







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
lek przeciwhistaminowy (5 mg dekschlorfenyraminy lub 25 mg difenhydraminy, albo lek
o równoważnej sile działania),
kortykosteroid (8 mg deksametazonu, albo lek o równoważnej sile działania),
antagonista receptora H2 (ranitydyna, albo lek o równoważnej sile działania) (patrz punkt 4.4).
Zaleca się profilaktycznie stosować leki przeciwwymiotne, które można podawać doustnie lub dożylnie, w zależności od potrzeby.
Podczas leczenia należy zapewnić odpowiednie nawodnienie pacjenta, aby zapobiec powikłaniom, takim jak niewydolność nerek.
Dawkowanie
Zalecana dawka produktu leczniczego Eleber wynosi 25 mg/m2 pc., podawana w 1-godzinnej infuzji dożylnej co 3 tygodnie, w skojarzeniu z doustnym prednizonem lub prednizolonem w dawce 10 mg podawanym codziennie podczas leczenia.
Dostosowanie dawki
Dawkę należy zmodyfikować, jeśli u pacjentów pojawią się następujące działania niepożądane (stopnie odnoszą się do klasyfikacji zdarzeń niepożądanych według Common Terminology Criteria for Adverse Events [CTCAE 4.0]):
Tabela 1: Zalecane modyfikacje dawki w przypadku wystąpienia działań niepożądanych u pacjentów otrzymujących kabazytaksel
Działania niepożądane
Modyfikacja dawki
Długotrwała (powyżej 1 tygodnia) neutropenia stopnia ≥3. pomimo zastosowania odpowiedniego leczenia, w tym G-CSF
Leczenie należy odroczyć do czasu osiągnięcia liczby granulocytów obojętnochłonnych
>1500/mm3, a następnie zmniejszyć dawkę kabazytakselu z 25 mg/m2 pc. do 20 mg/m2 pc.
Gorączka neutropeniczna lub zakażenie w przebiegu neutropenii
Leczenie należy odroczyć do czasu poprawy lub ustąpienia objawów oraz osiągnięcia liczby granulocytów obojętnochłonnych >1500/mm3,
a następnie zmniejszyć dawkę kabazytakselu z 25 mg/m2 pc. do 20 mg/m2 pc.
Biegunka stopnia ≥3., albo biegunka utrzymująca się pomimo zastosowania odpowiedniego leczenia, w tym uzupełnienia niedoborów płynu i
elektrolitów
Leczenie należy odroczyć do czasu poprawy lub ustąpienia objawów, a następnie zmniejszyć dawkę kabazytakselu z 25 mg/m2 pc. do 20 mg/m2 pc.
Neuropatia obwodowa stopnia >2.
Leczenie należy odroczyć do czasu poprawy,
a następnie zmniejszyć dawkę kabazytakselu
z 25 mg/m2 pc. do 20 mg/m2 pc.
Jeśli u pacjentów nadal występują którekolwiek z opisanych działań niepożądanych po zastosowaniu dawki 20 mg/m2 pc., należy rozważyć dalsze zmniejszenie dawki do 15 mg/m2 pc. lub przerwanie leczenia produktem leczniczym Eleber. Dane dotyczące pacjentów stosujących dawkę mniejszą niż 20 mg/m2 pc. są ograniczone.
Szczególne grupy pacjentów
Pacjenci z zaburzeniami czynności wątroby
Kabazytaksel jest w znacznym stopniu metabolizowany w wątrobie. U pacjentów z łagodnymi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej od >1 do ≤1,5 × Górna Granica Normy (GGN) lub aktywność AspAT >1,5 × GGN) należy zmniejszyć dawkę kabazytakselu do
20 mg/m2 pc. U pacjentów z łagodnymi zaburzeniami czynności wątroby należy zachować ostrożność i dokładnie monitorować bezpieczeństwo podczas podawania kabazytakselu.
U pacjentów z umiarkowanymi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej
od >1,5 do ≤3,0 × GGN) maksymalna tolerowana dawka to 15 mg/m2 pc. W przypadku leczenia
u pacjentów z umiarkowanymi zaburzeniami czynności wątroby dawka kabazytakselu nie powinna być większa niż 15 mg/m2 pc. Dane dotyczące skuteczności leczenia z zastosowaniem tej dawki są ograniczone.
Nie należy podawać kabazytakselu pacjentom z ciężkimi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej >3 × GGN) (patrz punkty 4.3, 4.4 i 5.2).
Pacjenci z zaburzeniami czynności nerek
Kabazytaksel jest w minimalnym stopniu wydalany przez nerki. Nie ma konieczności dostosowania dawki u pacjentów z zaburzeniami czynności nerek, niewymagających hemodializy. U pacjentów ze schyłkową niewydolnością nerek (klirens kreatyniny (CLCR <15 ml/min/1,73 m2), ze względu na ich stan oraz ograniczone dane, należy zachować ostrożność podczas leczenia i uważnie ich monitorować w trakcie leczenia (patrz punkty 4.4 i 5.2).
Pacjenci w podeszłym wieku
Brak specjalnych zaleceń dotyczących dostosowania dawki kabazytakselu u pacjentów w podeszłym wieku (patrz również punkty 4.4, 4.8 i 5.2).
Jednoczesne stosowanie innych produktów leczniczych
Należy unikać jednoczesnego stosowania produktów leczniczych będących silnymi induktorami lub silnymi inhibitorami CYP3A. Jednak jeśli pacjent wymaga jednoczesnego przyjmowania silnego inhibitora CYP3A, należy rozważyć zmniejszenie dawki kabazytakselu o 25% (patrz punkty 4.4 i 4.5).
Dzieci i młodzież
Kabazytaksel nie ma istotnego zastosowania u dzieci i młodzieży.
Nie określono dotychczas bezpieczeństwa stosowania i skuteczności kabazytakselu u dzieci
i młodzieży w wieku poniżej 18 lat (patrz punkt 5.1).
Sposób podawania
Produkt leczniczy Eleber jest przeznaczony do podawania dożylnego.
Instrukcja dotycząca przygotowania i podawania produktu leczniczego, patrz punkt 6.6. Nie należy używać worków infuzyjnych wykonanych z PVC i poliuretanowych zestawów do infuzji.
Produktu leczniczego Eleber nie wolno mieszać z innymi produktami leczniczymi oprócz wymienionych w punkcie 6.6.
Przeciwwskazania
Nadwrażliwość na kabazytaksel, inne taksany, na polisorbat 80 lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Liczba granulocytów obojętnochłonnych poniżej 1500/mm3.
Ciężkie zaburzenia czynności wątroby (stężenie bilirubiny całkowitej >3 × GGN).
Jednoczesne szczepienie szczepionką przeciwko żółtej gorączce (patrz punkt 4.5).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane Podsumowanie profilu bezpieczeństwa
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Koncentrat: 1,5 ml koncentratu w fiolce z bezbarwnego szkła typu I, zamkniętej korkiem z gumy chlorobutylowej pokrytym fluoropolimerem, z aluminiowym uszczelnieniem (zamknięcie typu filp off). Każda fiolka zawiera 60 mg kabazytakselu w 1,5 ml objętości nominalnej (objętość napełnienia: 73,2 mg kabazytakselu/1,83 ml). Objętość napełnienia została ustalona w trakcie rozwoju produktu leczniczego Eleber w celu wyrównania strat płynu podczas przygotowywania roztworu wstępnego. Nadmiar ten gwarantuje, że po rozcieńczeniu całą zawartością rozpuszczalnika towarzyszącego produktowi leczniczemu Eleber, w fiolce znajduje się minimalna ekstrahowalna objętość 6 ml roztworu wstępnego, zawierająca 10 mg/ml produktu leczniczego Eleber, co odpowiada oznakowanej ilości 60 mg na fiolkę.
Rozpuszczalnik: 4,5 ml rozpuszczalnika w fiolce z bezbarwnego szkła typu I, zamkniętej korkiem z gumy chlorobutylowej pokrytym fluoropolimerem, z aluminiowym uszczelnieniem (zamknięcie typu flip off). Każda fiolka zawiera 4,5 ml objętości nominalnej (objętość napełnienia: 5,67 ml). Objętość napełnienia została ustalona w trakcie rozwoju, a nadmiar zapewnia, że po dodaniu całej zawartości fiolki z rozpuszczalnikiem do zawartości fiolki produktu leczniczego Eleber z 60 mg koncentratu stężenie roztworu wstępnego produktu leczniczego Eleber wynosi 10 mg/ml.
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Eleber, 60 mg, koncentrat i rozpuszczalnik do sporządzania roztworu do infuzji
1 ml koncentratu zawiera 40 mg kabazytakselu.
Każda fiolka z 1,5 ml (objętość nominalna) koncentratu zawiera 60 mg kabazytakselu.
Po wstępnym rozcieńczeniu całą objętością rozpuszczalnika każdy ml roztworu zawiera 10 mg kabazytakselu.
Uwaga: zarówno fiolka z koncentratem produktu leczniczego Eleber 60 mg/1,5 ml (objętość napełnienia: 73,2 mg kabazytakselu/1,83 ml), jak i fiolka z rozpuszczalnikiem (objętość napełnienia: 5,67 ml) zawierają nadmiar płynu w celu wyrównania jego strat podczas przygotowania. Nadmiar ten zapewnia, że po rozcieńczeniu CAŁĄ zawartością dołączonego rozpuszczalnika otrzymany roztwór zawiera 10 mg/ml kabazytakselu.
Substancja pomocnicza o znanym działaniu
Każda fiolka z rozpuszczalnikiem zawiera 542,4 mg etanolu 100%.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Koncentrat i rozpuszczalnik do sporządzania roztworu do infuzji (jałowy koncentrat). Koncentrat jest przejrzystym, bezbarwnym do bladożółtego, lepkim roztworem.
Rozpuszczalnik jest przejrzystym, bezbarwnym roztworem.
Produkt leczniczy Eleber, w skojarzeniu z prednizonem lub prednizolonem, jest wskazany do leczenia pacjentów dorosłych z opornym na kastrację rakiem gruczołu krokowego z przerzutami, leczonych wcześniej schematem chemioterapii zawierającym docetaksel (patrz punkt 5.1).
Produkt leczniczy Eleber należy stosować wyłącznie w oddziałach wyspecjalizowanych w podawaniu leków cytotoksycznych i należy go podawać tylko pod kontrolą lekarza mającego odpowiednie kwalifikacje do stosowania chemioterapii przeciwnowotworowej. Produkt leczniczy można podawać jedynie w przypadku dysponowania odpowiednimi pomieszczeniami i wyposażeniem zapewniającym możliwość leczenia ciężkich reakcji nadwrażliwości, takich jak niedociśnienie i skurcz oskrzeli (patrz punkt 4.4).
Premedykacja
W celu zmniejszenia ryzyka wystąpienia i nasilenia reakcji nadwrażliwości zalecany schemat premedykacji powinien być wykonany przynajmniej 30 minut przed każdym podaniem produktu leczniczego Eleber poprzez dożylne podanie następujących produktów leczniczych:
Reakcje nadwrażliwości
Wszyscy pacjenci powinni otrzymać premedykację przed rozpoczęciem infuzji kabazytakselu
(patrz punkt 4.2).
Należy ściśle obserwować pacjentów w kierunku wystąpienia reakcji nadwrażliwości, zwłaszcza w trakcie pierwszej i drugiej infuzji dożylnej. Ponieważ reakcje nadwrażliwości mogą wystąpić w ciągu kilku minut po rozpoczęciu infuzji dożylnej kabazytakselu, należy zabezpieczyć dostępność
pomieszczeń i wyposażenia niezbędnego do leczenia niedociśnienia i skurczu oskrzeli. Mogą wystąpić ciężkie reakcje nadwrażliwości, które mogą obejmować uogólnioną wysypkę i (lub) rumień, niedociśnienie i skurcz oskrzeli. Ciężkie reakcje nadwrażliwości wymagają natychmiastowego przerwania infuzji kabazytakselu i wdrożenia odpowiedniego leczenia. Należy przerwać stosowanie produktu leczniczego Eleber u pacjentów, u których wystąpi reakcja nadwrażliwości (patrz punkt 4.3).
Zahamowanie czynności szpiku kostnego
Podczas stosowania produktu leczniczego może wystąpić zahamowanie czynności szpiku kostnego objawiające się neutropenią, niedokrwistością, małopłytkowością lub pancytopenią (patrz poniżej
„Ryzyko neutropenii” i „Niedokrwistość” w punkcie 4.4).
Ryzyko neutropenii
Pacjenci leczeni kabazytakselem mogą profilaktycznie otrzymywać G-CSF, zgodnie z zaleceniami Amerykańskiego Stowarzyszenia Onkologii Klinicznej (ang. ASCO, American Society of Clinical Oncology) i (lub) aktualnymi wytycznymi ośrodka prowadzącego leczenie, w celu zmniejszenia ryzyka lub opanowania powikłań neutropenii (gorączka neutropeniczna, przedłużona neutropenia lub zakażenie w przebiegu neutropenii).
Pierwotną profilaktykę przy użyciu G-CSF należy rozważyć u pacjentów z cechami klinicznymi wysokiego ryzyka (wiek >65 lat, zły stan czynnościowy, wcześniejsze epizody gorączki neutropenicznej, rozległe obszary ciała poddane wcześniej napromienianiu, zły stan odżywienia lub inne ciężkie choroby współistniejące), które predysponują do zwiększenia powikłań wynikających z przedłużonej neutropenii. Wykazano, że zastosowanie G-CSF ogranicza częstość występowania
i stopień ciężkości neutropenii.
Neutropenia jest najczęstszym działaniem niepożądanym występującym po zastosowaniu kabazytakselu (patrz punkt 4.8). Niezbędne jest wykonywanie badań pełnej morfologii krwi, co tydzień podczas 1. cyklu leczenia oraz przed każdym kolejnym cyklem, tak aby w razie potrzeby można było dostosować dawkę.
Należy zmniejszyć dawkę w przypadku wystąpienia gorączki neutropenicznej lub przedłużającej się neutropenii, pomimo zastosowania odpowiedniego leczenia (patrz punkt 4.2).
Ponowne leczenie pacjentów można rozpocząć jedynie w przypadku, gdy liczba neutrofilów powróci do poziomu ≥1500/mm3 (patrz punkt 4.3).
Zaburzenia żołądka i jelit
Objawy, takie jak ból i tkliwość brzucha, gorączka, uporczywe zaparcie, biegunka, z towarzyszącą neutropenią lub bez, mogą być wczesnymi objawami ciężkiej toksyczności układu pokarmowego, którą należy niezwłocznie ocenić oraz leczyć. Może zajść konieczność odroczenia lub zaprzestania leczenia kabazytakselem.
Ryzyko nudności, wymiotów, biegunki i odwodnienia
Jeśli u pacjentów wystąpi biegunka po podaniu kabazytakselu, można ich leczyć powszechnie stosowanymi lekami przeciwbiegunkowymi. Należy podjąć odpowiednie czynności w celu przywrócenia stanu nawodnienia pacjentów. Biegunka może występować częściej u pacjentów, którzy uprzednio poddani byli napromienianiu okolicy brzucha i miednicy. Odwodnienie dotyczy częściej pacjentów w wieku 65 lat i starszych. Należy podjąć odpowiednie kroki w celu ponownego nawodnienia pacjentów oraz monitorowania i skorygowania stężenia elektrolitów w surowicy krwi, szczególnie potasu. W przypadku biegunki stopnia ≥3. może być konieczne odroczenie leczenia, albo zmniejszenie dawki kabazytakselu (patrz punkt 4.2). Jeśli u pacjentów wystąpią nudności lub wymioty, można ich leczyć powszechnie stosowanymi lekami przeciwwymiotnymi.
Ryzyko ciężkich zaburzeń żołądka i jelit
U pacjentów leczonych kabazytakselem zgłaszano krwotok z przewodu pokarmowego oraz perforację przewodu pokarmowego, niedrożność porażenną jelit, zapalenie jelita grubego, w tym zakończone zgonem (patrz punkt 4.8). Zaleca się zachowanie ostrożności podczas leczenia pacjentów najbardziej zagrożonych wystąpieniem powikłań ze strony przewodu pokarmowego: u pacjentów z neutropenią, w podeszłym wieku, stosujących jednocześnie niesteroidowe leki przeciwzapalne, leczenie przeciwpłytkowe, leczenie przeciwzakrzepowe, u pacjentów z uprzednią radioterapią miednicy lub
u pacjentów z chorobą przewodu pokarmowego, taką jak owrzodzenie i krwawienie z przewodu pokarmowego.
Neuropatia obwodowa
U pacjentów otrzymujących kabazytaksel obserwowano przypadki neuropatii obwodowej, obwodowej neuropatii czuciowej (np. parestezje, dyzestezje) i obwodowej neuropatii ruchowej. Pacjentów leczonych kabazytakselem należy pouczyć o konieczności poinformowania lekarza przed kontynuowaniem leczenia o wystąpieniu objawów neuropatii, takich jak ból, pieczenie, mrowienie, drętwienie lub osłabienie. Lekarz powinien ocenić obecność lub pogorszenie neuropatii przed każdym leczeniem. Leczenie należy odroczyć do czasu poprawy objawów. Należy zmniejszyć
dawkę kabazytakselu z 25 mg/m2 pc. do 20 mg/m2 pc. w przypadku utrzymującej się neuropatii obwodowej stopnia >2. (patrz punkt 4.2).
Niedokrwistość
Obserwowano występowanie niedokrwistości u pacjentów przyjmujących kabazytaksel (patrz punkt 4.8). Należy skontrolować wartość hemoglobiny i hematokrytu przed rozpoczęciem stosowania kabazytakselu oraz u pacjentów z objawami przedmiotowymi i podmiotowymi niedokrwistości lub utraty krwi. Należy zachować ostrożność u pacjentów ze stężeniem hemoglobiny <10 g/dl
i zastosować odpowiednie środki wynikające ze wskazań klinicznych.
Ryzyko niewydolności nerek
Opisywano zaburzenia czynności nerek w połączeniu z sepsą, ciężkim odwodnieniem spowodowanym biegunką, wymiotami oraz zaporową uropatią. Odnotowano niewydolność nerek, w tym przypadki zgonów. Jeśli wystąpią powyższe objawy, należy podjąć odpowiednie środki w celu określenia ich przyczyny oraz intensywnie leczyć pacjentów.
Podczas stosowania kabazytakselu należy zapewnić odpowiednie nawodnienie pacjentów. Pacjentów należy pouczyć o konieczności natychmiastowego zgłaszania lekarzowi każdej znaczącej zmiany objętości moczu wydalanego w ciągu doby. Należy oznaczyć stężenie kreatyniny w surowicy przed rozpoczęciem leczenia, podczas wszystkich badań morfologii krwi oraz każdorazowo w przypadku zgłoszenia przez pacjenta zmiany objętości moczu. Należy przerwać stosowanie kabazytakselu
w przypadku jakiegokolwiek zaburzenia czynności nerek prowadzącego do wystąpienia niewydolności nerek stopnia ≥3. według CTCAE 4.0.
Zaburzenia układu oddechowego
Podczas stosowania produktu leczniczego zgłaszano przypadki występowania śródmiąższowego nieinfekcyjnego zapalenia płuc i (lub) zapalenia płuc oraz śródmiąższowych chorób płuc, które czasem kończyły się zgonem (patrz punkt 4.8).
Jeżeli u pacjenta wystąpią lub nasilą się działania niepożądane ze strony płuc, należy uważnie monitorować jego stan, a w razie konieczności niezwłocznie przeprowadzić odpowiednie badania i podjąć leczenie. Zalecane jest zaprzestanie leczenia kabazytakselem do czasu postawienia
rozpoznania. Wczesne rozpoczęcie leczenia wspomagającego może przynieść poprawę stanu pacjenta. Należy staranie rozważyć korzyści wynikające ze wznowienia leczenia kabazytakselem.
Ryzyko zaburzeń rytmu serca
Odnotowano zaburzenia rytmu serca, najczęściej tachykardię i migotanie przedsionków
(patrz punkt 4.8).
Pacjenci w podeszłym wieku
Osoby w podeszłym wieku (≥65 lat) są zazwyczaj narażone na zwiększone ryzyko pewnych działań niepożądanych, w tym neutropenii i gorączki neutropenicznej (patrz punkt 4.8).
Pacjenci z zaburzeniami czynności wątroby
Stosowanie produktu leczniczego Eleber jest przeciwwskazane u pacjentów z ciężkimi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej >3 × GGN) (patrz punkty 4.3 i 5.2). Należy zmniejszyć dawkę u pacjentów z łagodnymi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej >1 do ≤1,5 × GGN lub AspAT >1,5 × GGN) (patrz punkty 4.2 i 5.2).
Interakcje
Należy unikać jednoczesnego stosowania silnych inhibitorów CYP3A, ponieważ mogą zwiększać stężenie kabazytakselu w osoczu (patrz punkty 4.2 i 4.5). Jeśli nie można uniknąć jednoczesnego stosowania z silnym inhibitorem CYP3A, należy rozważyć uważne monitorowanie w celu wykrycia toksyczności oraz zmniejszenie dawki kabazytakselu (patrz punkty 4.2 i 4.5).
Należy unikać jednoczesnego stosowania silnych induktorów CYP3A4, ponieważ mogą zmniejszać stężenie kabazytakselu w osoczu (patrz punkty 4.2 and 4.5).
Substancje pomocnicze
Rozpuszczalnik zawiera 96% (13% v/v) etanolu (alkoholu), tj. 542,4 mg 100% etanolu na jedną dawkę, co odpowiada 10 ml piwa lub 4 ml wina.
Niewielka ilość alkoholu w tym produkcie leczniczym nie spowoduje żadnych zauważalnych efektów.
Badania in vitro wykazały, że kabazytaksel jest metabolizowany głównie za pośrednictwem CYP3A (80% do 90%) (patrz punkt 5.2).
Inhibitory CYP3A
Wielokrotne przyjmowanie ketokonazolu (400 mg raz na dobę), który jest silnym inhibitorem CYP3A, prowadziło do zmniejszenia klirensu kabazytakselu o 20%, co odpowiada zwiększeniu AUC
o 25%. Dlatego z uwagi na możliwość zwiększenia stężenia kabazytakselu w osoczu należy unikać jednoczesnego stosowania silnych inhibitorów CYP3A (np. ketokonazol, itrakonazol, klarytromycyna, indynawir, nefazodon, nelfinawir, rytonawir, sakwinawir, telitromycyna, worykonazol) (patrz punkty 4.2 i 4.4).
Jednoczesne stosowanie aprepitantu, umiarkowanego inhibitora CYP3A nie miało wpływu na klirens
kabazytakselu.
Induktory CYP3A
Wielokrotne przyjmowanie ryfampicyny (600 mg raz na dobę), która jest silnym induktorem CYP3A prowadziło do zwiększenia klirensu kabazytakselu o 21%, co odpowiada zmniejszeniu AUC o 17%. Dlatego z uwagi na możliwość zmniejszenia stężenia kabazytakselu w osoczu należy unikać jednoczesnego stosowania silnych induktorów CYP3A (np. fenytoina, karbamazepina, ryfampicyna, ryfabutyna, ryfapentyna, fenobarbital) (patrz punkty 4.2 i 4.4). Ponadto pacjenci nie powinni przyjmować produktów zawierających ziele dziurawca zwyczajnego.
OATP1B1
Wykazano również, że w warunkach in vitro kabazytaksel hamuje białka transportowe z grupy polipeptydów transportujących aniony organiczne OATP1B1. Ryzyko interakcji z substratami OATP1B1 (np. statyny, walsartan, repaglinid) jest możliwe, zwłaszcza w czasie trwania infuzji
(1 godzina) i aż do 20 minut po zakończeniu infuzji. Zalecany jest 12-godzinny odstęp czasu przed infuzją i przynajmniej 3-godzinny po zakończeniu infuzji przed podaniem substratu OATP1B1.
Szczepienia
Stosowanie żywych lub żywych atenuowanych szczepionek, u pacjentów z obniżoną odpornością na skutek podawania chemioterapii, może prowadzić do ciężkich lub śmiertelnych zakażeń. Należy unikać szczepienia za pomocą żywej atenuowanej szczepionki u pacjentów otrzymujących kabazytaksel. Można stosować martwe lub inaktywowane szczepionki, ale reakcja na takie szczepionki może być osłabiona.
Ciąża
Nie ma danych dotyczących stosowania kabazytakselu u kobiet w okresie ciąży. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję po zastosowaniu dawek toksycznych dla ciężarnych samic (patrz punkt 5.3) oraz przenikanie kabazytakselu przez barierę łożyskową (patrz punkt 5.3). Podobnie jak inne cytotoksyczne produkty lecznicze, kabazytaksel może powodować uszkodzenie płodu u narażonych ciężarnych kobiet.
Kabazytaksel nie powinien być stosowany w okresie ciąży oraz u kobiet w wieku rozrodczym niestosujących skutecznej metody antykoncepcji.
Karmienie piersią
Dostępne dane dotyczące farmakokinetyki u zwierząt wykazały przenikanie kabazytakselu i jego metabolitów do mleka samic (patrz punkt 5.3). Nie można wykluczyć zagrożenia dla karmionego piersią dziecka.
Nie należy stosować kabazytakselu podczas karmienia piersią.
Płodność
Badania na zwierzętach wykazały wpływ kabazytakselu na układ rozrodczy samców szczurów i psów, bez żadnego oddziaływania funkcjonalnego na płodność (patrz punkt 5.3). Biorąc jednak pod uwagę właściwości farmakologiczne taksanów, ich potencjał genotoksyczny i wpływ kilku związków z tej grupy na płodność w badaniach na zwierzętach, nie można wykluczyć wpływu kabazytakselu na płodność u mężczyzn.
Ze względu na potencjalny wpływ kabazytakselu na gamety męskie i możliwość wpływu na organizm za pośrednictwem nasienia, mężczyźni otrzymujący kabazytaksel powinni stosować skuteczną metodę antykoncepcji w trakcie leczenia i do 6 miesięcy od podania ostatniej dawki kabazytakselu. Mężczyźni otrzymujący kabazytaksel powinni zapobiegać kontaktowi innych osób ze swoim ejakulatem w trakcie leczenia, ze względu na potencjalny wpływ kabazytakselu na organizm za pośrednictwem nasienia.
Mężczyźni otrzymujący kabazytaksel powinni się poradzić w sprawie przechowania swojego nasienia przed rozpoczęciem leczenia.
Kabazytaksel wywiera umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn, ponieważ powoduje zmęczenie i zawroty głowy. Pacjentów należy pouczyć, aby nie prowadzili pojazdów i nie obsługiwali maszyn w przypadku pojawienia się powyższych działań niepożądanych w trakcie leczenia.
Bezpieczeństwo kabazytakselu w skojarzeniu z prednizonem lub prednizolonem oceniano w 3
randomizowanych, otwartych, kontrolowanych badaniach (TROPIC, PROSELICA i CARD), obejmujących łącznie 1092 pacjentów z opornym na kastrację rakiem gruczołu krokowego z przerzutami, którzy byli leczeni kabazytakselem w dawce 25 mg/m2 raz na 3 tygodnie. Średni czas leczenia kabazytakselem wynosił 6 do 7 cykli.
Częstość pochodzącą ze zbiorczej analizy tych 3 badań przedstawiono poniżej oraz w wykazie
tabelarycznym.
Najczęściej występującymi (≥10%) działaniami niepożądanymi wszystkich stopni były: niedokrwistość (99,0%), leukopenia (93,0%), neutropenia (87,9%), trombocytopenia (41,1%)
i biegunka (42,1%), zmęczenie (25.0%) i astenia (15,4%).. Najczęstszymi działaniami niepożądanymi
stopnia ≥3, występującymi u co najmniej 5% pacjentów były: neutropenia (73,1%), leukopenia
(59,5%), niedokrwistość (12,0%), gorączka neutropeniczna (8,0%), biegunka (4,7%).
Przerwanie leczenia z powodu działań niepożądanych wystąpiło z podobną częstością we wszystkich 3 badaniach (18,3% w TROPIC, 19,5% w PROSELICA i 19,8% w CARD) u pacjentów otrzymujących kabazytaksel. Najczęstszymi działaniami niepożądanym prowadzącym do przerwania stosowania kabazytakselu były krwiomocz, zmęczenie i neutropenia.
Tabelaryczne zestawienie działań niepożądanych
Działania niepożądane przedstawione w tabeli 2 wymieniono według klasyfikacji układów i narządów MedDRA i częstości występowania. W obrębie każdej grupy o określonej częstości występowania działania niepożądane wymieniono zgodnie ze zmniejszającym się nasileniem. Nasilenie działań niepożądanych sklasyfikowano wg CTCAE 4.0 (stopień ≥3. = G ≥3.). Częstość występowania dotyczy wszystkich stopni ciężkości i jest zdefiniowana następująco: bardzo często (≥1/10); często (≥1/100 do
<1/10); niezbyt często (≥1/1000 do <1/100); rzadko (≥1/10 000 do <1/1000); bardzo rzadko
(<1/10 000); częstość nieznana (nie może być określona na podstawie dostępnych danych).
Tabela 2: Zgłaszane działania niepożądane i zaburzenia hematologiczne u pacjentów otrzymujących kabazytaksel w skojarzeniu z prednizonem lub prednizolonem z analizy zbiorczej (n=1 092)
Klasyfikacja układów i narządów | Działanie niepożądane | Wszystkie stopnie n (%) | Stopień ≥3 n (%) | ||
Bardzo często | Często | Niezbyt często | |||
Zakażenia i zarażenia pasożytnicze | Zakażenie/sepsa z neutropenią* | 48 (4.4) | 42 (3.8) | ||
Wstrząs septyczny | 10 (0,9) | 10 (0,9) | |||
Sepsa | 13 (1,2) | 13 (1,2) | |||
Zapalenie tkanki łącznej | 8 (0,7) | 3 (0,3) | |||
Zakażenie układu moczowego | 103 (9,4) | 19 (1,7) | |||
Grypa | 22 (2,0) | 0 | |||
Zapalenie pęcherza moczowego | 22 (2,0) | 2 (0,2) | |||
Zakażenie górnych dróg oddechowych | 23 (2,1) | 0 | |||
Półpasiec | 14 (1,3) | 0 | |||
Kandydoza | 11 (1,0) | 1 (<0.1) | |||
Zaburzenia krwi i układu chłonnego | Neutropeniaa* | 950 (87,9) | 790 (73,1) | ||
Niedokrwistośća | 1073 (99,0) | 130 (12,0) | |||
Leukopeniaa | 1008 (93,0) | 645 (59,5) | |||
Małopłytkowośća | 478 (44,1) | 44 (4,1) | |||
Gorączka neutropeniczna | 87 (8,0) | 87 (8,0) | |||
Zaburzenia układu immunologicznego | Nadwrażliwość | 7 (0,6) | 0 | ||
Zaburzenia metabolizmu i odżywiania | Jadłowstręt | 192 (17,6) | 11 (1,0) | ||
Odwodnienie | 27 (2,5) | 11 (1,0) | |||
Hiperglikemia | 11 (1,0) | 7 (0,6) | |||
Hipokaliemia | 8 (0,7) | 2 (0,2) | |||
Zaburzenia psychiczne | Bezsenność | 45 (4,1) | 0 | ||
Lęk | 13 (1,2) | 0 | |||
Stan splątania | 12 (1,1) | 2 (0,2) | |||
Zaburzenia układu | Dysgeuzja | 64 (5,9) | 0 | ||
nerwowego | Zaburzenia smaku | 56 (5,1) | 0 | ||
Neuropatia obwodowa | 40 (3,7) | 2 (0,2) | |||
Obwodowa neuropatia czuciowa | 89 (8,2) | 6 (0,5) | |||
Polineuropatia | 9 (0,8) | 2 (0,2) | |||
Parestezje | 46 (4,2) | 1 (<0,1) | |||
Niedoczulica | 18 (1,6) | 0 | |||
Zawroty głowy | 63 (5,8) | 0 | |||
Ból głowy | 56 (5,1) | 1 (<0,1) | |||
Ospałość | 15 (1,4) | 1 (<0,1) | |||
Rwa kulszowa | 9 (0,8) | 1 (<0,1) | |||
Zaburzenia oka | Zapalenie spojówek | 11 (1,0) | 0 | ||
Zwiększone łzawienie | 22 (2,0) | 0 | |||
Zaburzenia ucha i błędnika | Szumy uszne | 7 (0,6) | 0 | ||
Zawroty głowy pochodzenia obwodowego (błędnikowego) | 15 (1,4) | 1 (<0,1) | |||
Zaburzenia serca* | Migotanie przedsionków | 14 (1,3) | 5 (0,5) | ||
Tachykardia | 11 (1,0) | 1 (<0,1) | |||
Zaburzenia naczyniowe | Niedociśnienie | 38 (3,5) | 5 (0,5) | ||
Zakrzepica żył głębokich | 12 (1,1) | 9 (0,8) | |||
Nadciśnienie tętnicze | 29 (2,7) | 12 (1,1) | |||
Niedociśnienie ortostatyczne | 6 (0,5) | 1 (<0.1) | |||
Uderzenia gorąca | 23 (2,1) | 1 (<0.1) | |||
Napadowe zaczerwienienie skóry | 9 (0,8) | 0 | |||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Duszność | 97 (8,9) | 9 (0,8) | ||
Kaszel | 79 (7,2) | 0 | |||
Ból jamy ustnej i gardła | 26 (2,4) | 1 (<0.1) | |||
Zapalenie płuc | 26 (2,4) | 16 (1,5) | |||
Zatorowość płucna | 30 (2,7) | 23 (2,1) | |||
Zaburzenia żołądka i jelit | Biegunka | 460 (42,1) | 51 (4,7) | ||
Nudności | 347 (31,8) | 14 (1,3) | |||
Wymioty | 207 (19,0) | 14 (1,3) | |||
Zaparcia | 202 (18,5) | 8 (0,7) | |||
Ból brzucha | 105 (9,6) | 15 (1,4) | |||
Niestrawność | 53 (4,9) | 0 | |||
Ból w nadbrzuszu | 46 (4,2) | 1 (<0,1) | |||
Guzki krwawnicze | 22 (2,0) | 0 | |||
Choroba refluksowa przełyku | 26 (2,4) | 1 (<0,1) | |||
Krwawienie z odbytnicy | 14 (1,3) | 4 (0,4 | |||
Suchość w ustach | 19 (1,7) | 2 (0,2) | |||
Wzdęcia | 14 (1,3) | 1 (<0,1) | |||
Zapalenie jamy ustnej | 46 (4,2) | 2 (0,2) | |||
Niedrożność porażenna jelit* | 7 (0,6) | 5 (0,5) | |||
Zapalenie żołądka | 10 (0,9) | 0 |
Zapalenie jelita grubego* | 10 (0,9) | 5 (0,5) | |||
Perforacja przewodu pokarmowego | 3 (0,3) | 1 (<0,1) | |||
Krwawienia z przewodu pokarmowego | 2 (0,2) | 1 (<0,1) | |||
Zaburzenia skóry i tkanki podskórnej | Łysienie | 80 (7,3) | 0 | ||
Suchość skóry | 23 (2,1) | 0 | |||
Rumień | 8 (0,7) | 0 | |||
Zaburzenia paznokci | 18 (1,6) | 0 | |||
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Ból pleców | 166 (15,2) | 24 (2,2) | ||
Ból stawów | 88 (8,1) | 9 (0,8) | |||
Ból kończyn | 76 (7,0) | 9 (0,8) | |||
Skurcz mięśni | 51 (4,7) | 0 | |||
Bóle mięśni | 40 (3,7) | 2 (0,2) | |||
Ból w klatce piersiowej | 34 (3,1) | 3 (0,3) | |||
Osłabienie mięśni | 31 (2,8) | 1 (0,2) | |||
Ból w bocznej części ciała | 17 (1,6) | 5 (0,5) | |||
Zaburzenia nerek i dróg moczowych | Ostra niewydolność nerek | 21 (1,9) | 14 (1,3) | ||
Niewydolność nerek | 8 (0,7) | 6 (0,5) | |||
Trudności w oddawaniu moczu | 52 (4,8) | 0 | |||
Kolka nerkowa | 14 (1,3) | 2 (0,2) | |||
Krwiomocz | 205 (18,8) | 33 (3,0) | |||
Częstomocz | 26 (2,4) | 2 (0,2) | |||
Wodonercze | 25 (2,3) | 13 (1,2) | |||
Zatrzymanie moczu | 36 (3,3) | 4 (0,4) | |||
Nietrzymanie moczu | 22 (2,0) | 0 | |||
Niedrożność moczowodów | 8 (0.7) | 6 (0,5) | |||
Zaburzenia układu rozrodczego i piersi | Ból miednicy | 20 (1,8) | 5 (0,5) | ||
Zaburzenia ogólne i stany w miejscu podania | Zmęczenie | 333 (30,5) | 42 (3,8) | ||
Astenia | 227 (20,8) | 32 (2,9) | |||
Gorączka | 90 (8,2) | 5 (0,5) | |||
Obrzęki obwodowe | 96 (8.8) | 2 (0,2) | |||
Zapalenie śluzówek | 23 (2,1) | 1 (<0,1) | |||
Ból | 36 (3,3) | 7 (0,6) | |||
Ból w klatce piersiowej | 11 (1,0) | 2 (0,2) | |||
Obrzęki | 8 (0,7) | 1 (<0,1) | |||
Dreszcze | 12 (1,1) | 0 | |||
Złe samopoczucie | 21 (1,9) | 0 | |||
Badania diagnostyczne | Zmniejszenie masy ciała | 81 (7,4) | 0 | ||
Zwiększenie aktywności aminotransferazy asparaginianowej | 13 (1,2) | 1 (<0,1) | |||
Zwiększenie aktywności transaminaz | 7 (0,6) | 1 (<0,1) |
a na podstawie badań laboratoryjnych
* szczegółowe informacje patrz punkt poniżej Opis wybranych działań niepożądanych
Neutropenia i towarzyszące jej zaburzenia kliniczne
Wykazano, że stosowanie G-CSF ogranicza częstość występowania i nasilenie neutropenii (patrz punkty 4.2 i 4.4). Częstość występowania neutropenii stopnia ≥3. na podstawie danych laboratoryjnych wahała się w zależności od stosowania G-CSF od 44,7% do 76,7%, przy czym najniższą częstość odnotowano w przypadku stosowania profilaktyki G-CSF. Podobnie częstość występowania gorączki neutropenicznej ≥3. stopnia wahała się od 3,2% do 8,6%. Powikłania z neutropenią (w tym gorączka neutropeniczna, zakażenie/posocznica z neutropenią i neutropeniczne zapalenie jelita grubego), które w niektórych przypadkach kończyły się zgonem, zgłoszono u 4,0% pacjentów stosujących pierwotną profilaktykę G-CSF i u 12,8% pacjentów w innych przypadkach
Zaburzenia czynności serca i zaburzenia rytmu serca
W analizie zbiorczej zdarzenia sercowe zgłoszono u 5,5% pacjentów, z których 1,1% miało zaburzenia rytmu serca ≥3. Częstość występowania tachykardii podczas stosowania kabazytakselu wynosiła 1,0%, z czego mniej niż 0,1% miało stopień ≥3. Częstość występowania migotania przedsionków wynosiła 1,3%. Zdarzenia niewydolności serca zgłoszono u 2 pacjentów (0,2%), z których jeden zakończył się zgonem. Migotanie komór zakończone zgonem zgłoszono u 1 pacjenta (0,3%), a zatrzymanie akcji serca u 3 pacjentów (0,5%). Żadne z powyższych nie zostało uznane przez badacza za związane ze stosowaniem kabazytakselu.
Krwiomocz
W analizie zbiorczej, krwiomocz we wszystkich stopniach zaobserwowano u 18,8% z zastosowaniem dawki 25 mg/m2 pc. (patrz punkt 5.1). W blisko połowie przypadków zidentyfikowano czynniki sprzyjające, jeśli zostały udokumentowane, takie jak progresja choroby, jej nasilenie, infekcja lub leczenie antykoagulantami/NLPZ/kwasem acetylosalicylowym.
Inne nieprawidłowości w wynikach badań laboratoryjnych
W analizie zbiorczej częstość występowania niedokrwistości stopnia ≥3., zwiększonej aktywności AspAT, AlAT i stężenia bilirubiny na podstawie wyników badań laboratoryjnych wynosiła odpowiednio 12,0%, 1,3%, 1,0% i 0,5%.
Zaburzenia żołądka i jelit
Obserwowano występowanie: zapalenia jelita grubego (w tym zapalenia jelit i neutropenicznego zapalenia jelit), zapalenia żołądka, . Zgłaszano także krwotok z przewodu pokarmowego, perforację przewodu pokarmowego oraz niedrożność porażenną jelit (niedrożność jelit) (patrz punkt 4.4).
Zaburzenia układu oddechowego
Podczas stosowania produktu leczniczego zgłaszano przypadki występowania (częstość występowania nieznana – nie można określić na podstawie dostępnych danych) śródmiąższowego nieinfekcyjnego zapalenia płuc i (lub) zapalenia płuc oraz śródmiąższowych chorób płuc, które czasem kończyły się zgonem (patrz punkt 4.4).
Zaburzenia nerek i dróg moczowych
Niezbyt często zgłaszano występowanie zapalenia pęcherza moczowego spowodowane nawrotem objawów popromiennych (ang. radiation recall phenomenon), w tym krwotoczne zapalenie pęcherza moczowego.
Dzieci i młodzież
Patrz punkt 4.2.
Inne szczególne grupy pacjentów
Pacjenci w podeszłym wieku
Spośród 1092 pacjentów leczonych kabazytakselem w dawce 25 mg/m2 w badaniach nad rakiem gruczołu krokowego, 755 pacjentów miało 65 lat lub więcej, w tym 238 pacjentów było starszych niż 75 lat. Następujące niehematologiczne działania niepożądane zgłaszano z częstością ≥5% większą u pacjentów w wieku 65 lat lub starszych w porównaniu z młodszymi pacjentami: zmęczenie (33,5% w porównaniu z 23,7%), astenia (23,7 w porównaniu z 14,2%), zaparcia (20,4%) vs 14,2%) i duszności
(10,3% vs 5,6%). Neutropenia (90,9% vs 81,2%) i trombocytopenia (48,8% vs 36,1%) były również o 5% wyższe u pacjentów w wieku 65 lat lub powyżej 12 lat w porównaniu z młodszymi pacjentami.
Neutropenię stopnia ≥3. i gorączkę neutropeniczną zgłaszano z największymi odsetkami różnic między obiema grupami wiekowymi, odpowiednio o 14% i 4% większą u pacjentów w wieku ≥65 lat w porównaniu z pacjentami w wieku <65 lat (patrz punkty 4.2 i 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Nie jest znane antidotum dla kabazytakselu. Potencjalne powikłania po przedawkowaniu mogą obejmować zaostrzenie działań niepożądanych w postaci supresji szpiku kostnego oraz zaburzenia żołądka i jelit.
W przypadku przedawkowania pacjenta należy umieścić na specjalistycznym oddziale i ściśle monitorować. W przypadku rozpoznania przedawkowania należy najszybciej jak to możliwe podać pacjentowi dawkę leczniczą G-CSF. Należy wdrożyć inne odpowiednie leczenie objawowe.
Grupa farmakoterapeutyczna: leki przeciwnowotworowe, taksany, kod ATC: L01CD04.
Mechanizm działania
Kabazytaksel jest lekiem przeciwnowotworowym działającym przez zakłócenie sieci połączeń mikrotubul w komórkach. Kabazytaksel wiąże się z tubuliną i pobudza proces odkładania się tubuliny do mikrotubul, hamując równocześnie ich rozpad. Prowadzi to do stabilizacji mikrotubul, co powoduje zahamowanie mitotycznych i interfazowych podziałów komórki.
Działanie farmakodynamiczne
Kabazytaksel wykazuje szerokie spektrum działania przeciwnowotworowego przeciwko zaawansowanym nowotworom ludzkim wszczepionym myszom. Kabazytaksel wykazuje aktywność wobec nowotworów podatnych na działanie docetakselu. Ponadto kabazytaksel wykazuje aktywność w modelach nowotworów niewrażliwych na chemioterapię zawierającą docetaksel.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność i bezpieczeństwo stosowania kabazytakselu w skojarzeniu z prednizonem lub prednizolonem oceniano w międzynarodowym, wieloośrodkowym badaniu III fazy (badanie EFC6193), prowadzonym z randomizacją metodą otwartej próby, w grupie pacjentów z opornym na kastrację rakiem gruczołu krokowego z przerzutami leczonych wcześniej schematem leczenia zawierającym docetaksel.
Pierwszorzędowym punktem końcowym skuteczności badania było przeżycie całkowite (OS, ang. overall survival).
Drugorzędowe punkty końcowe obejmowały przeżycie bez progresji choroby (PFS, ang. progression free survival) [definiowane jako czas od randomizacji do progresji guza, progresji wg stężenia swoistego antygenu sterczowego (PSA, ang. prostatic specific antigen), progresji bólu lub zgonu
z jakiejkolwiek przyczyny, bez względu na to, który pojawił się pierwszy], wskaźnik odpowiedzi terapeutycznej ze strony guza wg klasyfikacji RECIST (ang. response evaluation criteria in solid tumours), progresję wg PSA (definiowaną jako wzrost ≥25% u pacjentów, u których początkowo nie stwierdzono spadku PSA, lub >50% u pacjentów, u których stężenie antygenu zmniejszyło się), odpowiedź PSA na leczenie (zmniejszenie stężenia PSA w surowicy przynajmniej o 50%), progresję bólu [ocenianą przy użyciu skali aktualnego nasilenia bólu (PPI, ang. present pain intensity) według kwestionariusza McGilla i Melzacka oraz skali uwzględniającej rodzaj leków przeciwbólowych (AS, ang. analgesic score)] oraz odpowiedź bólowa (definiowana jako zmniejszenie nasilenia
w porównaniu z wartościami wyjściowymi o ponad 2 punkty według PPI bez równoczesnego zwiększenia AS, albo zmniejszenie stosowanych leków przeciwbólowych o ≥50% w porównaniu z wartościami wyjściowymi AS bez równoczesnego zwiększenia bólu).
W badaniu uczestniczyło łącznie 755 pacjentów, których losowo przydzielono do grupy otrzymującej kabazytaksel w dawce 25 mg/m2 pc. dożylnie co 3 tygodnie przez maksymalnie 10 cykli, z doustnym prednizonem lub prednizolonem w dawce dobowej 10 mg (n = 378), albo mitoksantron w dawce
12 mg/m2 pc. dożylnie co 3 tygodnie przez maksymalnie 10 cykli, z doustnym prednizonem lub
prednizolonem w dawce dobowej 10 mg (n = 377).
Do badania kwalifikowano pacjentów w wieku powyżej 18 lat z opornym na kastrację rakiem gruczołu krokowego z przerzutami, u których choroba była mierzalna wg kryteriów RECIST lub niemierzalna, z równoczesnym wzrostem stężenia PSA albo pojawieniem się nowych zmian,
i wskaźnikiem sprawności 0 do 2 wg skali ECOG (ang. Eastern Cooperative Oncology Group). Pacjenci musieli również spełnić następujące kryteria laboratoryjne: liczba granulocytów obojętnochłonnych >1500/mm3, płytek krwi >100 000/mm3, stężenie hemoglobiny >10 g/dl, kreatyniny <1,5 × GGN, bilirubiny całkowitej <1 × GGN, aktywność AspAT i AlAT <1,5 × GGN. Do badania nie kwalifikowano pacjentów z zastoinową niewydolnością serca w wywiadzie, zawałem mięśnia sercowego w ciągu ostatnich 6 miesięcy, pacjentów z nieleczonymi zaburzeniami rytmu serca, dusznicą bolesną i (lub) nadciśnieniem tętniczym.
Cechy demograficzne, w tym wiek, rasa i stan sprawności wg ECOG (0 do 2) były podobne w obydwu grupach. Średnia wieku w grupie pacjentów otrzymujących kabazytaksel wynosiła 68 lat, zakres (46-92) a dystrybucja rasowa wynosiła 83,9% osób rasy kaukaskiej, 6,9% rasy azjatyckiej i (lub) orientalnej, 5,3% rasy czarnej i 4% innej.
Mediana liczby cykli wynosiła 6 w grupie kabazytakselu i 4 w grupie mitoksantronu. Liczba pacjentów, którzy ukończyli badane leczenie (10 cykli) wynosiła odpowiednio 29,4% i 13,5% w grupach kabazytakselu i leku porównawczego.
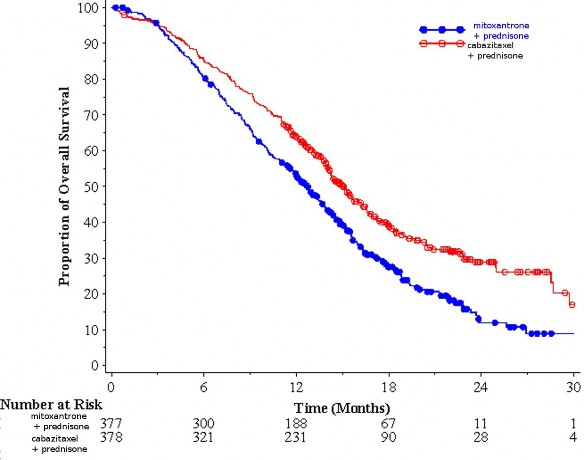
Całkowite przeżycie było znamiennie dłuższe u pacjentów przyjmujących kabazytaksel w porównaniu z mitoksantronem (odpowiednio 15,1 vs 12,7 miesiąca), z 30% zmniejszeniem ryzyka zgonu
w porównaniu z mitoksantronem (patrz tabela 3 i rycina 1).
Podgrupa 59 pacjentów otrzymywała wcześniej skumulowaną dawkę docetakselu <225 mg/m² pc.
(29 pacjentów w grupie kabazytakselu, 30 pacjentów w grupie mitoksantronu). Nie odnotowano
znamiennej różnicy w całkowitym przeżyciu [HR (95% CI) 0,96 (0,49 ‒ 1,86)].
Tabela 3: Skuteczność kabazytakselu w badaniu EFC6193, w leczeniu pacjentów z opornym na kastrację rakiem gruczołu krokowego z przerzutami
Kabazytaksel + prednizon n = 378 | Mitoksantron + prednizon n = 377 | |
Przeżycie całkowite | ||
Liczba zgonów (%) | 234 (61,9%) | 279 (74%) |
Mediana przeżycia (miesiące) (95% CI) | 15,1 (14,1 – 16,3) | 12,7 (11,6 – 13,7) |
Współczynnik hazardu (HR)1 (95% CI) | 0,70 (0,59 – 0,83) | |
Wartość p | <0,0001 | |
1HR oszacowany przy użyciu modelu Coxa; współczynnik ryzyka poniżej 1 przemawia na korzyść
kabazytakselu.
W grupie otrzymującej kabazytaksel odnotowano poprawę przeżycia bez progresji choroby (PFS) w porównaniu z grupą otrzymującą mitoksantron, odpowiednio 2,8 (2,4 ‒ 3,0) miesiąca
vs 1,4 (1,4 ‒ 1,7) miesiąca, HR (95% CI) 0,74 (0,64 ‒ 0,86), p < 0,0001.
Odnotowano znamiennie większy odsetek odpowiedzi guza wynoszący 14,4% (95% CI: 9,6 ‒ 19,3) u pacjentów otrzymujących kabazytaksel w porównaniu z 4,4% (95% CI: 1,6 ‒ 7,2) u pacjentów
w grupie mitoksantronu, p = 0,0005.
Drugorzędowe punkty końcowe dotyczące PSA były korzystne w grupie otrzymującej kabazytaksel. Mediana czasu progresji wg PSA wynosiła 6,4 miesiąca (95% CI: 5,1-7,3) u pacjentów w grupie kabazytakselu w porównaniu z 3,1 miesiąca (95% CI: 2,2-4,4) w grupie mitoksantronu, HR
0,75 miesiąca (95% CI: 0,63 ‒ 0,90), p = 0,0010. Odpowiedź według stężenia PSA wynosiła 39,2% u pacjentów w grupie kabazytakselu (95% CI: 33,9-44,5) vs 17,8% u pacjentów otrzymujących mitoksantron (95% CI: 13,7 ‒ 22,0), p = 0,0002.
Pomiędzy obiema grupami nie stwierdzono znamiennych statystycznie różnic w progresji bólu
i odpowiedzi bólowej.
W międzynarodowym, wieloośrodkowym badaniu III fazy (EFC11785) typu non-inferiority (badanie typu badana interwencja nie jest gorsza), prowadzonym z randomizacją metodą otwartej próby,
1200 pacjentów z opornym na kastrację rakiem gruczołu krokowego z przerzutami leczonych wcześniej schematem leczenia zawierającym docetaksel, przydzielono losowo do dwóch grup otrzymujących albo dawkę 25 mg/m2 pc. (n = 602) kabazytakselu lub dawkę 20 mg/m2 pc. (n = 598). Pierwszorzędowym punktem końcowym skuteczności było przeżycie całkowite (ang. OS).
W badaniu osiągnięto podstawowy cel wskazujący, że dawka 20 mg/m2 pc. kabazytakselu nie jest gorsza w porównaniu z dawką 25 mg/m2 pc. (patrz tabela 4). Statystycznie istotny, większy procent (p < 0,001) pacjentów wykazał odpowiedź PSA na leczenie w grupie otrzymującej dawkę 25 mg/m2 pc. (42,9%) w porównaniu z grupą otrzymującą dawkę 20 mg/m2 pc. (29,5%). Zaobserwowano statystycznie istotne, większe ryzyko progresji wg PSA u pacjentów otrzymujących dawkę 20 mg/m2 pc. w porównaniu z dawką 25 mg/m2 pc. (HR 1,195; 95% CI: 1,025 ‒ 1,393). Nie stwierdzono znamiennych statystycznych różnic w odniesieniu do innych drugorzędowych punktów końcowych (PFS, odpowiedzi guza i bólowej, progresji guza i bólu oraz czterech podkategorii FACTP
[ang. Functional Assessment of Cancer Therapy-Prostate]).
Tabela 4: Całkowite przeżycie w badaniu EFC11785 w grupie otrzymującej kabazytaksel w dawce 25 mg/m2 pc. vs grupa otrzymująca kabazytaksel w dawce 20 mg/m2 pc. (analiza intent-to- treat) – pierwszorzędowy punkt końcowy skuteczności
CBZ20 + PRED n = 598 | CBZ25 + PRED n = 602 | |
Przeżycie całkowite | ||
Liczba zgonów, n (%) | 497 (83,1%) | 501 (83,2%) |
Mediana przeżycia (95% CI) (miesiące) | 13,4 (12,19 - 14,88) | 14,5 (13,47 - 15,28) |
Współczynnik hazardua vs CBZ25+PRED | 1,024 | - |
1-stronny 98,89% UCI | 1,184 | - |
1-stronny 95% LCI | 0,922 | - |
CBZ20 = kabazytaksel 20 mg/m2, CBZ25 = kabazytaksel 25 mg/m2, PRED = prednizon/prednizolon, CI = przedział ufności, LCI = dolna granica przedziału ufności, UCI = górna granica przedziału ufności.
aWspółczynnik hazardu oszacowany przy użyciu modelu regresji proporcjonalnego hazardu Coxa. Współczynnik hazardu <1 wskazuje na niższe ryzyko kabazytakselu 20 mg/m2 pc. w odniesieniu do 25 mg/m2 pc.
Profil bezpieczeństwa kabazytakselu w dawce 25 mg/m2 pc. zaobserwowany w badaniu EFC11785 był jakościowo i ilościowo podobny do zaobserwowanego w badaniu EFC6193. W badaniu EFC11785 wykazano lepszy profil bezpieczeństwa kabazytakselu w dawce 20 mg/m2 pc.
Tabela 5: Podsumowanie danych dotyczących bezpieczeństwa stosowania w badaniu EFC11785 w grupie otrzymującej kabazytaksel w dawce 25 mg/m2 pc. vs grupa otrzymująca kabazytaksel w dawce 20 mg/m2 pc
CBZ20 + PRED n = 580 | CBZ25 + PRED n = 595 | |
Mediana liczby cykli/ mediana długości leczenia | 6/18 tygodni | 7/21 tygodni |
Liczba pacjentów z redukcją dawki n (%) | 25 - 20 mg/m2 pc.: 128 (21,5%) | |
20 - 15 mg/m2 pc.: 58 (10,0%) | 20 - 15 mg/m2 pc.: 19 (3,2%) | |
15 - 12 mg/m2 pc.: 9 (1,6%) | 15 - 12 mg/m2 pc.: 1 (0,2%) | |
Działania niepożądane wszystkich stopnia (%) | ||
Biegunka | 30,7 | 39,8 |
Nudności | 24,5 | 32,1 |
Zmęczenie | 24,7 | 27,1 |
Krwiomocz | 14,1 | 20,8 |
Astenia | 15,3 | 19,7 |
Zmniejszony apetyt | 13,1 | 18,5 |
Wymioty | 14,5 | 18,2 |
Zaparcia | 17,6 | 18,0 |
Ból pleców | 11,0 | 13,9 |
Kliniczna neutropenia | 3,1 | 10,9 |
Zakażenie układu moczowego | 6,9 | 10,8 |
Obwodowa neuropatia czuciowa | 6.6 | 10,6 |
Zaburzenia smaku | 7,1 | 10,6 |
Działania niepożądane stopnia ≥3.b (%) | ||
Kliniczna neutropenia | 2,4 | 9,6 |
Gorączka neutropeniczna | 2,1 | 9,2 |
Nieprawidłowości hematologicznec (%) | ||
Neutropenia stopnia ≥3. | 41,8 | 73,3 |
Niedokrwistość stopnia ≥3. | 9,9 | 13,7 |
Małopłytkowość stopnia ≥3. | 2,6 | 4,2 |
CBZ20 = kabazytaksel 20 mg/m2 pc., CBZ25 = kabazytaksel 25 mg/m2 pc., PRED = prednizon/prednizolon.
aDziałania niepożądane wszystkich stopni występujące z częstością większą niż 10%.
bDziałania niepożądane stopnia ≥3. występujące z częstością większą niż 5%.
cNa podstawie badań laboratoryjnych.
W prospektywnym, międzynarodowym, randomizowanym, aktywnie kontrolowanym i otwartym badaniu IV fazy (badanie LPS14201/CARD) 255 pacjentów z przerzutowym rakiem gruczołu krokowego opornym na kastrację (mCRPC), wcześniej leczonych w dowolnej kolejności schematem zawierającym docetaksel i lekiem ukierunkowanym na AR (abirateron lub enzalutamid, z progresją choroby w ciągu 12 miesięcy od rozpoczęcia leczenia) był randomizowany do grupy otrzymującej kabazytaksel 25 mg/m2 co 3 tygodnie plus prednizon/prednizolon 10 mg na dobę (n=129) lub leki ukierunkowane na AR (abirateron 1000 mg raz na dobę plus prednizon/prednizolon 5 mg dwa razy na dobę lub enzalutamid 160 mg raz na dobę) (n=126). Pierwszorzędowym punktem końcowym było przeżycie wolne od progresji radiograficznej (rPFS), zgodnie z definicją grupy roboczej 2 raka gruczołu krokowego (PCWG2).
Drugorzędowe punkty końcowe obejmowały przeżycie całkowite, przeżycie bez progresji, odpowiedź PSA i odpowiedź guza.
Dane demograficzne i charakterystyka choroby były zrównoważone w grupach leczenia. Na początku ogólna mediana wieku wynosiła 70 lat, 95% pacjentów miało PS w skali ECOG od 0 do 1, a mediana wyniku w skali Gleasona wynosiła 8. Sześćdziesiąt jeden procent (61%) pacjentów było wcześniej leczonych lekiem ukierunkowanym na AR po wcześniejszym podaniu docetakselu.
Badanie osiągnęło swój pierwszorzędowy punkt końcowy: rPFS był istotnie dłuższy w przypadku kabazytakselu w porównaniu z lekiem nakierowanym na AR (odpowiednio 8,0 miesiąca w porównaniu z 3,7 miesiąca), z 46% zmniejszeniem ryzyka progresji radiograficznej w porównaniu z lekiem nakierowanym na AR (patrz tabela 6 i ryc. 2).
Tabela 6 Skuteczność kabazytakselu w badaniu CARD w leczeniu pacjentów z przerzutami raka gruczołu krokowego opornego na kastrację (analiza ITT) – Przeżycie bez progresji radiograficznej (rPFS)
Kabazytaksel+prednizon /prednizolon + G-CSF | Lek ukierunkowany na AR: abirateron+ prednizon/prednizolon lub enzalutamid | |
n=129 | n=126 | |
Liczba zdarzeń w dacie granicznej (%) | 95 (73,6%) | 101 (80,2%) |
Mediana rPFS (miesiące) (95% CI) | 8.0 (5,7 do 9,2) | 3.7 (2,8 do 5,1) |
Współczynnik ryzyka (HR) (95% CI) | 0,54 (0,40 do 0,73) | |
Wartość p1 | <0,0001 |
1stratyfikowany test log-rank, próg istotności = 0,05
Rycina 2 Pierwszorzędowy punkt końcowy: wykres Kaplana-Meiera dla radiograficznego PFS
(populacja ITT)
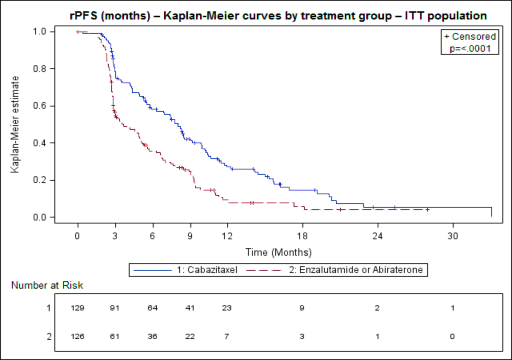
Znaczniki wskazują cenzurowane dane
Zaplanowane analizy podgrup pod kątem rPFS w oparciu o czynniki stratyfikacji przy randomizacji dały współczynnik ryzyka 0,61 (95% CI: 0,39 do 0,96) u pacjentów, którzy otrzymywali wcześniej lek ukierunkowany na AR przed docetakselem i współczynnik ryzyka 0,48 (95% CI: 0,32 do 0,70) u pacjentów, którzy po docetakselu otrzymywali wcześniej lek ukierunkowany na AR.
Kabazytaksel był statystycznie lepszy od komparatorów ukierunkowanych na AR dla każdego z kluczowych drugorzędowych punktów końcowych z ochroną alfa, w tym przeżycia całkowitego (13,6 miesiąca w ramieniu kabazytakselu vs. 11,0 miesiąca w ramieniu z lekiem ukierunkowanym na AR, HR 0,64, 95% CI: 0,46 do 0,89 p=0,008), przeżycie wolne od progresji (4,4 miesiąca dla ramienia kabazytakselu vs. 2,7 miesiąca dla ramienia leku ukierunkowanego na AR, HR 0,52; 95%CI: 0,40 do 0,68), potwierdzona odpowiedź PSA (36,3% dla ramienia kabazytakselu vs. 14,3% w ramieniu z lekiem ukierunkowanym na AR, p=0,0003 i najlepsza odpowiedź guza (36,5% w ramieniu kabazytakselu vs. 11,5% w ramieniu z lekiem ukierunkowanym na AR, p=0,004).
Profil bezpieczeństwa kabazytakselu 25 mg/m2 obserwowany w badaniu CARD był ogólnie zgodny z obserwowanym w badaniach TROPIC i PROSELICA (patrz punkt 4.8). Częstość występowania zdarzeń niepożądanych stopnia ≥3. wynosiła 53,2% w ramieniu kabazytakselu w porównaniu do 46,0% w ramieniu leków ukierunkowanych na AR. Częstość występowania ciężkich zdarzeń niepożądanych stopnia ≥3. wyniosła 31,7% w ramieniu kabazytakselu w porównaniu z 37,1% w ramieniu leków ukierunkowanych na AR. Częstość występowania pacjentów, którzy ostatecznie przerwali badane leczenie z powodu działań niepożądanych, wyniosła 19,8% w ramieniu kabazytakselu w porównaniu z 8,1% w ramieniu z lekiem nakierowanym na AR. Częstość występowania u pacjentów zdarzenia niepożądanego prowadzącego do zgonu wyniosła 5,6% w ramieniu kabazytakselu w porównaniu z 10,5% w ramieniu z lekiem ukierunkowanym na AR.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań kabazytakselu we wszystkich podgrupach populacji dzieci i młodzieży we wskazaniu rak gruczołu krokowego (patrz punkt 4.2, informacje na temat stosowania u dzieci i młodzieży).
Kabazytaksel oceniano w otwartym, wieloośrodkowym badaniu fazy I/II przeprowadzonym na grupie 39 pacjentów należących do populacji dzieci i młodzieży (w wieku pomiędzy 4 do 18 lat w fazie I. części badania oraz pomiędzy 3 do 16 lat w fazie II. części badania). W fazie II. nie wykazano skuteczności kabazytakselu w monoterapii u dzieci i młodzieży leczonych dawką 30 mg/m² pc. w nawracającym lub opornym na leczenie rozlanym glejaku pnia mózgu (DIPG, ang. diffuse intrinsic pontine glioma) oraz glejaku o wysokim stopniu złośliwości (HGG, ang. high grade glioma).
Analizę farmakokinetyki populacyjnej przeprowadzono w grupie 170 pacjentów, w tym pacjentów z zaawansowanymi nowotworami litymi (n = 69), rakiem piersi z przerzutami (n = 34) i rakiem gruczołu krokowego z przerzutami (n = 67). Pacjenci otrzymywali kabazytaksel w dawkach od
10 do 30 mg/m2 co tydzień lub co 3 tygodnie.
Wchłanianie
Po 1-godzinnej infuzji dożylnej kabazytakselu w dawce 25 mg/m2 pc. u pacjentów z rakiem gruczołu krokowego z przerzutami (n = 67) Cmax wynosiło 226 ng/ml (współczynnik zmienności [ang. CV, Coefficient of Variation]: 107%) i wartość ta została osiągnięta pod koniec trwania 1-godzinnej infuzji (Tmax). Średnia wartość AUC wynosiła 991 ng.h/ml (CV: 34%).
Nie obserwowano większych odchyleń od proporcjonalności dawki w zakresie dawek od
10 do 30 mg/m² pc. u pacjentów z zaawansowanymi nowotworami litymi (n = 126).
Dystrybucja
Objętość dystrybucji w stanie równowagi (Vss) wynosiła 4870 l (2640 l/m² pc. u pacjenta z medianą powierzchni ciała 1,84 m²).
W warunkach in vitro wiązanie kabazytakselu z ludzkimi białkami surowicy wynosiło 89-92%
i proces ten nie osiągnął stanu wysycenia do 50 000 ng/ml, co odpowiada najwyższemu stężeniu odnotowanemu w badaniach klinicznych. Kabazytaksel wiąże się głównie z ludzkimi albuminami surowicy (82,0%) i lipoproteinami (87,9% z HDL, 69,8% z LDL i 55,8% z VLDL). Współczynnik stężenia we krwi i osoczu w badaniach in vitro w ludzkiej krwi wahał się od 0,90 do 0,99, co wskazuje na równomierną dystrybucję kabazytakselu we krwi i osoczu.
Metabolizm
Kabazytaksel jest intensywnie metabolizowany w wątrobie (>95%), głównie za pośrednictwem izoenzymu CYP3A (od 80% do 90%). Kabazytaksel jest głównym związkiem krążącym w osoczu u ludzi. W osoczu stwierdzono obecność 7 metabolitów (w tym 3 aktywne metabolity powstałe
w wyniku O-demetylacji), z których główny stanowił 5% wpływu substancji czynnej na organizm. Około 20 metabolitów kabazytakselu jest wydalanych u ludzi z moczem i kałem.
Badania in vitro wskazują, że kabazytaksel w klinicznie istotnych stężeniach może potencjalnie hamować metabolizm produktów leczniczych, głównie będących substratami CYP3A. Jednakże badanie kliniczne wykazało, że kabazytaksel (w dawce 25 mg/m2 pc. podawany w pojedynczej infuzji trwającej 1 godzinę) nie zmieniał w osoczu stężenia midazolamu, który jest substratem wzorcowym dla CYP3A. Dlatego też u pacjentów otrzymujących w dawkach terapeutycznych jednocześnie substraty CYP3A razem z kabazytakselem nie jest spodziewany żaden wpływ kliniczny.
Nie istnieje potencjalne ryzyko zahamowania przemian metabolicznych produktów leczniczych będących substratami innych enzymów CYP (1A2, 2B6, 2C9, 2C8, 2C19, 2E1 i 2D6) ani indukcji przez kabazytaksel metabolizmu produktów leczniczych będących substratami CYP1A, CYP2C9 i CYP3A. Kabazytaksel nie hamował w warunkach in vitro głównego szlaku biotransformacji
warfaryny do 7-hydroksywarfaryny, zachodzącej za pośrednictwem CYP2C9. Dlatego nie przewiduje się interakcji farmakokinetycznych kabazytakselu i warfaryny w warunkach in vivo.
W badaniach in vitro kabazytaksel nie hamował białek oporności wielolekowej (ang. MRP, Multidrug-Resistant Proteins): MRP1 i MRP2 lub transporterów kationów organicznych (OCT1). Kabazytaksel hamował transport za pośrednictwem P-glikoproteiny (P-gp) (digoksyna, winblastyna), białek oporności raka piersi (ang. BCRP, Breast-Cancer-Resistant-Proteins) (metotreksat)
i polipeptydów transportujących aniony organiczne OATP1B3 (CCK8) w stężeniach przynajmniej 15- krotnie większych od obserwowanych w warunkach klinicznych, podczas gdy transport OATP1B1 (17β-glukuronid estradiolu) hamował w stężeniu tylko 5-krotnie wyższym od obserwowanych
w warunkach klinicznych. Z tego względu ryzyko interakcji z substratami MRP, OCT1, Pgp, BCRP oraz OATP1B3 w warunkach in vivo po podaniu dawki 25 mg/m2 pc. jest mało prawdopodobne.
Ryzyko interakcji z transporterami OATP1B1 jest możliwe, zwłaszcza w czasie trwania infuzji
(1 godzina) i aż do 20 minut po zakończeniu infuzji (patrz punkt 4.5).
Eliminacja
Po 1-godzinnej infuzji dożylnej [14C]-kabazytakselu w dawce 25 mg/m2 pc. około 80% podanej dawki zostało wydalone w ciągu 2 tygodni. Kabazytaksel jest głównie wydalany z kałem w postaci licznych metabolitów (76% dawki), podczas gdy wydalanie kabazytakselu i metabolitów przez nerki stanowi mniej niż 4% dawki (2,3% w postaci niezmienionego produktu leczniczego w moczu).
Kabazytaksel wykazuje wysoki klirens osoczowy wynoszący 48,5 l/h (26,4 l/h/m² pc. u pacjenta
o medianie powierzchni ciała wynoszącej 1,84 m²) i długi kres półtrwania w fazie końcowej wynoszący 95 godzin.
Szczególne grupy pacjentów
Pacjenci w podeszłym wieku
W populacyjnej analizie farmakokinetyki obejmującej 70 pacjentów w wieku 65 lat i starszych (57 pacjentów w wieku od 65 do 75 lat i 13 pacjentów powyżej 75 lat), nie stwierdzono wpływu wieku na farmakokinetykę kabazytakselu.
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania i skuteczności kabazytakselu u dzieci
i młodzieży w wieku poniżej 18 lat.
Zaburzenia czynności wątroby
Kabazytaksel jest wydalany głównie w wyniku metabolizmu wątrobowego.
W badaniu przeprowadzonym z udziałem 43 pacjentów nowotworowych z łagodnymi (stężenie bilirubiny całkowitej od >1 do ≤1,5 × GGN lub AspAT >1,5 × GGN) lub umiarkowanymi (stężenie bilirubiny całkowitej od >1,5 do ≤3 × GGN) zaburzeniami czynności wątroby nie wykazano, aby wywierały one wpływ na farmakokinetykę kabazytakselu. Maksymalna tolerowana dawka (MTD) kabazytakselu wynosiła odpowiednio 20 i 15 mg/m2 pc.
U 3 pacjentów z ciężkimi zaburzeniami czynności wątroby (stężenie bilirubiny całkowitej >3 × GGN), zaobserwowano zmniejszenie klirensu o 39% w porównaniu do pacjentów z łagodnymi zaburzeniami czynności wątroby, co wskazuje na pewien wpływ ciężkich zaburzeń czynności wątroby na farmakokinetykę kabazytakselu. Nie ustalono wielkości maksymalnej tolerowanej dawki kabazytakselu dla pacjentów z ciężkimi zaburzeniami czynności wątroby.
Na podstawie danych dotyczących bezpieczeństwa stosowania oraz tolerancji kabazytakselu jego dawka powinna zostać zmniejszona u pacjentów z łagodnymi zaburzeniami czynności wątroby (patrz punkty 4.2 i 4.4). Kabazytaksel jest przeciwskazany do stosowania u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkt 4.3).
Zaburzenia czynności nerek
Kabazytaksel jest w minimalnym stopniu wydalany przez nerki (2,3% dawki). Analiza farmakokinetyki populacyjnej przeprowadzona z udziałem 170 pacjentów, w tym 14 pacjentów
z umiarkowanymi zaburzeniami czynności nerek (klirens kreatyniny 30 – 50 ml/min) i 59 pacjentów z łagodnymi zaburzeniami czynności nerek (klirens kreatyniny 50 – 80 ml/min) wykazała, że łagodne do umiarkowanych zaburzenia czynności nerek nie wpływają znacząco na farmakokinetykę kabazytakselu. Zostało to potwierdzone w dedykowanych, porównawczych badaniach farmakokinetycznych u pacjentów z rakiem narządów miąższowych, z prawidłową czynnością nerek (8 pacjentów), umiarkowanymi zaburzeniami czynności nerek (8 pacjentów) i ciężkimi zaburzeniami czynności nerek (9 pacjentów), którzy otrzymali kilka cykli leczenia kabazytakselem w pojedynczych infuzjach, w dawce do 25 mg/m2 pc.
Działania niepożądane nieobserwowane w badaniach klinicznych, lecz obserwowane u psów po podaniu pojedynczej dawki, po zastosowaniu kabazytakselu przez 5 dni i w odstępach tygodniowych, przy poziomach ekspozycji niższych niż kliniczne poziomy ekspozycji i mogące mieć znaczenie
w stosowaniu klinicznym, obejmowały: zmiany martwicze tętniczek wątrobowych lub w ich sąsiedztwie, hiperplazję nabłonka kanalików żółciowych i (lub) martwicę hepatocytów (patrz punkt 4.2).
Działania niepożądane nieobserwowane w badaniach klinicznych, lecz obserwowane u szczurów w badaniach toksyczności po podaniu wielokrotnym, przy poziomach ekspozycji wyższych niż kliniczne poziomy ekspozycji i mogące mieć znaczenie przy stosowaniu klinicznym, obejmowały zaburzenia oka charakteryzujące się podtorebkowym obrzękiem i (lub) zwyrodnieniem włókien soczewki. Te zmiany były częściowo odwracalne po 8 tygodniach.
Dotychczas nie przeprowadzono badań dotyczących działania rakotwórczego kabazytakselu. Kabazytaksel nie indukował mutacji w teście rewersji mutacji u bakterii (Amesa). Nie stwierdzono działania klastogennego w badaniu in vitro przy użyciu limfocytów ludzkich (brak indukcji strukturalnych aberracji chromosomalnych, ale wzrosła liczby komórek poliploidalnych).
Odnotowano natomiast wzrost liczby mikrojąderek w teście in vivo u szczurów. Jednak ustalenia dotyczące genotoksyczności są naturalnym elementem aktywności farmakologicznej związku (hamowanie depolimeryzacji tubuliny) i były obserwowane także w przypadku produktów leczniczych wykazujących analogiczną aktywność farmakologiczną.
Kabazytaksel nie wpływał na kopulację i płodność u samców szczurów. Jednak w badaniu
toksyczności po podaniu wielokrotnym, obserwowano zwyrodnienie pęcherzyków nasiennych
i atrofię kanalików wyprowadzających jąder u szczurów oraz zwyrodnienie jąder (niewielkiego stopnia martwica pojedynczych komórek nabłonka najądrza) u psów. Ekspozycja u zwierząt była podobna lub niższa od obserwowanej u ludzi otrzymujących klinicznie odpowiadające dawki kabazytakselu.
Kabazytaksel podawany dożylnie raz dziennie w okresie od 6 do 17 dnia ciąży wywierał toksyczny wpływ na zarodki i płody u samic szczura, a także toksyczny wpływ na samice, prowadząc do obumarcia płodów i spadku średniej masy ciała płodu z towarzyszącym opóźnieniem procesu kostnienia szkieletu. Ekspozycja u zwierząt była niższa od obserwowanej u ludzi otrzymujących klinicznie odpowiadające dawki kabazytakselu. Kabazytaksel przenikał przez barierę łożyskową
u szczurów.
U szczurów kabazytaksel i jego metabolity przenikają do mleka samic w ilości do 1,5% dawki
podanej w ciągu 24 godzin.
Ocena ryzyka dla środowiska
Wyniki badań nad ryzykiem dla środowiska wykazały, że kabazytaksel nie będzie powodował znaczącego ryzyka dla środowiska wodnego (patrz punkt 6.6 dotyczący usuwania niezużytego produktu).
Koncentrat Polisorbat 80 Kwas cytrynowy
Rozpuszczalnik
Etanol 96%
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, oprócz wymienionych w punkcie 6.6.
Nie należy używać worków infuzyjnych wykonanych z PVC i poliuretanowych zestawów do infuzji.
Nieotwarta fiolka:
3 lata.
Po otwarciu
Fiolki z koncentratem i rozpuszczalnikiem należy zużyć natychmiast po otwarciu. W przeciwnym wypadku użytkownik ponosi odpowiedzialność za czas i warunki przechowywania.
Po wstępnym rozcieńczeniu koncentratu przy użyciu rozpuszczalnika
Wykazano stabilność chemiczną i fizyczną przez okres 30 minut w temperaturze otoczenia (15°C - 30°C). Z mikrobiologicznego punktu widzenia mieszaninę koncentratu i rozpuszczalnika należy zużyć natychmiast po przygotowaniu. W przeciwnym wypadku użytkownik ponosi odpowiedzialność za czas i warunki przechowywania.
Po końcowym rozcieńczeniu w worku i (lub) butelce do infuzji
Wykazano stabilność chemiczną i fizyczną roztworu do infuzji przez okres 8 godzin w temperaturze otoczenia (włączając czas infuzji wynoszący 1 godzinę) oraz przez 24 godziny w warunkach chłodniczych (włączając czas infuzji wynoszący 1 godzinę).
Z mikrobiologicznego punktu widzenia roztwór do infuzji należy zużyć natychmiast po przygotowaniu. W przeciwnym wypadku użytkownik ponosi odpowiedzialność za czas oraz warunki przechowywania i zwykle czas przechowywania nie powinien przekraczać 24 godzin w temperaturze 2°C – 8°C, chyba że rozcieńczenie miało miejsce w kontrolowanych i zwalidowanych warunkach aseptycznych.
Brak szczególnych środków ostrożności dotyczących przechowywania.
Warunki przechowywania produktu leczniczego po rozcieńczeniu, patrz punkt 6.3.
Jedno opakowanie zawiera jedną fiolkę z koncentratem i jedną fiolkę z rozpuszczalnikiem w tekturowym pudełku:
do stosowania
Produkt leczniczy Eleber powinien być przygotowywany i podawany jedynie przez personel przeszkolony w obchodzeniu się z substancjami cytotoksycznymi. Kobiety w ciąży należące do personelu nie powinny mieć kontaktu z produktem leczniczym. Podobnie jak w przypadku innych leków przeciwnowotworowych należy zachować ostrożność podczas obchodzenia się z produktem leczniczym Eleber i przygotowywania jego roztworów, biorąc pod uwagę użycie wyposażenia ograniczającego ekspozycję na produkt leczniczy, środków ochrony osobistej (np. rękawiczki) i procedur przygotowania produktu leczniczego do użycia. W przypadku kontaktu produktu leczniczego Eleber ze skórą na dowolnym etapie obchodzenia się z nim należy natychmiast dokładnie umyć zanieczyszczone miejsce wodą z mydłem. W przypadku kontaktu z błoną śluzową miejsce zanieczyszczone należy natychmiast dokładnie zmyć wodą.
Koncentrat do sporządzania roztworu do infuzji należy zawsze rozcieńczyć przy pomocy całego
załączonego rozpuszczalnika przed dodaniem do roztworu do infuzji.
Przed mieszaniem i rozcieńczeniem należy uważnie przeczytać CAŁY punkt. Przygotowanie produktu leczniczego Eleber przed podaniem wymaga DWÓCH etapów rozcieńczenia. Należy postępować zgodnie z zamieszczoną poniżej instrukcją.
Uwaga: zarówno fiolka z koncentratem produktu leczniczego Eleber 60 mg/1,5 ml (objętość napełnienia: 73,2 mg kabazytakselu/1,83 ml), jak i fiolka z rozpuszczalnikiem (objętość napełnienia: 5,67 ml) zawierają nadmiar płynu w celu wyrównania jego strat podczas przygotowania. Nadmiar ten zapewnia, że po rozcieńczeniu CAŁĄ zawartością dołączonego rozpuszczalnika otrzymany roztwór zawiera 10 mg/ml kabazytakselu.
Opisany poniżej dwuetapowy proces rozcieńczania musi być przeprowadzony w sposób aseptyczny w celu przygotowania roztworu do infuzji.
Etap 1: Wstępne rozcieńczenie koncentratu do sporządzania roztworu do infuzji przy użyciu załączonego rozpuszczalnika.
Etap 1.1
Należy uważnie obejrzeć fiolkę z koncentratem oraz fiolkę z dołączonym rozpuszczalnikiem. Roztwór koncentratu i rozpuszczalnik powinny być przejrzyste.
Fiolka
z rozpuszczalnikiem
Fiolka z koncentratem (60 mg – 1,5 ml)
Etap 1.2
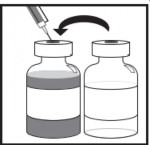
Używając strzykawki z przymocowaną igłą, z zachowaniem jałowości, należy pobrać całą zawartość dołączonego rozpuszczalnika, obracając częściowo fiolkę.









Fiolka
z rozpuszczalnikiem
Etap 1.3
Wstrzyknąć całą zawartość do odpowiedniej fiolki z koncentratem.
Aby podczas wstrzykiwania rozpuszczalnika w jak największym stopniu ograniczyć powstawanie piany, należy skierować igłę na wewnętrzną ścianę fiolki z roztworem koncentratu i wstrzykiwać powoli.
Po pierwszym rozcieńczeniu otrzymany roztwór zawiera 10 mg/ml kabazytakselu.
Fiolka
z rozpuszczalnikiem
Mieszanina koncentratu
z rozpuszczalnikiem 10 mg/ml
Etap 1.4
Usunąć strzykawkę i igłę oraz wymieszać delikatnie ręcznie poprzez wielokrotne odwracanie do momentu otrzymania przejrzystego i jednorodnego roztworu. Może to trwać około 45 sekund.

Mieszanina koncentratu
z rozpuszczalnikiem 10 mg/ml
Etap 1.5
Zostawić roztwór na około 5 minut i następnie sprawdzić, czy roztwór jest jednorodny i przejrzysty. Utrzymywanie się piany po tym czasie jest zjawiskiem normalnym.
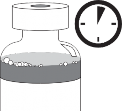
Mieszanina koncentratu
z rozpuszczalnikiem 10 mg/ml
Otrzymana mieszanina koncentratu i rozpuszczalnika zawiera kabazytaksel w stężeniu 10 mg/ml
(co najmniej 6 ml objętości do podania). Drugie rozcieńczenie należy wykonać natychmiast (w ciągu
1 godziny) zgodnie z opisem w punkcie Etap 2.
Do podania przepisanej dawki może być potrzebna więcej niż jedna fiolka mieszaniny koncentratu i rozpuszczalnika.
Etap 2: Drugie (ostatnie) rozcieńczenie roztworu do infuzji
Etap 2.1
Przenieść z zachowaniem jałowości wymaganą objętość mieszaniny koncentratu i rozpuszczalnika (stężenie kabazytakselu 10 mg/ml), używając strzykawki z podziałką i przymocowaną igłą. Na przykład dawka 45 mg produktu leczniczego Eleber będzie wymagać podania 4,5 ml mieszaniny koncentratu i rozpuszczalnika przygotowanej zgodnie z opisem w punkcie Etap 1.
Podczas ekstrakcji zaleca się umieścić igłę strzykawki wewnątrz, postępując zgodnie z opisem w punkcie Etap 1, ponieważ może utrzymywać się piana na ściankach fiolki z roztworem.
Mieszanina koncentratu
z rozpuszczalnikiem 10 mg/ml






Etap 2.2
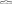
Wstrzyknąć do jałowego worka wykonanego z innego materiału niż PVC, zawierającego 5% roztwór glukozy lub 9 mg/ml (0,9%) roztwór chlorku sodu do infuzji.
Stężenie roztworu do infuzji powinno wynosić od 0,10 mg/ml do 0,26 mg/ml.
5% roztwór glukozy lub 9 mg/ml (0,9%) roztwór chlorku sodu do infuzji
Wymagana ilość mieszaniny
koncentratu z rozpuszczalnikiem


Etap 2.3

Usunąć strzykawkę i wymieszać ręcznie zawartość worka lub butelki do infuzji, wykonując ruch kołysania.
Etap 2.4
Tak jak w przypadku każdego produktu leczniczego do podawania pozajelitowego, otrzymany roztwór do infuzji należy obejrzeć przed użyciem. Ponieważ roztwór do infuzji jest przesycony, w miarę upływu czasu może krystalizować. W takim przypadku roztworu nie wolno używać i należy go usunąć.

Roztwór do infuzji należy zużyć natychmiast po przygotowaniu. Czas przechowywania przygotowanego roztworu może być jednak dłuższy w określonych warunkach opisanych w punkcie 6.3.
Podczas podawania produktu leczniczego zaleca się stosować filtr o nominalnej wielkości porów 0,22 mikrometra (określany także jako 0,2 mikrometra), założony na zestawie do infuzji.
Do przygotowywania i podawania produktu leczniczego Eleber nie należy używać worków do infuzji wykonanych z PVC lub poliuretanowych zestawów do infuzji.
Produktu leczniczego Eleber nie wolno mieszać z innymi produktami leczniczymi niż wymieniono.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami.
DOPUSZCZENIE DO OBROTU
Zentiva k.s.
U kabelovny 130 Dolní Měcholupy 102 37 Praga 10 Republika Czeska
Pozwolenie nr 26950
OBROTU / DATA PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 10.03.2022








