







Spis treści:
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Mediana (m)
95% CI dla mediany
HR
95% CI dla HR
wartość p dla równoważności efektu (HR)
Mediana
HR (95% CI)
Mediana
HR (95% CI)
Wskaźnik odpowiedzi (%) (95% CI)
Stabilizacja choroby (%)
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania produktu leczniczego
Pemetrexed EVER Pharma, 25 mg/ml, koncentrat do sporządzania roztworu do infuzji
Jeden mililitr koncentratu zawiera 25 mg pemetreksedu (w postaci pemetreksedu disodowego). Jedna fiolka 4 ml koncentratu zawiera 100 mg pemetreksedu (w postaci pemetreksedu disodowego).
Jedna fiolka 20 ml koncentratu zawiera 500 mg pemetreksedu (w postaci pemetreksedu disodowego). Jedna fiolka 40 ml koncentratu zawiera 1000 mg pemetreksedu (w postaci pemetreksedu disodowego).
Substancja pomocnicza o znanym działaniu Ten produkt leczniczy zawiera 2,7 mg/ml sodu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Koncentrat do sporządzania roztworu do infuzji.
Koncentrat to roztwór wodny, klarowny o barwie jasnożółtej lub zielonożółtej.
pH roztworu jest pomiędzy 7,5 a 8,1.
Złośliwy międzybłoniak opłucnej
Pemetreksed w skojarzeniu z cisplatyną jest przeznaczony do stosowania u nieleczonych wcześniej chemioterapią pacjentów z nieoperacyjnym, złośliwym międzybłoniakiem opłucnej.
Niedrobnokomórkowy rak płuca
Pemetreksed w skojarzeniu z cisplatyną wskazany jest jako leczenie pierwszego rzutu u pacjentów z niedrobnokomórkowym rakiem płuca w stadium miejscowo zaawansowanym lub z przerzutami,
o budowie histologicznej innej, niż w przeważającym stopniu płaskonabłonkowa (patrz punkt 5.1).
Pemetreksed w monoterapii jest wskazany do stosowania jako leczenie podtrzymujące u pacjentów
z niedrobnokomórkowym rakiem płuca w stadium miejscowo zaawansowanym lub z przerzutami,
o budowie histologicznej innej, niż w przeważającym stopniu płaskonabłonkowa, u których nie nastąpiła progresja choroby bezpośrednio po zakończeniu chemioterapii, opartej na pochodnych platyny (patrz punkt 5.1).
Pemetreksed w monoterapii jest wskazany do stosowania jako leczenie drugiego wyboru u pacjentów, z niedrobnokomórkowym rakiem płuca w stadium miejscowo zaawansowanym lub z przerzutami,
o budowie histologicznej innej, niż w przeważającym stopniu płaskonabłonkowa (patrz punkt 5.1).
Dawkowanie
Pemetreksed można podawać wyłącznie pod kontrolą lekarza wykwalifikowanego w stosowaniu chemioterapii przeciwnowotworowej.
Pemetreksed w skojarzeniu z cisplatyną
Zalecana dawka pemetreksedu wynosi 500 mg/m2 powierzchni ciała (pc.). Produkt należy podawać
w infuzji dożylnej w ciągu 10 minut, w pierwszym dniu każdego 21-dniowego cyklu. Zalecana dawka cisplatyny wynosi 75 mg/m2 pc. Cisplatynę należy podawać w infuzji w ciągu 2 godzin, rozpoczynając około 30 minut po zakończeniu infuzji pemetreksedu pierwszego dnia każdego 21-dniowego cyklu leczenia. Pacjent musi otrzymać leki przeciwwymiotne oraz płyny w odpowiedniej ilości, przed i (lub) po podaniu cisplatyny (szczegółowe dane na temat dawkowania cisplatyny można znaleźć
w Charakterystyce Produktu Leczniczego tego leku).
Pemetreksed w monoterapii
W przypadku pacjentów z niedrobnokomórkowym rakiem płuca, poddawanych wcześniej chemioterapii, zalecana dawka pemetreksedu wynosi 500 mg/m2 pc. Produkt należy podawać w infuzji dożylnej w ciągu 10 minut, w pierwszym dniu każdego 21-dniowego cyklu leczenia.
Premedykacja
W celu ograniczenia częstości występowania i nasilenia odczynów skórnych, w dniu poprzedzającym podanie pemetreksedu, jak również w dniu podania leku i następnego dnia pacjent powinien otrzymać lek z grupy kortykosteroidów. Dawka kortykosteroidu powinna odpowiadać dawce 4 mg deksametazonu, podawanego doustnie dwa razy na dobę (patrz punkt 4.4).
W celu ograniczenia objawów toksyczności, pacjenci leczeni pemetreksedem powinni również otrzymywać suplementację witaminową (patrz punkt 4.4). Pacjentom należy codziennie podawać doustnie kwas foliowy lub produkt multiwitaminowy zawierający ten związek (od 350 do 1000 mikrogramów).
W ciągu 7 dni poprzedzających podanie pierwszej dawki pemetreksedu pacjent powinien przyjąć co najmniej 5 dawek kwasu foliowego. Kwas foliowy należy także podawać przez cały cykl leczenia
i przez 21 dni po podaniu ostatniej dawki pemetreksedu. W tygodniu poprzedzającym przyjęcie pierwszej dawki pemetreksedu, a następnie co trzy cykle leczenia, pacjenci muszą także otrzymywać domięśniowo witaminę B12 (1000 mikrogramów). Kolejne wstrzyknięcia witaminy B12 można wykonywać w dniu podania pemetreksedu.
Kontrola stanu pacjenta
Przed podaniem każdej dawki pemetreksedu należy wykonać pełną morfologię krwi, w tym oznaczenie wzoru odsetkowego krwinek białych (rozmaz) i liczby płytek krwi. Przed każdym podaniem chemioterapeutyku należy wykonać badania krwi, oceniające czynności nerek i wątroby. Warunkiem umożliwiającym rozpoczęcie każdego cyklu chemioterapii są następujące wartości parametrów laboratoryjnych: bezwzględna liczba neutrofilów (ang. Absolute Neutrophil Count, ANC)
≥ 1500 komórek/mm3, liczba płytek krwi ≥100 000 komórek/mm3, klirens kreatyniny ≥ 45 ml/min, bilirubina całkowita ≤ 1,5 razy górna granica wartości uznanych za prawidłowe; fosfataza zasadowa, aminotransferaza asparaginianowa (AspAT), aminotransferaza alaninowa (AlAT) ≤ 3 razy górna granica wartości uznanych za prawidłowe. U pacjentów z przerzutami guza do wątroby dopuszczalne są wartości aktywności fosfatazy zasadowej, AspAT i AlAT nie większe, niż 5-krotne wartości uznane za prawidłowe.
Modyfikacja dawki
Decyzję o modyfikacji dawki przed rozpoczęciem kolejnego cyklu chemioterapii należy podejmować na podstawie najmniejszych wartości parametrów morfologii krwi oznaczonych podczas poprzedniego cyklu lub największego nasilenia objawów toksyczności, przy którym nie wystąpiły zmiany w obrazie krwi. Rozpoczęcie kolejnego cyklu można opóźnić, by w ten sposób umożliwić normalizację stanu zdrowia pacjenta. Po uzyskaniu odpowiedniej poprawy stanu pacjenta należy kontynuować leczenie
zgodnie z zasadami przedstawionymi w tabelach 1, 2 i 3, które odnoszą się zarówno do stosowania pemetreksedu w monoterapii, jak i w skojarzeniu pemetreksedu z cisplatyną.
Tabela 1 - Modyfikacja dawki pemetreksedu (w monoterapii lub leczeniu skojarzonym) i cisplatyny- zmiany w obrazie krwi | |
Najmniejsza bezwzględna liczba neutrofilów < 500/mm3 i najmniejsza liczba płytek ≥ 50 000/mm3 | 75% poprzedniej dawki (zarówno pemetreksedu jak i cisplatyny) |
Najmniejsza liczba płytek <50 000/mm3 bez względu na to, jaka jest najmniejsza liczba neutrofilów | 75% poprzedniej dawki (zarówno pemetreksedu jak i cisplatyny) |
Najmniejsza liczba płytek <50 000/mm3 oraz Krwawienie a bez względu na to, jaka jest najmniejsza liczba neutrofilów | 50% poprzedniej dawki (zarówno pemetreksedu jak i cisplatyny) |
a zgodnie z definicją krwawień stopnia 2. lub wyższego wg ogólnych kryteriów toksyczności
(ang. Common Toxicity Criteria, CTC, v2.0; NCI 1998) National Cancer Institute.
Jeżeli wystąpią działania niepożądane ≥ 3 stopnia, inne niż zmiany w obrazie krwi (bez objawów toksyczności neurologicznej), należy przerwać stosowanie pemetreksedu aż do powrotu ocenianych parametrów do wartości sprzed leczenia lub mniejszych. Ponowne leczenie należy rozpocząć zgodnie z wytycznymi z Tabeli 2.
Tabela 2 - Modyfikacja dawki pemetreksedu (w monoterapii lub leczeniu skojarzonym) i cisplatyny-działania toksyczne inne, niż zmiany w obrazie krwi a, b | ||
Dawka pemetreksedu (mg/m2) | Dawka cisplatyny (mg/m2) | |
Jakiekolwiek działania niepożądane stopnia 3 lub 4 z wyjątkiem zapalenia błon śluzowych. | 75% poprzedniej dawki | 75% poprzedniej dawki |
Biegunka wymagająca hospitalizacji (bez względu na nasilenie) lub biegunka stopnia 3 lub 4. | 75% poprzedniej dawki | 75% poprzedniej dawki |
Zapalenie błon śluzowych stopnia 3 lub 4. | 50% poprzedniej dawki | 100% poprzedniej dawki |
a Ogólne kryteria toksyczności (ang. Common Toxicity Criteria, CTC v2.0; NCI 1998) wg National Cancer Institute.
b bez objawów toksyczności neurologicznej
Jeżeli wystąpią objawy toksyczności neurologicznej, dawkę pemetreksedu i cisplatyny należy zmodyfikować zgodnie z danymi z Tabeli 3. W przypadku wystąpienia toksyczności neurologicznej stopnia 3 lub 4, leczenie należy przerwać.
Tabela 3 - Modyfikacja dawki pemetreksedu (w monoterapii lub leczeniu skojarzonym) i cisplatyny – toksyczność neurologiczna | ||
Nasilenie objawów wg CTCa | Dawka pemetreksedu (mg/m2) | Dawka cisplatyny (mg/m2) |
0-1 | 100% poprzedniej dawki | 100% poprzedniej dawki |
2 | 100% poprzedniej dawki | 50% poprzedniej dawki |
a CTC –Ogólne Kryteria Toksyczności (ang. Common Toxicity Criteria, CTC v2.0; NCI 1998) wg National Cancer Institute.
Leczenie pemetreksedem należy przerwać, jeżeli u pacjenta wystąpią objawy toksyczności hematologicznej lub innego rodzaju stopnia 3. lub 4. po dwukrotnym zmniejszeniu dawki. Leczenie należy przerwać natychmiast, jeżeli wystąpią objawy toksyczności neurologicznej stopnia 3. lub 4.
Szczególne grupy pacjentów
Osoby w podeszłym wieku
W badaniach klinicznych nie stwierdzono, by osoby w wieku 65 lat i starsze były w większym stopniu narażone na działania niepożądane w porównaniu z osobami w wieku do 65 lat. Brak szczególnych
zaleceń dotyczących zmniejszania dawki u osób w podeszłym wieku, z wyjątkiem zaleceń ustalonych dla wszystkich pacjentów.
Dzieci i młodzież
Stosowanie pemetreksedu u dzieci i młodzieży nie jest właściwe w leczeniu złośliwego międzybłoniaka opłucnej i niedrobnokomórkowego raka płuca.
Pacjenci z zaburzeniem czynności nerek (obniżenie GFR obliczane na podstawie standardowego wzoru Cockrofta i Gaulta lub przez pomiar przesączania kłębuszkowego metodą klirensu
Tc99m-DPTA z surowicy)
Pemetreksed jest wydalany głównie w postaci niezmienionej przez nerki. W badaniach klinicznych nie stwierdzono konieczności zmiany dawki (z wyjątkiem zaleceń ustalonych dla wszystkich pacjentów)
u osób z klirensem kreatyniny ≥ 45 ml/min. Nie zebrano dostatecznej ilości danych na temat stosowania pemetreksedu u osób z klirensem kreatyniny mniejszym niż 45 ml/min.
Z tego względu, nie zaleca się stosowania pemetreksedu u tych pacjentów (patrz punkt 4.4).
Pacjenci z zaburzeniem czynności wątroby
Nie wykazano związku między aktywnością AspAT, AlAT, całkowitym stężeniem bilirubiny
a farmakokinetyką pemetreksedu. Nie przeprowadzano jednak osobnych analiz dla podgrup pacjentów z objawami zaburzeń czynności wątroby, jak np. zwiększenie stężenia bilirubiny > 1,5 razy powyżej górnej granicy wartości uznanych za prawidłowe i (lub) zwiększenie aktywności aminotransferaz
> 3 razy powyżej górnej granicy wartości uznanych za prawidłowe (u pacjentów bez przerzutów nowotworu do wątroby) lub > 5 razy powyżej górnej granicy wartości uznanych za prawidłowe (u pacjentów z przerzutami nowotworowymi do wątroby).
Sposób podawania
Podanie dożylne po rozcieńczeniu.
Produkt leczniczy Pemetrexed EVER Pharma należy podawać w infuzji dożylnej w ciągu 10 minut w pierwszym dniu każdego 21-dniowego cyklu. Instrukcja dotycząca rozcieńczania produktu Pemetrexed EVER Pharma przed podaniem, patrz punkt 6.6.
Środki ostrożności, które należy podjąć przed użyciem lub podaniem pemetreksedu, patrz punkt 6.6.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Karmienie piersią (patrz punkt 4.6).
Jednoczesne podanie szczepionki przeciwko żółtej gorączce (patrz punkt 4.5).
Pemetreksed może wywoływać mielosupresję, objawiającą się neutropenią, trombocytopenią i niedokrwistością (lub pancytopenią) (patrz punkt 4.8). Wystąpienie tego powikłania oznacza
zazwyczaj konieczność zmniejszenia dawki. Podczas leczenia należy obserwować, czy nie występują objawy mielosupresji. Pemetreksedu nie należy podawać aż do momentu, gdy całkowita liczba neutrofilów wzrośnie co najmniej do poziomu 1500 komórek/mm3, a liczba płytek krwi wzrośnie co najmniej do poziomu 100 000 komórek/mm3. Decyzje o zmniejszeniu dawki podczas kolejnych cyklów chemioterapii należy podejmować na podstawie obserwowanych w poprzednim cyklu najmniejszych wartości liczby neutrofilów i płytek krwi i największego stopnia nasilenia objawów toksyczności niehematologicznej (patrz punkt 4.2).
U osób, które przyjmowały kwas foliowy i witaminę B12 przed rozpoczęciem leczenia pemetreksedem stwierdzono mniejszą toksyczność oraz zmniejszenie częstości występowania działań niepożądanych hematologicznych i niehematologicznych stopnia 3. lub 4., np. neutropenii, gorączki neutropenicznej i zakażeń z neutropenią stopnia 3. lub 4. Wszystkim pacjentom leczonym pemetreksedem należy zatem zalecić profilaktyczne stosowanie kwasu foliowego i witaminy B12, w celu ograniczenia działań niepożądanych, związanych z leczeniem (patrz punkt 4.2).
U pacjentów, którym przed leczeniem pemetreksedem nie podawano kortykosteroidów, obserwowano odczyny skórne. Częstość występowania i nasilenie reakcji skórnych może zmniejszyć premedykacja deksametazonem (lub innym równoważnym produktem leczniczym) (patrz punkt 4.2).
W badaniach produktu leczniczego nie uczestniczyła odpowiednio liczna grupa pacjentów z klirensem kreatyniny mniejszym niż 45 ml/min. Z tego względu, nie zaleca się stosowania pemetreksedu u osób z klirensem kreatyniny mniejszym niż 45 ml/min (patrz punkt 4.2).
Pacjenci z łagodną lub umiarkowaną niewydolnością nerek (klirens kreatyniny od 45 do 79 ml/min) powinni unikać przyjmowania niesteroidowych leków przeciwzapalnych (NLPZ), takich, jak ibuprofen i kwas acetylosalicylowy (> 1,3 g na dobę) na 2 dni przed podaniem pemetreksedu, w dniu podania leku i przez 2 dni po podaniu pemetreksedu (patrz punkt 4.5).
U pacjentów z łagodną lub umiarkowaną niewydolnością nerek, zakwalifikowanych do terapii pemetreksedem, należy przerwać stosowanie NLPZ o długim okresie półtrwania na co najmniej 5 dni przed podaniem pemetreksedu, w dniu podania leku i przez co najmniej 2 dni po podaniu pemetreksedu (patrz punkt 4.5).
Po podaniu pemetreksedu w monoterapii lub w skojarzeniu z innymi lekami chemioterapeutycznymi zgłaszano wystąpienie ciężkich zaburzeń czynności nerek, w tym ostrej niewydolności nerek.
U większości pacjentów, u których pojawiły się te zaburzenia stwierdzono obecność czynników ryzyka wiodących do rozwoju zaburzeń czynności nerek, w tym odwodnienia, wcześniej rozpoznanego nadciśnienia lub cukrzycy. Po wprowadzeniu produktu leczniczego do obrotu zarówno w monoterapii pemetreksedem, jak i w terapii skojarzonej z innymi chemioterapeutykami obserwowano nefrogenną moczówkę prostą i ostrą martwicę cewek nerkowych. Większość objawów ustępowała po zaprzestaniu terapii. Pacjenci powinni być monitorowani pod kątem pojawiania się objawów ostrej martwicy cewkowej, obniżenia wydolności nerek oraz oznak i objawów nefrogennej moczówki prostej (np. pojawienie się hypernatremii).
Obecność płynu w trzeciej przestrzeni, np. wysięku w opłucnej lub wodobrzusza na pemetreksed nie został w pełni określony. W badaniu 2 fazy u 31 pacjentów z guzami litymi i stabilną objętością płynu w trzeciej przestrzeni, po podaniu dawki pemetreksedu nie obserwowano różnic w znormalizowanym stężeniu w osoczu ani w klirensie leku, w porównaniu z wartościami obserwowanymi u pacjentów,
u których nie stwierdzono nagromadzenia płynu w trzeciej przestrzeni. Dlatego też, należy rozważyć wykonanie drenażu płynu nagromadzonego w trzeciej przestrzeni przed podaniem pemetreksedu, jednak może nie być to konieczne.
Obserwowano przypadki znacznego odwodnienia związanego z toksycznym działaniem pemetreksedu stosowanego w skojarzeniu z cisplatyną na układ pokarmowy. W związku z tym, przed podaniem leków należy podać pacjentowi leki przeciwwymiotne oraz przed i (lub) po ich podaniu zastosować odpowiednie nawodnienie.
Ciężkie zdarzenia niepożądane ze strony układu sercowo-naczyniowego, w tym zawał mięśnia sercowego i zdarzenia naczyniowo-mózgowe obserwowano niezbyt często podczas badań klinicznych z zastosowaniem pemetreksedu. Zdarzenia te występowały zwykle w przypadku, gdy pemetreksed był stosowany w skojarzeniu z innym lekiem cytotoksycznym. U większości pacjentów, u których wystąpiły opisywane zdarzenia niepożądane, występowały wcześniej czynniki ryzyka chorób układu krążenia (patrz punkt 4.8).
U pacjentów chorych na nowotwory złośliwe często stwierdza się osłabienie czynności układu odpornościowego. W związku z tym nie zaleca się jednoczesnego podawania szczepionek żywych atenuowanych (patrz punkty 4.3 i 4.5).
Pemetreksed może uszkadzać materiał genetyczny. Zaleca się, aby mężczyźni dojrzali płciowo nie decydowali się na poczęcie dziecka podczas leczenia i w okresie 3 miesięcy po jego zakończeniu. Zaleca się stosowanie środków antykoncepcyjnych lub wstrzemięźliwość płciową. Ze względu na możliwość wywołania przez pemetreksed trwałej niepłodności zaleca się, by przed rozpoczęciem leczenia mężczyźni zwrócili się o poradę dotyczącą kriokonserwacji nasienia.
Kobiety w wieku rozrodczym muszą stosować skuteczne metody antykoncepcji w okresie leczenia
pemetreksedem i przez 6 miesięcy po zakończeniu leczenia (patrz punkt 4.6).
U pacjentów poddawanych radioterapii przed, w trakcie lub po stosowaniu pemetreksedu zgłaszano przypadki zapalenia płuc po napromienianiu. Należy zwrócić szczególną uwagę podczas leczenia tych pacjentów oraz zachować ostrożność w przypadku stosowania innych środków promieniouwrażliwiających.
U pacjentów, którzy w ciągu poprzedzających tygodni lub lat poddawani byli radioterapii zgłaszano przypadki nawrotu objawów popromiennych.
Produkt leczniczy zawiera 96,6 mg sodu na dawkę [500 mg/m2 powierzchni ciała (na podstawie przeciętnej BSA (ang. Body Surface Area – powierzchnia ciała) 1,79 m2)], co odpowiada 4,8% zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych.
Pemetreksed jest wydalany głównie w postaci niezmienionej w wyniku wydzielania w kanalikach nerkowych, a w mniejszym stopniu w wyniku przesączania kłębuszkowego. Równoczesne stosowanie leków nefrotoksycznych (np. antybiotyków aminoglikozydowych, diuretyków pętlowych, związków platyny, cyklosporyny) może potencjalnie prowadzić do opóźnionego oczyszczania organizmu
z pemetreksedu. Należy zachować ostrożność, stosując pemetreksed z lekami z wyżej wymienionych grup. Jeżeli zachodzi taka konieczność, należy ściśle kontrolować klirens kreatyniny.
Równoczesne stosowanie substancji, które również są wydzielane w kanalikach nerkowych (np. probenecydu, penicyliny) może potencjalnie prowadzić do opóźnionego usuwania pemetreksedu
z organizmu. Należy zachować ostrożność, stosując pemetreksed z takimi lekami. Jeżeli zachodzi taka konieczność, należy ściśle kontrolować klirens kreatyniny.
U pacjentów z prawidłową czynnością nerek (klirens kreatyniny ≥ 80 ml/min), stosowanie dużych dawek niesteroidowych leków przeciwzapalnych (NLPZ, takich jak ibuprofen > 1600 mg na dobę) oraz kwasu acetylosalicylowego w większych dawkach (≥ 1,3 g na dobę) może spowodować zmniejszenie wydalania pemetreksedu, a w konsekwencji zwiększenie częstości występowania działań niepożądanych. Należy zwrócić szczególną uwagę w czasie stosowania dużych dawek NLPZ oraz kwasu acetylosalicylowego równocześnie z pemetreksedem u pacjentów z prawidłową czynnością nerek (klirens kreatyniny ≥ 80 ml/min).
U pacjentów z łagodną lub umiarkowaną niewydolnością nerek (klirens kreatyniny od 45 do
79 ml/min), należy unikać równoczesnego podawania pemetreksedu z NLPZ (np. ibuprofenem) lub z kwasem acetylosalicylowym w dużych dawkach 2 dni przed podaniem pemetreksedu, w dniu podania i przez 2 dni po podaniu pemetreksedu (patrz punkt 4.4).
Ze względu na brak danych dotyczących możliwych interakcji z NLPZ o dłuższym okresie półtrwania, takich, jak piroksykam lub rofekoksyb, u pacjentów z łagodną lub umiarkowaną niewydolnością nerek należy przerwać stosowanie tych leków w okresie co najmniej 5 dni przed podaniem pemetreksedu,
w dzień podania leku i co najmniej 2 dni po podaniu pemetreksedu (patrz punkt 4.4). Jeżeli konieczne jest jednoczesne stosowanie NLPZ, należy uważnie kontrolować stan pacjentów w celu wykrycia
objawów toksyczności, szczególnie zahamowania czynności szpiku i toksyczności na przewód pokarmowy.
Metabolizm pemetreksedu w wątrobie zachodzi w ograniczonym stopniu. Z badań in vitro
z zastosowaniem mikrosomów z ludzkiej wątroby wynika, że nie należy oczekiwać klinicznie istotnego hamowania przez pemetreksed metabolizmu leków z udziałem izoenzymów CYP3A, CYP2D6, CYP2C9 i CYP1A2.
Interakcje typowe dla wszystkich leków cytotoksycznych
Ze względu na zwiększone zagrożenie zakrzepami u pacjentów chorych na nowotwory złośliwe, często stosuje się leczenie przeciwzakrzepowe. Jeżeli podjęto decyzję o podawaniu pacjentowi leków przeciwzakrzepowych, ze względu na dużą zmienność osobniczą w czynności układu krzepnięcia,
u tego samego pacjenta, w różnych fazach choroby i możliwość interakcji doustnych leków przeciwzakrzepowych i chemioterapeutyków przeciwnowotworowych, konieczne jest częstsze oznaczanie wskaźnika INR (ang. International Normalised Ratio).
Jednoczesne stosowanie przeciwwskazane: szczepionka przeciwko żółtej gorączce - możliwość wystąpienia uogólnionego odczynu poszczepiennego prowadzącego do zgonu (patrz punkt 4.3).
Nie zaleca się jednoczesnego stosowania z: szczepionkami żywymi atenuowanymi (z wyjątkiem szczepionki przeciwko żółtej gorączce, której jednoczesne stosowanie jest przeciwwskazane) - możliwość wystąpienia ogólnoustrojowego odczynu, mogącego prowadzić do zgonu. Ryzyko wystąpienia odczynu jest większe u pacjentów ze zmniejszoną odpornością, spowodowaną chorobą podstawową. Należy stosować szczepionki inaktywowane, jeżeli takie istnieją (poliomyelitis) (patrz punkt 4.4).
Kobiety w wieku rozrodczym/Antykoncepcja u mężczyzn i kobiet
Pemetreksed może uszkadzać materiał genetyczny. Kobiety w wieku rozrodczym muszą stosować skuteczne metody antykoncepcji w okresie leczenia pemetreksedem i przez 6 miesięcy po zakończeniu leczenia.
Dojrzali płciowo mężczyźni powinni stosować skuteczne środki antykoncepcyjne i nie powinni decydować się na poczęcie dziecka podczas leczenia i w okresie 3 miesięcy po jego zakończeniu.
Ciąża
Brak danych dotyczących stosowania pemetreksedu u kobiet w ciąży, jednak należy podejrzewać, że pemetreksed stosowany w okresie ciąży, podobnie jak inne antymetabolity, powoduje ciężkie uszkodzenia płodu. W badaniach na zwierzętach wykazano szkodliwy wpływ leku na rozrodczość (patrz punkt 5.3). Pemetreksedu nie należy stosować w okresie ciąży, chyba że, po starannym rozważeniu konieczności leczenia matki i ryzyka dla płodu (patrz punkt 4.4).
Karmienie piersią
Nie wiadomo, czy pemetreksed przenika do mleka kobiecego. Nie można wykluczyć występowania objawów niepożądanych u dzieci karmionych mlekiem matki, leczonej pemetreksedem. W okresie leczenia pemetreksedem należy zaprzestać karmienia piersią (patrz punkt 4.3).
Płodność
Ze względu na możliwość wywołania przez pemetreksed trwałej niepłodności zaleca się, by przed rozpoczęciem leczenia mężczyźni zwrócili się o poradę do ośrodka specjalizującego się
w kriokonserwacji nasienia.
Nie przeprowadzano badań dotyczących wpływu leku na zdolność prowadzenia pojazdów mechanicznych i obsługiwania maszyn. Donoszono o występowaniu uczucia zmęczenia u osób
leczonych pemetreksedem. Należy ostrzec pacjentów, by nie prowadzili pojazdów i nie obsługiwali maszyn, jeżeli wystąpi ten objaw.
Podsumowanie profilu bezpieczeństwa
Do najczęściej zgłaszanych działań niepożądanych podczas stosowania pemetreksedu w monoterapii i w leczeniu skojarzonym należą: zahamowanie czynności szpiku objawiające się niedokrwistością,
neutropenią, leukopenią, trombocytopenią; objawy toksyczności w obrębie układu pokarmowego takie jak: jadłowstręt, nudności, wymioty, biegunka, zaparcie, zapalenie gardła, zapalenie błon śluzowych
i zapalenie jamy ustnej. Do innych działań niepożądanych należą: nefrotoksyczność, zwiększona aktywność aminotransferaz, łysienie, zmęczenie, odwodnienie, wysypka, zakażenie lub sepsa
i neuropatia. Rzadko obserwowane objawy to: zespół Stevensa-Johnsona i martwica rozpływna naskórka.
Tabelaryczne zestawienie działań niepożądanych
W tabeli 4. wymieniono niepożądane działania niezależnie od związku przyczynowo-skutkowego obserwowane podczas stosowania pemetreksedu w monoterapii lub w skojarzeniu z cisplatyną,
w głównych badaniach rejestracyjnych (JMCH, JMEI, JMBD, JMEN i PARAMOUNT) oraz po wprowadzeniu produktu do obrotu.
Niepożądane działania leku wymieniono zgodnie z klasyfikacją układów i narządów MedDRA. Częstość występowania określono w następujących kategoriach: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1 000), bardzo rzadko (<1/10 000), częstość nieznana (nie może być określona na podstawie dostępnych danych).
Tabela 4. Częstość występowania działań niepożądanych wszystkich stopni niezależnie od związku przyczynowo-skutkowego, obserwowanych w głównych badaniach rejestracyjnych: JMEI (pemetreksed w porównaniu z docetakselem), JMDB (pemetreksed i cisplatyna w porównaniu z gemcytabiną i cisplatyną), JMCH (pemetreksed łącznie z cisplatyną w porównaniu z cisplatyną), JMEN i PARAMOUNT (pemetreksed łącznie z najlepszym leczeniem objawowym w porównaniu z placebo stosowanym łącznie z najlepszym leczeniem objawowym) oraz po wprowadzeniu produktu do obrotu.
Klasyfikacja układów i narządów (MedDRA) | Bardzo często | Często | Niezbyt często | Rzadko | Bardzo rzadko | Częstość nieznana |
Zakażenia i zarażenia pasożytnicze | Zakażeniea Zapalenie gardła | Sepsab | Zapalenie skóry i tkanki podskórnej | |||
Zaburzenia hematologiczne (krwi i układu chłonnego) | Neutropenia Leukopenia Zmniejszenie stężenia hemoglobiny | Gorączka neutropeniczna Zmniejszenie liczby płytek krwi | Pancytopenia | Niedokrwistość hemolityczna o podłożu immunologicz- nym | ||
Zaburzenia układu immunologicznego | Nadwrażliwość | Wstrząs anafilaktyczny | ||||
Zaburzenia metabolizmu i odżywiania | Odwodnienie | |||||
Zaburzenia układu nerwowego | Zaburzenia smaku Neuropatia ruchowa Neuropatia nerwów | Udar naczyniowo- mózgowy Udar niedokrwienny Krwotok |
czuciowych Zawroty głowy | wewnątrzczasz- kowy | |||||
Uszkodzenia i zaburzenia funkcji gałki ocznej | Zapalenie spojówek Suchość oczu Wzmożone łzawienie Suche zapalenie rogówki i spojówki Obrzęk powiek, Choroba warstwy powierzchniowej gałki ocznej | |||||
Zaburzenia funkcji serca | Niewydolność serca Zaburzenia rytmu serca | Dusznica bolesna Zawał mięśnia sercowego, Choroba niedokrwienna serca Nadkomorowe zaburzenia rytmu serca | ||||
Zaburzenia układu naczyniowego | Niedokrwienie obwodowec | |||||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Zatorowość płucna, Śródmiąższowe zapalenie płucbd | |||||
Zaburzenia funkcji żołądka i jelit | Zapalenie jamy ustnej Jadłowstręt Wymioty Biegunka Nudności | Niestrawność Zaparcie Ból brzucha | Krwotok z odbytnicy, Krwotok z przewodu pokarmowego Perforacja jelit Zapalenie błony śluzowej przełyku Zapalenie okrężnicye | |||
Zaburzenia funkcji wątroby i dróg żółciowych | Zwiększenie aktywności aminotransfe- razy alaninowej Zwiększenie aktywności aminotransferazy asparaginiano- wej | Zapalenie wątroby | ||||
Powikłania dermatologiczne (dotyczące skóry i tkanki podskórnej) | Wysypka Łuszczenie skóry | Hiperpigmenta- cja Świąd Rumień, wielopostaciowy Łysienie Pokrzywka | Rumień | Zespół Stevensa- Johnsonab Toksyczna martwica rozpływna naskórkab Pemfigoid Pęcherzowe zapalenie skóry Nabyte pęcherzowe |
oddzielanie się naskórka Obrzęk rumieniowyf Rzekome zapalenie tkanki podskórnej Zapalenie skóry Wyprysk Świerzbiączka | ||||||
Zaburzenia nerek i dróg moczowych | Zmniejszenie klirensu kreatyniny Zwiększenie stężenia kreatyniny we krwi | Niewydolność nerek Zmniejszenie wartości wskaźnika filtracji kłębuszkowej | Nefrogenna moczówka prosta Martwica cewek nerkowych | |||
Zaburzenia ogólne i stany w miejscu podania | Uczucie zmęczenia | Gorączka Ból Obrzęk Ból w klatce piersiowej Zapalenie błon śluzowych | ||||
Badania diagnostyczne | Zwiększenie aktywności gamma- glutamylotrans- peptydazy | |||||
Urazy, zatrucia i powikłania po zabiegach | Popromienne zapalenie przełyku Popromienne zapalenie płuc | Nawroty objawów popromiennych |
a z towarzyszącą neutropenią i bez neutropenii
b śmiertelne w niektórych przypadkach
c czasami prowadzące do martwicy kończyny
d z niewydolnością oddechową
e obserwowane tylko w przypadku stosowania w skojarzeniu z cisplatyną
f głównie kończyn dolnych
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych.
Al. Jerozolimskie 181 C, 02-222 Warszawa
Tel.: +48 22 49 21 301, Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Objawy przedawkowania, o jakich donoszono, obejmują: neutropenię, niedokrwistość, trombocytopenię, zapalenie błon śluzowych, polineuropatię czuciową i wysypkę. Prawdopodobne powikłania przedawkowania leku to: mielosupresja, objawiająca się neutropenią, trombocytopenią i niedokrwistością. Mogą także wystąpić zakażenia z gorączką lub bez gorączki, biegunka i (lub)
zapalenie błon śluzowych. Jeżeli podejrzewane jest przedawkowanie leku należy monitorować stan pacjenta, wykonując badania morfologii krwi i w razie potrzeby wdrożyć leczenie podtrzymujące. Jeżeli doszło do przedawkowania pemetreksedu, należy rozważyć zastosowanie folanu wapnia lub kwasu foliowego.
5.1. Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: lek przeciwnowotworowy, analogi kwasu foliowego, kod ATC: L01BA04.
Pemetreksed to lek przeciwnowotworowy o wielokierunkowym działaniu, antagonista kwasu foliowego zaburzający podstawowe procesy metaboliczne wykorzystujące folany niezbędne dla podziału komórek.
W badaniach in vitro wykazano, że wielokierunkowe działanie pemetreksedu polega na hamowaniu syntazy tymidylowej (TS), reduktazy dihydrofolanowej (DHFR) i formylotransferazy rybonukleotydu glicynamidowego (GARFT), czyli podstawowych enzymów wykorzystujących folany, uczestniczących w biosyntezie de novo nukleotydów tymidynowych i purynowych.
Transport pemetreksedu do wnętrza komórek odbywa się z udziałem systemu nośnika zredukowanych folianów i białka błonowego wiążącego foliany. W komórce pemetreksed jest szybko i wydajnie przekształcany w poliglutaminiany przez enzym syntetazę folylpoliglutaminianową. Poliglutaminiany pozostają we wnętrzu komórki i wykazują jeszcze silniejsze działanie hamujące TS i GARFT. Proces poliglutaminizacji, którego intensywność zależy od czasu i stężenia, zachodzi w komórkach nowotworowych oraz w mniejszym stopniu, w prawidłowych tkankach organizmu. Metabolity powstające w wyniku poliglutaminizacji charakteryzują się przedłużonym okresem półtrwania wewnątrz komórki, co warunkuje dłuższe działanie leku w komórkach nowotworów złośliwych.
Skuteczność kliniczna
Złośliwy międzybłoniak opłucnej
Badanie EMPHACIS (wieloośrodkowe, randomizowane badanie III fazy z pojedynczą ślepą próbą, porównujące stosowanie pemetreksedu w skojarzeniu z cisplatyną i cisplatyny u nieleczonych wcześniej chemioterapią pacjentów ze złośliwym międzybłoniakiem opłucnej, wykazało, że mediana czasu przeżycia pacjentów leczonych pemetreksedem i cisplatyną była o 2,8 miesiąca większa (różnica istotna statystycznie), w porównaniu z osobami leczonymi tylko cisplatyną.
W okresie trwania badania stosowano suplementację małymi dawkami kwasu foliowego i witaminy B12 w celu ograniczenia objawów toksyczności. W głównej analizie wykorzystano dane o wszystkich pacjentach przydzielonych losowo do jednej z podgrup, którzy otrzymali badany lek (pacjenci randomizowani i leczeni). Do analizy podgrup wybrano dane o pacjentach, którzy otrzymywali kwas foliowy i witaminę B12 przez cały okres leczenia ocenianego w badaniu (pacjenci z pełną suplementacją). Wyniki tych analiz skuteczności przedstawiono w poniższej tabeli:
Skuteczność schematu pemetreksed + cisplatyna w porównaniu z monoterapią cisplatyną
w leczeniu złośliwego międzybłoniaka opłucnej
Pacjenci randomizowani i leczeni | Pacjenci z pełną suplementacją | |||
Parametr skuteczności | Pemetreksed/ Cisplatyna (N = 226) | Cisplatyna (N = 222) | Pemetreksed / Cisplatyna (N = 168) | Cisplatyna (N = 163) |
Mediana czasu przeżycia (miesiące)(95% CI) | 12,1 (10,0-14,4) | 9,3 (7,8-10,7) | 13,3 (11,4-14,9) | 10,0 (8,4-11,9) |
wartość p* w teście Log Rank | 0,020 | 0,051 | ||
Mediana czasu do progresji choroby (miesiące) (95% CI) | 5,7 (4,9-6,5) | 3,9 (2,8-4,4) | 6,1 (5,3-7,0) | 3,9 (2,8-4,5) |
wartość p* w teście Log Rank | 0,001 | 0,008 | ||
Czas do niepowodzenia leczenia (miesiące)(95% CI) | 4,5 (3,9-4,9) | 2,7 (2,1-2,9) | 4,7 (4,3-5,6) | 2,7 (2,2-3,1) |
wartość p* w teście Log Rank | 0,001 | 0,001 | ||
Całkowity odsetek odpowiedzi na leczenie**(95% CI) | 41,3% (34,8-48,1) | 16,7% (12,0-22,2) | 45,5% (37,8-53,4) | 19,6% (13,8-26,6) |
wartość p* w teście Fisher Exact | <0,001 | <0,001 | ||
Skróty: CI – przedział ufności * wartość p odnosi się do porównań pomiędzy podgrupami ** w podgrupie pemetreksed +cisplatyna: pacjenci randomizowani i leczeni (N = 225), pacjenci z pełną suplementacją (N = 167) | ||||
Stosując Skalę Objawów Raka Płuca (ang. Lung Cancer Symptom Scale) wykazano istotną statystycznie poprawę w odniesieniu do klinicznie istotnych objawów (ból i duszność) złośliwego międzybłoniaka opłucnej w grupie leczonej schematem pemetreksed + cisplatyna (212 pacjentów) w porównaniu z leczonymi cisplatyną w monoterapii (218 pacjentów). Stwierdzono także istotne statystycznie różnice parametrów czynności płuc. Obserwowane różnice między grupami były wynikiem poprawy stanu czynnościowego płuc w grupie leczonej schematem pemetreksed + cisplatyna, jak również z upływem czasu pogorszenia czynności płuc w grupie kontrolnej.
Istnieje niewiele danych na temat pacjentów ze złośliwym międzybłoniakiem opłucnej leczonych wyłącznie pemetreksedem. Badano stosowanie tego leku w dawce 500 mg/m2 pc. w monoterapii
i u 64 nie poddawanych wcześniej chemioterapii pacjentów ze złośliwym międzybłoniakiem opłucnej. Całkowity odsetek odpowiedzi na leczenie wyniósł 14,1%.
Niedrobnokomórkowy rak płuca, leczenie II rzutu
W wieloośrodkowym, randomizowanym, otwartym, badaniu klinicznym III fazy porównującym stosowanie pemetreksedu i docetakselu u pacjentów z miejscowo zaawansowanym lub dającym przerzuty niedrobnokomórkowym rakiem płuca wykazano mediany czasu przeżycia wynoszące 8,3 miesiąca w grupie leczonej pemetreksedem (populacja wyodrębniona zgodnie z zaplanowanym leczeniem, ang. Intent-To-Treat, ITT, n = 283) i 7,9 miesiąca w grupie leczonej docetakselem (populacja ITT, n = 288). W schemacie leczenia pierwszego rzutu nie stosowano pemetreksedu.
Analiza zależności między wynikami leczenia, określonymi jako czas całkowitego przeżycia (ang. Over all survival– OS), a typem histologicznym niedrobnokomórkowego raka płuca wykazała przewagę pemetreksedu nad docetakselem u pacjentów z niedrobnokomórkowym rakiem płuca
o histologii innej niż w przeważającym stopniu płaskonabłonkowa (n = 399, 9,3 vs.8,0 miesięcy, skorygowany współczynnik ryzyka (HR) = 0,78; 95% CI = 0,61-1,00, p = 0,047) i przewagę docetakselu u pacjentów z niedrobnokomórkowym rakiem płuca o histologii płaskonabłonkowej
(n = 172, 6,2 vs. 7,4 miesięcy, skorygowany współczynnik ryzyka (HR) = 1,56;95% CI = 1,08-2,26,
p = 0,018). W obrębie poszczególnych podgrup histologicznych nie obserwowano istotnych klinicznie różnic w profilu bezpieczeństwa pemetreksedu.
Ograniczone dane kliniczne pochodzące z pojedynczego, randomizowanego, kontrolowanego badania III fazy wskazują, że skuteczność pemetreksedu (mierzona jako czas całkowitego przeżycia – OS
i czas przeżycia wolny od progresji choroby nowotworowej - PFS) u pacjentów leczonych wcześniej
docetakselem (n=41) jest zbliżona do skuteczności obserwowanej u pacjentów, którzy nie byli wcześniej leczeni docetakselem (n=540).
Skuteczność pemetreksedu i docetakselu w niedrobnokomórkowym raku płuca –
populacja wyodrębniona zgodnie z zaplanowanym leczeniem (ITT)
Pemetreksed | Docetaksel | |
Przeżycie (miesiące) | (N = 283) 8,3 (7,0-9,4) | (N = 288) 7,9 (6,3-9,2) |
0,99 (0,82-1,20) 0,226 | ||
Czas przeżycia wolny od progresji choroby nowotworowej (miesiące) | (N = 283) 2,9 | (N = 288) 2,9 |
0,97 (0,82-1,16) | ||
Czas do niepowodzenia leczenia (miesiące) | (N = 283) 2,3 | (N = 288) 2,1 |
0,84 (0,71-0,997) | ||
Odpowiedź na leczenie (n: zakwalifikowani do analizy odpowiedzi na leczenie) | (N = 264) 9,1 (5,9-13,2) 45,8 | (N = 274) 8,8 (5,7-12,8) 46,4 |
Skróty: CI = przedział ufności, HR – wskaźnik ryzyka, ITT- populacja wyodrębniona zgodnie z zaplanowanym leczeniem n = całkowita liczebność populacji | ||
Niedrobnokomórkowy rak płuca, leczenie I wyboru
W wieloośrodkowym, randomizowanym, otwartym badaniu III fazy z udziałem pacjentów
z niedrobnokomórkowym rakiem płuca w stadium miejscowo zaawansowanym lub z przerzutami (stopień zaawansowania IIIB lub IV) niepoddawanych wcześniej chemioterapii, porównywano skuteczność pemetreksedu w skojarzeniu z cisplatyną oraz gemcytabiny w skojarzeniu z cisplatyną. W przypadku stosowania pemetreksedu w skojarzeniu z cisplatyną (populacja wyodrębniona zgodnie z zaplanowanym leczeniem, ang. Intent-To-Treat, ITT, n = 862) osiągnięto pierwszorzędowy punkt końcowy i uzyskano podobną skuteczność kliniczną, jak w przypadku stosowania gemcytabiny
w skojarzeniu z cisplatyną (ITT n = 863), w zakresie OS (skorygowany współczynnik ryzyka 0,94; 95% CI = 0,84-1,05). Stopień sprawności wszystkich pacjentów biorących udział w badaniu oceniano na 0 lub 1 w skali ECOG.
Pierwotną analizę skuteczności oparto na wynikach uzyskanych w populacji ITT. Analizy wrażliwości dla głównych punktów końcowych związanych ze skutecznością oceniano też w populacji wyodrębnionej zgodnie z protokołem (ang. Protocol Qualified- PQ). Analizy skuteczności oparte na wynikach uzyskanych w populacji PQ są zgodne z wynikami analizami dla populacji ITT
i potwierdzają nie mniejszą skuteczność (ang. non-inferiority) skojarzonej terapii pemetreksedem
z cisplatyną w porównaniu ze skojarzoną terapią gemcytabiną z cisplatyną.
Czas przeżycia wolny od progresji choroby nowotworowej (ang. Progression free survival– PFS) i całkowity odsetek odpowiedzi na leczenie (ORR) były podobne w obydwu ramionach badania.
Mediana PFS wynosiła 4,8 miesiąca w przypadku skojarzonego stosowania pemetreksedu z cisplatyną i 5,1 miesiąca w przypadku skojarzonego stosowania gemcytabiny z cisplatyną (skorygowany współczynnik ryzyka 1,04; 95% CI = 0,94-1,15). Całkowity odsetek odpowiedzi na leczenie wynosił 30,6% (95% CI = 27,3- 33,9) w przypadku skojarzonego stosowania pemetreksedu z cisplatyną
i 28,2% (95% CI = 25,0-31,4) w przypadku skojarzonego stosowania gemcytabiny z cisplatyną. Wyniki PFS zostały po części potwierdzone niezależną oceną (do kontroli w sposób losowy wybrano 400/1725 pacjentów).
Analiza wpływu typu histologicznego niedrobnokomórkowego raka płuca na OS wykazała istotne klinicznie różnice czasu przeżycia w zależności od typu histologicznego, patrz tabela poniżej.
Skuteczność pemetreksedu w skojarzeniu z cisplatyną w porównaniu ze skojarzoną terapią gemcytabiną z cisplatyną w leczeniu pierwszego wyboru niedrobnokomórkowego raka
płuca –populacja ITT (ang. intent-to-treat) i podgrupy histologiczne
Populacja ITT | Mediana czasu przeżycia całkowitego w | Skorygowany | Wartość p | |||
i podział na | miesiącach (95% CI) | wskaźnik | ||||
podgrupy | Pemetreksed + | Gemcytabina+ | ryzyka (HR) | |||
histologiczne | Cisplatyna | Cisplatyna | (95% CI) | |||
Populacja ITT | 10,3 | N = 862 | 10,3 | N = 863 | 0,94a | 0,259 |
(N = 1725) | (9,8 – 11,2) | (9,6 – 10,9) | (0,84 – 1,05) | |||
Gruczołowy | 12,6 | N = 436 | 10,9 | N = 411 | 0,84 | 0,033 |
(N=847) | (10,7 – 13,6) | (10,2 –11,9) | (0,71–0,99) | |||
Wielkokomórk | 10,4 | N = 76 | 6,7 | N = 77 | 0,67 | 0,027 |
owy (N=153) | (8,6 – 14,1) | (5,5 – 9,0) | (0,48–0,96) | |||
Inne (N=252) | 8,6 | N = 106 | 9,2 | N = 146 | 1,08 | 0,586 |
(6,8 – 10,2) | (8,1 – 10,6) | (0,81–1,45) | ||||
Płaskonabłonko | 9,4 | N = 244 | 10,8 | N = 229 | 1,23 | 0,050 |
wy (N=473) | (8,4 – 10,2) | (9,5 – 12,1) | (1,00–1,51) | |||
Skróty: CI = przedział ufności, ITT - populacja wyodrębniona zgodnie z zaplanowanym leczeniem, N= całkowita liczebność populacji a Statystycznie istotne, aby wykazać nie mniejszą skuteczność, przy całkowitym przedziale ufności dla wskaźnika ryzyka znacznie poniżej przyjętej granicy 1,17645 (p <0,001). | ||||||
Krzywe przeżywalności Kaplana Meiera w zależności od typu histologicznego
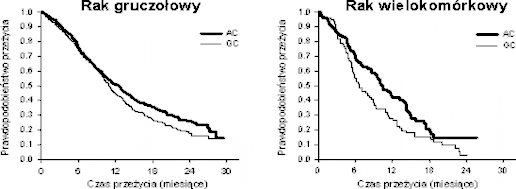
W obrębie poszczególnych podgrup histologicznych nie obserwowano istotnych klinicznie różnic w profilu bezpieczeństwa skojarzonej terapii pemetreksedem z cisplatyną.
Pacjenci leczeni pemetreksedem w skojarzeniu z cisplatyną rzadziej wymagali transfuzji
(16,4%w porównaniu z 28,9%, p<0,001), przetoczenia krwinek czerwonych (16,1% w porównaniu
z 27,3%, p< 0,001) i płytek krwi (1,8% w porównaniu z 4,5%, p=0,002). Pacjenci wymagali podania mniejszej ilości erytropoetyny lub darbepoetyny (10,4% w porównaniu z 18,1%, p<0,001),
G-CSF/GM-CSF (3,1% w porównaniu z 6,1%, p=0,004), i produktów zawierających żelazo (4,3% w porównaniu z 7,0%, p=0,021).
Niedrobnokomórkowy rak płuca, leczenie podtrzymujące
JMEN
W wieloośrodkowym randomizowanym, kontrolowanym placebo badaniu III fazy, z podwójnie ślepą próbą (JMEN) porównywano skuteczność i bezpieczeństwo leczenia podtrzymującego pemetreksedem, stosowanym jednocześnie z najlepszym leczeniem wspomagającym (BSC, ang. Best supportivecare) (n = 441) z terapią polegającą na podawaniu placebo, jednocześnie z najlepszym
leczeniem wspomagającym (n = 222) u pacjentów z miejscowo zaawansowanym (stadium zaawansowania IIIB) lub z przerzutami (stadium zaawansowania IV) niedrobnokomórkowym rakiem płuca, u których nie stwierdzono progresji choroby po 4 cyklach terapii dwulekowej pierwszego wyboru, zawierającej cisplatynę lub karboplatynę w skojarzeniu z gemcytabiną, paklitakselem lub docetakselem. W dwulekowym schemacie leczenia pierwszego wyboru nie stosowano pemetreksedu. Stopień sprawności wszystkich pacjentów biorących udział w badaniu oceniono na 0 lub 1 w skali ECOG. Pacjenci otrzymywali leczenie podtrzymujące do czasu stwierdzenia progresji choroby.
Skuteczność i bezpieczeństwo leczenia oceniano od momentu randomizacji po ukończeniu leczenia pierwszego wyboru. Mediana liczby cykli leczenia wyniosła odpowiednio 5 dla leczenia podtrzymującego pemetreksedem oraz 3,5 dla placebo. W sumie 213 pacjentów (48,3%) ukończyło
≥ 6 cykli leczenia, a 103 pacjentów ogółem (23,4%) ukończyło ≥ 10 cykli leczenia pemetreksedem.
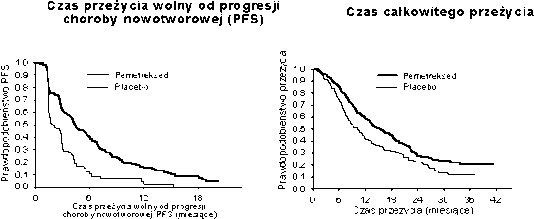
Uzyskano pierwszorzędowy punkt końcowy badania i wykazano statystycznie istotne wydłużenie PFS w grupie leczonej pemetreksedem w porównaniu z grupą placebo (n = 581, populacja analizowana niezależnie; mediana wyniosła odpowiednio 4,0 miesiące i 2,0 miesiące) (współczynnik ryzyka = 0,60; 95% CI = 0,49-0,73; p < 0,00001). Niezależna analiza wyników badań obrazowych pacjentów potwierdziła wyniki PFS uzyskane na podstawie oceny dokonanej przez badaczy. Mediana OS w całej populacji (n = 663) wyniosła 13,4 miesiąca w grupie leczonej pemetreksedem i 10,6 miesiąca w grupie placebo, współczynnik ryzyka = 0,79 (95% CI = 0,65-0,95; p = 0,01192).
Podobnie jak w przypadku innych badań z zastosowaniem pemetreksedu, w badaniu JMEN obserwowano różnice skuteczności leczenia zależne od typu histologicznego niedrobnokomórkowego raka płuca. W przypadku pacjentów z niedrobnokomórkowym rakiem płuca o budowie histologicznej innej niż w przeważającym stopniu płaskonabłonkowa (n= 430, populacja analizowana niezależnie), mediana PFS wyniosła 4,4 miesiąca w grupie leczonej pemetreksedem i 1,8 miesiąca w grupie placebo, współczynnik ryzyka = 0,47; 95% CI = 0,37-0,60; p = 0,00001. Mediana OS u pacjentów
z niedrobnokomórkowym rakiem płuca o histologii innej niż w przeważającym stopniu płaskonabłonkowa (n = 481), wyniosła 15,5 miesiąca w grupie leczonej pemetreksedem
i 10,3 miesiąca w grupie placebo (współczynnik ryzyka = 0,70; 95% CI = 0,56-0,88; p = 0,002). Mediana OS łącznie z okresem leczenia pierwszego wyboru u pacjentów z niedrobnokomórkowym rakiem płuca o histologii innej niż w przeważającym stopniu płaskonabłonkowa, wyniosła
18,6 miesiąca w grupie leczonej pemetreksedem i 13,6 miesiąca w grupie placebo (współczynnik ryzyka = 0,71; 95% CI = 0,56-0,88; p = 0,002).
Wyniki oceny PFS i OS pacjentów z niedrobnokomórkowym rakiem płuca o histologii płaskonabłonkowej wskazują na brak przewagi leczenia pemetreksedem w porównaniu z placebo.
W obrębie poszczególnych podgrup histologicznych nie obserwowano istotnych klinicznie różnic w profilu bezpieczeństwa pemetreksedu.
JMEN: Krzywe Kaplana Meiera czasu przeżycia wolnego od progresji choroby nowotworowej i czasu przeżycia całkowitego pacjentów z niedrobnokomórkowym rakiem płuca o histologii innej, niż w przeważającym stopniu płaskonabłonkowa, leczonych pemetreksedem oraz pacjentów otrzymujących placebo
PARAMOUNT
W wieloośrodkowym, randomizowanym, kontrolowanym placebo badaniu III fazy, z podwójnie ślepą próbą (PARAMOUNT) porównywano skuteczność i bezpieczeństwo leczenia podtrzymującego pemetreksedem w ramach kontynuacji leczenia tym produktem, stosowanym w skojarzeniu
z najlepszą terapią wspomagającą (BSC, ang. best supportive care) (n = 359), z terapią polegającą na podawaniu placebo w skojarzeniu z najlepszą terapią wspomagającą (n = 180) u pacjentów
z miejscowo zaawansowanym (stadium zaawansowania IIIB) lub uogólnionym (stadium zaawansowania IV) niedrobnokomórkowym rakiem płuca o budowie histologicznej innej, niż w przeważającym stopniu płaskonabłonkowa, u których nie stwierdzono progresji choroby po 4 cyklach terapii pierwszego wyboru pemetreksedem w skojarzeniu z cisplatyną. Spośród 939 pacjentów leczonych w fazie indukcji pemetreksedem w skojarzeniu z cisplatyną, 539 losowo
przydzielono do grupy otrzymującej leczenie podtrzymujące polegające na podawaniu pemetreksedu lub placebo. Wśród losowo przydzielonych pacjentów, u 44,9% obserwowano całkowitą lub częściową odpowiedź na leczenie pierwszego wyboru (w fazie indukcji) z zastosowaniem pemetreksedu w skojarzeniu z cisplatyną, a u 51,9% stwierdzono stabilizację choroby. Wymagano, aby stopień sprawności randomizowanych pacjentów wynosił 0 lub 1 w skali ECOG. Mediana czasu od rozpoczęcia leczenia pierwszego wyboru (indukcja) pemetreksedem w skojarzeniu z cisplatyną do rozpoczęcia leczenia podtrzymującego wynosiła 2,96 miesiąca w grupie leczonej pemetreksedem i w grupie placebo. Losowo przydzieleni pacjenci otrzymywali leczenie podtrzymujące do czasu progresji choroby. Skuteczność i bezpieczeństwo oceniano od czasu randomizacji po zakończeniu leczenia pierwszego wyboru. Mediana liczby cykli leczenia, które otrzymali pacjenci w ramach leczenia podtrzymującego pemetreksedem wyniosła odpowiednio 4 dla grupy otrzymującej pemetreksed oraz 4 dla grupy otrzymującej placebo. Łącznie 169 pacjentów (47,1%) otrzymało ≥ 6 cykle leczenia podtrzymującego pemetreksedem, co po uwzględnieniu fazy indukcji odpowiada łącznie co najmniej 10 cyklom leczenia pemetreksedem.
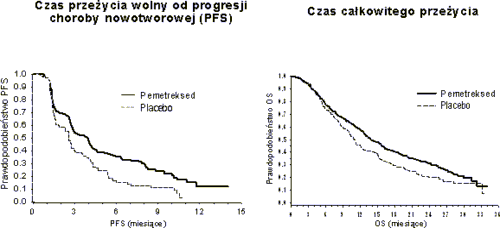
Osiągnięto pierwszorzędowy punkt końcowy badania i wykazano statystycznie istotne wydłużenie PFS w grupie leczonej pemetreksedem w porównaniu z grupą placebo (n = 472, niezależna analiza populacji; mediana wyniosła odpowiednio 3,9 miesiąca i 2,6 miesiąca) (współczynnik ryzyka = 0,64; 95% CI = 0,51-0,81; p = 0,0002). Niezależna analiza wyników badań obrazowych wykonanych
u pacjentów potwierdziła dokonaną przez badaczy ocenę PFS. Mediana PFS pacjentów zrandomizowanych do leczenia podtrzymującego oceniona przez badaczy od rozpoczęcia leczenia pierwszego wyboru pemetreksedem w skojarzeniu z cisplatyną wyniosła 6,9 miesiąca w grupie leczonej pemetreksedem i 5,6 miesiąca w grupie placebo (współczynnik ryzyka = 0,59 95%CI = 0,47-0,74).
Podczas kontynuacji terapii pemetreksedem po fazie indukcji z zastosowaniem pemetreksedu
w skojarzeniu z cisplatyną (4 cykle) wykazano istotne statystycznie wydłużenie czasu całkowitego przeżycia (ang. Overall survival– OS) w porównaniu z placebo (mediana 13,9 miesiąca vs. 11,0 miesięcy, współczynnik ryzyka = 0,78; 95% CI=0,64-0,96; p=0,0195). W momencie przeprowadzania końcowej analizy całkowitego przeżycia, 28,7% pacjentów z grupy otrzymującej pemetreksed pozostawało przy życiu lub nie było dalej obserwowanych pod kątem przeżycia (ang. lost to
follow-up) i 21,7% pacjentów z grupy otrzymującej placebo. Obiektywne wyniki leczenia pemetreksedem były zgodne wśród badanych podgrup (również w podgrupach uwzględniających stopień zaawansowania choroby, odpowiedź na leczenie pierwszego wyboru, stopień sprawności w skali ECOG, fakt palenia lub niepalenia tytoniu, płeć, typ histologiczny nowotworu oraz wiek)
i podobne do uzyskanych podczas analizy nieskorygowanych wartości OS i PFS. Wskaźnik przeżyć rocznych i dwuletnich w grupie pacjentów otrzymujących pemetreksed wyniósł odpowiednio 58%
i 32%, a w grupie placebo 45% i 21%. Mediana OS, mierzonego od rozpoczęcia leczenia pierwszego wyboru z zastosowaniem pemetreksedu w skojarzeniu z cisplatyną u pacjentów otrzymujących pemetreksed, wyniosła 16,9 miesięcy, a w grupie otrzymującej placebo 14,0 miesięcy (współczynnik ryzyka = 0,78; 95% CI= 0,64-0,96). Odsetek pacjentów, którzy otrzymali dalsze leczenie po zakończeniu udziału w badaniu wyniósł 64,3% w grupie otrzymującej pemetreksed i 71,7% w grupie placebo.
PARAMOUNT: krzywe Kaplana Meiera czasu przeżycia wolnego od progresji choroby nowotworowej (PFS) i czasu całkowitego przeżycia (OS) w przypadku leczenia podtrzymującego pemetreksedem w ramach kontynuacji leczenia tym lekiem w porównaniu z placebo, u chorych na niedrobnokomórkowego raka płuca o histologii innej, niż w przeważającym stopniu płaskonabłonkowa (od czasu randomizacji)
Profil bezpieczeństwa pemetreksedu stosowanego w leczeniu podtrzymującym w badaniach JMEN i PARAMOUT był podobny.
Właściwości farmakokinetyczne pemetreksedu oceniano po leczeniu w monoterapii u 426 pacjentów chorych na różne rodzaje złośliwych guzów litych. Wielkość dawki wahała się od 0,2 do
838 mg/m2 pc. Lek podawano w infuzji dożylnej przez 10 minut. Objętość dystrybucji pemetreksedu w stanie równowagi dynamicznej wynosi 9 l/m2. Z badań in vitro wynika, że stopień wiązania pemetreksedu z białkami osocza krwi wynosi około 81%. Nie stwierdzono znaczącego wpływu stopnia zaburzeń czynności nerek na wiązanie się leku z białkami osocza. Pemetreksed
w ograniczonym stopniu jest metabolizowany w wątrobie. Lek jest wydalany głównie w moczu: w ciągu pierwszej doby po podaniu leku w moczu znajduje się 70% - 90% dawki w postaci nie zmienionej. Z badań in vitro wynika, że pemetreksed jest czynnie wydzielany za pośrednictwem
transportera anionów organicznych-3 (OAT3, ang. organic anion transporter 3). Całkowity klirens układowy pemetreksedu wynosi 91,8 ml/min, a końcowy okres półtrwania u pacjentów z prawidłową czynnością nerek (klirens kreatyniny 90 ml/min) jest równy 3,5 godziny. Zmienność wartości klirensu oznaczanych u różnych pacjentów jest niewielka i wynosi 19%. Całkowita ekspozycja organizmu na pemetreksed (AUC) i największe stężenie w osoczu zmieniają się proporcjonalnie do dawki leku.
Właściwości farmakokinetyczne pemetreksedu u pacjentów poddawanych wielokrotnym cyklom leczenia pozostają takie same.
Podawana równocześnie cisplatyna nie zmienia właściwości farmakokinetycznych pemetreksedu, podobnie jak podawanie kwasu foliowego (doustnie) i witaminy B12 (domięśniowo).
Po podaniu pemetreksedu ciężarnym samicom myszy obserwowano zmniejszoną zdolność płodów do życia, zmniejszenie masy ciała płodów, niepełne kostnienie niektórych struktur kostnych i rozszczep podniebienia.
Po podaniu pemetreksedu samcom myszy obserwowano szkodliwy wpływ na reprodukcję, objawiający się zmniejszoną płodnością i zanikiem jąder. W badaniu, w którym psom rasy beagle przez 9 miesięcy podawano lek w dożylnym bolusie, obserwowano szkodliwy wpływ na jądra (zwyrodnienie lub martwicę nabłonka plemnikotwórczego). To wskazuje, że pemetreksed może zaburzać płodność osobników męskich. Nie badano płodności samic.
Pemetreksed nie wykazywał działania mutagennego ani w teście aberracji chromosomalnych w komórkach jajnika chomika chińskiego in vitro, ani w teście Amesa. In vivo w teście mikrojądrowym u myszy wykazano klastogenność pemetreksedu.
Nie badano potencjalnego działania rakotwórczego pemetreksedu.
Trometamol Monotioglicerol Kwas cytrynowy
Sodu wodorotlenek (do ustalenia pH)
Kwas solny (do ustalenia pH) Woda do wstrzykiwań
Pemetreksed jest fizycznie niezgodny z rozcieńczalnikami zawierającymi wapń, w tym roztworem Ringera do wstrzykiwań z mleczanami i roztworem Ringera do wstrzykiwań. Nie wolno mieszać tego produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
Pemetrexed EVER Pharma zawiera trometamol jako substancję pomocniczą. Trometamol wykazuje niezgodności z cisplatyną powodując rozpad cisplatyny. Nie wolno mieszać tego produktu leczniczego z innymi produktami leczniczymi. Linie do wlewów dożylnych powinny być przepłukane po podaniu produktu leczniczego Pemetrexed EVER Pharma.
Nieotwarta fiolka
2 lata.
Rozcieńczony roztwór
Wykazano stabilność chemiczną i fizyczną roztworu do infuzji przygotowanego do użycia przechowywanego w lodówce (od 2°C do 8°C) przez 28 dni oraz przechowywanego w temperaturze od 20°C do 30°C przez 7 dni.
Z mikrobiologicznego punktu widzenia produkt należy wykorzystać bezpośrednio po sporządzeniu. Jeżeli produkt nie zostanie natychmiast zużyty, odpowiedzialność za okres przechowywania i warunki przechowywania przed użyciem ponosi użytkownik: roztwór należy przechowywać w temperaturze 2ºC-8ºC przez okres nie dłuższy niż 24 godziny, chyba że rozcieńczenia dokonano w kontrolowanych i zwalidowanych warunkach aseptycznych.
Nie zamrażać.
Warunki przechowywania produktu leczniczego po rozcieńczeniu, patrz punkt 6.3.
Fiolki z bezbarwnego szkła (typu I), zamknięte korkiem z gumy bromobutylowej, powlekanym fluoropolimerem z aluminiowym wieczkiem typu flip-off, w ochronnej osłonce lub bez osłonki, w tekturowym pudełku.
Wielkość opakowań
1 fiolka x 4 ml (100 mg/4 ml)
1 fiolka x 20 ml (500 mg/20 ml)
1 fiolka x 40 ml (1000 mg/40 ml)
Nie wszystkie wielkości opakowań mogą znajdować się w obrocie.
Kobiety w ciąży nie powinny przygotowywać tego leku.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
EVER Valinject GmbH Oberburgau 3
4866 Unterach am Attersee Austria
26655
I DATA PRZEDŁUŻENIA POZWOLENIA
08.10.2021







