







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
z ostrym zespołem wieńcowym (OZW) lub
z zawałem mięśnia sercowego (zawał serca) w wywiadzie i wysokim ryzykiem zdarzeń sercowo-naczyniowych (patrz punkty 4.2 i 5.1).
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną
w punkcie 6.1 (patrz punkt 4.8).
Czynne krwawienie patologiczne.
Krwotok śródczaszkowy w wywiadzie (patrz punkt 4.8).
Ciężkie zaburzenie czynności wątroby (patrz punkty 4.2, 4.4 i 5.2).
Jednoczesne stosowanie tikagreloru i silnych inhibitorów enzymu CYP3A4 (np. ketokonazol, klarytromycyna, nefazodon, rytonawir i atazanawir), ponieważ może prowadzić do istotnego zwiększenia narażenia na tikagrelor (patrz punkt 4.5).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Pacjenci ze skłonnością do krwawień (np. ze względu na niedawne urazy, zabiegi chirurgiczne, zaburzenia krzepnięcia, czynne lub niedawne krwawienia z przewodu pokarmowego) lub u których występuje zwiększone ryzyko urazu. Stosowanie tikagreloru jest przeciwwskazane u pacjentów z czynnym, patologicznym krwawieniem, u pacjentów z krwotokiem śródczaszkowym w wywiadzie oraz u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkt 4.3).
Pacjenci stosujący jednocześnie leki, które mogą zwiększać ryzyko krwawień (np. niesteroidowe leki przeciwzapalne (NLPZ), doustne leki przeciwzakrzepowe i (lub) leki fibrynolityczne) zastosowane w ciągu 24 godzin przed zażyciem dawki tikagreloru.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Silne inhibitory CYP3A4 – jednoczesne stosowanie ketokonazolu z tikagrelorem spowodowało 2,4-krotne zwiększenie Cmax i 7,3-krotne zwiększenie AUC tikagreloru. Cmax i AUC czynnego metabolitu było zmniejszone odpowiednio o 89% i 56%. Przewiduje się, że inne silne inhibitory CYP3A4 (klarytromycyna, nefazodon, rytonawir i atazanawir) wywołują podobne działanie i dlatego jednoczesne stosowanie silnych inhibitorów CYP3A4 z tikagrelorem jest przeciwwskazane (patrz punkt 4.3).
Umiarkowane inhibitory CYP3A4 – jednoczesne stosowanie diltiazemu i tikagreloru powodowało zwiększenie Cmax tikagreloru o 69%, a AUC 2,7-krotnie oraz zmniejszenie Cmax czynnego metabolitu o 38%, bez wpływu na jego AUC. Tikagrelor nie wpłynął na stężenie diltiazemu w osoczu. Inne umiarkowane inhibitory CYP3A4 (np. amprenawir, aprepitant, erytromycyna i flukonazol) mogą wykazywać podobne działanie i również mogą być stosowane jednocześnie z tikagrelorem.
Obserwowano 2-krotne zwiększenie ekspozycji na tikagrelor po codziennym spożywaniu dużych ilości soku grejpfrutowego (3x200 ml). Nie należy spodziewać się, by ta wartość zwiększenia ekspozycji na tikagrelor była klinicznie istotna u większości pacjentów.
35%). Ta interakcja może mieć związek z obniżoną motoryką żołądkowo-jelitową i dotyczy także innych opioidów. Kliniczne znaczenie tego jest nieznane, ale dane wskazują na możliwość zmniejszenia skuteczności tikagreloru u pacjentów otrzymujących jednocześnie tikagrelor i morfinę. U pacjentów z ACS, u których nie można wstrzymać podawania morfiny a szybkie zahamowanie P2Y12 jest uważane za krytycznie istotne, można rozważyć stosowanie pozajelitowego inhibitora P2Y12.
Wpływ tikagreloru na działanie innych leków
Produkty lecznicze metabolizowane przez CYP3A4
Symwastatyna – jednoczesne stosowanie tikagreloru z symwastatyną powodowało zwiększenie Cmax symwastatyny o 81% i AUC o 56% oraz zwiększenie Cmax kwasu symwastatyny o 64% i jego AUC o 52% z pojedynczymi przypadkami zwiększenia 2- lub 3- krotnego. Jednoczesne stosowanie tikagrelolu i symwastatyny w dawce większej niż 40 mg na dobę mogłoby spowodować wystąpienie działań niepożądanych symwastatyny i dlatego należy je uwzględnić w ocenie potencjalnych korzyści tego skojarzenia. Nie stwierdzono wpływu symwastatyny na stężenie tikagreloru w osoczu. Tikagrelor może mieć podobny wpływ na stosowanie lowastatyny. Nie zaleca się jednoczesnego stosowania tikagreloru
z symwastatyną lub lowastatyną w dawkach większych niż 40 mg.
Atorwastatyna – jednoczesne stosowanie atorwastatyny i tikagreloru powoduje zwiększenie Cmax i AUC kwasu atorwastatyny odpowiednio o 23% i 36%. Podobne zwiększenie AUC
Nie można wykluczyć podobnego wpływu na inne statyny metabolizowane przez CYP3A4. W badaniu PLATO leczeni tikagrelorem pacjenci przyjmowali jednak różne statyny i u 93% spośród wszystkich pacjentów biorących udział w tym badaniu nie było zastrzeżeń co do bezpieczeństwa wynikającego ze stosowania statyn.
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
z badania PLATO [PLATelet Inhibition and Patient Outcomes], w którym tikagrelor porównywano z klopidogrelem, przy czym oba te leki podawano w skojarzeniu z ASA (kwas acetylosalicylowy)
i z innymi standardowymi sposobami leczenia;
z badania PEGASUS TIMI-54 [PrEvention with TicaGrelor of SecondAry Thrombotic Events in High-RiSk AcUte Coronary Syndrome Patients], w którym tikagrelor w skojarzeniu z ASA porównywano z ASA w monoterapii.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
blister z folii OPA/Aluminium/PVC/Aluminium lub PVC/PE/PVDC/Aluminium (symbole: słońce/księżyc) w tekturowych pudełkach zawierających 14, 56 lub 60 tabletek;
opakowania zbiorcze zawierające 168 (3 opakowania po 56) tabletek lub 180 (3 opakowania po 60) tabletek w blistrze z folii OPA/Aluminium/PVC/Aluminium lub PVC/PE/PVDC/Aluminium (symbole: słońce/księżyc)
blister jednodawkowy z folii OPA/Aluminium/PVC/Aluminium lub PVC/PE/PVDC/Aluminium w tekturowych pudełkach zawierających 100 x 1 tabletek;
Specjalne środki ostrożności dotyczące usuwania produktu leczniczego
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Tilobrastil, 90 mg, tabletki powlekane
Każda tabletka powlekana zawiera 90 mg tikagreloru (Ticagrelorum).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana (tabletka).
Okrągłe, obustronnie wypukłe, różowe tabletki z nadrukiem „90” na jednej stronie i gładkie po drugiej
stronie, o średnicy około 9,6 mm ± 5%.
Produkt leczniczy Tilobrastil, w skojarzeniu kwasem acetylosalicylowym (ASA), jest wskazany w celu zapobiegania zdarzeniom sercowo-naczyniowym u dorosłych pacjentów:
Dawkowanie
Pacjenci przyjmujący produkt leczniczy Tilobrastil powinni codziennie przyjmować również małą dawkę podtrzymującą kwasu acetylosalicylowego (ASA) 75-150 mg, jeśli nie jest to indywidualnie przeciwwskazane.
Ostre zespoły wieńcowe
Stosowanie produktu leczniczego Tilobrastil należy rozpocząć od pojedynczej dawki nasycającej 180 mg (2 tabletki o mocy 90 mg) i kontynuować leczenie dawką 90 mg dwa razy na dobę. U pacjentów z OZW czas trwania leczenia produktem Tilobrastil 90 mg dwa razy na dobę, powinien wynosić 12 miesięcy, chyba, że istnieją wskazania kliniczne do przerwania leczenia (patrz punkt 5.1).
Zawał mięśnia sercowego w wywiadzie
Zalecaną dawką produktu Tilobrastil jest 60 mg dwa razy na dobę, jeśli potrzebne jest przedłużone leczenie pacjentów z przebytym co najmniej rok temu, zawałem serca w wywiadzie i z wysokim ryzykiem zdarzeń sercowo-naczyniowych (patrz punkt 5.1). Leczenie można zacząć bez przerywania, jako kontynuację początkowego rocznego leczenia produktem leczniczym Tilobrastil 90 mg lub innym lekiem hamującym receptory difosforanu adenozyny (ADP) u pacjentów z OZW i wysokim ryzykiem zdarzeń sercowo-naczyniowych. Leczenie można również rozpocząć do 2 lat po zawale serca lub w ciągu roku od zaprzestania leczenia poprzednim inhibitorem receptora ADP. Dane kliniczne dotyczące skuteczności i bezpieczeństwa stosowania tikagreloru ponad 3 lata długotrwałego leczenia są ograniczone.
Jeżeli potrzebna jest zmiana leku, pierwszą dawkę produktu Tilobrastil należy podać 24 godziny po ostatniej dawce innego leku przeciwpłytkowego.
Pominięcie dawki
Należy także unikać błędów w dawkowaniu. W przypadku pominięcia dawki produktu Tilobrastil pacjent powinien zastosować tylko jedną tabletkę (następną dawkę) zgodnie z przyjętym schematem dawkowania.
Szczególne grupy pacjentów Osoby w podeszłym wieku
U osób w podeszłym wieku nie jest wymagane dostosowanie dawki (patrz punkt 5.2).
Zaburzenia czynności nerek
Dostosowanie dawki nie jest konieczne u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2).
Zaburzenia czynności wątroby
Nie prowadzono badań dotyczących stosowania tikagreloru u pacjentów z ciężkimi zaburzeniami czynności wątroby i z tego powodu jego stosowanie u tych pacjentów jest przeciwwskazane (patrz punkt 4.3). Dostępne są jedynie ograniczone informacje na temat stosowania produktu u pacjentów z umiarkowanymi zaburzeniami czynności wątroby. Dostosowanie dawki nie jest konieczne, jednak
tikagrelor należy stosować ostrożnie (patrz punkty 4.4 i 5.2). U pacjentów z łagodnymi zaburzeniami czynności wątroby dostosowanie dawki nie jest konieczne (patrz punkt 5.2).
Dzieci i młodzież
Nie ustalono bezpieczeństwa stosowania i skuteczności tikagreloru u dzieci w wieku poniżej 18 lat. Stosowanie tikagreloru u dzieci nie jest właściwe we wskazaniu niedokrwistość sierpowatokrwinkowa (patrz punkty 5.1 i 5.2).
Sposób podawania Podanie doustne.
Produkt Tilobrastil może być stosowany podczas posiłku lub niezależnie od posiłku.
W przypadku pacjentów, którzy mają trudność z połknięciem tabletki/tabletek w całości, tabletki można rozgnieść na drobny proszek, zmieszać z połową szklanki wody i natychmiast wypić. Szklankę należy następnie przepłukać wodą (kolejne pół szklanki wody) i ponownie wypić zawartość.
Zmieszaną z wodą rozgniecioną tabletkę (lub tabletki) można również podać przez zgłębnik nosowo-żołądkowy (CH8 lub większy). Ważne jest, aby przepłukać zgłębnik nosowo-żołądkowy wodą po podaniu mieszaniny.
Ryzyko krwawień
U pacjentów, u których stwierdzono zwiększone ryzyko wystąpienia krwawień, należy rozważyć stosunek zagrożeń do korzyści związanych z zapobieganiem zdarzeniom sercowo-naczyniowym (patrz punkty 4.8 i 5.1). W przypadku istnienia wskazań klinicznych do stosowania tikagreloru należy stosować go ostrożnie u następujących grup pacjentów:
Transfuzja płytek nie powodowała odwrócenia działania przeciwpłytkowego tikagreloru u zdrowych ochotników i jest mało prawdopodobne, aby była korzystna klinicznie u pacjentów z krwawieniami. Ponieważ zastosowanie desmopresyny wraz z tikagrelorem nie skraca standardowego czasu krwawienia, wątpliwe jest, aby desmopresyna była skuteczna w leczeniu klinicznych incydentów krwawienia (patrz punkt 4.5).
Leczenie przeciwfibrynolityczne (kwas aminokapronowy lub kwas traneksamowy) i (lub) leczenie rekombinowanym czynnikiem VIIa mogą zwiększać hemostazę. Tikagrelor może być ponownie zastosowany, jeśli przyczyna krwawienia została zidentyfikowana i opanowana.
Zabiegi chirurgiczne
Należy poinstruować pacjentów, aby przed planowanymi zabiegami chirurgicznymi i zastosowaniem jakichkolwiek nowych leków informowali lekarzy i lekarzy stomatologów o stosowaniu tikagreloru.
U pacjentów biorących udział w badaniu PLATO, którzy byli poddawani pomostowaniu aortalno- wieńcowemu (ang. coronary artery bypass grafting, CABG), w grupie leczonej tikagrelorem wystąpiło więcej krwawień niż w grupie leczonej klopidogrelem, jeśli stosowanie leku przerwano na jeden dzień przed zabiegiem, ale jeśli stosowanie leku przerwano na dwa lub więcej dni przed zabiegiem, liczba ciężkich krwawień była podobna w obu grupach (patrz punkt 4.8). Jeśli pacjent ma być poddany planowemu zabiegowi chirurgicznemu i działanie przeciwpłytkowe nie jest pożądane, tikagrelor należy odstawić na 5 dni przed zabiegiem (patrz punkt 5.1).
Pacjenci po przebytym niedokrwiennym udarze mózgu
Pacjenci z OZW po przebytym niedokrwiennym udarze mózgu mogą być leczeni tikagrelorem przez maksymalnie 12 miesięcy (badanie PLATO).
Do badania PEGASUS nie włączano pacjentów z zawałem serca w wywiadzie i z przebytym niedokrwiennym udarem mózgu. Dlatego, z uwagi na brak danych, nie zaleca się leczenia tych pacjentów powyżej roku.
Zaburzenia czynności wątroby
Stosowanie tikagreloru u pacjentów z ciężkimi zaburzeniami czynności wątroby jest przeciwwskazane (patrz punkty 4.2 i 4.3). Istnieją jedynie ograniczone doświadczenia ze stosowaniem tikagreloru
u pacjentów z umiarkowaną niewydolnością wątroby, w związku z tym zaleca się zachowanie ostrożności u tych chorych (patrz punkty 4.2 i 5.2).
Pacjenci z ryzykiem wystąpienia incydentów bradykardii
Monitorowanie parametrów EKG w badaniu Holtera wykazało zwiększoną częstość występowania w większości bezobjawowych pauz komorowych podczas leczenia tikagrelorem w porównaniu
z klopidogrelem. Pacjenci ze zwiększonym ryzykiem incydentów bradykardii (np. pacjenci bez rozrusznika z zespołem chorego węzła zatokowego, z blokiem przedsionkowo-komorowym II lub III stopnia, lub u których występują omdlenia związane z bradykardią) zostali wykluczeni z głównych badań oceniających bezpieczeństwo i skuteczność stosowania tikagreloru. Dlatego też, ze względu na ograniczone doświadczenie kliniczne, tikagrelor powinien być stosowany w tej grupie pacjentów
z zachowaniem ostrożności (patrz punkt 5.1).
Dodatkowo należy zachować ostrożność podczas jednoczesnego stosowania tikagreloru z produktami leczniczymi wywołującymi bradykardię. Jednak nie było dowodów na klinicznie znaczące działania
niepożądane obserwowane w badaniu PLATO po jednoczesnym podaniu z jednym lub więcej produktami leczniczymi wywołującymi bradykardię (tj. 96% beta-adrenolityki, 33% antagoniści wapnia diltiazem i werapamil oraz 4% digoksyna), patrz punkt 4.5.
W badaniu PLATO, w podgrupie poddanej badaniu Holtera, u pacjentów stosujących tikagrelor, częściej niż u pacjentów przyjmujących klopidogrel, obserwowano pauzy komorowe ≥3 sekundy w ostrej fazie ostrego zespołu wieńcowego (OZW). Zwiększenie liczby wykrytych dzięki badaniu
Holtera pauz komorowych podczas leczenia tikagrelorem było wyraźniejsze u pacjentów z przewlekłą niewydolnością serca niż w populacji ogólnej w ostrej fazie OZW, ale nie w obserwacji jednomiesięcznej stosowania tikagreloru, ani nie w porównaniu z klopidogrelem. Nie stwierdzono żadnych niepożądanych konsekwencji klinicznych towarzyszących tej dysproporcji (w tym omdleń lub wszczepień rozrusznika serca) w tej grupie pacjentów (patrz punkt 5.1).
Po wprowadzeniu produktu do obrotu u pacjentów przyjmujących tikagrelor zgłaszano przypadki bradyarytmii i bloków AV (patrz punkt 4.8), głównie u pacjentów z OZW, gdzie niedokrwienie serca i stosowane jednocześnie leki obniżające częstość rytmu serca lub wpływające na przewodzenie w sercu są potencjalnymi czynnikami zakłócającymi. Kliniczny stan pacjenta oraz przyjmowane leki powinny być ocenione, jako potencjalne przyczyny przed dostosowaniem leczenia.
Duszność
Pacjenci leczeni tikagrelorem zgłaszali występowanie duszności. Duszność jest zwykle łagodna do umiarkowanej i często ustępuje bez konieczności odstawienia leku. U pacjentów z astmą/przewlekłą obturacyjną chorobą płuc (POChP) może dojść do zwiększenia bezwzględnego ryzyka duszności podczas stosowania tikagreloru. Tikagrelor powinien być stosowany ostrożnie u pacjentów z astmą
i (lub) POChP w wywiadzie. Mechanizm występowania duszności nie został wyjaśniony. Jeśli pacjent zgłosi nowe incydenty duszności, wydłuży się czas ich trwania lub pogorszą się objawy duszności podczas leczenia tikagrelorem, należy przeprowadzić pełną diagnostykę i, jeśli pacjent nie toleruje leczenia tikagrelorem, należy je przerwać. Dalsze informacje znajdują się w punkcie 4.8.
Ośrodkowy bezdech senny
Po wprowadzeniu produktu do obrotu u pacjentów przyjmujących tikagrelor zgłaszano występowanie ośrodkowego bezdechu sennego, w tym oddychanie Cheyne’a-Stokesa. Jeśli podejrzewa się wystąpienie ośrodkowego bezdechu sennego, należy rozważyć dalszą ocenę kliniczną.
Zwiększenie stężenia kreatyniny
Podczas leczenia tikagrelorem może zwiększyć się stężenie kreatyniny. Mechanizm tego zjawiska nie został ustalony. Należy wykonywać badania kontrolne czynności nerek zgodnie ze stosowaną praktyką kliniczną. U pacjentów z OZW zaleca się kontrolę czynności nerek również po miesiącu od rozpoczęcia leczenia tikagrelorem, ze zwróceniem szczególnej uwagi na pacjentów w wieku ≥75 lat, pacjentów z umiarkowanymi do ciężkich zaburzeniami czynności nerek i tych, którzy stosują leki
z grupy antagonistów receptora angiotensyny (ang. angiotensin receptor blocker, ARB).
Zwiększenie stężenia kwasu moczowego
W trakcie leczenia tikagrelorem może się rozwinąć hiperurykemia (patrz punkt 4.8). Należy zachować ostrożność w przypadku pacjentów z hiperurykemią lub dnawym zapaleniem stawów w wywiadzie.
Jako środek ostrożności odradza się stosowanie tikagreloru u pacjentów z nefropatią moczanową.
Zakrzepowa plamica małopłytkowa (ang. thrombotic thrombocytopenic purpura, TTP)
W trakcie leczenia tikagrelorem bardzo rzadko zgłaszano zakrzepową plamicę małopłytkową (TTP). Charakteryzuje się ona małopłytkowością i mikroangiopatyczną niedokrwistością hemolityczną związaną z objawami neurologicznymi, zaburzeniami czynności nerek lub gorączką. TTP jest potencjalnie śmiertelnym schorzeniem wymagającym szybkiego leczenia, w tym plazmaferezy.
Zakłócenia testów czynnościowych płytek krwi wykonywanych w celu zdiagnozowania małopłytkowości zależnej od heparyny (ang. heparin induced thrombocytopenia, HIT).
W czynnościowym teście aktywacji płytek indukowanej heparyną (ang. heparin induced platelet
activation, HIPA) stosowanym do diagnozowania HIT, przeciwciała przeciwko kompleksowi czynnik
płytkowy 4/heparyna w surowicy pacjenta aktywują płytki krwi zdrowych dawców w obecności heparyny.
U pacjentów przyjmujących tikagrelor zgłaszano fałszywie ujemne wyniki testów czynnościowych płytek krwi (w tym m.in. testu HIPA) mających na celu zdiagnozowanie HIT. Jest to związane
z hamowaniem receptora P2Y12 na zdrowych płytkach dawcy przez tikagrelor obecny
w surowicy/osoczu pacjenta. Informacje na temat równoczesnego leczenia tikagrelorem są wymagane do interpretacji wyników testów czynnościowych płytek krwi stosowanych do diagnozy HIT.
U pacjentów, u których rozwinęła się małopłytkowość zależna od heparyny, należy ocenić stosunek korzyści do ryzyka dalszego leczenia tikagrelorem, biorąc pod uwagę zarówno prozakrzepowy stan HIT, jak i zwiększone ryzyko wystąpienia krwawienia podczas jednoczesnego leczenia antykoagulantem i tikagrelorem.
Inne
Na podstawie zaobserwowanej w badaniu PLATO zależności pomiędzy dawką podtrzymującą kwasu acetylosalicylowego a względną skutecznością tikagreloru w porównaniu do klopidogrelu, nie zaleca się jednoczesnego stosowania tikagreloru i kwasu acetylosalicylowego w dużych dawkach podtrzymujących (>300 mg), patrz punkt 5.1.
Przedwczesne przerwanie leczenia
Przedwczesne przerwanie jakiegokolwiek leczenia przeciwpłytkowego, również produktem Tilobrastil, może skutkować zwiększonym ryzykiem zgonu z przyczyn sercowo-naczyniowych, zawału serca lub udaru spowodowanego chorobą podstawową. Dlatego należy unikać przedwczesnego przerywania leczenia.
Tilobrastil zawiera sód
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na tabletkę, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
Tikagrelor jest przede wszystkim substratem izoenzymu CYP3A4, a także jego łagodnym inhibitorem. Tikagrelor jest również substratem glikoproteiny P (P-gp) i słabym inhibitorem P-gp i może zwiększać narażenie na substraty P-gp.
Wpływ innych produktów leczniczych na działanie tikagreloru
Inhibitory CYP3A4
Induktory CYP3A
Jednoczesne zastosowanie ryfampicyny i tikagreloru zmniejszyło Cmax i AUC tikagreloru odpowiednio o 73% i 86%. Cmax czynnego metabolitu nie uległo zmianie, a jego AUC zmniejszyło się o 46%.
Przewiduje się, że inne induktory CYP3A (np. fenytoina, karbamazepina i fenobarbital) również
zmniejszają narażenie na tikagrelor. Jednoczesne stosowanie tikagreloru i silnych induktorów CYP3A może zmniejszyć stężenie i skuteczność tikagreloru, dlatego jednoczesne stosowanie ich
z tikagrelorem nie jest zalecane.
Cyklosporyna (inhibitor P-gp i CYP3A)
Jednoczesne stosowanie cyklosporyny (600 mg) i tikagreloru podwyższyło Cmax tikagreloru 2,3- krotnie, a AUC – 2,8-krotnie. W obecności cyklosporyny AUC czynnego metabolitu tikagreloru wzrosło o 32%, a Cmax zmalało o 15%.
Brak danych dotyczących jednoczesnego stosowania tikagreloru i innych substancji czynnych będących silnymi inhibitorami glikoproteiny P (P-gp) i umiarkowanymi inhibitorami CYP3A4 (np. werapamil, chinidyna), które mogą zwiększać narażenie na tikagrelor. Jeśli nie można uniknąć leczenia skojarzonego, ich jednoczesne stosowanie wymaga zachowania ostrożności.
Inne
Badania dotyczące farmakologii klinicznej interakcji wykazały, że jednoczesne stosowanie tikagreloru z heparyną, enoksaparyną i ASA lub desmopresyną nie wpływało na farmakokinetykę tikagreloru lub jego czynnego metabolitu, lub indukowaną przez ADP agregację płytek w porównaniu ze stosowaniem samego tikagreloru. Jeśli jest to klinicznie wskazane, produkty lecznicze zmieniające hemostazę powinny być stosowane ostrożnie w skojarzeniu z tikagrelorem.
U pacjentów z ACS leczonych morfiną obserwowano opóźnienie i zmniejszenie ekspozycji na doustne inhibitory P2Y12, w tym tikagrelor i jego aktywny metabolit (zmniejszenie ekspozycji na tikagrelor
i Cmax obserwowano dla wszystkich metabolitów kwasu atorwastatyny. Uważa się, że nie jest
to istotne klinicznie.
Tikagrelor jest łagodnym inhibitorem CYP3A4. Nie zaleca się jednoczesnego stosowania tikagreloru i substratów CYP3A4 o wąskim indeksie terapeutycznym (tj. cyzapryd lub alkaloidy sporyszu), ponieważ tikagrelor może powodować zwiększenie narażenia na te produkty lecznicze.
Substraty P-gp (w tym digoksyna, cyklosporyna)
Jednoczesne stosowanie tikagreloru zwiększa Cmax i AUC digoksyny odpowiednio o 75% i 28%.
Średnie stężenia digoksyny w fazie eliminacji zwiększyły się o około 30% po jednoczesnym stosowaniu z tikagrelorem, z pojedynczymi przypadkami maksymalnie 2-krotnego zwiększenia. Obecność digoksyny nie wpływa na Cmax i AUC tikagreloru i jego czynnego metabolitu. Dlatego zaleca się odpowiednią kontrolę kliniczną i (lub) monitorowanie parametrów laboratoryjnych podczas jednoczesnego stosowania produktów leczniczych o wąskim indeksie terapeutycznym, zależnych od P-gp, takich jak digoksyna i tikagrelor.
Tikagrelor nie wpływał na stężenie cyklosporyny we krwi. Nie badano wpływu tikagreloru na inne substraty P-gp.
Produkty lecznicze metabolizowane przez CYP2C9
Jednoczesne stosowanie tikagreloru i tolbutamidu nie spowodowało zmiany stężenia w osoczu żadnego z tych produktów leczniczych, co sugeruje, że tikagrelor nie jest inhibitorem CYP2C9 i jest mało prawdopodobne, aby zaburzał metabolizm produktów, takich jak warfaryna czy tolbutamid związany z izoenzymem CYP2C9.
Rozuwastatyna
Tikagrelor może wpływać na nerkowe wydalanie rozuwastatyny, zwiększając ryzyko akumulacji rozuwastatyny. Chociaż właściwy mechanizm nie jest znany, w niektórych przypadkach, równoczesne stosowanie tikagreloru i rozuwastatyny prowadziło do pogorszenia czynności nerek, wzrostu aktywności CPK (kinaza fosfokreatynowa) i rabdomiolizy.
Doustne leki antykoncepcyjne
Jednoczesne stosowanie tikagreloru i lewonorgestrelu oraz etynyloestradiolu spowodowało około 20% zwiększenie narażenia na etynyloestradiol, ale nie wpływało na farmakokinetykę lewonorgestrelu. Nie przewiduje się klinicznie znaczącego wpływu na skuteczność doustnych środków antykoncepcyjnych, w przypadku jednoczesnego stosowania lewonorgestrelu
i etynyloestradiolu z tikagrelorem.
Produkty lecznicze wywołujące bradykardię
W związku z obserwowanymi, zwykle bezobjawowymi, pauzami komorowymi i bradykardią, należy zachować ostrożność podczas jednoczesnego stosowania tikagreloru z produktami leczniczymi wywołującymi bradykardię (patrz punkt 4.4). W badaniu PLATO nie zaobserwowano jednak dowodów na występowanie znaczących klinicznie działań niepożądanych po jednoczesnym zastosowaniu z jednym lub więcej produktami leczniczymi wywołującymi bradykardię
(tj. 96% leki beta-adrenolityczne, 33% antagoniści wapnia diltiazem i werapamil oraz 4% digoksyna).
Jednoczesne stosowanie z innymi produktami leczniczymi
W badaniach klinicznych tikagrelor był stosowany jednocześnie z ASA, inhibitorami pompy protonowej, statynami, lekami beta-adrenolitycznymi, inhibitorami konwertazy angiotensyny (ACE)
i antagonistami receptora angiotensyny, stosowanymi długotrwale ze względu na konieczność leczenia schorzeń współistniejących jak również z heparyną, heparyną drobnocząsteczkową i dożylnymi inhibitorami GpIIb/IIIa przez krótki czas (patrz punkt 5.1). Nie zaobserwowano żadnych istotnych klinicznie interakcji podczas stosowania tych produktów leczniczych.
Jednoczesne stosowanie tikagreloru i heparyny, enoksaparyny lub desmopresyny nie wpływało na czas częściowej tromboplastyny po aktywacji (aPTT), aktywowany czas krzepnięcia (ACT) ani na oznaczanie aktywności czynnika Xa. Jednak ze względu na potencjalne interakcje farmakodynamiczne, należy zachować ostrożność podczas jednoczesnego stosowania tikagreloru z lekami zmieniającymi hemostazę.
W związku z obserwowanymi podczas stosowania selektywnych inhibitorów wychwytu zwrotnego serotoniny (SSRIs) (tj. paroksetyny, sertraliny i cytalopram), nieprawidłowymi krwawieniami skórnymi należy zachować ostrożność w przypadku stosowania SSRIs razem z tikagrelorem, ponieważ może to zwiększyć ryzyko krwawienia.
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym powinny stosować odpowiednie środki antykoncepcyjne, aby zapobiec zajściu w ciążę podczas leczenia tikagrelorem.
Ciąża
Nie ma lub jest ograniczona ilość danych dotyczących stosowania tikagreloru w czasie ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na rozrodczość (patrz punkt 5.3). Nie zaleca się stosowania tikagreloru w czasie ciąży.
Karmienie piersią
Dostępne dane farmakodynamiczno-toksykologiczne z badań na zwierzętach wykazały, że tikagrelor i jego czynne metabolity przenikają do mleka (patrz punkt 5.3). Nie można wykluczyć ryzyka dla noworodków lub niemowląt. Należy podjąć decyzję, czy przerwać karmienie piersią, czy zakończyć/przerwać terapię tikagrelorem, biorąc pod uwagę korzyści karmienia piersią dla dziecka oraz korzyści terapii dla kobiety.
Płodność
U zwierząt tikagrelor nie wpływa na płodność samców ani samic (patrz punkt 5.3)
Tikagrelor nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Zgłaszano występowanie zawrotów głowy i splątania u pacjentów leczonych tikagrelorem. W związku z tym pacjenci, u których wystąpią te objawy, powinni zachować ostrożność podczas prowadzenia pojazdów lub obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Profil bezpieczeństwa tikagreloru był oceniany w ramach wyników dwóch dużych badań fazy 3 (PLATO i PEGASUS), obejmujących ponad 39 000 pacjentów (patrz punkt 5.1).
W badaniu PLATO u pacjentów otrzymujących tikagrelor stwierdzono większą częstość przerwania leczenia z powodu zdarzeń niepożądanych niż w grupie otrzymującej klopidogrel (7,4% wobec 5,4%). W badaniu PEGASUS u pacjentów otrzymujących tikagrelor stwierdzono większą częstość przerwania leczenia z powodu zdarzeń niepożądanych w porównaniu
z pacjentami leczonymi ASA w monoterapii (16,1% w grupie leczonej tikagrelorem w dawce 60 mg w skojarzeniu z ASA wobec 8,5% w grupie otrzymującej ASA w monoterapii). Najczęściej zgłaszanymi działaniami niepożądanymi u pacjentów leczonych tikagrelorem były krwawienie
i duszność (patrz punkt 4.4).
Tabelaryczne zestawienie działań niepożądanych
Poniższe działania niepożądane rozpoznano w wyniku badań lub zgłoszono po wprowadzeniu tikagreloru do obrotu (tabela 1).
Działania niepożądane wymieniono zgodnie z klasyfikacją układów i narządów (ang. System Organ Class, SOC) MedDRA. W obrębie każdej grupy SOC działania niepożądane uporządkowano według częstości występowania. Częstość określono następująco: bardzo często (≥1/10), często (≥1/100 do<1/10), niezbyt często (≥1/1000 do <1/100), rzadko (≥1/10 000 do
<1/1000), bardzo rzadko (<10 000), nieznana (nie może być określona na podstawie dostępnych danych).
Tabela 1 – Działania niepożądane przedstawione według częstości występowania oraz klasyfikacji układów i narządów (SOC)
Klasyfikacja układów i narządów | Bardzo często | Często | Niezbyt często | Częstość nieznana |
Łagodne, złośliwe i nieokreślone nowotwory (w tym torbiele i polipy) | Krwawienia z guza2 | |||
Zaburzenia krwi i układu chłonnego | Zaburzenia krwi, krwawieniab | Zakrzepowa plamica małopłytkowac | ||
Zaburzenia układu immunologicznego | Nadwrażliwość, w tym obrzęk naczynioruchowyc | |||
Zaburzenia metabolizmu i odżywiania | Hiperurykemiad | Dna moczanowa/Dnawe zapalenie stawów | ||
Zaburzenia psychiczne | Splątanie | |||
Zaburzenia układu nerwowego | Zawroty głowy, omdlenia, bóle głowy | Krwotok śródczaszkowy | ||
Zaburzenia oka | Krwotok do okae | |||
Zaburzenia ucha i błędnika | Zawroty głowy pochodzenia błędnikowego | Krwotok do ucha | ||
Zaburzenia serca | Bradyarytmia, blok AVc | |||
Zaburzenia naczyń | Niedociśnienie | |||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Duszność | Krwawienia z układu oddechowegof | ||
Zaburzenia żołądka i jelit | Krwotok z przewodu pokarmowegog, biegunka, nudności, niestrawność, zaparcia | Krwotok zaotrzewnowy | ||
Zaburzenia skóry i tkanki podskórnej | Krwawienia podskórne lub do skóry właściwejh, wysypka, świąd | |||
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Krwawienia do mięśnii | |||
Zaburzenia nerek i dróg moczowych | Krwawienie z układu moczowegoj | |||
Zaburzenia układu rozrodczego i piersi | Krwawienia z układu rozrodczegok | |||
Badania | Zwiększone |
diagnostyczne | stężenie kreatyniny we krwid | |||
Urazy, zatrucia i powikłania po zabiegach | Krwotok po zabiegu, krwawienia pourazowel |
a np. krwawienie z nowotworu pęcherza moczowego, wrzodu żołądka, nowotworu okrężnicy b np. zwiększona skłonność do powstawania siniaków, krwiak samoistny, skaza krwotoczna c Zaobserwowane po wprowadzeniu leku do obrotu
d Dane dotyczące częstości pochodzą z obserwacji laboratoryjnych (zwiększenie stężenia kwasu moczowego powyżej górnej granicy normy w stosunku do stanu wyjściowego poniżej lub zakresie normy. Zwiększenie stężenia kreatyniny o >50% w stosunku do stanu wyjściowego.) i nie stanowią ogólnej częstości ze zgłoszeń wszystkich zdarzeń niepożądanych.
e np. krwawienie do spojówki, siatkówki, gałki ocznej
f np. krwotok z nosa, krwioplucie
g np. krwawienie z dziąseł, krwotok z odbytu, krwotok z wrzodu żołądka
h np. siniaki, krwotok do skóry, wybroczyny krwawe
i np. krwawienie do stawu, krwotok mięśniowy
j np. krwiomocz, krwotoczne zapalenie pęcherza
k np. krwotok z pochwy, hematospermia, krwotok pomenopauzalny
l np. stłuczenie, krwiak urazowy, krwotok urazowym tj. spontaniczny, związany z zabiegiem lub urazowy krwotok śródczaszkowy
Opis wybranych działań niepożądanych
Krwawienia
Wyniki badania PLATO dotyczące krwawień
Ogólny wynik dotyczący częstości krwawień w badaniu PLATO przedstawiono w tabeli 2.
Tabela 2 – Analiza wszystkich zdarzeń krwotocznych, wartości oszacowane metodą
Kaplana-Meiera po 12 miesiącach (PLATO)
Tikagrelor 90 mg dwa razy na dobę N=9235 | Klopidogrel N=9186 | Wartość p* | |
Ciężkie krwawienia ogółem, PLATO | 11,6 | 11,2 | 0,4336 |
Ciężkie prowadzące do zgonu/zagrażające życiu, PLATO | 5,8 | 5,8 | 0,6988 |
Ciężkie, niezwiązane z CABG, PLATO | 4,5 | 3,8 | 0,0264 |
Ciężkie, niezwiązane z zabiegami, PLATO | 3,1 | 2,3 | 0,0058 |
Ciężkie + niewielkie ogółem, PLATO | 16,1 | 14,6 | 0,0084 |
Ciężkie + niewielkie, niezwiązane z zabiegami, PLATO | 5,9 | 4,3 | <0,0001 |
Ciężkie, definicja TIMI | 7,9 | 7,7 | 0,5669 |
Ciężkie + niewielkie, definicja TIMI | 11,4 | 10,9 | 0,3272 |
Definicje kategorii krwawień:
Ciężkie krwawienie prowadzące do zgonu/zagrażające życiu: krwawienie jawne klinicznie, ze zmniejszeniem o >50 g/l stężenia hemoglobiny lub z przetoczeniem ≥4 jednostek masy erytrocytarnej; lub prowadzące do zgonu; lub śródczaszkowe; lub do worka osierdziowego z tamponadą serca; lub ze wstrząsem hipowolemicznym lub ciężkim niedociśnieniem wymagającym podania leków wazopresyjnych lub wykonania zabiegu chirurgicznego.
Ciężkie inne: jawne klinicznie, ze zmniejszeniem stężenia hemoglobiny o 30 – 50 g/l lub z przetoczeniem 2 –
3 jednostek masy erytrocytarnej; lub prowadzące do znacznej niepełnosprawności.
Niewielkie krwawienie: wymaga interwencji medycznej w celu jego zatrzymania lub wyleczenia. Ciężkie krwawienie zdefiniowane wg TIMI: jawne klinicznie, ze zmniejszeniem stężenia hemoglobiny o >50 g/l lub z krwotokiem śródczaszkowym.
Niewielkie krwawienie zdefiniowane wg TIMI: jawne klinicznie, ze zmniejszeniem stężenia hemoglobiny
o 30 – 50 g/l.
*Wartość p obliczono z użyciem modelu proporcjonalnych hazardów Coxa z grupą badaną, jako jedyną zmienną wyjaśniającą.
Tikagrelor i klopidogrel nie różniły się pod względem częstości występowania ciężkich prowadzących do zgonu/zagrażających życiu krwawień wg PLATO, ciężkich krwawień ogółem wg PLATO, poważnych krwawień wg TIMI czy niewielkich krwawień wg TIMI (tabela 2). Jednak więcej poważnych i niewielkich krwawień ogółem wg kryteriów badania PLATO występowało
w grupie tikagreloru w porównaniu z klopidogrelem. U jedynie niewielkiej liczby pacjentów uczestniczących w badaniu PLATO wystąpiły krwawienia prowadzące do zgonu: 20 (0,2%)
w grupie otrzymującej tikagrelor i 23 (0,3%) w grupie otrzymującej klopidogrel (patrz punkt 4.4).
Wiek, płeć, masa ciała, rasa, region geograficzny, schorzenia współistniejące, równocześnie stosowane leczenie i przebyty udar mózgu lub przemijający atak niedokrwienny w wywiadzie, nie stanowiły czynników predykcyjnych poważnych krwawień ogółem lub poważnych krwawień niezwiązanych z zabiegami wg kryteriów badania PLATO. W związku z tym nie zidentyfikowano żadnej grupy, w której istniałoby zwiększone ryzyko jakiejś podgrupy krwawień.
Krwawienie związane z CABG:
W badaniu PLATO u 42% z 1584 pacjentów (12% kohorty), u których wykonano zabieg CABG, wystąpiło ciężkie prowadzące do zgonu/zagrażające życiu krwawienie wg kryteriów badania PLATO, przy czym nie stwierdzono różnicy pomiędzy leczonymi grupami. Zakończone zgonem krwawienie po CABG wystąpiło u 6 pacjentów w każdej grupie badanej (patrz punkt 4.4).
Krwawienia niezwiązane z CABG i krwawienia niezwiązane z zabiegami:
Tikagrelor i klopidogrel nie różniły się pod względem ciężkich, niezwiązanych z CABG, prowadzących do zgonu/zagrażających życiu krwawień wg definicji krwawień PLATO, jednak ciężkie krwawienia ogółem wg PLATO, ciężkie krwawienia wg TIMI oraz ciężkie + niewielkie krwawienia wg TIMI występowały częściej w grupie otrzymującej tikagrelor. Analogicznie, gdy wyeliminowano wszystkie krwawienia związane z zabiegami, okazało się, że więcej krwawień występowało w grupie otrzymującej tikagrelor niż klopidogrel (tabela 2). Do przerwania leczenia z powodu krwawień niezwiązanych z zabiegami dochodziło częściej w grupie tikagreloru (2,9%) niż w grupie klopidogrelu (1,2%; p<0,001).
Krwawienia śródczaszkowe:
Stwierdzono większą liczbę krwawień śródczaszkowych niezwiązanych z zabiegami w przypadku tikagreloru (n=27 krwawień u 26 pacjentów, 0,3%) niż w przypadku klopidogrelu (n=14 krwawień, 0,2%), w tym 11 krwawień w przypadku tikagreloru i 1 w przypadku klopidogrelu prowadziło do zgonu. Nie stwierdzono różnicy pod względem ogólnej liczby krwawień prowadzących do zgonu.
Wyniki badania PEGASUS dotyczące krwawień
Ogólny wynik dotyczący zdarzeń krwotocznych w badaniu PEGASUS przedstawiono w tabeli 3.
Tabela 3 – Analiza wszystkich zdarzeń krwotocznych, wartości oszacowane metodą
Kaplana-Meiera po 36 miesiącach (PEGASUS)
Tikagrelor 60 mg dwa razy na dobę + ASA N=6958 | ASA w monoterapii N=6996 | |||
Punkty końcowe oceny bezpieczeństwa | KM% | Współczynnik ryzyka (95% CI) | KM% | Wartość p |
Kategorie krwawień zdefiniowane wg TIMI | ||||
Ciężkie krwawienie wg TIMI | 2,3 | 2,32 (1,68, 3,21) | 1,1 | <0,0001 |
Prowadzące do zgonu | 0,3 | 1,00 (0,44, 2,27) | 0,3 | 1,0000 |
Krwawienie śródczaszkowe (ICH) | 0,6 | 1,33 (0,77, 2,31) | 0,5 | 0,3130 |
Inne ciężkie krwawienie wg TIMI | 1,6 | 3,61 (2,31, 5,65) | 0,5 | <0,0001 |
Ciężkie lub niewielkie krwawienie wg TIMI | 3,4 | 2,54 (1,93, 3,35) | 1,4 | <0,0001 |
Ciężkie lub niewielkie krwawienie wymagające pomocy medycznej wg TIMI | 16,6 | 2,64 (2,35, 2,97) | 7,0 | <0,0001 |
Kategorie krwawień zdefiniowane w badaniu PLATO | ||||
Ciężkie krwawienie w badaniu PLATO | 3,5 | 2,57 (1,95, 3,37) | 1,4 | <0,0001 |
Prowadzące do zgonu/zagrażające życiu | 2,4 | 2,38 (1,73, 3,26) | 1,1 | <0,0001 |
Inne ciężkie krwawienie w badaniu PLATO | 1,1 | 3,37 (1,95, 5,83) | 0,3 | <0,0001 |
Ciężkie lub niewielkie krwawienie w badaniu PLATO | 15,2 | 2,71 (2,40, 3,08) | 6,2 | <0,0001 |
Definicje kategorii krwawień:
Ciężkie krwawienie wg TIMI: śmiertelne krwawienie, LUB dowolne krwawienie śródczaszkowe, LUB jawne klinicznie oznaki krwawienia związanego ze zmniejszeniem stężenia hemoglobiny (Hb) o ≥50 g/l, lub jeśli brak jest danych na temat stężenia Hb, zmniejszenie hematokrytu (Hct) o 15%.
Krwawienie prowadzące do zgonu: incydent krwotoczny, który bezpośrednio doprowadził do zgonu w ciągu 7 dni.
ICH: krwawienie śródczaszkowe.
Inne ciężkie krwawienie wg TIMI: poważne krwawienie wg TIMI nieprowadzące do zgonu i niebędące ICH.
Niewielkie krwawienie wg TIMI: jawne klinicznie, ze zmniejszeniem stężenia hemoglobiny o 30 – 50 g/l. Krwawienie wymagające pomocy medycznej wg TIMI: krwawienie wymagające interwencji, LUB prowadzące do hospitalizacji, LUB wymagające oceny.
Ciężkie prowadzące do zgonu/zagrażające życiu wg badania PLATO: krwawienie prowadzące do zgonu, LUB krwawienie śródczaszkowe, LUB krwawienie do worka osierdziowego z tamponadą serca, LUB ze wstrząsem hipowolemicznym lub z ciężkim niedociśnieniem wymagającym podania leków wazopresyjnych/inotropowych lub wykonania zabiegu chirurgicznego LUB jawne klinicznie ze zmniejszeniem stężenia hemoglobiny o >50 g/l lub przetoczeniem ≥4 jednostek masy erytrocytarnej.
Inne ciężkie krwawienie wg badania PLATO: prowadzące do istotnej niepełnosprawności, LUB jawne klinicznie, ze zmniejszeniem stężenia hemoglobiny o 30 – 50 g/l, LUB z przetoczeniem 2 – 3 jednostek masy erytrocytarnej.
Niewielkie krwawienie wg badania PLATO: wymaga interwencji medycznej w celu jego zatrzymania lub wyleczenia.
W badaniu PEGASUS częstość występowania poważnych krwawień wg TIMI podczas stosowania tikagreloru w dawce 60 mg dwa razy na dobę była większa niż podczas stosowania ASA
w monoterapii. Nie stwierdzono zwiększenia ryzyka krwawień w przypadku krwawień prowadzących do zgonu i jego jedynie niewielkie zwiększenie obserwowano w przypadku krwotoków śródczaszkowych, w porównaniu ze stosowaniem ASA w monoterapii. W badaniu obserwowano nieliczne krwawienia prowadzące do zgonu, 11 (0,3%) podczas stosowania tikagreloru w dawce 60 mg i 12 (0,3%) podczas stosowania ASA w monoterapii. Obserwowane zwiększenie ryzyka ciężkich krwawień wg TIMI podczas stosowania tikagreloru w dawce 60 mg wynikało przede wszystkim z większej częstości innych ciężkich krwawień wg TIMI, związanych ze zdarzeniami niepożądanymi ze strony przewodu pokarmowego.
Stwierdzono zwiększenie częstości krwawień, podobne do zwiększenia ciężkich krwawień wg TIMI, w przypadku kategorii poważnych lub niewielkich krwawień wg TIMI i poważnych krwawień w badaniu PLATO i ciężkich lub niewielkich krwawień w badaniu PLATO (patrz tabela 3). Do
przerwania leczenia z powodu krwawień dochodziło częściej podczas stosowania tikagreloru w
dawce 60 mg niż podczas stosowania ASA w monoterapii (odpowiednio 6,2% oraz 1,5%). Większość z tych krwawień miała mniejsze nasilenie (klasyfikowano je jako krwawienia wymagające pomocy medycznej wg TIMI), np. krwotoki z nos, siniaki i krwiaki.
Profil krwawień związanych ze stosowaniem tikagreloru w dawce 60 mg był spójny w szeregu wyodrębnionych wcześniej podgrup (np. wg wieku, płci, masy ciała, rasy, regionu geograficznego, stanów współistniejących, równocześnie stosowanego leczenia i historii choroby) w przypadku ciężkich krwawień wg TIMI, poważnych lub niewielkich krwawień wg TIMI i ciężkich krwawień wg PLATO.
Krwawienia śródczaszkowe:
Samoistne krwawienia śródczaszkowe (ICH) obserwowano z podobną częstością u pacjentów otrzymujących tikagrelor w dawce 60 mg i ASA w monoterapii (n=13, 0,2% w obu badanych grupach). W przypadku ICH urazowych i związanych z zabiegami zaobserwowano niewielkie zwiększenie ich częstości występowania u pacjentów leczonych tikagrelorem w dawce 60 mg (n=15, 0,2%) w porównaniu z ASA w monoterapii (n=10, 0,1%). Wystąpiło 6 przypadków krwawienia śródczaszkowego prowadzącego do zgonu podczas leczenia tikagrelorem w dawce 60 mg i
5 przypadków krwawienia śródczaszkowego prowadzącego do zgonu podczas stosowania ASA
w monoterapii. Częstość występowania krwawień śródczaszkowych była niewielka w obu leczonych grupach, biorąc pod uwagę znaczne obciążenie badanej populacji chorobami współistniejącymi
i czynnikami ryzyka chorób sercowo-naczyniowych.
Duszność
Pacjenci leczeni tikagrelorem zgłaszają duszność, uczucie braku tchu. W badaniu PLATO zdarzenia niepożądane zgłaszane jako duszność (duszność, duszność spoczynkowa, duszność powysiłkowa, duszność napadowa nocna lub nocna duszność), gdy zestawione łącznie, zgłaszało 13,8% pacjentów leczonych tikagrelorem i 7,8% pacjentów leczonych klopidogrelem. U 2,2% pacjentów leczonych tikagrelorem i u 0,6% leczonych klopidogrelem prowadzący badanie PLATO uznali duszność za przyczynowo związaną z leczeniem i było kilka przypadków ciężkiej duszności (0,14% tikagrelor; 0,02% klopidogrel), (patrz punkt 4.4). Większość zdarzeń niepożądanych zgłaszanych jako duszność miała nasilenie łagodne do umiarkowanego i większość była zgłaszana jako pojedynczy epizod wcześnie na początku leczenia.
W porównaniu z kopidogrelem, pacjenci z astmą/POChP leczeni tikagrelorem mogą mieć zwiększone ryzyko pojawienia się nie-ciężkiej duszności (3,29 % tikagrelor vs 0,53% klopidogrel) i ciężkiej duszności (0,38% tikagrelor vs 0,00% klopidogrel). W wartościach bezwzględnych, to ryzyko jest wyższe niż dla całej populacji badania PLATO. Należy zachować ostrożność stosując tikagrelor u pacjentów z astmą i (lub) POChP w wywiadzie (patrz punkt 4.4).
Około 30% epizodów duszności ustępowało w ciągu 7 dni. W badaniu PLATO brali udział pacjenci z zastoinową niewydolnością serca, POChP albo astmą w wywiadzie; ci pacjenci, i pacjenci
w podeszłym wieku, częściej zgłaszali duszność. 0,9% pacjentów w grupie leczonej
tikagrelorem zrezygnowało z leczenia z powodu duszności w porównaniu do 0,1% w grupie leczonej klopidogrelem. Zwiększona częstość epizodów duszności w trakcie stosowania tikagreloru nie jest związana z nową lub pogarszającą się chorobą serca lub płuc (patrz punkt 4.4). Tikagrelor nie wpływa na testy czynnościowe płuc.
W badaniu PEGASUS duszność odnotowano u 14,2% pacjentów otrzymujących tikagrelor w dawce 60 mg dwa razy na dobę i u 5,5% pacjentów otrzymujących ASA w monoterapii. Podobnie jak
w badaniu PLATO, większość przypadków zgłoszonej duszności miała nasilenie lekkie do umiarkowanego (patrz punkt 4.4). Pacjenci, którzy zgłaszali duszność, byli na ogół starsi i częściej mieli duszność, POChP lub astmę w wywiadzie.
Badania diagnostyczne
Zwiększenie stężenia kwasu moczowego: w badaniu PLATO zwiększenie stężenia kwasu moczowego w surowicy powyżej górnej granicy normy wystąpiło u 22% pacjentów otrzymujących
tikagrelor w porównaniu do 13% pacjentów stosujących klopidogrel. Wartości w badaniu PEGASUS wynosiły 9,1%, 8,8% i 5,5% odpowiednio dla tikagreloru w dawce 90 mg, tikagreloru w dawce 60 mg i placebo. Średnie stężenie kwasu moczowego w surowicy wzrosło o około 15% u osób stosujących tikagrelor w porównaniu do wzrostu o około 7,5% wśród leczonych
klopidogrelem. Po zakończeniu leczenia zaobserwowano zmniejszenie stężenia kwasu moczowego do około 7% u chorych leczonych tikagrelorem, ale nie stwierdzono zmniejszenia w przypadku klopidogrelu. W badaniu PEGASUS stwierdzono odwracalne zwiększenie średniego stężenia kwasu moczowego w surowicy o 6,3% i 5,6% w przypadku odpowiednio tikagreloru w dawce 90 mg i 60 mg, wobec zmniejszenia go o 1,5% w grupie placebo. W badaniu PLATO częstość występowania dnawego zapalenia stawów wynosiła 0,2% w grupie tikagreloru wobec 0,1% w grupie klopidogrelu. Odpowiednie częstości występowania dny/dnawego zapalenia stawów w badaniu PEGASUS wynosiły 1,6%, 1,5% i 1,1% odpowiednio w przypadku tikagreloru w dawce 90 mg, tikagreloru
w dawce 60 mg i placebo.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301/faks: + 48 22 49 21 309/strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Tikagrelor jest dobrze tolerowany po zastosowaniu pojedynczej dawki do 900 mg. W badaniu,
w którym stosowano pojedynczą, zwiększającą się dawkę, toksyczność w obrębie żołądka i jelit
była zależna od dawki. Do innych, klinicznie znaczących działań niepożądanych, które mogą wystąpić
po przedawkowaniu, zaliczają się duszność i pauzy komorowe (patrz punkt 4.8).
W przypadku przedawkowania mogą wystąpić powyższe potencjalne działania niepożądane i
należy rozważyć monitorowanie elektrokardiogramu (EKG).
Obecnie nie jest znane antidotum, które niweluje działanie tikagreloru, a tikagrelor nie jest usuwany podczas dializy (patrz punkt 5.2). Leczenie przedawkowania należy prowadzić zgodne
z miejscową standardową praktyką medyczną. Spodziewanym efektem przedawkowania tikagreloru jest ryzyko przedłużającego się czasu trwania krwawienia, związanego z zahamowaniem płytek krwi.
Jest mało prawdopodobne, aby transfuzja płytek krwi była korzystna klinicznie u pacjentów
z krwawieniami (patrz punkt 4.4). Jeśli wystąpi krwawienie, należy podjąć inne odpowiednie leczenie wspomagające.
Grupa farmakoterapeutyczna: leki przeciwzakrzepowe. leki hamujące agregację płytek z wyłączeniem heparyny.
Kod ATC: B01AC24.
Mechanizm działania
Tilobrastil zawiera tikagrelor, należący do chemicznej grupy cyklopentylotriazolopirymidyn (CPTP).
Tikagrelor jest doustnym, bezpośrednio działającym, selektywnym i wiążącym się
odwracalnie antagonistą receptora P2Y12, który zapobiega ADP-zależnej aktywacji i agregacji płytek związanej z receptorem P2Y12. Tikagrelor nie zapobiega wiązaniu ADP, ale po przyłączeniu się do receptora P2Y12 zapobiega stymulowanemu przez ADP przekazywaniu sygnału. Ponieważ płytki krwi
uczestniczą w inicjowaniu i (lub) progresji zakrzepowych powikłań miażdżycy, wykazano, że hamowanie czynności płytek krwi zmniejsza ryzyko zdarzeń sercowo-naczyniowych, takich jak zgon, zawał serca czy udar mózgu.
Tikagrelor zwiększa także lokalne stężenia endogennej adenozyny na skutek hamowania równowagowego transportera nukleozydów-1 (ENT-1, ang. equilibrative nucleoside transporter 1).
Wykazano, że tikagrelor nasila następujące, zależne od adenozyny, działania u zdrowych ochotników oraz pacjentów z OZW: rozszerzenie naczyń krwionośnych (mierzone jako wzrost przepływu wieńcowego u zdrowych ochotników oraz u pacjentów z OZW; ból głowy), hamowanie czynności płytek krwi (w pełnej krwi ludzkiej w warunkach in vitro) oraz duszność. Jednak związek pomiędzy obserwowanym zwiększeniem ilości adenozyny a skutkami klinicznymi (np. zachorowalność- śmiertelność) nie został wyraźnie określony.
Działania farmakodynamiczne
Początek działania
U pacjentów ze stabilną chorobą wieńcową stosujących kwas acetylosalicylowy tikagrelor wykazuje szybki początek działania farmakologicznego, czego przejawem jest średnie zahamowanie agregacji płytek (IPA) równe około 41% po 30 minutach od zastosowania tikagreloru w dawce nasycającej
180 mg z maksymalnym wpływem IPA równym 89% po upływie 2 do 4 godzin od zastosowania leku, utrzymującym się od 2 do 8 godzin. U 90% pacjentów największy stopień zahamowania płytek, przewyższający 70%, obserwowany jest po 2 godzinach od zastosowania leku.
Koniec działania
Jeśli jest planowane wykonanie zabiegu CABG, ryzyko krwawienia związane ze stosowaniem tikagreloru jest większe w porównaniu z klopidogrelem po zaprzestaniu stosowania na krócej niż 96 godzin przed zabiegiem.
Dane dotyczące zmiany terapii
Zmiana leczenia z klopidogrelu w dawce 75 mg na tikagrelor w dawce 90 mg dwa razy na dobę skutkuje zwiększeniem IPA o 26,4% w liczbach bezwzględnych, a zmiana z tikagreloru na klopidogrel powoduje zmniejszenie IPA o 24,5% w liczbach bezwzględnych. Pacjenci mogą być przestawiani z klopidogrelu na tikagrelor bez zaburzenia działania przeciwpłytkowego (patrz punkt 4.2).
Skuteczność i bezpieczeństwo stosowania w badaniach klinicznych
Dane kliniczne potwierdzające skuteczność i bezpieczeństwo stosowania tikagreloru pochodzą z dwóch badań fazy 3:
Badanie PLATO (ostre zespoły wieńcowe)
Badanie PLATO objęło 18 624 pacjentów z ostrym zespołem wieńcowym, którzy zgłaszali się
w ciągu 24 godzin od wystąpienia objawów niestabilnej dusznicy (UA), zawału mięśnia sercowego bez uniesienia odcinka ST (NSTEMI) lub zawału mięśnia sercowego z uniesieniem odcinka ST (STEMI) i którzy byli wstępnie leczeni farmakologicznie, lub mieli wykonaną przezskórną interwencję wieńcową (PCI), lub mieli wykonane CABG.
Skuteczność kliniczna
W połączeniu z dobową dawką ASA tikagrelor w dawce 90 mg dwa razy na dobę był lepszy
niż klopidogrel w dawce 75 mg na dobę w zapobieganiu wystąpieniu złożonego punktu końcowego (zgon z przyczyn sercowo-naczyniowych, zawał mięśnia sercowego lub udar), przy czym różnica
wynikała głównie z liczby zgonów z przyczyn sercowo-naczyniowych i zawałów mięśnia sercowego. Pacjenci otrzymywali klopidogrel w dawce początkowej 300 mg (u pacjentów poddawanych przezskórnej interwencji wieńcowej możliwa była dawka wynosząca 600 mg) lub tikagrelor w dawce 180 mg.
Taki wynik uzyskano wcześnie (bezwzględna redukcja ryzyka [ARR] 0,6% i względna
redukcja ryzyka [RRR] 12% w 30. dniu), a skuteczność leczenia utrzymywała się nadal przez cały okres 12 miesięcy, osiągając ARR 1,9% w ciągu roku i RRR 16%. Te wyniki wskazują, że odpowiedni czas leczenia pacjentów tikagrelolem 90 mg dwa razy na dobę wynosi 12 miesięcy (patrz punkt 4.2). Leczenie 54 pacjentów z OZW tikagrelolem zamiast klopidogrelem zapobiega 1 incydentowi sercowo-naczyniowemu; leczenie 91 pacjentów zapobiega 1 zgonowi z przyczyn sercowo-naczyniowych (patrz wykres 1 i tabela 4).
Lepsze wyniki leczenia tikagrelorem w porównaniu z klopidogrelem są w sposób spójny widoczne w wielu podgrupach pacjentów, włączając masę ciała, płeć, cukrzycę w wywiadzie, przemijające napady niedokrwienne lub udar niezwiązany z krwotokiem lub rewaskularyzację; jednoczesne leczenie z zastosowaniem heparyny, inhibitorów GpIIb/IIIa i inhibitorów pompy protonowej (patrz punkt 4.5); ostateczne rozpoznanie kliniczne (STEMI, NSTEMI czy UA) i planowany w czasie randomizacji sposób leczenia (leczenie inwazyjne lub zachowawcze).
Z niewielką znamiennością efekt leczenia różnił się w zależności od regionu, przez co współczynnik ryzyka (HR) dla pierwszorzędowego punktu końcowego wskazuje na korzyści ze
stosowania tikagreloru na całym świecie z wyjątkiem Ameryki Północnej, która reprezentuje około 10% ogółu badanej populacji, gdzie wynik HR jest korzystniejszy dla klopidogrelu (obecność interakcji p=0,045). Analizy czynnikowe wskazują na możliwość istnienia związku z dawką ASA, co oznacza, że obserwowano zmniejszenie skuteczności tikagreloru wraz ze zwiększeniem dawek ASA. Dawki ASA do przewlekłego stosowania z tikagrelorem powinny wynosić 75 – 150 mg (patrz punkty 4.2 i 4.4).
Wykres 1 pokazuje szacunkowe ryzyko pierwszego wystąpienia któregokolwiek zdarzenia złożonego punktu końcowego do oceny skuteczności.
Wykres 1 – Analiza pierwszorzędowego klinicznego złożonego punktu końcowego zgonu z przyczyn sercowo-naczyniowych, zawału mięśnia sercowego i udaru mózgu (PLATO)
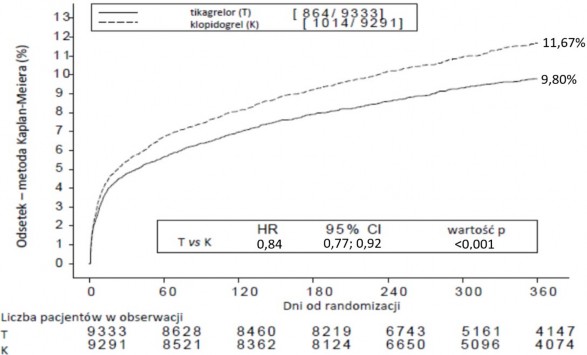
Tikagrelor zmniejszał częstość występowania pierwszorzędowego złożonego punktu końcowego
w porównaniu z klopidogrelem, w obu grupach pacjentów UA/NSTEMI i STEMI (tabela 4). Z tego względu produkt Tilobrastil w dawce 90 mg dwa razy na dobę łącznie z ASA w małej dawce można stosować u pacjentów z OZW (niestabilna dusznica bolesna, zawał mięśnia sercowego bez uniesienia odcinka [NSTEMI] lub zawał mięśnia sercowego z uniesieniem odcinka ST [STEMI]), w tym
u pacjentów leczonych farmakologicznie oraz u pacjentów poddawanych zabiegowi przezskórnej interwencji wieńcowej (PCI) lub pomostowania aortalno-wieńcowemu (CABG).
Tabela 4 - Analiza pierwszorzędowych i drugorzędowych punktów końcowych oceny skuteczności (PLATO)
Tikagrelor 90 mg dwa razy na dobę (% pacjentów, u których wystąpiło zdarzenie) N=9333 | Klopidogrel 75 mg raz na dobę (% pacjentów, u których wystąpiło zdarzenie) N=9291 | ARRa (%/rok ) | RRRa (%) (95% CI) | Wartość p | |
Zgon z przyczyn CV (sercowo- naczyniowych), MI (zawał serca z wyjątkiem niemego MI) lub udar | 9,3 | 10,9 | 1,9 | 16 (8, 23) | 0,0003 |
Plan leczenia inwazyjnego | 8,5 | 10,0 | 1,7 | 16 (6, 25) | 0,0025 |
Plan leczenia zachowawczego | 11,3 | 13,2 | 2,3 | 15 (0,3, 27) | 0,0444d |
Zgon z przyczyn CV | 3,8 | 4,8 | 1,1 | 21 (9, 31) | 0,0013 |
MI (z wyjątkiem niemego MI)b | 5,4 | 6,4 | 1,1 | 16 (5, 25) | 0,0045 |
Udar | 1,3 | 1,1 | -0,2 | -17 (-52, 9) | 0,2249 |
Zgon z jakiejkolwiek przyczyny, MI (z wyjątkiem niemego MI) lub udar | 9,7 | 11,5 | 2,1 | 16 (8, 23) | 0,0001 |
Zgon z przyczyn CV, ogółem MI, udar, SRI, RI, TIA lub inne ATE | 13,8 | 15,7 | 2,1 | 12 (5, 19) | 0,0006 |
Zgon z jakiejkolwiek przyczyny | 4,3 | 5,4 | 1,4 | 22 (11, 31) | 0,0003d |
Skrzeplina zamykająca stent | 1,2 | 1,7 | 0,6 | 32 (8, 49) | 0,0123d |
aARR = bezwzględna redukcja ryzyka; RRR = względna redukcja ryzyka = (1-ryzyko względne) x
100%. Ujemna wartość RRR wskazuje na względny wzrost ryzyka.
b Z wyłączeniem niemego MI.
c SRI = poważne nawracające niedokrwienie; RI = nawracające niedokrwienie; TIA = przemijający napad niedokrwienny; ATE = tętniczy incydent zakrzepowy. Ogółem MI obejmuje zawał niemy, za
datę incydentu przyjęto datę wykrycia.
d Nominalna wartość istotności; wszystkie pozostałe wartości są formalnie istotne statystycznie zgodnie z predefiniowanym testowaniem hierarchicznym.
Subanaliza genetyczna w badaniu PLATO
Genotypowanie pod kątem CYP2C19 i ABCB1, wykonane w badaniu PLATO u 10 285 pacjentów, pozwoliło na określenie relacji między grupami genotypowymi a wynikami badania PLATO. Wyższość tikagreloru nad klopidogrelem w zmniejszaniu liczby ciężkich incydentów sercowo- naczyniowych nie była znamiennie zależna od genotypu CYP2C19 lub ABCB1. Podobnie jak
w całym badaniu PLATO, całkowita liczba ciężkich krwawień wg definicji PLATO nie różniła się
w grupie tikagreloru i klopidogrelu, niezależnie od genotypu CYP2C19 lub ABCB1. Krwawienia ciężkie zgodnie z definicją PLATO, niezwiązane z CABG występowały częściej w grupie tikagrelolu w porównaniu z klopidogrelem u pacjentów z utratą jednego lub więcej funkcyjnych alleli CYP2C19, ale podobnie do grupy klopidogrelu u pacjentów bez utraty alleli funkcyjnych.
Łączna ocena skuteczności i bezpieczeństwa stosowania
Łączna ocena skuteczności i bezpieczeństwa stosowania (zgon z przyczyn sercowo-naczyniowych, zawał serca, udar lub ciężkie krwawienie wg definicji PLATO) wskazuje, że korzyści wynikające ze skuteczności tikagreloru, w porównaniu z klopidogrelem, nie są utracone z powodu liczby ciężkich krwawień (ARR 1,4%, RRR 8%, HR 0,92; p=0,0257) przez okres 12 miesięcy od wystąpienia OZW.
Bezpieczeństwo kliniczne
Podgrupa z badaniem Holtera
W celu zbadania występowania pauz komorowych i innych arytmii w trakcie badania PLATO badacze monitorowali metodą Holtera podgrupę blisko 3000 pacjentów, z których u około 2000 wykonano zapisy w ostrej fazie OZW i po upływie miesiąca. Podstawową obserwowaną zmienną było występowanie pauz komorowych ≥3 sekundy. Większą liczbę pauz komorowych obserwowano
w grupie tikagreloru (6,0%) niż w grupie klopidogrelu (3,5%) w ostrej fazie OZW; i po upływie miesiąca – odpowiednio 2,2% i 1,6% (patrz punkt 4.4). Zwiększona częstość pauz komorowych
w ostrej fazie OZW obserwowana była wyraźniej u pacjentów leczonych tikagrelorem z zastoinową niewydolnością serca w wywiadzie (9,2% wobec 5,4% pacjentów bez zastoinowej niewydolności serca w wywiadzie; w przypadku klopidogrelu, 4,0% pacjentów z zastoinową niewydolnością serca w wywiadzie i 3,6% pacjentów bez zastoinowej niewydolności serca). Ta dysproporcja nie wystąpiła po 1 miesiącu: 2% vs 2,1% w przypadku pacjentów stosujących tikagrelor, odpowiednio z lub bez zastoinowej niewydolności serca; i 3,8% wobec 1,4% w przypadku stosowania klopidogrelu. Nie stwierdzono niekorzystnych konsekwencji klinicznych towarzyszących tym nieprawidłowościom (włączając zastosowanie rozrusznika) w tej grupie pacjentów.
Badanie PEGASUS (zawał mięśnia sercowego w wywiadzie)
Badanie PEGASUS TIMI-54 było obejmującym 21 162 pacjentów, randomizowanym, prowadzonym metodą podwójnie ślepej próby w grupach równoległych, kontrolowanym placebo, międzynarodowym badaniem wieloośrodkowym o przebiegu zależnym od punktów końcowych, oceniającym zapobieganie zdarzeniom sercowo-naczyniowym dzięki stosowaniu tikagreloru w 2 dawkach (albo 90 mg dwa razy na dobę, albo 60 mg dwa razy na dobę) w skojarzeniu z ASA w małej dawce
(75-150 mg) w porównaniu z ASA w monoterapii u pacjentów z zawałem serca w wywiadzie
i z dodatkowymi czynnikami ryzyka wystąpienia takich zdarzeń.
Pacjentów kwalifikowano do udziału w badaniu, gdy byli w wieku co najmniej 50 lat, przebyli
w przeszłości zawał serca (w okresie od 1 do 3 lat przed randomizacją) i mieli co najmniej jeden
z następujących czynników ryzyka zdarzeń zakrzepowych o podłożu miażdżycowym: wiek ≥65 lat, cukrzycę wymagającą leczenia farmakologicznego, drugi przebyty zawał serca, cechy wielonaczyniowej choroby wieńcowej lub przewlekłą nie krańcową niewydolność nerek.
Pacjenci nie kwalifikowali się do udziału w badaniu, gdy planowano u nich stosowanie
antagonisty receptora P2Y12, dipirydamolu, cylostazolu lub leczenia przeciwzakrzepowego w okresie
badania; jeśli występowało u nich zaburzenie krwotoczne lub mieli udar niedokrwienny mózgu albo krwawienie śródczaszkowe, guz ośrodkowego układ nerwowego lub nieprawidłową budowę naczyń śródczaszkowych w wywiadzie; jeśli wystąpiło u nich krwawienie z przewodu pokarmowego
w okresie ostatnich 6 miesięcy lub przebyli poważny zabieg chirurgiczny w okresie ostatnich 30 dni.
Skuteczność kliniczna
Wykres 2 – Analiza pierwszorzędowego klinicznego złożonego punktu końcowego zgonu z przyczyn sercowo-naczyniowych, zawału mięśnia sercowego i udaru mózgu (PEGASUS)
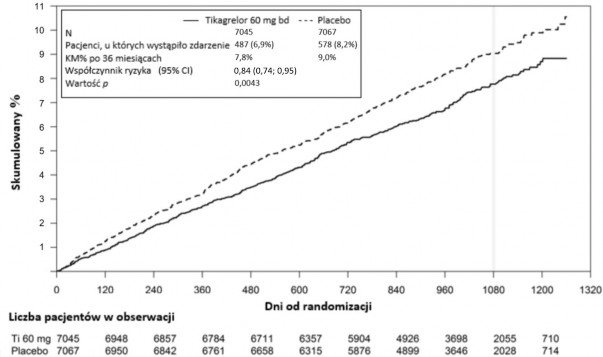
Tabela 5 - Analiza pierwszorzędowych i drugorzędowych punktów końcowych oceny skuteczności (PEGASUS)
Tikagrelor 60 mg dwa razy na dobę + ASA N = 7045 | ASA w monoterapii N = 7067 | Wartość p | ||||
Cecha | Pacjenci, u których wystąpiło zdarzenie | KM % | HR (95% CI) | Pacjenci, u których wystąpiło zdarzenie | KM % | |
Pierwszorzędowy punkt końcowy | ||||||
Złożony punkt końcowy zgonu z przyczyn CV/MI/udaru mózgu | 487 (6,9%) | 7,8% | 0,84 (0,74, 0,95) | 578 (8,2%) | 9,0% | 0,0043 (s) |
Zgon z przyczyn CV | 174 (2,5%) | 2,9% | 0,83 (0,68, 1,01) | 210 (3,0%) | 3,4% | 0,0676 |
Zawał serca | 285 (4,0%) | 4,5% | 0,84 (0,72, 0,98) | 338 (4,8%) | 5,2% | 0,0314 |
Udar mózgu | 91 (1,3%) | 1,5% | 0,75 (0,57, 0,98) | 122 (1,7%) | 1,9% | 0,0337 |
Drugorzędowy punkt końcowy | ||||||
Zgon z przyczyn CV | 174 (2,5%) | 2,9% | 0,83 (0,68, 1,01) | 210 (3,0%) | 3,4% | - |
Zgon z jakiejkolwiek przyczyny | 289 (4,1%) | 4,7% | 0,89 (0,76, 1,04) | 326 (4,6%) | 5,2% | - |
Współczynnik ryzyka i wartości p obliczono oddzielnie w odniesieniu do tikagreloru w porównaniu z ASA w monoterapii z użyciem modelu proporcjonalnych hazardów Cox z grupą badaną, jako jedyną zmienną wyjaśniającą.
Procent obliczony metodą KM po 36 miesiącach.
Uwaga: liczby pierwszych zdarzeń w przypadku komponentów: zgon z przyczyn CV, MI i udar mózgu są rzeczywistą liczbą pierwszych zdarzeń stanowiących każdy z komponentów i nie sumują się one do liczby zdarzeń stanowiących złożony punkt końcowy.
(s) wskazuje na istotność statystyczną
CI = przedział ufności; CV = sercowo-naczyniowe; HR = współczynnik ryzyka; KM = Kaplan-Meier; MI = zawał serca; N = liczba pacjentów.
Oba schematy leczenia tikagrelorem w skojarzeniu z ASA (z zastosowaniem dawek tikagreloru 60 mg dwa razy na dobę i 90 mg dwa razy na dobę) wykazywały przewagę nad ASA w monoterapii pod względem zapobiegania zdarzeniom sercowo-naczyniowym (złożony punkt końcowy: zgon
z przyczyn sercowo-naczyniowych, zawał serca i udar mózgu), z niezmiennym skutkiem leczenia w całym okresie badania, co dało wskaźniki 16% RRR i 1,27% ARR w grupie tikagreloru w dawce 60 mg oraz 15% RRR i 1,19% ARR w grupie tikagreloru w dawce 90 mg.
Chociaż profile skuteczności dawek 90 mg i 60 mg były podobne, istnieją dane świadczące o tym, że mniejsza dawka wykazuje korzystniejszy profil tolerancji i bezpieczeństwa w stosunku do ryzyka krwawień i duszności. W związku z tym, tylko produkt leczniczy Tilobrastil w dawce 60 mg dwa razy na dobę w skojarzeniu z ASA jest zalecany w zapobieganiu zdarzeniom sercowo-naczyniowym (zgon z przyczyn sercowo-naczyniowych, zawał serca i udar mózgu) u pacjentów z zawałem serca
w wywiadzie i z wysokim ryzykiem zdarzeń zakrzepowych o podłożu miażdżycowym.
W porównaniu z ASA w monoterapii, stosowanie tikagreloru w dawce 60 mg dwa razy na dobę prowadziło do istotnego statystycznie zmniejszenia częstości występowania pierwszorzędowego złożonego punktu końcowego w postaci zgonu z przyczyn sercowo-naczyniowych, zawału serca i udaru mózgu. Każdy z komponentów przyczynił się do zmniejszenia częstości występowania złożonego punktu końcowego (zgon z przyczyn sercowo-naczyniowych 17% RRR, zawał serca 16% RRR i udar mózgu 25% RRR).
Współczynnik RRR złożonego punktu końcowego w okresie od 1. do 360. dnia (17% RRR)
i począwszy od 361. dnia (16% RRR) był podobny. Dane dotyczące skuteczności i bezpieczeństwa użycia tikagreloru powyżej 3 lat przedłużonego leczenia są ograniczone.
Nie ma danych wskazujących na korzyści stosowania (brak redukcji pierwszorzędowego złożonego punktu końcowego dotyczącego śmierci sercowo-naczyniowej, zawału serca i udaru przy wzroście poważnych krwawień) tikagreloru 60 mg dwa razy na dobę u pacjentów klinicznie stabilnych powyżej 2 lat od zawału serca lub kiedy więcej niż rok upłynął od zaprzestania przyjmowania poprzedniego ADP inhibitora (patrz punkt 4.2)
Bezpieczeństwo kliniczne
Częstość przerywania leczenia tikagrelorem 60 mg z powodu krwawień i duszności była wyższa u pacjentów w wieku powyżej 75 lat (42%) niż u pacjentów młodszych (od 23 do 31% ) z różnicą w stosunku do placebo większą niż 10% (42% vs 29%) u pacjentów w wieku >75 lat.
Dzieci i młodzież
W randomizowanym badaniu III fazy prowadzonym w grupach równoległych metodą podwójnie ślepej próby (HESTIA 3) 193 dzieci i młodzieży (w wieku od 2 do mniej niż 18 lat)
z niedokrwistością sierpowatokrwinkową zostało losowo przydzielonych do grupy otrzymującej placebo lub tikagrelor w dawkach od 15 mg do 45 mg dwa razy na dobę, w zależności od masy ciała. Tikagrelor powodował zahamowanie płytek krwi o medianie wynoszącej 35% przed podaniem dawki
i 56% po 2 godzinach od podania dawki w stanie stacjonarnym.
W porównaniu z placebo nie stwierdzono korzyści z leczenia tikagrelorem w odniesieniu do częstości występowania przełomów naczyniowo-okluzyjnych.
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego tikagrelor wszystkich podgrupach populacji dzieci i młodzieży w ostrym zespole wieńcowym (OZW) oraz z zawałem mięśnia sercowego (zawałem serca) w wywiadzie (informacje na temat stosowania produktu u dzieci i młodzieży przedstawiono w punkcie 4.2).
Tikagrelor wykazuje liniową farmakokinetykę, a ekspozycja na tikagrelor i jego czynny metabolit (AR-C124910XX) jest w przybliżeniu zależna od dawki, w przedziale do 1260 mg.
Wchłanianie
Wchłanianie tikagreloru jest szybkie, mediana czasu tmax wynosi około 1,5 godziny. Tworzenie się głównego krążącego metabolitu AR-C124910XX (również czynnego) z tikagreloru jest szybkie, mediana tmax wynosi około 2,5 godziny. Po podaniu pojedynczej dawki doustnej 90 mg tikagreloru na czczo zdrowym ochotnikom Cmax wynosi 529 ng/ml, a AUC 3451 ng*h/ml. Dla metabolitu powiązane z substancją wyjściową współczynniki wynoszą 0,28 dla Cmax i 0,42 dla AUC. Farmakokinetyka tikagreloru i AR-C124910XX u chorych z zawałem serca w wywiadzie była zasadniczo podobna do obserwowanej w populacji pacjentów z OZW. Według analizy farmakokinetyki populacyjnej w badaniu PEGASUS mediana tikagreloru Cmax wynosiła 391 ng/ml i AUC wynosiło 3801 ng*h/ml w stanie stacjonarnym po podaniu tikagreloru w dawce 60 mg.
W przypadku tikagreloru w dawce 90 mg Cmax wynosiło 627 ng/ml i AUC wynosiło 6255 ng*h/ml w stanie stacjonarnym.
Średnią bezwzględną biodostępność tikagreloru oszacowano na 36%. Spożycie wysokotłuszczowego posiłku skutkuje zwiększeniem AUC tikagreloru o 21% i zmniejszeniem Cmax
czynnego metabolitu o 22%, ale nie spowodowało zmian Cmax tikagreloru i AUC czynnego metabolitu. Uważa się, że te niewielkie zmiany mają minimalne znaczenie kliniczne, dlatego też tikagrelor może być zażywany w trakcie posiłków lub niezależnie od nich. Zarówno tikagrelor, jak i czynny metabolit są substratami glikoproteiny P (P-gp).
Tikagrelor w postaci rozgniecionych tabletek wymieszanych z wodą, podanych doustnie lub przez zgłębnik nosowo-żołądkowy ma biodostępność porównywalną do tabletki podawanej w całości
w zakresie AUC i Cmax dla tikagreloru i aktywnego metabolitu. Ekspozycja początkowa
(0,5 i 1 godzina po podaniu) tikagreloru zastosowanego w postaci rozgniecionej tabletki wymieszanej z wodą była większa niż zastosowanego w postaci całej (niepokruszonej) tabletki, z zasadniczo identycznym profilem stężeń w późniejszym czasie (od 2 do 48 godzin).
Dystrybucja
Objętość dystrybucji w stanie stacjonarnym wynosi 87,5 l. Tikagrelor i czynny metabolit w znacznym stopniu wiążą się z białkami osocza ludzkiego (>99,0%).
Metabolizm
CYP3A4 jest głównym enzymem odpowiedzialnym za metabolizm tikagreloru i tworzenie czynnego metabolitu a ich interakcje z innymi substratami izoenzymu CYP3A obejmują zarówno aktywację jak i hamowanie.
Główny metabolit tikagreloru, AR-C124910XX, jest także czynny, co określono w badaniach in vitro, w których wiąże się on z receptorem płytkowym ADP P2Y12. Ogólnoustrojowe narażenie na czynny metabolit stanowi około 30 – 40% narażenia na tikagrelor.
Wydalanie
Podstawowa droga eliminacji tikagreloru to metabolizm wątrobowy. Po podawaniu znakowanego
radioaktywnie tikagreloru, średni wychwyt zwrotny radioaktywności wynosił około 84% (57,8% w kale
i 26,5% w moczu). Odzyskany tikagrelor i czynny metabolit w moczu w obydwu przypadkach stanowiły mniej niż 1% zastosowanej dawki. Główną drogą eliminacji czynnego metabolitu jest najprawdopodobniej wydzielanie z żółcią. Średni okres półtrwania wynosił około 7 godzin dla tikagreloru i 8,5 godziny dla czynnego metabolitu.
Szczególne grupy pacjentów
Osoby w podeszłym wieku
W trakcie analiz farmakokinetycznych w populacjach, u osób w podeszłym wieku (≥75 lat) z OZW obserwowano większe narażenie na tikagrelor (o około 25% dla Cmax i AUC) i na czynny metabolit w porównaniu do młodszych pacjentów. Uważa się, że różnice te nie są istotne klinicznie (patrz punkt 4.2).
Dzieci i młodzież
Dostępne są ograniczone dane dotyczące dzieci i młodzieży z niedokrwistością sierpowatokrwinkową (patrz punkt 4.2 i 5.1).
W badaniu HESTIA 3 pacjentom w wieku od 2 do mniej niż 18 lat, ważącym od ≥ 12 do ≤ 24 kg, od
>24 do ≤ 48 kg i > 48 kg podawano tikagrelor w postaci 15 mg tabletek ulegających rozpadowi w jamie ustnej przeznaczonych dla dzieci, w dawkach wynoszących odpowiednio 15, 30 i 45 mg dwa razy na dobę. Analiza farmakokinetyki populacyjnej wykazała, że średnie AUC wahało się od 1095 ng*h/ml do 1458 ng*h/ml, a średnie Cmax wynosiło od 143 ng/ml do 206 ng/ml w stanie stacjonarnym.
Płeć
U kobiet obserwowano większe narażenie na tikagrelor i na czynny metabolit niż u mężczyzn. Uważa się, że różnice te nie są istotne klinicznie.
Zaburzenia czynności nerek
U pacjentów z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny <30 ml/min) narażenie na tikagrelor było o około 20% mniejsze zaś narażenie na czynny metabolit było około 17% większe niż w przypadku pacjentów z prawidłową funkcją nerek.
U pacjentów z krańcową chorobą nerek poddawanych hemodializie, wartości AUC i Cmax tikagreloru w dawce 90 mg podawanego w dniu bez dializy były o 38% i 51% większe niż u pacjentów z prawidłową czynnością nerek. Podobne zwiększenie ekspozycji obserwowano, gdy tikagrelor był podawany bezpośrednio przed dializą (odpowiednio o 49% i 61%), co wskazuje, że tikagrelor nie ulega dializie.
Ekspozycja na aktywny metabolit wzrosła w mniejszym stopniu (AUC o 13-14%, a Cmax o 17-36%). Działanie tikagreloru polegające na hamowaniu agregacji płytek krwi było niezależne od dializy u pacjentów z krańcową chorobą nerek i podobne, jak u pacjentów z prawidłową czynnością nerek (patrz punkt 4.2).
Zaburzenia czynności wątroby
Cmax i AUC tikagreloru było odpowiednio 12% i 23% większe u pacjentów z łagodnymi zaburzeniami czynności wątroby w porównaniu do odpowiadających im zdrowych osobników, jednak działanie tikagreloru hamujące agregację płytek było podobne w obu grupach. Nie ma konieczności korygowania dawek u pacjentów z umiarkowanymi zaburzeniami czynności wątroby. Nie prowadzono badań dotyczących stosowania tikagreloru u pacjentów z ciężką niewydolnością wątroby i nie są dostępne informacje na temat jego farmakokinetyki u pacjentów z umiarkowanymi zaburzeniami czynności wątroby. U pacjentów z wyjściowym umiarkowanym lub ciężkim zwiększeniem wyników jednej lub dwóch prób wątrobowych stężenie tikagreloru w osoczu było średnio podobne lub nieco większe do mierzonego u pacjentów bez wyjściowego zwiększenia tych parametrów. Nie jest konieczna korekta dawkowania u pacjentów z umiarkowanymi zaburzeniami czynności wątroby (patrz punkty 4.2 i 4.4).
Różnice rasowe
U pacjentów pochodzenia azjatyckiego obserwuje się o 39% większą średnią biodostępność
w porównaniu z pacjentami rasy kaukaskiej. U pacjentów, którzy określają swoją rasę jako czarną, biodostępność tikagreloru jest o 18% mniejsza niż u pacjentów rasy kaukaskiej. W badaniach
farmakologii klinicznej wśród Japończyków obserwowano większe o mniej więcej 40% (a o 20% po
dostosowaniu do masy ciała) narażenie na tikagrelor (Cmax i AUC), w porównaniu do osób rasy
Dane przedkliniczne pochodzące z konwencjonalnych badań farmakologicznych tikagreloru i jego głównego metabolitu, które dotyczyły bezpieczeństwa farmakoterapii, badań toksyczności po podaniu pojedynczym i wielokrotnym oraz potencjalnej genotoksyczności nie wykazały niedopuszczalnego ryzyka wystąpienia działań niepożądanych u ludzi.
Przy odpowiadającym warunkom klinicznym narażeniu u kilku gatunków zwierząt zaobserwowano podrażnienie przewodu pokarmowego (patrz punkt 4.8).
U samic szczurów, którym podawano tikagrelor w dużych dawkach, zaobserwowano zwiększenie liczby przypadków guzów macicy (gruczolakoraki) i zwiększenie liczby przypadków gruczolaków wątroby. Mechanizm powstawania guzów macicy u szczurów polega prawdopodobnie na zaburzeniu równowagi hormonalnej, która może prowadzić do powstania guzów u szczurów. Mechanizm powstawania gruczolaków wątroby to prawdopodobnie specyficzne dla gryzoni zwiększenie aktywności enzymatycznej w wątrobie. Dlatego uważa się za mało prawdopodobne, aby te przypadki rakotwórczości miały znaczenie dla ludzi.
U szczurów obserwowano niewielkie nieprawidłowości rozwojowe przy podawaniu ciężarnym samicom dawek toksycznych (margines bezpieczeństwa 5,1). U płodów królików obserwowano niewielkie opóźnienie dojrzewania wątroby i rozwoju układu szkieletowego, gdy ciężarnym samicom podawano duże dawki bez oznak toksyczności u ciężarnych samic (margines bezpieczeństwa 4,5).
Badania na szczurach i królikach wykazały toksyczne działanie na rozmnażanie, z niewielkim zmniejszeniem przyrostu masy ciała ciężarnych samic oraz zmniejszoną przeżywalnością noworodków i mniejszą wagą urodzeniową oraz opóźnionym wzrostem. Tikagrelor powodował u samic szczurów nieregularne cykle (w większości wydłużone), ale nie wpływał na całkowitą
płodność u samców i samic szczurów. Badania farmakokinetyczne przeprowadzone ze znakowanym radioaktywnie tikagrelorem wykazały, że zarówno sama substancja czynna jak i jej metabolity przenikają do mleka szczurów (patrz punkt 4.6).
Rdzeń tabletki
Mannitol
Wapnia wodorofosforan dwuwodny Skrobia kukurydziana
Skrobia żelowana (kukurydziana) Talk
Sodu stearylofumaran
Otoczka:
Alkohol poliwinylowy
Talk
Tytanu dwutlenek (E 171) Glicerolu monokaprylokapronian Sodu laurylosiarczan
Żelaza tlenek żółty (E 172)
Nie dotyczy.
2 lata
Bez specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Nie wszystkie rodzaje i wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Sandoz GmbH
Biochemiestrasse 10
6250 Kundl Austria
Pozwolenie nr 26209
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 22.01.2021
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
11.01.2023








