobserwowania klinicznie istotnego obniżenia stężenia hemoglobiny lub hematokrytu, po wykluczeniu innych przyczyn, należy rozważyć zmniejszenie dawki lub przerwanie leczenia. Częstość występowania niedokrwistości była większa w przypadku leczenia ambrisentanem w skojarzeniu z tadalafilem (częstość zdarzenia niepożądanego 15%), w porównaniu do częstości występowania niedokrwistości w przypadku stosowania ambrisentanu oraz tadalafilu w monoterapii (odpowiednio: 7% i 11%).
Zatrzymanie płynów
Po zastosowaniu ERA, w tym również ambrisentanu obserwowano obrzęki obwodowe. W większości przypadków obrzęki obwodowe występujące w badaniach klinicznych były łagodne do umiarkowanych, chociaż mogą one występować częściej i być bardziej nasilone u pacjentów w wieku
≥65 lat. W krótkoterminowych badaniach klinicznych obrzęki obwodowe obserwowano częściej po zastosowaniu dawki 10 mg ambrisentanu (patrz punkt 4.8).
Po wprowadzeniu ambrisentanu do obrotu zgłaszano przypadki zatrzymania płynów, które występowały w ciągu kilku tygodni od rozpoczęcia leczenia ambrisentanem, w niektórych przypadkach konieczne było zastosowanie diuretyków lub hospitalizacja lub zgłaszano przypadki niewyrównanej niewydolności serca. Jeżeli u pacjentów występuje istniejące uprzednio zatrzymanie płynów, powinno być leczone zgodnie z zasadami leczenia przed rozpoczęciem stosowania ambrisentanu.
W przypadku wystąpienia klinicznie istotnego zatrzymania płynów, związanego lub niezwiązanego ze zwiększeniem masy ciała, należy ustalić przyczynę tego objawu, którą może być stosowanie ambrisentanu lub współistniejąca niewydolność serca oraz określić potrzebę zastosowania konkretnego leczenia lub odstawienia ambrisentanu. Częstość występowania obrzęków obwodowych była większa w przypadku leczenia ambrisentanem w skojarzeniu z tadalafilem (częstość zdarzenia niepożądanego 45%), w porównaniu do częstości występowania obrzęków obwodowych w przypadku stosowania ambrisentanu oraz tadalafilu w monoterapii (odpowiednio: 38% i 28%). Częstość występowania obrzęków obwodowych była największa w ciągu pierwszego miesiąca od rozpoczęcia leczenia.
Kobiety w wieku rozrodczym
Nie wolno rozpoczynać leczenia ambrisentanem u kobiet w wieku rozrodczym dopóki nie zostanie uzyskany ujemny wynik testu ciążowego oraz dopóki nie są stosowane skuteczne metody antykoncepcji. W przypadku jakichkolwiek wątpliwości, jaka skuteczna metoda antykoncepcji powinna być stosowana u pacjentki, należy rozważyć konsultację u ginekologa. Zaleca się comiesięczne wykonywanie testów ciążowych podczas stosowania ambrisentanu (patrz punkty 4.3 i 4.6).
Choroba żylno-okluzyjna płuc
Stwierdzono przypadki obrzęku płuc u pacjentów z chorobą żylno-okluzyjną płuc podczas stosowania leków rozszerzających naczynia, takich jak ERA (antagoniści receptora endoteliny). W rezultacie, jeżeli u pacjentów rozwija się ostry obrzęk płuc podczas stosowania ambrisentanu, należy rozważyć możliwość wystąpienia u nich choroby żylno-okluzyjnej płuc.
Jednoczesne stosowanie z innymi produktami leczniczymi
Należy dokładnie monitorować pacjentów stosujących ambrisentan, u których rozpoczęto leczenie ryfampicyną (patrz punkty 4.4 i 5.2).
Substancje pomocnicze
Tabletki Ambrisentan Zentiva zawierają laktozę jednowodną. Pacjenci z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy nie powinni stosować tego produktu leczniczego.
Ten produkt leczniczy zawiera mniej niż 1 mmol sodu (23 mg) na jedną tabletkę, więc jest on zasadniczo
„wolny od sodu”.
Ten produkt leczniczy zawiera lecytynę sojową (patrz punkt 4.3). Lecytyna sojowa może zawierać pozostałości białka sojowego i dlatego pacjenci, u których występuje nadwrażliwość na orzeszki ziemne albo soję nie powinni stosować tego leku.
Ten produkt leczniczy zawiera barwnik czerwień Allura AC, lak aluminiowy (E129), który może powodować reakcje uczuleniowe.
Ambrisentan nie hamuje ani nie indukuje enzymów fazy I lub II metabolizujących leki w stężeniach istotnych klinicznie w badaniach nieklinicznych in vitro oraz in vivo, co wskazuje, że jest mało prawdopodobne, aby ambrisentan zmieniał profil produktów leczniczych metabolizowanych w tych mechanizmach.
Możliwość indukowania aktywności CYP3A4 przez ambrisentan była oceniana u zdrowych ochotników, przy czym wyniki sugerują brak działania indukującego ambrisentanu na izoenzym CYP3A4.
Cyklosporyna A
Podczas badań u zdrowych ochotników, jednoczesne stosowanie cyklosporyny A i ambrisentanu w stanie stacjonarnym powodowało dwukrotne zwiększenie ekspozycji na ambrisentan. Może być to związane z hamowaniem przez cyklosporynę A nośników i enzymów metabolizujących, mających wpływ na farmakokinetykę ambrisentanu. Z tego względu, podczas jednoczesnego stosowania z cyklosporyną A, dawkę ambrisentanu u pacjentów dorosłych oraz dzieci i młodzieży o masie ciała ≥50 kg należy ograniczyć do 5 mg raz na dobę; u dzieci i młodzieży o masie ciała ≥20 do <50 kg dawka powinna być ograniczona do 2,5 mg raz na dobę (patrz punkt 4.2).Wielokrotne dawki ambrisentanu nie wywierały wpływu na ekspozycje na cyklosporynę A i dostosowywanie dawki cyklosporyny A nie jest uzasadnione.
Ryfampicyna
U zdrowych ochotników jednoczesne stosowanie ryfampicyny (inhibitora OATP, będącego silnym induktorem CYP3A i 2C19 oraz induktorem P-gp i urydyno-difosfo-glukonylotransferaz [UGTs]), wiązało sie z przemijającym (około dwukrotnym), zwiększeniem ekspozycji na ambrisentan po podaniu początkowych dawek leku. Tym niemniej, do 8. dnia nie stwierdzono żadnego, istotnego klinicznie wpływu wielokrotnych dawek ryfampicyny na ekspozycję na ambrisentan. Należy dokładnie monitorować pacjentów stosujących ambrisentan, u których rozpoczęto leczenie ryfampicyną (patrz punkty 4.4 i 5.2).
Inhibitory fosfodiesterazy
Równoczesne podawanie ambrisentanu z inhibitorem fosfodiesterazy, zarówno sildenafilem, jak i tadalafilem (substraty CYP3A4) u zdrowych ochotników nie zmieniało w istotny sposób farmakokinetyki ambrisentanu lub inhibitora fosfodiesterazy (patrz punkt 5.2).
Inne metody leczenia PAH
Skuteczność i bezpieczeństwo produktu leczniczego Ambrisentan Zentiva, stosowanego u pacjentów jednocześnie z innymi metodami leczenia PAH (np. stosowaniem prostanoidów czy stymulatorów rozpuszczalnej cyklazy guanylowej), nie były przedmiotem kontrolowanych badań klinicznych u pacjentów z PAH (patrz punkt 5.1). Na podstawie dostępnych danych dotyczących przemiany metabolicznej (patrz punkt 5.2), nie przewiduje się interakcji między ambrisentanem a stymulatorami rozpuszczalnej cyklazy guanylowej lub prostanoidami. Niemniej jednak, nie przeprowadzono żadnych badań dotyczących interakcji z tymi produktami leczniczymi. Dlatego też w przypadku równoczesnego stosowania tych leków zaleca się zachowanie ostrożności.
Doustne środki antykoncepcyjne
W badaniu klinicznym, prowadzonym u zdrowych ochotników, ambrisentan (10 mg raz na dobę) w stanie stacjonarnym nie wpływał w istotny sposób na farmakokinetykę pojedynczych dawek etynyloestradiolu i noretyndronu wchodzących w skład złożonych, doustnych środków antykoncepcyjnych (patrz punkt 5.2). Na podstawie wyników tego farmakokinetycznego badania można oczekiwać, że ambrisentan nie będzie istotnie wpływał na estrogenowe lub progestagenowe środki antykoncepcyjne.
Warfaryna
Ambrisentan nie wykazywał wpływu na farmakokinetykę stanu stacjonarnego ani na działanie przeciwzakrzepowe warfaryny w badaniu z udziałem zdrowych ochotników (patrz punkt 5.2). Warfaryna nie wykazuje także klinicznie istotnego wpływu na farmakokinetykę ambrisentanu. Ponadto, u pacjentów ambrisentan nie wykazywał ogólnego wpływu na tygodniową dawkę leków przeciwzakrzepowych typu warfaryny, czas protrombinowy (PT) oraz wskaźnik INR.
Ketokonazol
Podawanie ketokonazolu (silnego inhibitora CYP3A4) w stanie stacjonarnym nie powodowało klinicznie istotnego zwiększenia ekspozycji na ambrisentan (patrz punkt 5.2).
Wpływ ambrisentanu na nośniki ksenobiotyków
W warunkach in vitro, w istotnych klinicznie stężeniach, ambrisentan nie wykazuje hamującego działania na białka transportujące u ludzi, w tym transport zależny od glikoproteiny-P (Pgp), proteiny oporności na nowotwór piersi (BCRP), proteiny oporności wielolekowej 2 (MRP2), pompę eksportu soli kwasów żółciowych (BSEP), polipeptydy transportujące aniony organiczne (OATP1B1 i OATP1B3) i zależny od sodu polipeptyd kotransportujący taurocholan (NTCP).
Ambrisentan jest substratem dla reakcji zachodzących z udziałem Pgp.
Badania in vitro hepatocytów szczurzych wykazały ponadto, że ambrisentan nie indukował ekspresji protein Pgp, BSEP ani MRP2.
Ambrisentan w stanie stacjonarnym u zdrowych ochotników nie wywierał klinicznie istotnego wpływu na farmakokinetykę pojedynczej dawki digoksyny, będącej substratem dla Pgp (patrz punkt 5.2).
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono tylko z udziałem osób dorosłych.
Kobiety w wieku rozrodczym
Nie wolno rozpoczynać leczenia ambrisentanem u kobiet w wieku rozrodczym dopóki nie zostanie uzyskany ujemny wynik testu ciążowego oraz dopóki nie są stosowane skuteczne metody antykoncepcji. Podczas leczenia ambrisentanem zaleca się wykonywanie kontrolnego testu ciążowego co miesiąc.
Ciąża
Ambrisentan jest przeciwwskazany w czasie ciąży (patrz punkt 4.3). Badania na zwierzętach wykazały, że ambrisentan wykazuje działania teratogenne. Brak doświadczenia u ludzi.
Kobiety otrzymujące ambrisentan należy poinformować o ryzyku uszkodzenia płodu, a w przypadku zajścia w ciążę należy u nich rozpocząć leczenie alternatywne (patrz punkty 4.3, 4.4 i 5.3).
Karmienie piersią
Nie wiadomo, czy ambrisentan przenika do mleka kobiet karmiących piersią. Nie badano czy ambrisentan przenika do mleka u zwierząt. Dlatego karmienie piersią jest przeciwwskazane u pacjentek przyjmujących ambrisentan (patrz punkt 4.3).
Rozrodczość mężczyzn
Długotrwałe stosowanie ERA, w tym również ambrisentanu wiązało się z występowaniem zaniku kanalików nasiennych u zwierząt eksperymentalnych (patrz punkt 5.3). Pomimo, że w badaniu ARIES-E nie udowodniono szkodliwego wpływu długotrwałej ekspozycji na ambrisentan na liczbę
plemników, wykazano że długotrwałe przyjmowanie ambrisentanu wiązało się ze zmianami markerów spermatogenezy. Obserwowano zmniejszenie stężenia osoczowego inhibiny-B i zwiększenie stężenia FSH w osoczu. Wpływ tego zjawiska na rozrodczość u mężczyzn nie jest znany, ale nie można wykluczyć pogorszenia spermatogenezy. W badaniach klinicznych długotrwałe stosowanie ambrisentanu nie wiązało się ze zmianami stężenia testosteronu w osoczu.
Ambrisentan ma niewielki do średniego wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Należy brać pod uwagę stan kliniczny pacjenta i profil działań niepożądanych ambrisentanu (takich jak niedociśnienie, zawroty głowy, astenia, zmęczenie) oceniając zdolność pacjenta do wykonywania czynności wymagających oceny, zdolności motorycznych i poznawczych (patrz punkt 4.8). Pacjenci powinni mieć świadomość tego jaki wpływ może mieć na nich ambrisentan przed podjęciem decyzji o prowadzeniu pojazdów lub obsługiwaniu maszyn.
Podsumowanie profilu bezpieczeństwa
Najczęstszymi działaniami niepożądanymi obserwowanymi po podaniu ambrisentanu były obrzęki obwodowe (37%) i ból głowy (28%). Częstsze występowanie tych działań niepożądanych obserwowano po zastosowaniu dawki 10 mg ambrisentanu. W krótkotrwałych badaniach klinicznych obrzęki obwodowe były bardziej nasilone u pacjentów powyżej ≥65 lat (patrz punkt 4.4).
Ciężkie działania niepożądane związane ze stosowaniem ambrisentanu obejmują niedokrwistość (zmniejszenie stężenia hemoglobiny, obniżenie hematokrytu) i hepatotoksyczność.
Zmniejszenie stężenia hemoglobiny i obniżenie hematokrytu (10%) było związane ze stosowaniem anta- gonistów receptora endoteliny, w tym ambrisentanu. Większość z tych zmian stwierdzano w ciągupierw- szych 4 tygodni leczenia, a następnie poziom hemoglobiny ogólnie stabilizował się (patrz punkt 4.4).
W przypadku ambrisentanu obserwowano zwiększenie aktywności enzymów wątrobowych (2%), uszkodzenie wątroby i autoimmunologiczne zapalenie wątroby (w tym zaostrzenie choroby podsta- wowej) (patrz punkty 4.4 i 5.1).
Tabelaryczne zestawienie działań niepożądanych
Częstości występowania są zdefiniowane następująco: bardzo często (≥ 1/10); często (≥ 1/100 i
<1/10); niezbyt często (≥ 1/1 000 i <1/100); rzadko (≥ 1/10 000 i <1/1 000); bardzo rzadko (<1/10 000) i częstość nieznana (nie można jej określić na podstawie dostępnych danych). W przypadku działań niepożądanych zależnych od dawki, kategoria częstości dotyczy wyższej dawki ambrisentanu. W obrębie każdej grupy o określonej częstości występowania działania niepożądane są wymienione zgodnie ze zmniejszającą się ciężkością.
Klasyfikacja układów i narządów | Częstość | Działania niepożądane |
Zaburzenia krwi i układu chłonnego | Bardzo często | Niedokrwistość (zmniejszone stężenie hemoglobiny, obniżony hematokryt)1 |
Zaburzenia układu immunologicznego | Często | Reakcje nadwrażliwości (np. obrzęk naczy- niowo-ruchowy, wysypka, świąd) |
Zaburzenia układu nerwowego | Bardzo często | Ból głowy (w tym ból zatok, migrena)2, zawroty głowy |
Zaburzenia oka | Często | Nieostre widzenie, zaburzenia widzenia |
Zaburzenia ucha i błędnika | Często | Szumy uszne3 |
Niezbyt często | Nagła utrata słuchu3 | |
Zaburzenia serca | Bardzo często | Kołatanie serca |
Często | Niewydolność serca4 | |
Zaburzenia naczyniowe | Bardzo często | Zaczerwienienie skóry (zwłaszcza twarzy)5 |
Często | Niedociśnienie,omdlenie | |
Zaburzenia układu oddecho- wego, klatki piersioweji śród- piersia | Bardzo często | Duszność6, przekrwienie błony śluzowej (niedrożność) gór- nych dróg oddechowych (np. nosa, zatok)7,zapa- lenie nosa i gardła 7 |
Często | Krwawienie z nosa, | |
Nieżyt nosa7, zapalenie zatok7 | ||
Zaburzenia żołądka i jelit | Bardzo często | Nudności,biegunka,wymioty5 |
Często | Ból brzucha,zaparcie | |
Zaburzenia wątroby i dróg żółciowych | Często | Zwiększenie aktywności aminotransferaz wątrobowych |
Niezbyt często | Uszkodzenie wątroby (patrz punkt 4.4), autoim- munologiczne zapalenie wątroby (patrzpunkt 4.4) | |
Zaburzenia skóry i tkanki podskórnej | Często | Wysypka8 |
Zaburzenia ogólne i stany w miejscu podania | Bardzo często | Obrzęki obwodowe, zatrzymanie płynów, bóle/dyskomfort w klatce piersiowej5, zmęczenie |
Często | Astenia |
1 Patrz punkt 'Opis wybranych działań niepożądanych'
2 Częstość występowania bólu głowy wydaje się większa po zastosowaniu dawki 10 mg ambrisentanu
3 Przypadki zaobserwowano wyłącznie w kontrolowanym za pomocą placebo badaniu klinicznym dotyczącym stosowania ambrisentanu w skojarzeniu z tadalafilem.
4 W większości odnotowanych przypadków niewydolność serca związana była z zatrzymaniem płynów.
5 Częstość obserwowana w kontrolowanym za pomocą placebo badaniu klinicznym dotyczącym stosowania ambrisentanu w skojarzeniu z tadalafilem. Podczas monoterapii ambrisentanem częstość występowania była rzadsza.
6 Przypadki nasilenia duszności o nieznanej etiologii obserwowano wkrótce po rozpoczęciu podawania ambrisentanu.
7 Występowanie przekrwienia błony śluzowej (niedrożności) nosa podczas leczenia ambrisentanem było zależne od dawki.
8 Wysypka obejmuje wysypkę rumieniową, wysypkę uogólnioną, wysypkę plamistą i wysypkę ze świądem.
Opis wybranych działań niepożądanych
Zmniejszenie stężenia hemoglobiny
Po wprowadzeniu do obrotu obserwowano przypadki niedokrwistości wymagającej podania komórek krwi (patrz punkt 4.4). Zmniejszenie stężenia hemoglobiny (niedokrwistość) występowało częściej po zastosowaniu dawki 10 mg ambrisentanu. W trakcie 12 tygodniowych, kontrolowanych za pomocą placebo badaniach klinicznych fazy 3, średnie stężenie hemoglobiny zmniejszało się w grupie pacjentów otrzymujących produkt leczniczy Ambrisentan Zentiva, co stwierdzano już w tygodniu 4 (zmniejszenie o 0,83 g/dl); wydaje się, że średnia zmiana poziomu wyjściowego ulegała stabilizacji w okresie kolejnych 8 tygodni. Łącznie u 17 pacjentów (6,5%) w grupach leczonych ambrisentanem wystąpiło zmniejszenie stężenia hemoglobiny wynoszące ≥15% w odniesieniu do wartości wyjściowej, prowadzące do stężenia hemoglobiny poniżej dolnej granicy normy.
Dzieci i młodzież
Bezpieczeństwo stosowania ambrisentanu u dzieci i młodzieży z PAH w wieku od 8 lat do ukończenia
18. roku życia oceniano w trwającym 24 tygodnie otwartym badaniu klinicznym fazy 2b, w grupie 41 pacjentów, którym podawano raz na dobę ambrisentan 2,5 mg lub 5 mg (grupa pacjentów otrzymują- cych małą dawkę) lub ambrisentan 2,5 mg lub 5 mg w dawce dobowej stopniowo zwiększanej do 5 mg, 7,5 mg lub 10 mg, zależnie od masy ciała (grupa pacjentów otrzymujących dużądawkę), w monoterapii lub w skojarzeniu z innymi produktami leczniczymi stosowanymi w leczeniu PAH. Dalsza ocena bez- pieczeństwa została przeprowadzona w ramach długoterminowego przedłużenia badania u 38 spośród 41 pacjentów. Zaobserwowane działania niepożądane, ocenione jako związane ze stosowaniem ambri- sentanu, były zgodne z działaniami niepożądanymi zaobserwowanymi w kontrolowanych badaniach klinicznych przeprowadzonych u osób dorosłych, w tym z występującymi najczęściej bólem głowy (15%, 6 z 41 pacjentów podczas 24-tygodniowego otwartego badania fazy 2b i 8%, 3 z 38 pacjentów podczas długoterminowego przedłużenia badania klinicznego) i przekrwienie błony śluzowej (niedroż- ność) nosa (8%, 3 z 41 pacjentów podczas 24- tygodniowego otwartego badania fazy 2b).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301, faks: + 48 22 49 21 309, strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu lub przedstawicielowi podmiotu odpowiedzialnego w Polsce.
U zdrowych ochotników stosowanie pojedynczej dawki 50 i 100 mg (5-10 razy większe, niż maksymalna zalecana dawka terapeutyczna) wiązało się z występowaniem bólu głowy, uderzeń gorąca, zawrotami głowy, nudnościami i obrzękiem błony śluzowej nosa.
Ze względu na mechanizm działania ambrisentanu, jego przedawkowanie może prowadzić do hipotonii (patrz punkt 5.3). W przypadku znacznej hipotonii może być konieczne aktywne wspomaganie układu sercowo-naczyniowego. Nie jest dostępna swoista odtrutka.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Ambrisentan jest antagonistą ETA (około 4000- krotnie bardziej selektywnym w stosunku do ETA niż do ETB).
Ambrisentan blokuje podtyp receptorów ETA, występujący głównie na komórkach mięśniówki gładkiej naczyń krwionośnych i miocytach mięśnia sercowego. Zapobiega to zachodzącej przy udziale endoteliny aktywacji drugiego układu przekaźników, która prowadzi do skurczu naczyń i proliferacji komórek mięśni gładkich.
Oczekuje się, że selektywność ambrisentanu wobec receptorów ETA w porównaniu z receptorami ETB umożliwi zachowanie zachodzącego z udziałem receptorów ETB wytwarzania substancji powodujących rozkurcz naczyń – tlenku azotu i prostacykliny.
śmierć lub
hospitalizacja z powodu zaostrzenia PAH,
progresja choroby,
niesatysfakcjonująca długoterminowa odpowiedź kliniczna.
Właściwości farmakokinetyczne
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODULKTU LECZNICZEGO
Grupa farmakoterapeutyczna: leki przeciwnadciśnieniowe, inne leki przeciwnadciśnieniowe, kod ATC: C02KX02
Mechanizm działania
Ambrisentan jest aktywnym po podaniu doustnym, należącym do klasy pochodnych kwasu propionowego, selektywnym antagonistą receptora endoteliny A (ETA). Endotelina odgrywa istotną rolę w patofizjologii PAH.
Skuteczność kliniczna i bezpieczeństwo
Przeprowadzono dwa randomizowane, podwójnie zaślepione, wieloośrodkowe, kontrolowane placebo, kluczowe badania fazy 3 (ARIES-1 i 2). W badaniu ARIES-1 uczestniczyło 201 pacjentów i w badaniu tym porównywano dawki 5 mg i 10 mg ambrisentanu z placebo. W badaniu ARIES-2 uczestniczyło 192 pacjentów i w badaniu tym porównywano dawki 2,5 mg i 5 mg ambrisentanu z placebo. W obydwu badaniach ambrisentan dołączano do stosowanego przez pacjentów leczenia wspomagającego/podstawowego, które mogło obejmować skojarzenie digoksyny, leków przeciwzakrzepowych, diuretyków, tlenu i leków rozkurczających naczynia (antagonistów kanału wapniowego, inhibitorów konwertazy). Do badań tych kwalifikowano pacjentów z idiopatycznym PAH lub z PAH związanym z chorobami tkanki łącznej (PAH-CTD). U większości pacjentów występowały objawy odpowiadające klasie czynnościowej II (38,4%) lub klasie III (55,0%) wg WHO. Pacjenci z rozpoznaną wcześniej chorobą wątroby (marskość lub klinicznie istotne zwiększenie aktywności aminotransferaz) oraz pacjenci stosujący inne leczenie PAH (np. prostanoidy) byli wykluczeni z badania. W badaniach tych nie oceniano parametrów hemodynamicznych.
Pierwszorzędowym punktem końcowym zdefiniowanym w badaniach fazy 3 była poprawa wydolności wysiłkowej oceniana na podstawie zmiany odległości przebywanej podczas 6 minutowego marszu (6MWD) po 12 tygodniach w odniesieniu do wyniku wyjściowego. W obu badaniach leczenie każdą z dawek ambrisentanu powodowało istotną poprawę 6MWD.
W badaniach ARIES-1 i 2 poprawa średniego wyniku 6MWD względem placebo w grupie otrzymującej dawkę 5 mg w tygodniu 12 wynosiła odpowiednio 30,6 m (95% CI: od 2,9 do 58,3; p=0,008) oraz 59,4 m (95% CI: 29,6 to 89,3; p<0,001). W badaniu ARIES-1 poprawa średniego wyniku 6MWD względem placebo w grupie otrzymującej dawkę 10 mg w tygodniu 12 wynosiła 51,4
m (95% CI: od 26,6 do 76,2; p<0,001).
Przeprowadzono połączoną analizę (pre-specified) badań III fazy (ARIES-C). W porównaniu do placebo średnia poprawa wyniku 6MWD wynosiła 44,6 m (95% CI: od 24,3 do 64,9; p<0,0001) w grupie dawki 5 mg oraz 52,5 m (95% CI: od 28,8 do 76,2 m; p<0,001) w grupie dawki 10 mg.
W badaniu ARIES-2 (analiza dla wszystkich dawek) leczenie ambrisentanem było związane z istotnym wydłużeniem czasu do wystąpienia klinicznego pogorszenia przebiegu PAH w porównaniu z placebo (p<0,001) oraz ze zmniejszeniem ryzyka względnego o 80% (95% CI: 47% to 92%). Kryteria oceny obejmowały: zgon, konieczność przeszczepienia płuc, hospitalizację z powodu PAH, zabieg septostomii przedsionkowej, konieczność dołączenia innych leków z powodu PAH i tzw. wczesne kryteria przerwania terapii. Analizując wpływ wszystkich stosowanych dawek stwierdzono statystycznie istotną (3,41 ± 6,96) poprawę czynnościową względem placebo (-0,20 ± 8.14, p=0,005), ocenianą na podstawie skali funkcjonowania fizycznego w kwestionariuszu oceny stanu zdrowia SF-
36. Stosowanie ambrisentanu istotnie poprawiało wynik w skali oceny duszności wg Borga (ang. Borg Dyspnea Index, BDI) po 12 tygodniach leczenia (BDI -1,1 vs placebo; 95% CI: -1,8 to -0,4; p=0,019; dane dla wszystkich dawek).
Dane z obserwacji długoterminowej
Pacjentów uczestniczących w badaniach ARIES-1 i 2 włączano do długoterminowego badania ARIES-E, prowadzonego na zasadzie próby otwartej (n=383). Całkowita średnia ekspozycja wynosiła około 145 ± 80 tygodni a maksymalna ekspozycja około 295 tygodni. Głównymi punktami końcowymi tego badania była częstość występowania i nasilenie działań niepożądanych związanych z długoterminową ekspozycją na ambrisentan u pacjentów, w tym na wyniki testów czynnościowych wątroby. Informacje dotyczące bezpieczeństwa uzyskane po długotrwałej ekspozycji na ambrisentan w tym badaniu były zgodne z tymi, które obserwowano w 12-tygodniowych badaniach kontrolowanych placebo.
Obserwowane prawdopodobieństwo przeżycia pacjentów otrzymujących ambrisentan (łącznie dla grupy otrzymującej ambrisentan w różnych dawkach) po 1, 2 i 3 latach wynosiło odpowiednio 93%, 85% i 79%.
W prowadzonym na zasadzie otwartej próby badaniu (AMB222) oceniano wpływ stosowania ambrisentanu na zwiększenie aktywności aminotransferaz u 36 pacjentów, u których uprzednio przerwano leczenie innymi lekami z grupy ERA z powodu nieprawidłowości w aktywnościach tych enzymów. W okresie leczenia ambrisentanem trwającym średnio 53 tygodnie u żadnego z pacjentów zakwalifikowanych do tego badania nie wystąpiła potwierdzona aktywność AlAT w surowicy
>3xGGN, które wymagałoby trwałego odstawienia leczenia. U 50% pacjentów w tym czasie dawkę ambrisentanu zwiększono z 5 mg do 10 mg.
Skumulowana częstość występowania nieprawidłowych wyników aktywności aminotransferaz
>3 x GGN we wszystkich badaniach fazy II i III (w tym w otwartych badaniach długoterminowych) wynosiła 17 z 483 pacjentów ze średnim okresem ekspozycji wynoszącym 79,5 tygodni. Odpowiada to częstości 2,3 zdarzeń na 100 lat ekspozycji na ambrisentan na pacjenta. W długoterminowym prowadzonym na zasadzie otwartej próby rozszerzeniu badania ARIES-E, 2-letnie ryzyko wystąpienia zwiększenia aktywności aminotransferaz w osoczu >3xGGN u pacjentów leczonych ambrisentanem wynosiło 3,9%.
Dodatkowe informacje kliniczne
W badaniu fazy 2 (AMB220) u pacjentów z PAH po 12 tygodniach zaobserwowano poprawę parametrów hemodynamicznych (n=29). Leczenie ambrisentanem było związane ze wzrostem średniego wskaźnika sercowego, obniżeniem średniego ciśnienia w tętnicy płucnej oraz zmniejszeniem średniego oporu naczyniowego w tętnicy płucnej.
Podczas leczenia ambrisentanem obserwowano zmniejszenie wartości ciśnienia skurczowego i rozkurczowego. W kontrolowanych placebo badaniach klinicznych trwających 12 tygodni, średnie
obniżenie ciśnienia skurczowego i rozkurczowego w chwili zakończenia badania, oceniane względem wartości wyjściowych wynosiło odpowiednio 3mm Hg i 4,2 mmHg. W długoterminowym badaniu ARIES-E średnie obniżenie ciśnienia skurczowego i rozkurczowego utrzymywało się podczas leczenia ambrisentanem do 4 lat.
W badaniu interakcji przeprowadzonym z udziałem zdrowych ochotników nie zaobserwowano klinicznie istotnego wpływu na farmakokinetykę ambrisentanu lub sildenafilu, a skojarzenie to było dobrze tolerowane. Liczba pacjentów, którzy otrzymywali równocześnie ambrisentan i sildenafil w badaniach ARIES-E i AMB222, wynosiła odpowiednio 22 (5,7%) i 17 (47%). U pacjentów tych nie występowały żadne dodatkowe problemy dotyczące bezpieczeństwa.
Skuteczność kliniczna terapii skojarzonej z tadalafilem
Przeprowadzone zostało wieloośrodkowe badanie kliniczne 3 fazy z aktywnym komparatorem, podwójnie ślepą próbą i o przebiegu zależnym od zdarzeń (AMB112565/AMBITION), oceniające skuteczność terapii początkowej ambrisentanem w skojarzeniu z tadalafilem w porównaniu do monoterapii samym ambrisentanem oraz samym tadalafilem. Badanie to przeprowadzono u 500 pacjentów z nieleczonym wcześniej PAH, z randomizacją odpowiednio 2:1:1. Żaden z pacjentów nie otrzymywał samego placebo. Główna analiza dotyczyła grupy poddanej terapii skojarzonej w porównaniu do połączonych grup poddanych monoterapiom. Dokonano również dodatkowych porównań grupy z terapią skojarzoną z poszczególnymi grupami poddanymi monoterapiom. Zgodnie z kryteriami badania, z badania zostali wyłączeni pacjenci ze znaczną niedokrwistością, zatrzymaniem płynów oraz rzadkimi chorobami siatkówki. Wyłączeni zostali również pacjenci, u których wyjściowa wartość AlAT i AspAT była większa niż dwukrotność górnej granicy normy.
W momencie rozpoczęcia badania, 96% pacjentów stanowiły osoby nie poddane wcześniej jakiejkolwiek terapii właściwej dla PAH, a średni czas od postawienia diagnozy do włączenia pacjenta do badania wynosił 22 dni. Pacjenci początkowo otrzymywali ambrisentan w dawce 5 mg i tadalafil w dawce 20 mg, a następnie zwiększano dawkę do 40 mg tadalafilu w 4. tygodniu oraz do 10 mg ambrisentanu w 8. tygodniu, o ile nie wystąpiły problemy z tolerancją. Okres trwania terapii w ramach badania z podwójnie ślepą próbą wynosił ponad 1,5 roku.
Pierwszorzędowy punkt końcowy stanowiło wystąpienie pierwszego klinicznego przypadku niepowodzenia, zdefiniowanego jako:
Średni wiek wszystkich pacjentów wynosił 54 lata (SD 15; przedział 18 – 75 lat). W momencie rozpoczęcia udziału w badaniu pacjenci byli sklasyfikowani według WHO do klasy czynnościowej II (31%) i III (69%). W grupie poddanej badaniu, PAH najczęściej spowodowane było uwarunkowaniem genetycznym lub miało przyczynę idiopatyczną (56%), rzadziej występowało w związku z zaburzeniami tkanki łącznej (37%), w powiązaniu z lekami lub toksynami (3%), ze skorygowaną wrodzoną prostą wadą serca (2%) oraz z zakażeniem HIV (2%). Średni wynik przeprowadzonego początkowo testu 6-minutowego chodu (6MWD) u pacjentów z II i III klasy czynnościowej WHO wynosił 353 metry.
Wyniki w punkcie końcowym
Leczenie w terapii skojarzonej skutkowało 50-procentową redukcją ryzyka (ryzyko względne [en. hazard ratio, HR] 0,502; 95% CI: 0,348 do 0,724; p=0,0002) wystąpienia złożonego punktu końcowego, związanego z niepowodzeniem klinicznym przed końcową wizytą oceniającą, w porównaniu do połączonej grupy poddanej monoterapii (wykres nr 1 i tabela nr 1). Efekt terapeutyczny spowodowany 63-procentową redukcją liczby hospitalizacji po stosowaniu terapii skojarzonej, został osiągnięty wcześnie i utrzymany. Skuteczność terapii skojarzonej w pierwszorzędowym punkcie końcowym była spójna w porównaniu z poszczególnymi grupami poddanymi monoterapii oraz między poszczególnymi podgrupami różniącymi się wiekiem, pochodzeniem etnicznym, regionem geograficznym oraz etiologią choroby (IPAH/hPAH i PAH-
CTD). Efekt był znaczący zarówno dla pacjentów z II jak i III klasy czynnościowej WHO. Wykres nr 1
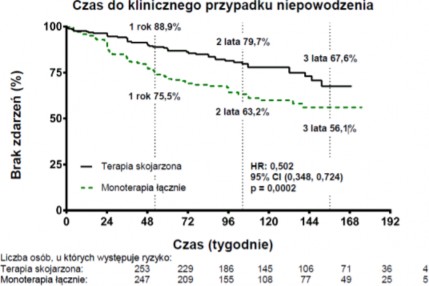
Tabela nr 1
Ambrisentan + Tadalafil (N=253) | Monoterapia łącznie (N=247) | Ambrisentan monoterapia (N=126) | Tadalafil monoterapia (N=121) | |
Czas do pierwszego klinicznego przypadku niepowodzenia (stwierdzonego) | ||||
Niepowodzenie kliniczne, liczba (%) | 46 (18) | 77 (31) | 43 (34) | 34 (28) |
Współczynnik ryzyka (95% CI) | 0,502 (0,348; 0,724) | 0,477 (0,314; 0,723) | 0,528 (0,338; 0,827) | |
Wartość p, test log-rank | 0,0002 | 0,0004 | 0,0045 | |
Składowe pierwszego klinicznego przypadku niepowodzenia (stwierdzone) | ||||
Śmierć (wszystkie przyczyny) | 9 (4%) | 8 (3%) | 2 (2%) | 6 (5%) |
Hospitalizacja z powodu zaostrzenia PAH | 10 (4%) | 30 (12%) | 18 (14%) | 12 (10%) |
Progresja choroby | 10 (4%) | 16 (6%) | 12 (10%) | 4 (3%) |
Niesatysfakcjonująca długoterminowa odpowiedź kliniczna | 17 (7%) | 23 (9%) | 11 (9%) | 12 (10%) |
Czas do pierwszej hospitalizacji z powodu zaostrzenia PAH (stwierdzonej) | ||||
Pierwsza hospitalizacja, liczba (%) | 19 (8%) | 44 (18%) | 27 (21%) | 17 (14%) |
Współczynnik ryzyka (95% CI) | 0,372 | 0,323 | 0,442 | |
Wartość p, test log-rank | 0,0002 | <0,0001 | 0,0124 |
Drugorzędowe punkty końcowe
Badano drugorzędowe punkty końcowe:
Tabela nr 2
Drugorzędowe punkty końcowe (zmiana od stanu wyjściowego do 24. tygodnia) | Ambrisentan + Tadalafil | Monoterapia łącznie | Różnica i przedział ufności | Wartość p |
NT-proBNP (% redukcji) | -67,2 | -50,4 | Różnica % -33,8; 95% CI: -44,8, -20,7 | p<0,0001 |
% pacjentów z satysfakcjonującą odpowiedzią kliniczną w 24. tygodniu | 39 | 29 | Współczynnik ryzyka 1,56; 95% CI: 1,05, 2,32 | p=0,026 |
6MWD (metry, mediana zmiany) | 49,0 | 23,8 | 22,75m; 95% CI: 12,00, 33,50 | p<0,0001 |
Idiopatyczne zwłóknienie płuc
Badanie przeprowadzane u 492 pacjentów (ambrisentan N=329, placebo N=163) chorych na idiopatycze zwłóknienie płuc z których 11% miało również PAH (grupa 3 zgodnie z klasyfikacją WHO) zostało przerwane na wczesnym etapie ze względu na fakt, iż niemożliwe okazało się osiągnięcie pierwotnego punktu końcowego dotyczącego skuteczności (badanie ARTEMIS-IPF). W grupie leczonej ambrisentanem stwierdzono dziewięćdziesiąt przypadków (27%) postępującego przebiegu idiopatycznego zwłóknienia płuc (z włączeniem hospitalizacji ze względu na problemy z oddychaniem) lub śmierci w porównaniu z 28 takimi przypadkami (17%) w grupie pacjentów którym podawano placebo. W związku z tym ambrisentan jest przeciwwskazany do stosowania u pacjentów z idiopatycznym zwłóknieniem płuc z lub bez PAH. (patrz punkt 4.3).
Dzieci i młodzież
Badanie AMB112529
Bezpieczeństwo stosowania i tolerancję ambrisentanu stosowanego raz na dobę przez 24 tygodnie oce- niono w otwartym, niekontrolowanym badaniu klicznicznym u 41 pacjentów z PAH, w wieku od 8lat do ukończenia 18. roku życia (mediana: 13 lat). Etiologia PAH była: idiopatyczna (n=26; 63%), prze- trwałe wrodzone PAH pomimo korekcji chirurgicznej (n=11; 27%), PAH wtórne do choroby tkanki łącznej (n=1; 2%) lub występujące rodzinnie (n=3 7,3%). Spośród 11 pacjentów z wrodzoną wadą serca, 9 miało wady przegrody międzykomorowej, 2 miało wady przegrody międzyprzedsionkowej i 1 przetrwały przewód tętniczy. W momencie rozpoczynania badania pacjenci byli zaklasyfikowani do klasy czynnościowej WHO II (n=32, 78%) lub III (n=9; 22%). W momencie włączenia do badania pa- cjenci przyjmowali produkty lecznicze stosowane w leczeniu PAH (najczęściej monoterapią PDE5i [n=18; 44%], terapia skojarzona PDE5i i prostanoidami [n=8; 20%]) lub monoterapia prostanoidami [n=1; 2%] i kontynuowali tę terapię w czasie trwania badania. Pacjencizostali podzieleni na dwie grupy pod względem otrzymywanej dawki ambrisentanu: grupę otrzymującą raz na dobę ambrisentan
2,5 mg lub 5 mg (mała dawka, n=21) oraz grupę otrzymującą razna dobę ambrisentan 2,5 mg lub 5 mg w dawce stopniowo zwiększanej do 5 mg, 7,5 mg lub 10 mg, w zależności od masy ciała (duża dawka, n=20). Po dwóch tygodniach u łącznie 20 pacjentów z obu grup dostosowano dawkę na podstawie od- powiedzi klinicznej i tolerancji. Badanie ukończyło 37 pacjentów, 4 pacjentów wycofało się z badania.
Nie zaobserwowano wpływu dawki na działanie ambrisentanu na główny wynik skuteczności doty- czący wydolności wysiłkowej (6MWD). Średnia zmiana w stosunku do wartości wyjściowej w
24. tygodniu w 6MWD dla pacjentów w grupach otrzymujących małe i duże dawki z pomiarem na po- czątku i po 24 tygodniach leczenia wyniosła odpowiednio: +55,14 m (95% CI: 4,32 do 105,95) u 18 pacjentów i +26,25 m (95 % CI: 4,59 do 57,09) u 18 pacjentów. Średnia zmiana w stosunku do warto- ści wyjściowej w 24. tygodniu w 6MWD dla wszystkich 36 pacjentów (obie dawki zbiorczo) wyniosła
+40,69 m (95% CI: 12,08 do 69,31). Wyniki te były spójne z wynikami obserwowanymi u osób doro- słych. W 24. tygodniu 95% i 100% pacjentów w grupach otrzymujących odpowiednio małą idużą dawkę pozostało ustabilizowanych (klasa czynnościowa niezmieniona lub poprawiona). Szacunkowa ocena przeżycia Kaplana-Meiera wolna od zdarzeń dotycząca pogorszenia PAH (zgon [wszystkie przyczyny], przeszczep płuca lub hospitalizacja z powodu pogorszenia PAH lub pogorszenia związa- nego z PAH) w 24. tygodniu wyniosła 86% i 85% w przypadku grupy otrzymującej odpowiednio: małą i dużą dawkę.
Hemodynamikę zmierzono u 5 pacjentów (z grupy otrzymującej małą dawkę). Średnie zwiększenie wskaźnika sercowego w stosunku do wartości początkowej wyniosło +0,94 l/min/m2, średnie obniże- nie średniego ciśnienia w tętnicy płucnej wyniosło 2,2 mmHg, a średnie obniżenie płucnego oporu na- czyniowego (ang. pulmonary vascular resistance, PVR) wyniosło -277 dyn·s/cm5 (-
3,46 mmHg/l/min).
U pacjentów pediatrycznych z PAH, którzy otrzymywali ambrisentan przez 24 tygodnie, obniżenie średniej geometrycznej NT-pro-BNP w stosunku do wartości początkowej wyniosło 31% w grupie otrzymującej małą dawkę (2,5 i 5 mg) i 28% w grupie otrzymującej dużą dawkę (5, 7,5 i 10 mg).
Badanie AMB112588
Dane długoterminowe uzyskano dla 38 spośród 41 pacjentów, którzy otrzymywali ambrisentan w ra- mach 24-tygodniowego randomizowanego badania klinicznego. Średni czas trwania ekspozycji na le- czenie ambrisentanem wynosił 3,4 ± 1,8 roku (do 6,4 roku), przy czym 63% pacjentów leczono przez co najmniej 3 lata, a 42% przez co najmniej 4 lata. W ramach przedłużonej, otwartej fazy badania pa- cjenci mogli otrzymać dodatkową, zgodną z wymaganiami terapię PAH. U większości pacjentów roz- poznano PAH idiopatyczne lub o podłożu dziedzicznym (68%). Ogółem 46% pacjentówpozostało w II klasie czynnościowej WHO. Szacunkowa ocena przeżycia Kaplana-Meiera wyniosła 94,42% i 90,64% odpowiednio po 3 i 4 latach od rozpoczęcia terapii. W tych samych punktach czasowych 77,09% i 73,24% pacjentów pozostało bez pogorszenia PAH, gdzie pogorszenie zdefiniowano jako: zgon (wszystkie przyczyny), skierowanie do przeszczepienia płuca lub septostomiiprzedsionkowej lub po- gorszenie PAH prowadzące do hospitalizacji, zmiany dawki ambrisentanu, dodania lub zmiany dawki istniejącego ukierunkowanego środka terapeutycznego stosowanego w PAH, wzrostu klasy czynno- ściowej WHO; zmniejszenia 6MWD lub wystąpienia objawów przedmiotowych lub podmiotowych prawokomorowej niewydolności serca.
Wchłanianie
Ambrisentan jest szybko wchłaniany u ludzi. Po podaniu doustnym maksymalne stężenie ambrisentanu w osoczu (Cmax) występuje zazwyczaj około 1,5 godziny po podaniu leku, zarówno na czczo, jak i po spożyciu posiłku. Cmax i pole pod krzywą stężenia w czasie (AUC) wzrastają w sposób proporcjonalny do dawki w zakresie dawek terapeutycznych. Stan stacjonarny zazwyczaj uzyskuje się po 4 dniach powtarzanego podawania.
Badanie wpływu pokarmu po podaniu ambrisentanu zdrowym ochotnikom na czczo lub po posiłku wysokotłuszczowym wykazało, że Cmax ulega zmniejszeniu o 12%, natomiast AUC nie ulega zmianie. Takie zmniejszenie maksymalnego stężenia nie jest istotne kliniczne, w związku z tym ambrisentan można przyjmować podczas posiłków lub niezależnie od posiłków.
Dystrybucja
Ambrisentan w znacznym stopniu wiąże się z białkami osocza. W warunkach in vitro stopień wiązania ambrisentanu z białkami osocza wynosi średnio 98,8% i jest niezależny od stężenia w zakresie od 0,2 do 20 mikrogramów/ml. Ambrisentan wiąże się głównie z albuminami (96,5%), a w mniejszym stopniu także z glikoproteiną kwaśną alfa1.
Dystrybucja ambrisentanu w erytrocytach jest mała, ze średnim wskaźnikiem krew: osocze wynoszącym odpowiednio 0,57 u mężczyzn i 0,61 u kobiet.
Metabolizm
Ambrisentan jest niesulfonamidowym (należącym do pochodnych kwasu propionowego) antagonistą receptora endoteliny (ERA).
Ambrisentan jest glukuronidowany przez kilka izoenzymów UGT (UGT1A9S, UGT2B7S i UGT1A3S), tworząc glukuronid ambrisentanu (13%). Ambrisentan jest także metabolizowany przez utlenianie, głównie przez CYP3A4, a w mniejszym stopniu przez CYP3A5 i CYP2C19, tworząc
4- hydroksymetyloambrisentan (21%), który jest następnie glukuronidowany do glukuronidu
4- hydroksymetyloambrisentanu (5%). Powinowactwo wiązania 4-hydroksymetyloambrisentanu z ludzkim receptorem endoteliny jest 65 razy mniejsze niż w przypadku ambrisentanu. Dlatego w stężeniach występujących w osoczu (około 4% w stosunku do macierzystego ambrisentanu),
4-hydroksymetyloambrisentan nie wydaje się mieć udziału w aktywności farmakologicznej ambrisentanu.
Z danych in vitro wynika, że ambrisentan w stężeniu 300 µM powodował, w stopniu mniejszym niż 50%, zahamowanie UGT1A1, UGT1A6, UGT1A9, UGT2B7 (do 30%) i enzymów cytochromu P450: 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 i 3A4 (do 25%). W warunkach in vitro ambrisentan nie wykazuje hamującego działania, w stężeniach istotnych klinicznie, na białka transportujące u ludzi, w tym transport zależny od glikoproteiny-P (Pgp), BCRP, MRP2, BSEP, OATP1B1, OATP1B3 i NTCP. Ponadto ambrisentan nie indukował ekspresji protein Pgp, BSEP ani MRP2 w hepatocytach szczurzych.
Uwzględniając wszystkie dane, z danych in vitro wynika, że ambrisentan, w klinicznie istotnych stężeniach (Cmax w surowicy do 3,2 µM) nie powinien mieć wpływu na UGT1A1, UGT1A6, UGT1A9, UGT2B7 lub enzymy cytochromu P450: 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4
lub transport za pośrednictwem BSEP, BCRP, Pgp, MRP2, OATP1B1/3 lub NTCP.
Wpływ ambrisentanu w stanie stacjonarnym (10 mg raz na dobę) na farmakokinetykę i farmakodynamikę pojedynczej dawki warfaryny (25 mg), mierzony na podstawie czasu protrombinowego (PT) i wskaźnika INR, oceniano u 20 zdrowych ochotników. Ambrisentan nie wykazywał żadnego istotnego klinicznie wpływu na farmakokinetykę lub farmakodynamikę warfaryny. Podobnie też, równoczesne podawanie warfaryny nie miało wpływu na farmakokinetykę ambrisentanu (patrz punkt 4.5).
Wpływ 7 dniowego podawania syldenafilu (w dawce 20 mg trzy razy na dobę) na farmakokinetykę pojedynczej dawki ambrisentanu oraz wpływ 7 dniowego podawania ambrisentanu (w dawce 10 mg raz na dobę) na farmakokinetykę pojedynczej dawki syldenafilu oceniano u 19 zdrowych ochotników. Z wyjątkiem 13 procentowego wzrostu Cmax syldenafilu po równoczesnym podawaniu z ambrisentanem, nie zaobserwowano innych zmian parametrów farmakokinetycznych syldenafilu,
N- desmetylosildenafilu ani ambrisentanu. Uważa się, że ten niewielki wzrost Cmax syldenafilu nie jest istotny klinicznie (patrz punkt 4.5).
Wpływ ambrisentanu w stanie stacjonarnym (10 mg raz na dobę) na farmakokinetykę pojedynczej dawki tadalafilu oraz wpływ tadalafilu w stanie stacjonarnym (40 mg raz na dobę) na farmakokinetykę
pojedynczej dawki ambrisentanu oceniano u 23 zdrowych ochotników. Ambrisentan nie wywierał jakiegokolwiek klinicznie istotnego wpływu na farmakokinetykę tadalafilu. Jednocześnie stosowany tadalafil nie wpływał również na farmakokinetykę ambrisentanu (patrz punkt 4.5).
Wpływ powtarzanych dawek ketokonazolu (w dawce 400 mg raz na dobę) na farmakokinetykę dawki pojedynczej 10 mg ambrisentanu oceniano u 16 zdrowych ochotników. Ekspozycja na ambrisentan, oceniana na podstawie AUC(0-inf) i Cmax, była większa o odpowiednio 35% i 20%. Nie wydaje się, aby taka zmiana ekspozycji miała jakiekolwiek znaczenie kliniczne, w związku z czym ambrisentan można podawać równocześnie z ketokonazolem.
U zdrowych ochotników badano wpływ powtarzanych dawek cyklosporyny A (100 - 150 mg dwa razy na dobę) na farmakokinetykę ambrisentanu (w dawce 5 mg raz na dobę) w stanie stacjonarnym oraz wpływ powtarzanych dawek ambrisentanu (5 mg raz na dobę) na farmakokinetykę cyklosporyny A (100 - 150 mg dwa razy na dobę) w stanie stacjonarnym. Po podaniu wielokrotnych dawek cyklosporyny A, wartości Cmax i AUC(0- τ) dla ambrisentanu zwiększyły się (odpowiednio o 48% i 121%). W związku z tym, podczas jednoczesnego stosowania z cyklosporyną A, u pacjentów dorosłych oraz dzieci i młodzieży o masie ciała ≥50 kg dawkę ambrisentanu należy ograniczyć do 5 mg raz na dobę, a u dzieci i młodzieży o masie ciała ≥20 do <50 kg dawkę ambrisentanu należy ograniczyć do 2,5 mg raz na dobę (patrz punkt 4.2). Jednakże, wielokrotne dawki ambrisentanu nie wywierały wpływu na ekspozycje na cyklosporynę A i dostosowywanie dawki cyklosporyny A nie jest wymagane.
W grupie zdrowych ochotników badano wpływ natychmiastowego stosowania powtarzanych dawek ryfampicyny (600 mg raz na dobę) na farmakokinetykę ambrisentanu w stanie stacjonarnym (podawanego 10 mg raz na dobę). Po podaniu początkowych dawek ryfampicyny stwierdzono przemijające zwiększenie stężenia ambrisentanu (AUC(0-τ)) (121% i 116% odpowiednio po pierwszej i drugiej dawce ryfampicyny), prawdopodobnie z powodu hamowania OATP przez ryfampicynę. Tym niemniej, do 8. dnia nie stwierdzono żadnego, istotnego klinicznie wpływu wielokrotnych dawek ryfampicyny na ekspozycję na ambrisentan. Należy dokładnie monitorować pacjentów stosujących ambrisentan, u których rozpoczęto leczenie ryfampicyną (patrz punkty 4.4 i 4.5).
Wpływ powtarzanych dawek ambrisentanu (10 mg) na farmakokinetykę dawki pojedynczej digoksyny oceniano u 15 zdrowych ochotników. Powtarzane dawki ambrisentanu powodowały niewielkie zwiększenie stężeń digoksyny w osoczu (AUC0-last) i zwiększenia stężenia maksymalnego Cmax digoksyny o 29%. Zwiększenie ekspozycji na digoksynę podczas stosowania powtarzanych dawek ambrisentanu nie zostało uznane za klinicznie istotne i nie wymaga dostosowywania dawek digoksyny (patrz punkt 4.5).
Wpływ 12 dniowego podawania ambrisentanu (10 mg raz na dobę) na farmakokinetykę pojedynczej dawki doustnego środka antykoncepcyjnego, zawierającego etynyloestradiol (35 µg) i noretindron (1 mg) był przedmiotem badania prowadzonego u zdrowych ochotniczek. Wartości Cmax i AUC(0–∞) były nieco zmniejszone dla etynyloestradiolu (odpowiednio o 8% i 4%) oraz nieco zwiększone dla noretindronu (odpowiednio o 13% i 14%). Opisane zmiany w warunkach ekspozycji na etynyloestradiol i noretyndron były niewielkie i jest mało prawdopodobne, aby miały znaczenie kliniczne (patrz punkt 4.5).
Eliminacja
Ambrisentan i jego metabolity są wydalane głównie z żółcią po metabolizmie wątrobowym i (lub) pozawątrobowym. Około 22% podanej dawki jest wydalane z moczem po podaniu doustnym, przy czym 3,3% stanowi niezmieniony ambrisentan. Okres półtrwania w osoczu u ludzi wynosi od 13,6 do 16,5 godziny.
Szczególne grupy pacjentów
Dorośli (płeć, wiek)
Na podstawie wyników populacyjnych analiz farmakokinetycznych u zdrowych ochotników oraz u pacjentów z PAH wykazano, że farmakokinetyka ambrisentanu nie jest w istotny sposób uzależniona od płci lub wieku (patrz punkt 4.2).
Dzieci i młodzież
Dostępne są ograniczone dane farmakokinetyczne dotyczące stosowania ambrisentanu u dzieci i mło- dzieży. Farmakokinetykę oceniano u pacjentów pediatrycznych w wieku od 8 lat do ukończenia 18. roku życia w jednym badaniu klinicznym (AMB112529).
Farmakokinetyka ambrisentanu po podaniu doustnym pacjentom z PAH w wieku od 8 lat do ukończenia 18. roku życia była zasadniczo zgodna z farmakokinetyką u dorosłych, po uwzględnieniu masy ciała. Oparte na modelu ekspozycje u dzieci i młodzieży w stanie stacjonarnym (AUCss) dla małych dawek i dużych dawek, dla wszystkich przedziałów wagowych pacjentów mieściły się między
Ze względu na działanie typowe dla tej klasy leków, duża, pojedyncza dawka ambrisentanu (np. w przypadku przedawkowania) może obniżać ciśnienie tętnicze powodując niedociśnienie i objawy związane z rozszerzeniem naczyń krwionośnych.
Ambrisentan nie jest inhibitorem transportu kwasów żółciowych i nie wykazano, aby powodował jawną hepatotoksyczność.
Po długotrwałym podawaniu ambrisentanu gryzoniom w dawkach mniejszych niż dawki terapeutyczne stosowane u ludzi, obserwowano stan zapalny i zmiany nabłonka jamy nosowej.
U psów, po przewlekłym stosowaniu dużych dawek ambrisentanu i ekspozycji przekraczającej 20 razy ekspozycję u ludzi obserwowano niewielkie reakcje zapalne.
Zaobserwowano przerost małżowin kości sitowej w jamie nosowej szczurów podczas ekspozycji na ambrisentan prowadzącej do trzykrotnego przekroczenia AUC w porównaniu do zastosowań klinicznych. Przerostu kości nosa nie obserwowano podczas badań u myszy i u psów. Na podstawie doświadczeń z innymi cząsteczkami przyjęto, że przerost małżowin nosowych u szczurów był powodem zmian zapalnych w tym obszarze.
Ambrisentan w wysokich stężeniach wykazywał działania klastogenne na komórki ssaków in vitro. Ambrisentan nie wykazywał działań mutagennych i genotoksycznych u bakterii, jak również nie powodował takich działań podczas dwóch badań in vivo prowadzonych na gryzoniach.
W 2-letnich badaniach u szczurów i myszy, w których lek był podawany doustnie, nie stwierdzono właściwości rakotwórczych. Występowało niewielkie zwiększenie częstości występowania gruczolakowłókniaków sutka - łagodnych guzów - jedynie u samców szczurów, które otrzymywały największą dawkę. Ekspozycja układowa na ambrisentan u samców szczurów po tej dawce (w oparciu o AUC w stanie stacjonarnym) była 6-krotnie większa niż występująca po dawce klinicznej 10 mg/dzień.
W badaniach toksyczności i płodności, po zastosowaniu doustnych dawek wielokrotnych, u samców szczurów i myszy obserwowano zanik kanalików nasiennych, czasem związany z brakiem wydzielania nasienia bez marginesu bezpieczeństwa. Zmiany w kanalikach nasiennych nie zawsze całkowicie ustępowały w okresie po odstawieniu leczenia. Nie stwierdzono jednak zmian w jądrach w badaniach prowadzonych na psach trwających do 39 tygodni i związanych z zastosowaniem dawek 35 razy przekraczających dawki stosowane u ludzi (na podstawie AUC). U samców szczurów ambrisentan nie miał wpływu na ruchliwość plemników, niezależnie od badanej dawki (do 300 mg/kg/dobę). Po podaniu dawki 300 mg/kg/dobę występowało niewielkie (<10%) zmniejszenie odsetka morfologicznie prawidłowych plemników po podaniu dawki 300 mg/kg/dobę, jednak nie występowało ono po podaniu dawki 100 mg/kg/dobę (odpowiednik ponad 9-krotnie większej ekspozycji na lek niż po podaniu dawki 10 mg/dobę w warunkach klinicznych). Wpływ tego zjawiska na rozrodczość u mężczyzn nie jest znany.
Wykazano, że ambrisentan wywiera działanie teratogenne u myszy i królików. Po zastosowaniu każdej stosowanej dawki obserwowano nieprawidłowości żuchwy, języka i (lub) podniebienia. Ponadto, w badaniu prowadzonym na szczurach obserwowano zwiększenie częstości występowania wady przegrody międzykomorowej, wady pnia naczyniowego, nieprawidłowości tarczycy i grasicy, kostnienia trzonu kości klinowej oraz występowanie tętnicy pępkowej po lewej stronie pęcherza moczowego zamiast po prawej. Teratogenność jest podejrzewanym skutkiem działania klasy leków ERA.
Podawanie ambrisentanu u samic szczurów począwszy od okresu późnej ciąży oraz w okresie laktacji powodowało niepożądane zachowania matki, zmniejszone przeżycie noworodków i upośledzenie zdolności rozrodczej potomstwa (stwierdzano zmniejszenie jąder podczas badań sekcyjnych) po ekspozycji na lek powodującej trzykrotne przekroczenie AUC uzyskiwanego podczas stosowania maksymalnych dawek u ludzi.
U młodych szczurów, którym podawano ambrisentan drogą pokarmową raz na dobę od 7. do 26., 36. lub 62. dnia po urodzeniu, (co odpowiada w przybliżeniu ludziom w wieku od noworodka do starszej młodzieży), stwierdzono zmniejszenie masy mózgu (-3% do -8%) bez zmian morfologicznych lub neurobehawioralnych po wystąpieniu szmerów oddechowych, bezdechu i niedotlenienia.
Efekty te występowały przy wartości AUC wynoszącej od 1,8 do 7-krotności ekspozycji u pacjentów pediatrycznych po dawce 10 mg. W innym badaniu, w którym terapii poddawano 5-tygodniowe szczury (odpowiadające wiekowi około 8 lat u ludzi), zmniejszenie masy mózgu obserwowano tylkopo bardzo dużych dawkach i wyłącznie u samców. Dostępne dane niekliniczne nie pozwalają na zrozumienie znaczenia klinicznego tej obserwacji dla dzieci w wieku poniżej 8 lat.
Rdzeń tabletki
Celuloza mikrokrystaliczna Laktoza jednowodna Kroskarmeloza sodowa Magnezu stearynian
Otoczka
Alkohol poliwinylowy Tytanu dwutlenek (E 171)
Makrogol 3350 (Glikol polietylenowy) (E 1521)
Talk
Czerwień Allura AC (E 129) Lecytyna sojowa (E 322)
Nie dotyczy.
3 lata.
Białe blistry z folii PVC/PVDC/Aluminium
Brak specjalnych zaleceń dotyczących temperatury przechowywania produktu leczniczego. Przechowywać w oryginalnym blistrze w celu ochrony przed światłem.
Przezroczyste blistry z folii PVC/PE/PVDC/Aluminium
Brak specjalnych zaleceń dotyczących temperatury przechowywania produktu leczniczego. Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Białe blistry z folii PVC/PVDC/Aluminium i/lub przezroczyste blistry z folii PVC/PE/PVDC/Aluminium
Wielkości opakowań: pudełka tekturowe zawierające blistry po 10 lub 30 tabletek powlekanych lub blistry jednodawkowe po 10x1 lub 30x1 tabletek powlekanych.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Zentiva k.s., Dolní Měcholupy, U Kabelovny 130, 102 37 Praga 10, Republika Czeska
Ambrisentan Zentiva 5 mg: Pozwolenie nr 26227 Ambrisentan Zentiva 10 mg: Pozwolenie nr 26228
DATA PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 05.02.2021
03/2022








