







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przed rozpoczęciem leczenia Dabisanem należy ocenić czynność nerek poprzez obliczenie klirensu kreatyniny (CrCL) w celu wykluczenia pacjentów z ciężkimi zaburzeniami czynności nerek (np. CrCL<30 mL/min) (patrz punkty 4.3, 4.4 i 5.2).
Czynność nerek należy również ocenić, gdy podejrzewa się pogorszenie czynności nerek podczas leczenia (np. hipowolemia, odwodnienie oraz w przypadku jednoczesnego stosowania wybranych produktów leczniczych).
Metodą przeznaczoną do oceny czynności nerek (CrCL w mL/min) jest metoda Cockcroft-Gault.
Pominięcie dawki
Zaleca się kontynuację stosowania pozostałych dawek dobowych dabigatranu eteksylanu o tej samej porze następnego dnia.
Nie należy stosować dawki podwójnej w celu uzupełniania pominiętej dawki.
Przerwanie stosowania dabigatranu eteksylanu
Nie należy przerywać leczenia dabigatranem eteksylanem bez wcześniejszej konsultacji z lekarzem. Należy pouczyć pacjentów, aby skontaktowali się z lekarzem prowadzącym w przypadku wystąpienia objawów ze strony układu pokarmowego, takich jak niestrawność (patrz punkt 4.8).
Zmiana leczenia
Z dabigatranu eteksylanu na lek przeciwzakrzepowy podawany pozajelitowo:
Po podaniu ostatniej dawki dabigatranu eteksylanu zaleca się odczekać 24 godziny przed zmianą na lek przeciwzakrzepowy podawany pozajelitowo (patrz punkt 4.5).
Z pozajelitowych leków przeciwzakrzepowych na dabigatran eteksylan:
Należy przerwać podawanie pozajelitowego leku przeciwzakrzepowego i rozpocząć podawanie dabigatranu eteksylanu od 0 do 2 godzin przed zaplanowanym terminem podania następnej dawki pozajelitowego leku przeciwzakrzepowego, lub w czasie przerwania stosowania w przypadku leczenia ciągłego (np. dożylnego podawania niefrakcjonowanej heparyny (ang. UFH – Unfractionated Heparin)) (patrz punkt 4.5).
Szczególne grupy pacjentów
Zaburzenia czynności nerek
Stosowanie dabigatranu eteksylanu u pacjentów z ciężkimi zaburzeniami czynności nerek (CrCL
<30 mL/min) jest przeciwwskazane (patrz punkt 4.3).
U pacjentów z umiarkowanymi zaburzeniami czynności nerek (CrCL 30-50 mL/min) zaleca się zmniejszenie dawki (patrz tabela 1 powyżej oraz punkty 4.4 i 5.1).
Stosowanie dabigatranu eteksylanu jednocześnie ze słabo/umiarkowanie działającymi inhibitorami P-glikoproteiny (P-gp), np. amiodaronem, chinidyną lub werapamilem
Dawkę produktu leczniczego należy zmniejszyć w sposób wskazany w tabeli 1 (patrz również punkty 4.4 i 4.5). W takim przypadku dabigatran eteksylan oraz inne produkty lecznicze powinny być przyjmowane jednocześnie.
U pacjentów z umiarkowanymi zaburzeniami czynności nerek jednocześnie leczonych werapamilem należy rozważyć zmniejszenie dawki dabigatranu eteksylanu do 75 mg na dobę (patrz punkty 4.4 i 4.5).
Pacjenci w podeszłym wieku
U pacjentów w podeszłym wieku (>75 lat) zaleca się zmniejszenie dawki (patrz tabela 1 powyżej oraz punkty 4.4 i 5.1).
Masa ciała
Istnieje bardzo ograniczone doświadczenie kliniczne dotyczące stosowania produktu leczniczego u pacjentów o masie ciała mniejszej niż 50 kg lub większej niż 110 kg w zalecanej dawce. Na podstawie dostępnych danych klinicznych i właściwości farmakokinetycznych nie jest konieczna modyfikacja dawkowania (patrz punkt 5.2), jednak zalecana jest ścisła obserwacja kliniczna pacjenta (patrz
punkt 4.4).
Płeć
Nie jest konieczna modyfikacja dawki (patrz punkt 5.2).
Dzieci i młodzież
Stosowanie dabigatranu eteksylanu u dzieci i młodzieży nie jest właściwe we wskazaniu w prewencji pierwotnej ŻChZZ u pacjentów po przebytej planowej alloplastyce całkowitej stawu biodrowego lub kolanowego.
Leczenie ŻChZZ i prewencja nawrotów ŻChZZ u dzieci i młodzieży
W przypadku leczenia ŻChZZ u dzieci i młodzieży leczenie należy rozpocząć po terapii lekiem przeciwzakrzepowym podawanym pozajelitowo przez przynajmniej 5 dni. W przypadku prewencji nawrotów ŻChZZ leczenie należy rozpocząć po uprzedniej terapii.
Dabigatran eteksylan w postaci kapsułek należy przyjmować dwa razy na dobę, jedną dawkę rano i jedną dawkę wieczorem, mniej więcej o tej samej porze każdego dnia. Odstęp między dawkami powinien wynosić w miarę możliwości 12 godzin.
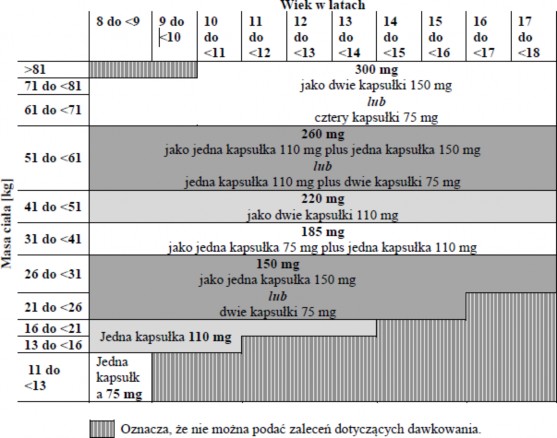
Zalecana dawka dabigatranu eteksylanu w postaci kapsułek zależy od wieku i masy ciała pacjenta zgodnie z tabelą 2. W tabeli podano pojedyncze dawki, które należy podawać dwa razy na dobę. W trakcie leczenia dawkę należy dostosowywać do wieku i masy ciała.
Tabela 2: Pojedyncza dawka dabigatranu eteksylanu w miligramach (mg) w zależności od masy ciała pacjenta w kilogramach (kg) i wieku w latach, podawana dwa razy na dobę
Ocena czynności nerek przed i w trakcie leczenia
Przed rozpoczęciem leczenia należy wyliczyć szacunkowy współczynnik przesączania kłębuszkowego (eGFR) na podstawie wzoru Schwartza.
Stosowanie dabigatranu eteksylanu u dzieci i młodzieży z eGFR <50 mL/min/1,73m2 jest przeciwwskazane (patrz punkt 4.3).
Pacjentów z eGFR ≥50 mL/min/1,73 m2 należy leczyć dawką zgodnie z tabelą 2.
Podczas leczenia czynność nerek należy oceniać w wybranych sytuacjach klinicznych, gdy
podejrzewa się osłabienie lub pogorszenie czynności nerek (takie jak hipowolemia, odwodnienie oraz w przypadku jednoczesnego stosowania wybranych produktów leczniczych itp.).
Czas stosowania
Czas trwania terapii powinien być ustalany indywidualnie na podstawie oceny stosunku korzyści i ryzyka.
Pominięcie dawki
Pominiętą dawkę dabigatranu eteksylanu można przyjąć do 6 godzin przed kolejną zaplanowaną dawką. Jeśli do kolejnej zaplanowanej dawki pozostało mniej niż 6 godzin, należy pominąć ominiętą dawkę.
Nigdy nie wolno stosować dawki podwójnej w celu uzupełnienia pominiętej dawki.
Przerwanie stosowania dabigatrananu eteksylanu
Nie należy przerywać leczenia dabigatranem eteksylanem bez wcześniejszej konsultacji z lekarzem. Należy pouczyć pacjentów lub ich opiekunów, aby skontaktowali się z lekarzem prowadzącym w przypadku wystąpienia objawów ze strony układu pokarmowego, takich jak niestrawność (patrz punkt 4.8).
Zmiana leczenia
Z dabigatranu eteksylanu na lek przeciwzakrzepowy podawany pozajelitowo:
Po podaniu ostatniej dawki dabigatranu eteksylanu zaleca się odczekać 12 godzin przed zmianą na lek przeciwzakrzepowy podawany pozajelitowo (patrz punkt 4.5).
Z pozajelitowych leków przeciwzakrzepowych na dabigatran eteksylan:
Należy przerwać podawanie pozajelitowego leku przeciwzakrzepowego i rozpocząć podawanie dabigatranu eteksylanu od 0 do 2 godzin przed zaplanowanym terminem podania następnej dawki pozajelitowego leku przeciwzakrzepowego, lub w czasie przerwania stosowania w przypadku leczenia ciągłego (np. dożylnego podawania niefrakcjonowanej heparyny (ang. UFH – Unfractionated Heparin)) (patrz punkt 4.5).
Z dabigatranu eteksylanu na antagonistę witaminy K (ang. VKA - Vitamin K Antagonists): Pacjenci powinni rozpocząć stosowanie VKA 3 dni przed przerwaniem leczenia dabigatranem eteksylanem .
Dabigatran eteksylan może mieć wpływ na wartości międzynarodowego współczynnika znormalizowanego (INR), dlatego pomiar INR lepiej odzwierciedli działanie VKA wyłącznie wówczas, gdy zostanie wykonany po przerwaniu terapii dabigatranem eteksylanem na przynajmniej 2 dni. Do tego czasu wartości pomiaru INR powinny być interpretowane z ostrożnością.
Z VKA na eteksylan dabigatranu:
Należy przerwać stosowanie VKA. Podawanie dabigatranu eteksylanu należy rozpocząć, jak tylko INR wyniesie <2,0.
Sposób podawania
Ten produkt leczniczy jest przeznaczony do stosowania doustnego.
Kapsułki mogą być przyjmowane z posiłkiem lub bez posiłku. Kapsułki należy połykać w całości, popijając szklanką wody w celu ułatwienia przedostania się do żołądka.
Należy pouczyć pacjentów, aby nie otwierali kapsułek, ponieważ może to zwiększyć ryzyko krwawienia (patrz punkty 5.2 i 6.6).
Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
Ciężkie zaburzenia czynności nerek (CrCL <30 mL/min) u dorosłych pacjentów
eGFR <50 mL/min/1,73 m2 u dzieci i młodzieży
Czynne, istotne klinicznie krwawienie
Zmiana lub schorzenie uważane za istotny czynnik ryzyka poważnego krwawienia, w tym owrzodzenie w obrębie przewodu pokarmowego obecnie lub w przeszłości, nowotwory złośliwe obarczone wysokim ryzykiem krwawienia, niedawny uraz mózgu lub rdzenia kręgowego, niedawny zabieg chirurgiczny mózgu, rdzenia kręgowego lub okulistyczny,
niedawny krwotok śródczaszkowy, stwierdzone lub podejrzewane żylaki przełyku, malformacje tętniczo-żylne, tętniaki naczyniowe lub istotne nieprawidłowości naczyniowe w obrębie rdzenia kręgowego lub mózgu
Leczenie skojarzone z jakimikolwiek produktami przeciwzakrzepowymi np. niefrakcjonowana heparyna (UHF), heparyny drobnocząsteczkowe (enoksaparyna, dalteparyna, itp.) pochodne heparyny (fondaparynuks itp.) doustne antykoagulanty (warfaryna, rywaroksaban, apiksaban itp.) z wyjątkiem szczególnych okoliczności. Należą do nich zamiana terapii przeciwzakrzepowej (patrz punkt 4.2), kiedy UHF jest podawana w dawkach niezbędnych do podtrzymania drożności cewników w naczyniach centralnych żylnych lub naczyniach
tętniczych lub kiedy UHF jest podawana podczas ablacji cewnikowej w migotaniu przedsionków (patrz punkt 4.5)
Zaburzenia czynności wątroby lub choroba wątroby o potencjalnym niekorzystnym wpływie na przeżycie
Leczenie skojarzone z następującymi silnymi inhibitorami P-gp: stosowanymi układowo
ketokonazolem, cyklosporyną, itrakonazolem, dronedaronem oraz lekiem złożonym o ustalonej dawce zawierającym glekaprewir i pibrentaswir (patrz punkt 4.5).
Stan po wszczepieniu sztucznej zastawki serca wymagający leczenia przeciwzakrzepowego (patrz punkt 5.1).
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Umiarkowane zaburzenia czynności nerek u dorosłych pacjentów (30-50 mL/min CrCL)
Silne inhibitory P-gp (patrz punkt 4.3 i 4.5)
Jednoczesne stosowanie słabo do umiarkowanie działającego inhibitora P-gp (np. amiodaron, werapamil, chinidyna i tikagrelor; patrz punkt 4.5)
Niska masa ciała (<50 kg) u dorosłych pacjentów
ASA i inne leki hamujące agregację płytek krwi, takie jak klopidogrel
NLPZ
SSRI lub SNRI
Inne produkty lecznicze, które mogą zaburzać hemostazę
Wrodzone lub nabyte zaburzenia krzepliwości
Małopłytkowość lub zaburzenia czynności płytek krwi
Niedawna biopsja lub duży uraz
Bakteryjne zapalenie wsierdzia
Zapalenie błony śluzowej przełyku, zapalenie błony śluzowej żołądka lub
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
wartość 90 percentyla stężenia dabigatranu w osoczu wynosiła 67 ng/mL (pomiar w
stężeniu minimalnym, 20-28 godzin po przyjęciu wcześniejszej dawki) (patrz punkt 4.4 i 4.9),
wartość 90 percentyla APTT (pomiar w stężeniu minimalnym, 20-28 godzin po przyjęciu wcześniejszej dawki) wynosił 51 sekund, co odpowiada 1,3-krotności górnego limitu normy.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Celuloza mikrokrystaliczna
Kroskarmeloza sodowa
Krospowidon
Kwas winowy (w postaci peletek)
Hydroksypropyloceluloza
Mannitol
Magnezu stearynian
Talk
Otoczka kapsułki
Żelaza tlenek czerwony (E 172)
Tytanu dwutlenek (E 171)
Hypromeloza
Czarny tusz do nadruków
Szelak
Glikol propylenowy
Amonowy wodorotlenek stężony
Żelaza tlenek czarny (E172)
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
Aby wyjąć kapsułkę, należy odkleić folię zabezpieczającą blister.
Nie należy wypychać kapsułek twardych przez folię blistra.
Należy odkleić folię blistra tylko przed wymaganym przyjęciem kapsułki twardej.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Wasedoc, 75 mg, kapsułki twarde
Każda kapsułka twarda zawiera 75 mg dabigatranu eteksylanu (w postaci mezylanu). Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Kapsułka twarda.
Kapsułki z różowym, nieprzezroczystym wieczkiem i różowym, nieprzezroczystym korpusem rozmiaru 2 (około 18,3 mm), z nadrukowanym czarnym tuszem napisem „DA75”, zawierające białe lub białawe peletki i jasnożółty granulat.
Prewencja pierwotna żylnej choroby zakrzepowo-zatorowej - ŻChZZ (ang. VTE – venous thromboembolic events) u dorosłych pacjentów po przebytej planowej alloplastyce całkowitej stawu biodrowego lub kolanowego.
Leczenie ŻChZZ i zapobieganie nawrotom ŻChZZ u dzieci i młodzieży od 8 lat do wieku poniżej 18 lat.
Postaci farmaceutyczne odpowiednie dla wieku, patrz punkt 4.2.
Dawkowanie
Produkt leczniczy Wasedoc w postaci kapsułek może być stosowany u dorosłych oraz dzieci i młodzieży w wieku 8 lat lub starszych, którzy potrafią połykać kapsułki w całości.
Dawkę podaną w odpowiedniej tabeli dawkowania danej postaci farmaceutycznej należy przepisać na podstawie wieku i masy ciała dziecka.
Prewencja pierwotna ŻChZZ po zabiegach ortopedycznych
Informacje na temat zalecanych dawek dabigatranu eteksylanu i czasu trwania leczenia w prewencji pierwotnej ŻChZZ po zabiegach ortopedycznych przedstawiono w tabeli 1.
Tabela 1: Zalecenia dotyczące dawkowania i czas trwania leczenia w prewencji pierwotnej ŻChZZ po zabiegach ortopedycznych
Rozpoczęcie leczenia w dniu zabiegu chirurgicznego w ciągu 1 do 4 godzin od zakończenia zabiegu chirurgicznego | Rozpoczęcie leczenia dawką podtrzymującą w pierwszym dniu po zabiegu chirurgicznym | Czas trwania leczenia dawką podtrzymującą | |
Pacjenci po przebytej planowej | 220 mg | 10 dni | |
alloplastyce stawu kolanowego | dabigatranu | ||
jedna kapsułka dabigatranu eteksylanu o mocy 110 mg | eteksylanu raz na dobę w postaci 2 kapsułek o | ||
Pacjenci po przebytej planowej alloplastyce stawu biodrowego | 28-35 dni | ||
mocy 110 mg | |||
Zalecane zmniejszenie dawki | |||
Pacjenci z umiarkowanymi | jedna kapsułka dabigatranu eteksylanu o mocy 75 mg | 150 mg dabigatranu eteksylanu raz na dobę w postaci 2 kapsułek o mocy 75 mg | 10 dni (alloplastyka stawu kolanowego) lub 28-35 dni (alloplastyka stawu biodrowego) |
zaburzeniami czynności nerek | |||
(klirens kreatyniny, CrCL | |||
30-50 mL/min) | |||
Pacjenci jednocześnie przyjmujący | |||
werapamil*, amiodaron, chinidynę | |||
Pacjenci w wieku 75 lat lub starsi |
* Pacjenci z umiarkowanymi zaburzeniami czynności nerek jednocześnie leczeni werapamilem, patrz
„Szczególne grupy pacjentów”
W przypadku obu zabiegów chirurgicznych należy odsunąć w czasie rozpoczęcie leczenia, jeżeli nie zostanie zapewniona hemostaza. Jeżeli leczenie nie zostanie rozpoczęte w dniu zabiegu
chirurgicznego, wówczas należy je rozpocząć od podania 2 kapsułek raz na dobę.
Ocena czynności nerek przed i w trakcie leczenia dabigatranem eteksylanem
U wszystkich pacjentów, a szczególnie u pacjentów w podeszłym wieku (>75 lat), ponieważ w tej grupie wiekowej zaburzenia czynności nerek mogą być częste:
Ryzyko krwotoku
Należy zachować ostrożność podczas stosowania dabigatranu eteksylanu w przypadku chorób związanych ze zwiększonym ryzykiem krwawienia lub w przypadku jednoczesnego stosowania produktów leczniczych wpływających na hemostazę poprzez zahamowanie agregacji płytek krwi.
Podczas leczenia krwawienie może wystąpić w każdym miejscu. Niewyjaśniony spadek stężenia hemoglobiny i (lub) hematokrytu lub ciśnienia tętniczego krwi powinien prowadzić do poszukiwania miejsca krwawienia.
U dorosłych pacjentów w razie zagrażającego życiu lub nieopanowanego krwawienia, w sytuacjach, w których konieczne jest szybkie odwrócenie działania przeciwzakrzepowego dabigatranu, dostępny jest swoisty czynnik odwracający, idarucyzumab. Wasedoc nie jest przeznaczony do stosowania jako zestaw. Nie określono skuteczności ani bezpieczeństwa stosowania idarucyzumabu u dzieci i
młodzieży. Dabigatran można usunąć na drodze hemodializy. U dorosłych pacjentów inne możliwe opcje to świeża krew pełna lub osocze świeżo mrożone, koncentrat czynników krzepnięcia (aktywowanych lub nieaktywowanych), koncentraty rekombinowanego czynnika VIIa lub płytek krwi (patrz również punkt 4.9).
Stosowanie leków hamujących agregację płytek krwi, takich jak klopidogrel i kwas acetylosalicylowy (ASA) lub niesteroidowe leki przeciwzapalne (NLPZ), jak również występowanie zapalenia przełyku, żołądka lub refluksu żołądkowo-przełykowego zwiększa ryzyko krwawienia z przewodu pokarmowego.
Czynniki ryzyka
W tabeli 3 podsumowano czynniki mogące zwiększać ryzyko krwotoku. Tabela 3: Czynniki mogące zwiększać ryzyko krwotoku.
Czynniki farmakodynamiczne i farmakokinetyczne | Wiek ≥75 lat |
Czynniki zwiększające stężenia osoczowe dabigatranu | Główne: Dodatkowe: |
Interakcje farmakodynamiczne | |
Choroby/zabiegi o szczególnym ryzyku krwotoku | refluks żołądkowo-przełykowy |
Dane dotyczące dorosłych pacjentów o masie ciała <50 kg są ograniczone (patrz punkt 5.2).
Jednoczesne stosowanie dabigatranu eteksylanu z inhibitorami P-gp nie zostało przebadane u dzieci i młodzieży, ale może zwiększać ryzyko krwawienia (patrz punkt 4.5).
Środki ostrożności i postępowanie w przypadku ryzyka krwotoku
Postępowanie w przypadku powikłań krwawienia, patrz również punkt 4.9.
Ocena stosunku korzyści do ryzyka
Uszkodzenia, schorzenia, zabiegi i (lub) leczenie farmakologiczne (takie jak NPLZ, leki przeciwpłytkowe, SSRI i SNRI, patrz punkt 4.5), które istotnie zwiększają ryzyko dużego krwawienia, wymagają starannej oceny stosunku korzyści do ryzyka. Dabigatran eteksylan należy podawać tylko wtedy, jeśli korzyść z leczenia przewyższa ryzyko krwawienia.
Dostępne są ograniczone dane kliniczne dla dzieci i młodzieży z czynnikami ryzyka, w tym pacjentów z czynnym zapaleniem opon mózgowo-rdzeniowych, zapaleniem mózgu i ropniem śródczaszkowym (patrz punkt 5.1). U tych pacjentów dabigatran eteksylan można podawać tylko wtedy, jeśli
oczekiwane korzyści przewyższają ryzyko krwawienia.
Ścisłe monitorowanie kliniczne
Ścisła obserwacja w kierunku objawów krwawienia lub niedokrwistości jest zalecana przez cały okres leczenia, szczególnie w przypadku występujących jednocześnie czynników ryzyka (patrz tabela 3 powyżej). Należy zachować szczególną ostrożność w przypadku podawania dabigatranu eteksylanu jednocześnie z werapamilem, amiodaronem, chinidyną lub klarytromycyną (inhibitorami P-gp) oraz szczególnie w przypadku wystąpienia krwawienia, zwłaszcza u pacjentów ze zmniejszoną czynnością nerek (patrz punkt 4.5).
Ścisłe monitorowanie w kierunku objawów krwawienia jest zalecane u pacjentów jednocześnie leczonych NLPZ (patrz punkt 4.5).
Przerwanie leczenia dabigatranem eteksylanem
U pacjentów, u których wystąpi ostra niewydolność nerek, należy przerwać leczenie dabigatranem eteksylanem (patrz również punkt 4.3).
W przypadku wystąpienia silnego krwawienia leczenie musi zostać przerwane, źródło krwawienia musi zostać określone i można rozważyć zastosowanie swoistego czynnika odwracającego (idarucyzumab) u dorosłych pacjentów. Nie określono skuteczności ani bezpieczeństwa stosowania idarucyzumabu u dzieci i młodzieży. Dabigatran można usunąć na drodze hemodializy.
Stosowanie inhibitorów pompy protonowej
Można rozważyć podanie inhibitora pompy protonowej (PPI) w celu uniknięcia krwawienia z przewodu pokarmowego. W przypadku dzieci i młodzieży należy stosować się do lokalnych zaleceń podanych na oznakowaniu opakowań inhibitorów pompy protonowej.
Parametry krzepnięcia w badaniach laboratoryjnych
Mimo że stosowanie tego produktu leczniczego nie wiąże się na ogół z koniecznością rutynowego monitorowania działania przeciwzakrzepowego, oznaczenie działania przeciwzakrzepowego dabigatranu może być pomocne w wykryciu nadmiernej ekspozycji na dabigatran w przypadku występowania dodatkowych czynników ryzyka.
Czas trombinowego krzepnięcia w rozcieńczonym osoczu (dTT), ekarynowy czas krzepnięcia (ECT) i czas kaolinowo-kefalinowy (aPTT) mogą dostarczyć przydatnych informacji, jednak uzyskane wyniki należy interpretować z zachowaniem ostrożności ze względu na zmienność wyników między badaniami (patrz punkt 5.1).
U pacjentów stosujących dabigatran eteksylan badanie międzynarodowego współczynnika znormalizowanego (INR) nie daje wiarygodnych wyników i zgłaszano przypadki uzyskania wyników fałszywie podwyższonych. Dlatego nie należy wykonywać badania INR.
Tabela 4 przedstawia najniższe progowe wartości badań krzepnięcia u dorosłych pacjentów, które mogą wskazywać na zwiększone ryzyko krwawienia. Odpowiednie wartości progowe u dzieci
i młodzieży nie są znane (patrz punkt 5.1).
Tabela 4: Najniższe progowe wartości badań krzepnięcia u dorosłych pacjentów, które mogą wskazywać na zwiększone ryzyko krwawienia.
Badanie (najniższa wartość) | Wartość progowa |
dTT [ng/mL] | >67 |
ECT [x-krotność górnego limitu normy] | Brak danych |
aPTT [x-krotność górnego limitu normy] | >1,3 |
INR | Nie należy wykonywać |
Stosowanie produktów leczniczych fibrynolitycznych w leczeniu ostrego udaru niedokrwiennego mózgu
Stosowanie produktów leczniczych fibrynolitycznych w leczeniu ostrego udaru niedokrwiennego mózgu może być wzięte pod uwagę w przypadku, gdy wyniki badań dTT, ECT lub aPTT nie
przekraczają górnej granicy normy (GGN) zgodnie z lokalnym zakresem wartości referencyjnych. Zabiegi chirurgiczne i procedury inwazyjne
Pacjenci leczeni dabigatranem eteksylanem , poddawani zabiegom chirurgicznym lub procedurom inwazyjnym są w grupie zwiększonego ryzyka krwawienia. Zabiegi chirurgiczne mogą zatem wymagać doraźnego przerwania leczenia dabigatranem eteksylanem .
Należy zachować ostrożność w przypadku doraźnego przerwania leczenia z powodu zabiegów inwazyjnych, konieczne jest wówczas monitorowanie przeciwzakrzepowe. U pacjentów z
niewydolnością nerek klirens dabigatranu może być wydłużony (patrz punkt 5.2). Należy to
uwzględnić przed każdym zabiegiem. W takich przypadkach test krzepliwości (patrz punkty 4.4 i 5.1) może być pomocny w celu określenia, czy hemostaza jest wciąż nieprawidłowa.
Zabieg chirurgiczny w trybie nagłym lub zabiegi pilne
Należy doraźnie przerwać stosowanie dabigatranu eteksylanu . W przypadku, gdy konieczne jest szybkie odwrócenie działania przeciwzakrzepowego, dla dorosłych pacjentów dostępny jest swoisty czynnik odwracający działanie dabigatranu (idarucyzumab). Nie określono skuteczności ani
bezpieczeństwa stosowania idarucyzumabu u dzieci i młodzieży. Dabigatran można usunąć na drodze hemodializy.
Odwrócenie terapii dabigatranem naraża pacjenta na ryzyko powstania zakrzepu spowodowanego chorobą podstawową. Leczenie dabigatranem eteksylanem może być wznowione 24 godziny po
podaniu idarucyzumabu, pod warunkiem, że pacjent jest stabilny klinicznie i osiągnięto odpowiednią hemostazę.
Zabiegi chirurgiczne/procedury inwazyjne w stanach podostrych
Należy doraźnie przerwać stosowanie dabigatranu eteksylanu . Zabieg chirurgiczny lub interwencję należy w miarę możliwości opóźnić co najmniej 12 godzin po podaniu ostatniej dawki. Jeśli zabiegu chirurgicznego nie można opóźnić, ryzyko krwawienia może być zwiększone. Należy rozważyć ryzyko krwawienia w stosunku do stopnia pilności zabiegu.
Planowe zabiegi chirurgiczne
W miarę możliwości stosowanie dabigatranu eteksylanu należy przerwać co najmniej 24 godziny przed zabiegami inwazyjnymi lub chirurgicznymi. U pacjentów z podwyższonym ryzykiem krwawienia lub poddawanych dużym zabiegom chirurgicznym, w przypadku których może być wymagana pełna hemostaza, należy rozważyć przerwanie stosowania dabigatranu eteksylanu na 2-4 dni przed zabiegiem chirurgicznym.
W Tabeli 5 podsumowano zasady dotyczące przerywania leczenia przed zabiegami inwazyjnymi lub chirurgicznymi u dorosłych pacjentów.
Tabela 5: Zasady dotyczące przerywania leczenia przed zabiegami inwazyjnymi lub chirurgicznymi u dorosłych pacjentów
Czynność nerek (CrCL w mL/min) | Szacowany okres półtrwania (godziny) | Należy przerwać stosowanie dabigatranu eteksylanu przed planowym zabiegiem chirurgicznym | |
Wysokie ryzyko krwawienia lub duży zabieg chirurgiczny | Ryzyko standardowe | ||
≥80 | ~ 13 | 2 dni przed | 24 godziny przed |
≥50-<80 | ~ 15 | 2-3 dni przed | 1-2 dni przed |
≥30-<50 | ~ 18 | 4 dni przed | 2-3 dni przed (>48 godzin) |
Zasady przerywania leczenia przed zabiegami inwazyjnymi lub chirurgicznymi u dzieci i młodzieży podsumowano w tabeli 6.
Tabela 6: Zasady przerywania leczenia przed zabiegami inwazyjnymi lub chirurgicznymi u dzieci i młodzieży
Czynność nerek (eGFR w mL/min/1,73 m2) | Należy przerwać stosowanie dabigatranu przed planowanym zabiegiem |
>80 | 24 godziny przed |
50–80 | 2 dni przed |
<50 | Nie przebadano tych pacjentów (patrz punkt 4.3). |
Znieczulenie rdzeniowe/znieczulenie zewnątrzoponowe/nakłucie lędźwiowe
Zabiegi takie jak znieczulenie rdzeniowe wymagają pełnej czynności hemostatycznej.
Ryzyko krwiaków rdzeniowych lub zewnątrzoponowych może być zwiększone w przypadku urazowego lub wielokrotnego nakłucia oraz przez długotrwałe stosowanie cewnika
zewnątrzoponowego. Po usunięciu cewnika należy odczekać co najmniej 2 godziny przed podaniem pierwszej dawki dabigatranu eteksylanu . Pacjenci tacy wymagają częstej obserwacji w kierunku neurologicznych objawów przedmiotowych i podmiotowych występowania krwiaków rdzeniowych lub zewnątrzoponowych.
Faza pooperacyjna
Leczenie dabigatranem eteksylanem należy wznowić po inwazyjnym zabiegu lub interwencji
chirurgicznej tak szybko, jak to możliwe, pod warunkiem, że pozwala na to sytuacja kliniczna i uzyskano odpowiednią hemostazę.
Należy zachować ostrożność (patrz punkty 4.4 i 5.1) podczas leczenia pacjentów z grupy ryzyka wystąpienia krwawienia lub pacjentów narażonych na nadmierną ekspozycję na produkt, a zwłaszcza pacjentów z zaburzoną czynnością nerek (patrz również tabela 3).
Pacjenci z grupy wysokiego ryzyka zgonu na skutek zabiegu chirurgicznego oraz z wewnętrznymi czynnikami ryzyka występowania zdarzeń zakrzepowo-zatorowych
Dostępne dane dotyczące skuteczności i bezpieczeństwa stosowania dabigatranu eteksylanu u tych pacjentów są ograniczone, dlatego należy zachować ostrożność podczas leczenia.
Zabieg chirurgiczny z powodu złamania szyjki kości udowej
Brak danych dotyczących stosowania dabigatranu eteksylanu u pacjentów poddawanych zabiegom chirurgicznym z powodu złamania szyjki kości udowej. W związku z tym stosowanie tego produktu leczniczego nie jest zalecane.
Zaburzenia czynności wątroby
Z udziału w głównych badaniach wykluczono pacjentów ze zwiększoną aktywnością enzymów wątrobowych ponad 2-krotnie powyżej górnej granicy normy. Brak dostępnego doświadczenia w
leczeniu tej subpopulacji pacjentów i dlatego nie zaleca się stosowania dabigatranu eteksylanu w tej grupie pacjentów. Przeciwwskazaniami do stosowania produktu leczniczego są niewydolność wątroby lub schorzenia tego narządu, które mogą wpływać na czas przeżycia (patrz punkt 4.3).
Interakcja z induktorami P-gp
Skojarzone stosowanie induktorów P-gp może zmniejszać stężenie dabigatranu w osoczu, dlatego też należy unikać ich podawania (patrz punkty 4.5 i 5.2).
Pacjenci z zespołem antyfosfolipidowym
Nie zaleca się stosowania doustnych antykoagulantów o działaniu bezpośrednim, takich jak dabigatran eteksylan, u pacjentów z zakrzepicą ze zdiagnozowanym zespołem antyfosfolipidowym. Zwłaszcza u pacjentów z trzema wynikami pozytywnymi (antykoagulant toczniowy, przeciwciała antykardiolipinowe oraz przeciwciała przeciwko β2 glikoproteinie-I) leczenie z zastosowaniem
doustnych antykoagulantów o działaniu bezpośrednim może być związane z większą liczbą nawrotów incydentów zakrzepowych niż podczas terapii antagonistami witaminy K.
Pacjenci z czynną chorobą nowotworową (dzieci i młodzież z ŻChZZ)
Dane dotyczące skuteczności i bezpieczeństwa stosowania u dzieci i młodzieży z czynną chorobą nowotworową są ograniczone.
Dzieci i młodzież
W przypadku niektórych bardzo specyficznych grup dzieci i młodzieży, np. pacjentów z chorobą jelita cienkiego, w przebiegu której wchłanianie może być zaburzone, należy rozważyć stosowanie leku przeciwzakrzepowego podawanego pozajelitowo.
Sód
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu w kapsułce, to znaczy produkt leczniczy uznaje się za „wolny od sodu”
Interakcje za pośrednictwem białek transportowych
Dabigatran eteksylan jest substratem transportera błonowego P-gp. Oczekuje się, że jednoczesne podawanie inhibitorów P-gp (patrz tabela 7) spowoduje zwiększone stężenie osoczowe dabigatranu.
Jeśli nie podano inaczej, podczas jednoczesnego podawania dabigatranu z silnymi inhibitorami P-gp zaleca się ścisłe monitorowanie kliniczne (w kierunku objawów krwawienia lub niedokrwistości). W przypadku jednoczesnego stosowania niektórych inhibitorów P-gp może być konieczne zmniejszenie dawki (patrz punkty 4.2, 4.3, 4.4 i 5.1).
Tabela 7: Interakcje za pośrednictwem białek transportowych
Inhibitory P-gp | |
Jednoczesne stosowanie jest przeciwwskazane (patrz punkt 4.3). | |
Ketokonazol | Ketokonazol zwiększał całkowite wartości AUC0-∞ i Cmax dabigatranu odpowiednio o 2,38 razy i 2,35 razy po podaniu doustnej jednorazowej dawki 400 mg oraz odpowiednio o 2,53 razy i 2,49 razy po doustnym podaniu wielokrotnym 400 mg ketokonazolu raz na dobę. |
Dronedaron | Jednoczesne podawanie dabigatranu eteksylanu i dronedaronu spowodowało odpowiednio około 2,4-krotny i 2,3-krotny wzrost wartości AUC0-∞ i Cmax dabigatranu w przypadku wielokrotnego podania 400 mg dronedaronu dwa razy na dobę oraz odpowiednio około 2,1-krotny i 1,9-krotny w przypadku podania pojedynczej dawki 400 mg. |
Itrakonazol, cyklosporyna | Na podstawie wyników badań in vitro można spodziewać się podobnego efektu jak w przypadku stosowania ketokonazolu. |
Glekaprewir i pibrentaswir | Wykazano, że jednoczesne stosowanie dabigatranu eteksylanu w skojarzeniu z inhibitorami P-gp glekaprewirem i pibrentaswirem w ustalonej dawce zwiększa ekspozycję na dabigatran i może zwiększać ryzyko krwawienia. |
Jednoczesne stosowanie nie jest zalecane. | |
Takrolimus | W badaniach in vitro stwierdzono, że hamujący wpływ takrolimusu na P-gp jest zbliżony do obserwowanego dla itrakonazolu i cyklosporyny. Nie przeprowadzono badań klinicznych dotyczących podawania dabigatranu eteksylanu z takrolimusem. Jednakże ograniczone dane kliniczne dotyczące innego substratu P-gp (ewerolimusu) wskazują, że takrolimus hamuje P-gp słabiej niż silne inhibitory P-gp. |
Należy zachować ostrożność w przypadku jednoczesnego stosowania (patrz punkty 4.2 i 4.4). | |
Werapamil | W trakcie jednoczesnego podawania dabigatranu eteksylanu (150 mg) z doustnym werapamilem Cmax i AUC dabigatranu uległy zwiększeniu, lecz zakres tych zmian różni się w zależności od momentu podawania werapamilu i jego postaci farmaceutycznej (patrz punkty 4.2 i 4.4). Największy wzrost ekspozycji na dabigatran obserwowano po pierwszej dawce werapamilu w postaci o natychmiastowym uwalnianiu podanej godzinę przed podaniem dabigatranu eteksylanu (zwiększenie Cmax o około 2,8 razy i AUC o około 2,5 razy). Wynik ten ulegał stopniowemu zmniejszaniu po podawaniu postaci o przedłużonym uwalnianiu (zwiększenie Cmax o około 1,9 razy i AUC o około 1,7 razy) i po podawaniu dawek wielokrotnych werapamilu (zwiększenie Cmax o około 1,6 razy i AUC około 1,5 razy). Nie stwierdzono żadnych istotnych interakcji podczas podawania werapamilu 2 godziny po eteksylanie dabigatranu (wzrost Cmax o około 1,1 razy i AUC o około 1,2 razy). Tłumaczy się to pełnym wchłonięciem dabigatranu po 2 godzinach. |
Amiodaron | W trakcie jednoczesnego podawania dabigatranu eteksylanu z amiodaronem w dawce pojedynczej wynoszącej 600 mg zasadniczo nie stwierdzano zmian stopnia i szybkości wchłaniania amiodaronu i jego czynnego metabolitu DEA. Stwierdzono zwiększenie wartości AUC i Cmax dabigatranu odpowiednio o około 1,6 razy i 1,5 razy. Ze względu na długi okres półtrwania amiodaronu możliwość |
wystąpienia interakcji istnieje przez kilka tygodni po odstawieniu amiodaronu (patrz punkty 4.2 i 4.4). | |
Chinidyna | Chinidynę podawano w dawce 200 mg co 2 godziny do całkowitej dawki wynoszącej 1 000 mg. Dabigatran eteksylan podawano dwa razy na dobę przez 3 kolejne dni, trzeciego dnia z chinidyną lub bez. AUCτ,ss i Cmax,ss dabigatranu były zwiększone średnio, odpowiednio, o około 1,53 razy i 1,56 razy w przypadku jednoczesnego podawania chinidyny (patrz punkty 4.2 i 4.4). |
Klarytromycyna | W trakcie jednoczesnego podawania zdrowym ochotnikom klarytromycyny (500 mg dwa razy na dobę) z dabigatranem eteksylanem stwierdzono około 1,19-krotny wzrost AUC i około 1,15-krotny wzrost Cmax. |
Tikagrelor | Po jednoczesnym podaniu pojedynczej dawki 75 mg dabigatranu eteksylanu i dawki nasycającej 180 mg tikagreloru wartości AUC i Cmax dla dabigatranu wzrastały odpowiednio 1,73 razy i 1,95 razy. Po wielokrotnym podawaniu tikagreloru w dawce 90 mg dwa razy na dobę ekspozycja na dabigatran wyrażona wartościami Cmax i AUC wzrastała odpowiednio 1,56 i 1,46 razy. Jednoczesne podawanie dawki nasycającej 180 mg tikagreloru i 110 mg dabigatranu eteksylanu (w stanie stacjonarnym) zwiększało wartość AUCτ,ss i Cmax,ss dla dabigatranu o 1,49 razy i 1,65 razy odpowiednio w porównaniu z dabigatranem eteksylanem w monoterapii. Kiedy dawka nasycająca 180 mg tikagreloru była podana 2 godziny po dawce 110 mg dabigatranu eteksylanu (w stanie stacjonarnym), wzrost wartości AUCτ,ss i Cmax,ss dla dabigatranu został obniżony do 1,27 razy i 1,23 razy odpowiednio w porównaniu z dabigatranem eteksylanem w monoterapii. To naprzemienne podawanie jest zalecaną metodą rozpoczęcia leczenia tikagrelorem w dawce nasycającej. Jednoczesne podawanie 90 mg tikagreloru dwa razy na dobę (dawka podtrzymująca) z 110 mg dabigatranu eteksylanu zwiększało skorygowaną wartość AUCτ,ss i Cmax,ss dla dabigatranu odpowiednio o 1,26 razy i 1,29 razy w porównaniu z dabigatranem eteksylanem w monoterapii. |
Pozakonazol | Pozakonazol również wykazuje w pewnym stopniu działanie hamujące P-gp, lecz nie został on przebadany klinicznie. Należy zachować ostrożność podczas jednoczesnego stosowania dabigatranu eteksylanu z pozakonazolem. |
Induktory P-gp | |
Należy unikać jednoczesnego stosowania. | |
np. ryfampicyna lub ziele dziurawca (Hypericum perforatum), karbamazepina lub fenytoina | Jednoczesne podawanie tych leków może zmniejszać stężenia dabigatranu. Wcześniejsze podanie induktora ryfampicyny w dawce 600 mg raz na dobę przez 7 dni zmniejszyło całkowite największe stężenie dabigatranu i całkowitą ekspozycję, odpowiednio, o 65,5% i 67%. Efekt indukcyjny został zmniejszony, co przełożyło się na ekspozycję bliską wartościom referencyjnym 7. dnia po zakończeniu leczenia ryfampicyną. Po kolejnych 7 dniach nie zaobserwowano kolejnego zwiększenia biodostępności. |
Inhibitory proteazy, takie jak rytonawir | |
Nie zaleca się jednoczesnego stosowania. | |
np. rytonawir i jego połączenie z innymi inhibitorami proteazy | Wywierają wpływ na P-gp (jako inhibitor lub jako induktor). Ich jednoczesne stosowanie nie było badane, dlatego nie zaleca się ich jednoczesnego stosowania z produktem leczniczym Wasedoc. |
Substrat P-gp | |
Digoksyna | Gdy dabigatran eteksylan podawano jednocześnie z digoksyną w badaniu z udziałem 24 zdrowych uczestników, nie obserwowano zmian ekspozycji na digoksynę ani istotnych klinicznie zmian ekspozycji na dabigatran. |
Produkty lecznicze przeciwzakrzepowe i produkty lecznicze hamujące agregację płytek
Brak lub istnieje jedynie ograniczone doświadczenie z następującymi produktami leczniczymi, które mogą zwiększać ryzyko krwawienia w przypadku jednoczesnego stosowania z dabigatranem eteksylanem : produkty lecznicze przeciwzakrzepowe takie jak niefrakcjonowane heparyny (ang.
UFH - Unfractionated Heparin), heparyny niskocząsteczkowe (ang. LMWH - Low Molecular Weight Heparins) i pochodne heparyny (fondaparynuks, desyrudyna), produkty lecznicze trombolityczne i
antagoniści witaminy K, rywaroksaban lub inne doustne antykoagulanty (patrz punkt 4.3) i produkty lecznicze hamujące agregację płytek krwi takie jak antagoniści receptora GPIIb/IIIa, tyklopidyna, prasugrel, tikagrelor, dekstran i sulfinpirazon (patrz punkt 4.4).
Niefrakcjonowaną heparynę można podawać w dawkach niezbędnych do utrzymania drożnego centralnego cewnika żylnego lub tętniczego lub podczas ablacji cewnikowej w migotaniu przedsionków (patrz punkty 4.3).
Tabela 8: Interakcje z produktami leczniczymi przeciwzakrzepowymi i produktami leczniczymi hamującymi agregację płytek
NLPZ | W trakcie jednoczesnego podawania NLPZ w krótkotrwałym leczeniu bólu z dabigatranem eteksylanem nie obserwowano zwiększonego ryzyka krwawienia. Podczas stosowania przewlekłego w badaniu klinicznym fazy III porównującym dabigatran z warfaryną w zapobieganiu udarom u pacjentów z migotaniem przedsionków (RE–LY) leki z grupy NLPZ zwiększały ryzyko krwawienia o około 50% zarówno w przypadku dabigatranu eteksylanu , jak i warfaryny. |
Klopidogrel | U zdrowych ochotników płci męskiej skojarzone podawanie dabigatranu eteksylanu i klopidogrelu nie powodowało dalszego wydłużania czasu krzepnięcia krwi metodą kapilarową w porównaniu do monoterapii klopidogrelem. Ponadto wartości AUCτ,ss oraz Cmax,ss dla dabigatranu i pomiary krzepliwości jako oddziaływania dabigatranu lub hamowania agregacji płytek jako oddziaływania klopidogrelu pozostawały zasadniczo niezmienione porównując leczenie skojarzone do odpowiadających mu monoterapii. Po użyciu dawki nasycającej 300 mg lub 600 mg klopidogrelu |
Kwas acetylosalicylowy | Skojarzone podawanie kwasu acetylosalicylowego oraz dabigatranu eteksylanu 150 mg dwa razy na dobę może zwiększać ryzyko każdego krwawienia od 12% do 18% oraz do 24% w przypadku dawki kwasu acetylosalicylowego wynoszącej odpowiednio 81 mg i 325 mg (patrz punkt 4.4). |
Heparyny niskocząsteczkowe (LMWH) | Nie badano skojarzonego stosowania LMWH, takich jak enoksaparyna i eteksylan dabigatranu. Po zmianie trzydniowego leczenia, w trakcie którego podawano podskórnie 40 mg enoksaparyny raz na dobę, 24 godziny po ostatniej dawce enoksaparyny, ekspozycja na dabigatran była nieco niższa niż po podaniu samej dawki dabigatranu eteksylanu (pojedyncza dawka 220 mg). Wyższą aktywność anty-FXa/FIIa obserwowano po podaniu dabigatranu eteksylanu po wstępnym leczeniu enoksaparyną w porównaniu do aktywności po leczeniu tylko dabigatranem eteksylanem . Uważa się, że jest to spowodowane efektem przeniesienia leczenia enoksaparyną i nie jest uznawane za znaczące klinicznie. Wyniki pozostałych testów działania przeciwzakrzepowego związanego z dabigatranem nie były znamiennie różne w przypadku leczenia wstępnego enoksaparyną. |
Inne interakcje
Tabela 9: Inne interakcje
Selektywne inhibitory wychwytu zwrotnego serotoniny (SSRI) lub selektywne inhibitory wychwytu zwrotnego noradrenaliny (SNRI) | |
SSRI, SNRI | SSRI i SNRI spowodowały wzrost ryzyka krwawień w badaniu klinicznym fazy III porównującym dabigatran z warfaryną w zapobieganiu udarom mózgu u pacjentów z migotaniem przedsionków (RE-LY) we wszystkich leczonych grupach. |
Substancje wpływające na pH żołądka | |
Pantoprazol | W trakcie jednoczesnego podawania dabigatranu eteksylanu z pantoprazolem stwierdzono zmniejszenie AUC dabigatranu o około 30%. Pantoprazol i inne inhibitory pompy protonowej (PPI) podawano jednocześnie z dabigatranem eteksylanem w badaniach klinicznych. Nie zaobserwowano wpływu tego skojarzenia na skuteczność leczenia dabigatranem eteksylanem . |
Ranitydyna | Podawanie ranitydyny jednocześnie z dabigatranem eteksylanem nie wywierało istotnego klinicznie wpływu na stopień wchłaniania dabigatranu. |
Interakcje związane z właściwościami metabolicznymi dabigatranu eteksylanu i dabigatranu
Dabigatran eteksylan i dabigatran nie są metabolizowane przez układ cytochromu P450
i w badaniach in vitro nie wpływały na enzymy ludzkiego cytochromu P450. Dlatego nie należy się spodziewać związanych z tym mechanizmem interakcji dabigatranu z innymi lekami.
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono wyłącznie u dorosłych.
Kobiety w wieku rozrodczym
Kobiety w wieku rozrodczym powinny unikać zajścia w ciążę podczas leczenia dabigatranem eteksylanem .
Ciąża
Istnieją tylko ograniczone dane dotyczące stosowania dabigatranu eteksylanu u kobiet w okresie ciąży.
Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Potencjalne zagrożenie dla człowieka nie jest znane.
Produktu leczniczego Wasedoc nie należy stosować w okresie ciąży, jeśli nie jest to bezwzględnie konieczne.
Karmienie piersią
Nie ma danych klinicznych dotyczących wpływu dabigatranu na niemowlęta podczas karmienia piersią. Podczas leczenia produktem leczniczym Wasedoc należy przerwać karmienie piersią.
Płodność
Brak danych dotyczących ludzi.
W badaniach na zwierzętach obserwowano wpływ produktu leczniczego na płodność samic w postaci zmniejszenia liczby zagnieżdżeń zapłodnionego jaja i zwiększenia częstości utraty zapłodnionego jaja przed zagnieżdżeniem po dawce 70 mg/kg (5-krotnie większej od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów). Nie obserwowano innego wpływu na płodność u samic. Nie obserwowano wpływu na płodność u samców. Po dawkach toksycznych dla matek (od 5- do 10-krotnie większych od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów) u szczurów i królików stwierdzono zmniejszenie masy ciała
i przeżywalności płodów, łącznie ze zwiększeniem liczby wad rozwojowych płodów. W badaniach pre- i postnatalnych zaobserwowano zwiększenie umieralności płodów po dawkach toksycznych dla samic (4-krotnie większych od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów).
Dabigatran eteksylan nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa stosowania
Dabigatran eteksylan oceniano w badaniach klinicznych łącznie u około 64 000 pacjentów, spośród nich około 35 000 pacjentów było leczonych dabigatranem eteksylanem .
W badaniach kontrolowanych, z zastosowaniem czynnego leczenia w grupie kontrolnej, dotyczących zapobiegania ŻChZZ 6 684 pacjentów było leczonych dabigatranem eteksylanem w dawce 150 mg lub 220 mg na dobę.
Najczęściej obserwowanymi zdarzeniami były krwawienia, występujące u około 14% pacjentów; częstość występowania dużych krwawień (w tym krwawień z ran) wynosiła poniżej 2%.
Chociaż w badaniach klinicznych przypadki krwawienia zdarzały się rzadko, nie można wykluczyć wystąpienia dużego lub silnego krwawienia, które niezależnie od lokalizacji może zagrażać życiu pacjenta lub prowadzić do kalectwa, a nawet zgonu.
Tabularyczne zestawienie działań niepożądanych
W tabeli 10 przedstawiono działania niepożądane według klasyfikacji układów i narządów (ang. SOC
- System Organ Classes) oraz częstości występowania zgodnie z następującą konwencją: bardzo
często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do
<1/1 000), bardzo rzadko (<1/10 000); nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 10: Działania niepożądane
Klasyfikacja układów i narządów / Zalecany termin | Częstość |
Zaburzenia krwi i układu chłonnego | |
Spadek stężenia hemoglobiny | Często |
Niedokrwistość | Niezbyt często |
Spadek hematokrytu | Niezbyt często |
Małopłytkowość | Rzadko |
Neutropenia | Nieznana |
Agranulocytoza | Nieznana |
Zaburzenia układu immunologicznego | |
Nadwrażliwość na lek | Niezbyt często |
Reakcja anafilaktyczna | Rzadko |
Obrzęk naczynioruchowy | Rzadko |
Pokrzywka | Rzadko |
Wysypka | Rzadko |
Świąd | Rzadko |
Skurcz oskrzeli | Nieznana |
Zaburzenia układu nerwowego | |
Krwotok wewnątrzczaszkowy | Rzadko |
Zaburzenia naczyniowe | |
Krwiak | Niezbyt często |
Krwotok z rany | Niezbyt często |
Krwotok | Rzadko |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Krwawienie z nosa | Niezbyt często |
Krwioplucie | Rzadko |
Zaburzenia żołądka i jelit | |
Krwotok do przewodu pokarmowego | Niezbyt często |
Krwotok z odbytnicy | Niezbyt często |
Krwotok z żylaków odbytu | Niezbyt często |
Biegunka | Niezbyt często |
Nudności | Niezbyt często |
Wymioty | Niezbyt często |
Wrzód żołądka lub jelit, w tym owrzodzenie przełyku | Rzadko |
Zapalenie żołądka i przełyku | Rzadko |
Refluks żołądkowo-przełykowy | Rzadko |
Ból brzucha | Rzadko |
Niestrawność | Rzadko |
Dysfagia | Rzadko |
Zaburzenia wątroby i dróg żółciowych | |
Nieprawidłowa czynność wątroby / Nieprawidłowe wyniki badań czynności wątroby | Często |
Wzrost aktywności aminotransferazy alaninowej | Niezbyt często |
Wzrost aktywności aminotransferazy asparaginianowej | Niezbyt często |
Wzrost aktywności enzymów wątrobowych | Niezbyt często |
Hiperbilirubinemia | Niezbyt często |
Zaburzenia skóry i tkanki podskórnej | |
Krwotok do skóry | Niezbyt często |
Łysienie | Nieznana |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Krwiak wewnątrzstawowy | Niezbyt często |
Zaburzenia nerek i dróg moczowych | |
Krwotok w obrębie układu moczowo-płciowego, w tym krwiomocz | Niezbyt często |
Zaburzenia ogólne i stany w miejscu podania | |
Krwotok w miejscu wstrzyknięcia | Rzadko |
Krwotok w miejscu cewnikowania | Rzadko |
Krwisty wyciek | Rzadko |
Urazy, zatrucia i powikłania po zabiegach | |
Krwotok urazowy | Niezbyt często |
Krwiak po zabiegu | Niezbyt często |
Krwotok po zabiegu | Niezbyt często |
Wyciek po zabiegu | Niezbyt często |
Wydzielina z rany | Niezbyt często |
Krwotok w miejscu nacięcia | Rzadko |
Niedokrwistość pooperacyjna | Rzadko |
Procedury medyczne i chirurgiczne | |
Drenaż rany | Rzadko |
Drenaż po zabiegu | Rzadko |
Opis wybranych działań niepożądanych
Reakcje w postaci krwawień
Ze względu na farmakologiczny mechanizm działania stosowanie dabigatranu eteksylanu może wiązać się ze zwiększonym ryzykiem utajonego lub jawnego krwawienia z dowolnej tkanki lub
narządu. Objawy przedmiotowe, objawy podmiotowe i nasilenie (w tym możliwość zgonu) różnią się w zależności od miejsca i stopnia lub rozległości krwawienia i (lub) niedokrwistości. W badaniach klinicznych w trakcie długotrwałego leczenia dabigatranem eteksylanem w porównaniu z leczeniem VKA częściej obserwowano krwawienia z błon śluzowych (np. układu pokarmowego, układu moczowo-płciowego). Dlatego też, oprócz odpowiedniego nadzoru klinicznego, badania laboratoryjne hemoglobiny/hematokrytu mogą być przydatne do wykrywania utajonego krwawienia. W niektórych grupach pacjentów ryzyko krwawienia może być większe, np. u pacjentów z umiarkowanymi
zaburzeniami czynności nerek i (lub) jednocześnie przyjmujących leki wpływające na hemostazę lub silne inhibitory P-gp (patrz punkt 4.4 Ryzyko krwotoku). Objawami powikłań krwotocznych mogą być osłabienie, bladość, zawroty głowy, ból głowy lub niewyjaśniony obrzęk, duszność i niewyjaśniony wstrząs.
Dla dabigatranu eteksylanu zgłaszano znane powikłania krwawienia, takie jak zespół ciasnoty międzypowięziowej i ostra niewydolność nerek z powodu obniżonej perfuzji, oraz nefropatię związaną z leczeniem przeciwzakrzepowym u pacjentów z predysponującymi czynnikami ryzyka. Oceniając stan każdego pacjenta, u którego stosowano leki przeciwzakrzepowe, należy zatem uwzględnić możliwość wystąpienia krwotoku. U dorosłych pacjentów w przypadku
niekontrolowanego krwawienia dostępny jest swoisty czynnik odwracający działanie dabigatranu, idarucyzumab (patrz punkt 4.9).
W tabeli 11 przedstawiono liczbę pacjentów (%), u których wystąpiło działanie niepożądane w postaci krwawienia w okresie leczenia we wskazaniu pierwotnego zapobiegania żylnej chorobie zakrzepowo- zatorowej po alloplastyce stawu biodrowego lub kolanowego w dwóch głównych badaniach klinicznych, zgodnie z dawką.
Tabela 11: Liczba pacjentów (%), u których wystąpiło działanie niepożądane w postaci krwawienia
Dabigatran eteksylan 150 mg N (%) | Dabigatran eteksylan 220 mg N (%) | Enoksaparyna N (%) | |
Pacjenci leczeni | 1 866 (100,0) | 1 825 (100,0) | 1 848 (100,0) |
Duże krwawienie | 24 (1,3) | 33 (1,8) | 27 (1,5) |
Każde krwawienie | 258 (13,8) | 251 (13,8) | 247 (13,4) |
Agranulocytoza i neutropenia
W okresie po wprowadzeniu dabigatranu eteksylanu do obrotu bardzo rzadko zgłaszano
agranulocytozę i neutropenię. Ponieważ działania niepożądane są zgłaszane w ramach systemu kontroli po wprowadzeniu do obrotu w populacji o nieokreślonej wielkości, dokładne określenie częstości ich występowania nie jest możliwe. Częstość zgłaszania oszacowano na 7 zdarzeń na 1 milion pacjento-lat w przypadku agranulocytozy oraz 5 zdarzeń na 1 milion pacjento-lat
w przypadku neutropenii.
Dzieci i młodzież
Bezpieczeństwo stosowania dabigatranu eteksylanu w leczeniu ŻChZZ i prewencji nawrotów ŻChZZ u dzieci i młodzieży badano w dwóch badaniach fazy III (DIVERSITY i 1160.108). Dabigatranem eteksylanem leczono łącznie 328 dzieci i młodzieży. Pacjenci otrzymywali dostosowane do wieku
i masy ciała dawki dabigatranu eteksylanu w postaci farmaceutycznej odpowiedniej dla wieku. Ogółem oczekuje się, że profil bezpieczeństwa stosowania u dzieci jest taki sam jak u dorosłych.
Łącznie u 26% dzieci i młodzieży leczonych dabigatranem eteksylanem z powodu ŻChZZ i w prewencji nawrotów ŻChZZ wystąpiły działania niepożądane.
Tabelaryczne zestawienie działań niepożądanych
W tabeli 12 przedstawiono działania niepożądane zidentyfikowane podczas badań w leczeniu ŻChZZ i prewencji nawrotów ŻChZZ u dzieci i młodzieży według klasyfikacji układów i narządów (ang. SOC – System Organ Class) oraz częstości występowania zgodnie z następującą konwencją: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1 000 do <1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000); nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 12: Działania niepożądane
Częstość | |
Klasyfikacja układów i narządów / Zalecany termin | Leczenie ŻChZZ i prewencja nawrotów ŻChZZ u dzieci i młodzieży |
Zaburzenia krwi i układu chłonnego | |
Niedokrwistość | Często |
Spadek stężenia hemoglobiny | Niezbyt często |
Małopłytkowość | Często |
Spadek hematokrytu | Niezbyt często |
Neutropenia | Niezbyt często |
Agranulocytoza | Nieznana |
Zaburzenia układu immunologicznego | |
Nadwrażliwość na lek | Niezbyt często |
Wysypka | Często |
Świąd | Niezbyt często |
Reakcja anafilaktyczna | Nieznana |
Obrzęk naczynioruchowy | Nieznana |
Pokrzywka | Często |
Skurcz oskrzeli | Nieznana |
Zaburzenia układu nerwowego | |
Krwotok wewnątrzczaszkowy | Niezbyt często |
Zaburzenia naczyniowe | |
Krwiak | Często |
Krwotok | Nieznana |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Krwawienie z nosa | Często |
Krwioplucie | Niezbyt często |
Zaburzenia żołądka i jelit | |
Krwotok do przewodu pokarmowego | Niezbyt często |
Ból brzucha | Niezbyt często |
Biegunka | Często |
Niestrawność | Często |
Nudności | Często |
Krwotok z odbytnicy | Niezbyt często |
Krwotok z żylaków odbytu | Nieznana |
Wrzód żołądka lub jelit, w tym owrzodzenie przełyku | Nieznana |
Zapalenie żołądka i przełyku | Niezbyt często |
Refluks żołądkowo-przełykowy | Często |
Wymioty | Często |
Dysfagia | Niezbyt często |
Zaburzenia wątroby i dróg żółciowych | |
Nieprawidłowa czynność wątroby / Nieprawidłowe wyniki badań czynności wątroby | Nieznana |
Wzrost aktywności aminotransferazy alaninowej | Niezbyt często |
Wzrost aktywności aminotransferazy asparaginianowej | Niezbyt często |
Wzrost aktywności enzymów wątrobowych | Często |
Hiperbilirubinemia | Niezbyt często |
Zaburzenia skóry i tkanki podskórnej | |
Krwotok do skóry | Niezbyt często |
Łysienie | Często |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Krwiak wewnątrzstawowy | Nieznana |
Zaburzenia nerek i dróg moczowych | |
Krwotok w obrębie układu moczowo- płciowego, w tym krwiomocz | Niezbyt często |
Zaburzenia ogólne i stany w miejscu podania | |
Krwotok w miejscu wstrzyknięcia | Nieznana |
Krwotok w miejscu cewnikowania | Nieznana |
Urazy, zatrucia i powikłania po zabiegach | |
Krwotok urazowy | Niezbyt często |
Krwotok w miejscu nacięcia | Nieznana |
Reakcje w postaci krwawień
W dwóch badaniach fazy III we wskazaniu leczenia ŻChZZ i prewencji nawrotów ŻChZZ u dzieci i młodzieży łącznie u 7 pacjentów (2,1%) wystąpił incydent dużego krwawienia, u 5 pacjentów (1,5%) klinicznie istotny inny niż duży incydent krwawienia, a u 75 pacjentów (22,9%) incydent małego krwawienia. Częstość występowania incydentów krwawień była ogółem większa w starszej grupie wiekowej (od 12 do <18 lat: 28,6%) niż w młodszych grupach wiekowych (od urodzenia do
<2 lat: 23,3%; od 2 do <12 lat: 16,2%). Duże lub ciężkie krwawienie, niezależnie od lokalizacji, może zagrażać życiu pacjenta lub prowadzić do kalectwa, a nawet zgonu.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań
niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem
Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych
Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C, 02-222 Warszawa, tel.+ 48 22 49-21-301, fax: +48 22 49-21-309, Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Większe niż zalecane dawki dabigatranu eteksylanu narażają pacjentów na zwiększone ryzyko krwawienia.
W przypadku podejrzenia przedawkowania testy krzepliwości mogą pomóc w określeniu ryzyka krwawienia (patrz punkty 4.4 i 5.1). Kalibrowany test ilościowy dTT lub powtarzane pomiary dTT umożliwiają określenie czasu osiągnięcia określonego stężenia dabigatranu (patrz punkt 5.1), również w przypadku podjęcia innych środków, np. dializy.
Nadmierne działanie przeciwzakrzepowe może wymagać przerwania leczenia dabigatranem eteksylanem . Ponieważ dabigatran wydala się przede wszystkim przez nerki, należy utrzymać wystarczającą diurezę. Ze względu na niski stopień wiązania z białkami dabigatran może być usuwany z organizmu za pomocą dializy, istnieją ograniczone dane kliniczne uzasadniające przydatność tej metody w badaniach klinicznych (patrz punkt 5.2).
Postępowanie w przypadku powikłań krwawienia
W przypadku powikłań krwotocznych konieczne jest przerwanie leczenia dabigatranem eteksylanem i zbadanie źródła krwawienia. W zależności od sytuacji klinicznej należy wdrożyć właściwe leczenie podtrzymujące, takie jak hemostaza chirurgiczna i przetoczenie objętości krwi, w
zależności od decyzji lekarza.
U dorosłych pacjentów w sytuacjach wymagających szybkiego odwrócenia działania przeciwzakrzepowego dabigatranu dostępny jest swoisty czynnik odwracający (idarucyzumab), antagonizujący działanie farmakodynamiczne dabigatranu. Nie określono skuteczności ani
bezpieczeństwa stosowania idarucyzumabu u dzieci i młodzieży (patrz punkt 4.4)
Można uwzględnić zastosowanie koncentratów czynników krzepnięcia (aktywowanych lub nieaktywowanych) lub rekombinowanego czynnika VIIa. Dostępne są eksperymentalne dane opisujące rolę tych produktów leczniczych w odwracaniu działania przeciwzakrzepowego dabigatranu, jednakże dane na temat ich przydatności w warunkach klinicznych, jak również
możliwości ryzyka nawrotu choroby zakrzepowo-zatorowej są ograniczone. Badania krzepnięcia wykonywane po podaniu sugerowanych koncentratów czynników krzepnięcia mogą nie dawać wiarygodnych wyników. Należy zachować ostrożność podczas ich interpretacji. Podanie koncentratów płytek należy rozważyć również w przypadku małopłytkowości lub stosowania długodziałających produktów leczniczych przeciwpłytkowych. Leczenie objawowe powinno być stosowane według uznania lekarza.
W przypadku poważnych krwawień należy rozważyć możliwość konsultacji z ekspertem, w zależności od lokalnych możliwości.
Grupa farmakoterapeutyczna: leki przeciwzakrzepowe, bezpośredni inhibitor trombiny, kod ATC: B01AE07.
Mechanizm działania
Dabigatran eteksylan jest niskocząsteczkowym prolekiem pozbawionym działania farmakologicznego. Po podaniu doustnym dabigatran eteksylan szybko się wchłania i ulega przemianie do dabigatranu w drodze katalizowanej przez esterazę hydrolizy w osoczu i w wątrobie. Dabigatran jest silnie działającym, kompetycyjnym, odwracalnym, bezpośrednim inhibitorem trombiny i główną substancją czynną znajdującą się w osoczu.
Zahamowanie trombiny (proteazy serynowej) zapobiega powstawaniu zakrzepu, ponieważ umożliwia ona przemianę fibrynogenu w fibrynę w trakcie kaskady krzepnięcia. Dabigatran hamuje wolną trombinę, trombinę związaną z fibryną i agregację płytek indukowaną trombiną.
Działanie farmakodynamiczne
Badania na zwierzętach prowadzone in vivo i ex vivo wykazały skuteczność i aktywność
przeciwzakrzepową dabigatranu po podaniu dożylnym i dabigatranu eteksylanu po podaniu doustnym wobec różnych modeli zwierzęcych zakrzepicy.
Istnieje ścisły związek pomiędzy stężeniem dabigatranu w osoczu a działaniem przeciwzakrzepowym na podstawie badań klinicznych fazy II. Dabigatran powoduje wydłużenie czasu trombinowego (TT), ECT i APTT.
Skalibrowane ilościowe badanie czasu trombinowego krzepnięcia w rozcieńczonym osoczu TT (dTT) pozwala oszacować stężenie dabigatranu w osoczu, które można porównać do stężeń
przewidywanych. Jeśli w skalibrowanym teście ilościowym dTT stężenie dabigatranu w osoczu znajduje się na granicy kwantyfikacji lub poniżej, należy rozważyć oznaczenie innych testów krzepnięcia, takich jak TT, ECT czy APTT.
ECT umożliwia bezpośredni pomiar aktywności bezpośrednich inhibitorów trombiny.
Badanie APTT jest powszechnie dostępne i stanowi przybliżony wskaźnik nasilenia działania przeciwzakrzepowego dabigatranu. Badanie to ma jednak ograniczoną czułość i nie nadaje się do dokładnego ilościowego określania działania przeciwzakrzepowego, szczególnie w dużym stężeniu
dabigatranu w osoczu. Wysokie wartości APTT należy interpretować ostrożnie, jednakże wysoki wynik APTT oznacza, że pacjent jest antykoagulowany.
Można założyć, że powyższe badania działania przeciwzakrzepowego odzwierciedlają stężenie dabigatranu i dają wskazówki dotyczące oceny ryzyka krwawienia. Wskaźnikiem podwyższonego ryzyka krwawienia jest m.in. przekroczenie 90 percentyla minimalnego stężenia dabigatranu lub badanie krzepnięcia (np. APTT) (wartości graniczne APTT podano w punkcie 4.4, w tabeli 3) mierzonego w stężeniu minimalnym.
Prewencja pierwotna ŻChZZ po zabiegach ortopedycznych
Średnia geometryczna maksymalnego stężenia osoczowego dabigatranu w stanie nasycenia (po 3 dniach), zmierzonego około 2 godziny po podaniu 220 mg dabigatranu eteksylanu , wynosiła 70,8 ng/mL i znajdowała się w przedziale od 35,2 ng/mL do 162 ng/mL (25-75 centyl). Średnia
geometryczna najniższego stężenia dabigatranu, mierzonego na końcu przedziału dawkowania (tj.
24 godziny po dawce 220 mg dabigatranu), wynosiła średnio 22,0 ng/mL i znajdowała się w przedziale od 13,0 ng/mL do 35,7 ng/mL (25-75 centyl).
W badaniu, do którego włączono wyłącznie pacjentów z zaburzeniami czynności nerek o nasileniu umiarkowanym (klirens kreatyniny, CrCL 30-50 mL/min) leczonych dabigatranem eteksylanem w dawce 150 mg raz na dobę, średnia geometryczna najniższego stężenia dabigatranu mierzonego na końcu przedziału dawkowania wynosiła przeciętnie 47,5 ng/mL, w zakresie od 29,6 ng/mL do 72,2 ng/mL
(25-75 percentyl).
U pacjentów, którym w ramach profilaktyki żylnych zdarzeń zakrzepowo-zatorowych po operacji alloplastyki stawu biodrowego lub alloplastyki stawu kolanowego podawano 220 mg dabigatranu eteksylanu raz na dobę,
U pacjentów, którym w ramach profilaktyki żylnych zdarzeń zakrzepowo-zatorowych po operacji alloplastyki stawu biodrowego lub alloplastyki stawu kolanowego podawano 220 mg dabigatranu eteksylanu raz na dobę, nie wykonywano pomiaru ECT.
Skuteczność kliniczna i bezpieczeństwo stosowania
Pochodzenie etniczne
Nie stwierdzono klinicznie znaczących różnic pomiędzy pacjentami rasy białej, rasy czarnej, pochodzenia latynoamerykańskiego, rasy żółtej.
Badania kliniczne dotyczące zapobiegania ŻChZZ po rozległym zabiegu chirurgicznym wszczepienia endoprotezy stawowej
W dwóch dużych, randomizowanych badaniach prowadzonych w grupach równoległych metodą podwójnie ślepej próby, potwierdzających wielkość dawek, pacjenci poddawani planowej rozległej operacji ortopedycznej (w pierwszym badaniu - wszczepienia endoprotezy stawu kolanowego, w drugim badaniu - wszczepienia endoprotezy stawu biodrowego) otrzymywali dabigatran eteksylan w dawce 75 mg lub 110 mg w ciągu od 1 do 4 godzin od zakończenia operacji, a następnie 150 mg lub 220 mg raz na dobę, o ile zapewniono hemostazę, lub enoksaparynę w dawce 40 mg w dniu przed zabiegiem chirurgicznym, a następnie codziennie.
W badaniu RE-MODEL (alloplastyka stawu kolanowego) leczenie prowadzono przez 6 do 10 dni, a w badaniu RE-NOVATE (alloplastyka stawu biodrowego) przez 28 do 35 dni. Leczenie otrzymywało
łącznie odpowiednio 2 076 pacjentów (kolano) i 3 494 pacjentów (biodro).
W obu badaniach złożony pierwszorzędowy punkt końcowy obejmował wszystkie epizody żylnej choroby zakrzepowo-zatorowej (ŻChZZ) [w tym zatorowość płucna (ZP), proksymalna i dystalna
zakrzepica żył głębokich (ZŻG), zarówno objawowa i bezobjawowa wykrywana za pomocą rutynowej flebografii] oraz zgon z jakiejkolwiek przyczyny. Drugorzędowy punkt końcowy składał się z dużego epizodu żylnej choroby zakrzepowo- zatorowej (w tym zatorowość płucna, proksymalna i dystalna
zakrzepica żył głębokich, zarówno objawowa i bezobjawowa wykrywana za pomocą rutynowej
flebografii) oraz zgonów związanych z żylną chorobą zakrzepowo-zatorową i był uznawany za bardziej znaczący klinicznie.
Wyniki obu badań wykazały, że działanie przeciwzakrzepowe dabigatranu eteksylanu 220 mg i 150 mg było statystycznie nie gorsze niż działanie enoksaparyny pod względem całkowitych
epizodów ŻChZZ i umieralności całkowitej. Estymacja punktowa częstości występowania dużych epizodów ŻChZZ i umieralności z powodu żylnej choroby zakrzepowo-zatorowej (ŻChZZ) w przypadku stosowania dawki 150 mg była nieco gorsza od tej dla enoksaparyny (tabela 13). Lepsze wyniki obserwowano dla dawki 220 mg, w przypadku której estymacja punktowa występowania dużych epizodów ŻChZZ była nieco lepsza niż ta dla enoksaparyny (tabela 13).
Badania kliniczne przeprowadzono w grupie pacjentów o średniej wieku >65 lat.
W fazie 3 badań klinicznych nie obserwowano żadnych różnic pod względem skuteczności i bezpieczeństwa pomiędzy mężczyznami a kobietami.
W populacji badanej w badaniach RE-MODEL i RE-NOVATE (5 539 leczonych pacjentów)
u 51% pacjentów występowało nadciśnienie tętnicze, u 9% cukrzyca, u 9% choroba wieńcowa i u 20% niewydolność żylna w wywiadzie. Żadne z tych schorzeń nie miało wpływu na wynik działania dabigatranu w zapobieganiu ŻChZZ lub krwawieniom.
Dane dotyczące punktu końcowego dużego epizodu ŻChZZ i zgonów związanych z ŻChZZ były jednorodne pod względem pierwszorzędowego punktu końcowego skuteczności i zostały przedstawione w tabeli 13.
Dane dla punktów końcowych ogólnej liczby epizodów ŻChZZ i zgonów z jakiejkolwiek przyczyny przedstawiono w tabeli 14.
Dane dla potwierdzonych punktów końcowych dla dużych krwawień przedstawiono w tabeli 15 poniżej.
Tabela 13: Analiza dużego epizodu ŻChZZ i śmiertelności związanej z ŻChZZ w trakcie leczenia w ramach badań RE-MODEL i RE-NOVATE dotyczących zabiegów ortopedycznych.
Badanie | Dabigatran eteksylan 220 mg | Dabigatran eteksylan 150 mg | Enoksaparyna 40 mg |
RE-NOVATE (biodro) | |||
N | 909 | 888 | 917 |
Zdarzenia (%) | 28 (3,1) | 38 (4,3) | 36 (3,9) |
Współczynnik ryzyka w porównaniu do enoksaparyny | 0,78 | 1,09 | |
95% CI | 0,48; 1,27 | 0,70; 1,70 | |
RE-MODEL (kolano) | |||
N | 506 | 527 | 511 |
Zdarzenia (%) | 13 (2,6) | 20 (3,8) | 18 (3,5) |
Współczynnik ryzyka w porównaniu do enoksaparyny | 0,73 | 1,08 | |
95% CI | 0,36; 1,47 | 0,58; 2,01 | |
Tabela 14:Analiza łącznych epizodów ŻChZZ i zgonów z jakiejkolwiek przyczyny w okresie leczenia w ramach badań dotyczących zabiegów ortopedycznych RE-NOVATE i RE- MODEL
Badanie | Dabigatran eteksylan 220 mg | Dabigatran eteksylan 150 mg | Enoksaparyna 40 mg |
RE-NOVATE (biodro) | |||
N | 880 | 874 | 897 |
Częstość występowania (%) | 53 (6,0) | 75 (8,6) | 60 (6,7) |
Współczynnik ryzyka w porównaniu do enoksaparyny | 0,9 | 1,28 | |
95% CI | (0,63; 1,29) | (0,93; 1,78) | |
RE-MODEL (kolano) | |||
N | 503 | 526 | 512 |
Częstość występowania (%) | 183 (36,4) | 213 (40,5) | 193 (37,7) |
Współczynnik ryzyka w porównaniu do enoksaparyny | 0,97 | 1,07 | |
95% CI | (0,82; 1,13) | (0,92; 1,25) | |
Tabela 15: Incydenty dużych krwawień w zależności od rodzaju leczenia w badaniach RE- MODEL i RE-NOVATE
Badanie | Dabigatran eteksylan 220 mg | Dabigatran eteksylan 150 mg | Enoksaparyna 40 mg |
RE-NOVATE (biodro) | |||
Liczba leczonych pacjentów N | 1 146 | 1 163 | 1 154 |
Liczba incydentów większych krwawień N (%) | 23 (2,0) | 15 (1,3) | 18 (1,6) |
RE-MODEL (kolano) | |||
Liczba leczonych pacjentów N | 679 | 703 | 694 |
Liczba incydentów większych krwawień N (%) | 10 (1,5) | 9 (1,3) | 9 (1,3) |
Badania kliniczne dotyczące prewencji powikłań zakrzepowo-zatorowych u pacjentów ze sztucznymi zastawkami serca
Przedmiotem badania fazy II było stosowanie dabigatranu eteksylanu i warfaryny u 252 pacjentów po niedawno przebytej operacji wszczepienia mechanicznej zastawki serca (tj. podczas obecnej hospitalizacji) oraz u pacjentów, u których od wszczepienia mechanicznej zastawki serca minęły ponad trzy miesiące. Więcej zdarzeń zakrzepowo-zatorowych (głównie udarów mózgu
i objawowych/bezobjawowych przypadków obecności skrzepliny na sztucznej zastawce serca) i więcej przypadków krwawienia było obserwowane podczas podawania dabigatranu eteksylanu niż warfaryny. U pacjentów po niedawno przebytej operacji przypadki dużego krwawienia miały
przeważnie postać krwotocznego wysięku osierdziowego; dotyczyło to zwłaszcza pacjentów, którzy wcześnie rozpoczęli przyjmowanie dabigatranu eteksylanu (tj. w 3. dniu) po operacji wszczepienia zastawki (patrz punkt 4.3).
Dzieci i młodzież
Badania kliniczne dotyczące zapobiegania ŻChZZ po rozległym zabiegu chirurgicznym wszczepienia endoprotezy stawowej
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań dabigatranu eteksylanu we wszystkich podgrupach populacji dzieci i młodzieży w prewencji zdarzeń zakrzepowo- zatorowych we wskazaniu prewencji pierwotnej ŻChZZ u pacjentów po przebytej planowej alloplastyce całkowitej stawu biodrowego lub kolanowego (patrz punkt 4.2 – informacje dotyczące stosowania u dzieci i młodzieży).
Leczenie ŻChZZ i prewencja nawrotów ŻChZZ u dzieci i młodzieży
Badanie DIVERSITY przeprowadzono w celu wykazania skuteczności i bezpieczeństwa stosowania dabigatranu eteksylanu w porównaniu ze standardowym leczeniem w zakresie leczenia ŻChZZ
u dzieci i młodzieży od urodzenia do wieku poniżej 18 lat. Badanie zaplanowano jako otwarte, randomizowane, prowadzone w grupach równoległych mające na celu potwierdzenie nie mniejszej skuteczności (ang. „non-inferiority”). Pacjentów włączonych do badania przydzielono losowo zgodnie ze schematem 2:1 do grupy przyjmującej dabigatran eteksylan w postaci farmaceutycznej odpowiedniej dla wieku (kapsułki, granulat powlekany lub roztwór doustny) (dawki dostosowane do wieku i masy ciała) lub standardowe leczenie obejmujące heparyny niskocząsteczkowe (ang.
LMWH – low molecular weight heparins) lub antagonistów witaminy K (ang. VKA – vitamin K antagonists) lub fondaparynuks (1 pacjent w wieku 12 lat). Pierwszorzędowym punktem końcowym był złożony punkt końcowy obejmujący pacjentów, u których nastąpiło całkowite rozpuszczenie się skrzepliny i nie występowały nawroty ŻChZZ oraz u których nie obserwowano zgonów związanych z ŻChZZ. Kryteria wykluczenia obejmowały czynne zapalenie opon mózgowo-rdzeniowych, zapalenie mózgu i ropień śródczaszkowy.
Randomizacji poddano łącznie 267 pacjentów. Spośród nich 176 pacjentów leczono dabigatranem eteksylanem , a 90 pacjentów zgodnie ze standardowym leczeniem (1 poddany randomizacji pacjent nie był leczony). 168 pacjentów było w wieku od 12 do poniżej 18 lat, 64 pacjentów było w wieku od
2 do poniżej 12 lat, a 35 pacjentów było w wieku poniżej 2 lat.
Spośród 267 poddanych randomizacji pacjentów, 81 pacjentów (45,8%) w grupie przyjmującej dabigatran eteksylan i 38 pacjentów (42,2%) w grupie leczonej w ramach standardowego leczenia spełniało kryteria złożonego pierwszorzędowego punktu końcowego (całkowite rozpuszczenie się skrzepliny, brak nawrotów ŻChZZ oraz brak zgonów związanych z ŻChZZ). Odpowiednia różnica w zakresie częstości występowania wykazała nie mniejszą skuteczność dabigatranu eteksylanu
względem standardowego leczenia. Spójne wyniki były również obserwowane ogółem w podgrupach: nie było żadnych znaczących różnic w efekcie leczenia w podgrupach w zależności od wieku, płci, regionu i obecności niektórych czynników ryzyka. W zakresie 3 różnych grup wiekowych odsetek pacjentów, którzy osiągnęli pierwszorzędowy punkt końcowy skuteczności w grupie przyjmującej dabigatran eteksylan oraz w grupie otrzymującej standardowe leczenie, wynosił odpowiednio 13/22 (59,1%) i 7/13 (53,8%) dla pacjentów w wieku od urodzenia do <2 lat, 21/43 (48,8%) i 12/21 (57,1%) u pacjentów w wieku od 2 do <12 lat oraz 47/112 (42,0%) i 19/56 (33,9%) u pacjentów w wieku od 12 do <18 lat.
Potwierdzone duże krwawienie zgłoszono u 4 pacjentów (2,3%) w grupie przyjmującej dabigatran eteksylan i u 2 pacjentów (2,2%) w grupie otrzymującej standardowe leczenie. Nie było
statystycznie istotnej różnicy dotyczącej czasu do wystąpienia pierwszego incydentu dużego krwawienia. U trzydziestu ośmiu pacjentów (21,6%) w grupie przyjmującej dabigatran eteksylan oraz 22 pacjentów (24,4%) w grupie otrzymującej standardowe leczenie wystąpił potwierdzony incydent krwawienia, przy czym większość z nich została sklasyfikowana jako małe krwawienia. Złożony punkt końcowy obejmujący potwierdzony incydent dużego krwawienia lub krwawienia klinicznie istotnego innego niż duże (występujące w trakcie leczenia) zgłoszono u 6 (3,4%) pacjentów z grupy przyjmującej dabigatran eteksylan oraz 3 (3,3%) pacjentów w grupie otrzymującej standardowe leczenie.
Przeprowadzono otwarte, prowadzone na jednej grupie, prospektywne kohortowe, wieloośrodkowe badanie fazy III (1160,108) w celu oceny bezpieczeństwa stosowania dabigatranu eteksylanu
w zapobieganiu nawrotom ŻChZZ u dzieci i młodzieży od urodzenia do wieku poniżej 18 lat. Do tego badania mogli zostać włączeni pacjenci, którzy wymagali dalszego leczenia przeciwzakrzepowego z powodu występowania klinicznego czynnika ryzyka po zakończeniu wstępnego leczenia potwierdzonego ŻChZZ (przez co najmniej 3 miesiące) lub po zakończeniu badania DIVERSITY.
Kwalifikujący się pacjenci otrzymywali dostosowane do wieku i masy ciała dawki produktu leczniczego w postaci farmaceutycznej odpowiedniej dla wieku (kapsułki, granulat powlekany lub roztwór doustny) dabigatranu eteksylanu do momentu ustąpienia klinicznego czynnika ryzyka lub przez maksymalnie 12 miesięcy. Pierwszorzędowe punkty końcowe badania obejmowały nawrót ŻChZZ, incydenty dużych i małych krwawień oraz śmiertelność (całkowitą i związaną z incydentami zakrzepowymi lub zakrzepowo-zatorowymi) w 6. i 12 miesiącu. Zdarzenia te były oceniane przez niezależną komisję rozstrzygającą, która nie wiedziała, jaki lek badany otrzymywał pacjent.
Ogółem do badania włączono 214 pacjentów; spośród nich 162 pacjentów było w grupie wiekowej 1 (od 12 do poniżej 18 lat), 43 pacjentów w grupie wiekowej 2 (od 2 do poniżej 12 lat), a 9 pacjentów w grupie wiekowej 3 (od urodzenia do wieku poniżej 2 lat). W okresie leczenia u 3 pacjentów (1,4%) wystąpił potwierdzony nawrót ŻChZZ w ciągu pierwszych 12 miesięcy od rozpoczęcia leczenia.
Potwierdzone incydenty krwawień w okresie leczenia zgłaszano u 48 pacjentów (22,5%) w ciągu pierwszych 12 miesięcy. Większość incydentów krwawień stanowiły małe krwawienia. U 3 pacjentów (1,4%) potwierdzony incydent dużego krwawienia wystąpił w ciągu pierwszych 12 miesięcy.
U 3 pacjentów (1,4%) potwierdzone klinicznie istotne inne niż duże krwawienie zgłaszano w
ciągu pierwszych 12 miesięcy. W trakcie leczenia nie wystąpił żaden zgon. W okresie leczenia u 3 pacjentów (1,4%) wystąpił zespół pozakrzepowy lub nasilenie zespołu pozakrzepowego w
ciągu pierwszych 12 miesięcy.
Po podaniu doustnym dabigatran eteksylan ulega szybkiej i całkowitej przemianie do dabigatranu, który stanowi czynną postać leku w osoczu. Główną reakcją metaboliczną jest rozszczepienie proleku dabigatranu eteksylanu w drodze hydrolizy katalizowanej przez esterazę do substancji czynnej, dabigatranu. Bezwzględna dostępność biologiczna dabigatranu po podaniu doustnym dabigatranu eteksylanu wynosiła około 6,5%.
Po doustnym podaniu dabigatranu eteksylanu u zdrowych ochotników profil farmakokinetyczny dabigatranu w osoczu charakteryzuje się szybkim zwiększeniem jego stężenia osoczowego z uzyskaniem Cmax w ciągu 0,5 do 2,0 godzin po podaniu.
Wchłanianie
W badaniu oceniającym pooperacyjne wchłanianie dabigatranu eteksylanu po upływie 1-3 godzin od zabiegu chirurgicznego wykazano względnie powolne wchłanianie produktu w porównaniu do zdrowych ochotników, z jednostajnym przebiegiem zmian stężenia w osoczu w czasie, bez dużych wartości maksymalnego stężenia w osoczu. Produkt leczniczy osiąga maksymalne stężenie w osoczu w ciągu 6 godzin od podania w okresie pooperacyjnym ze względu na oddziaływanie takich czynników, jak znieczulenie ogólne, porażenie mięśniówki przewodu pokarmowego i skutki zabiegu chirurgicznego, niezależnie od postaci, w jakiej występuje doustnie podawany produkt leczniczy.
W innym badaniu wykazano, że spowolnienie i opóźnienie wchłaniania ma miejsce na ogół wyłącznie w dniu operacji. W późniejszych dniach dabigatran szybko się wchłaniania, osiągając
maksymalne stężenie w osoczu w ciągu 2 godzin po podaniu produktu leczniczego.
Pokarm nie wpływa na dostępność biologiczną dabigatranu eteksylanu , jednak wydłuża czas do uzyskania maksymalnego stężenia leku w osoczu o 2 godziny.
Cmax i AUC były proporcjonalne do dawki.
W wyniku przyjęcia peletek bez otoczki kapsułki z hydroksypropylometylocelulozy (HPMC) biodostępność produktu leczniczego po podaniu doustnym może ulec zwiększeniu o 75% po podaniu dawki pojedynczej i 37% w stanie stacjonarnym, w porównaniu z preparatem referencyjnym w postaci kapsułek. Z tego powodu w warunkach klinicznych należy zawsze
zachować integralność kapsułek HPMC, aby uniknąć niezamierzonego zwiększenia biodostępności dabigatranu eteksylanu (patrz punkt 4.2).
Dystrybucja
Zaobserwowano, że dabigatran wiąże się z ludzkimi białkami osocza w małym stopniu (34-35%), niezależnie od stężenia. Objętość dystrybucji dabigatranu wynosząca od 60 do 70 l przekraczała objętość całkowitej ilości wody zawartej w organizmie, co wskazuje na umiarkowaną dystrybucję tkankową dabigatranu.
Metabolizm
Badano metabolizm i wydalanie dabigatranu po podaniu pojedynczej dawki dożylnej dabigatranu znakowanego radioaktywnie u zdrowych mężczyzn. Po podaniu dożylnym wykryto, że znakowany radioaktywnie dabigatran wydala się przede wszystkim z moczem (85%). Z kałem uległo wydaleniu 6% podanej dawki. Stopień odzysku radioaktywności całkowitej wahał się od 88 do 94% podanej dawki w ciągu 168 godzin od jej podania.
Dabigatran ulega sprzęganiu, z powstaniem czynnych farmakologicznie acyloglukuronidów. Istnieją cztery izomery pozycyjne: 1-O, 2-O, 3-O i 4-O-acyloglukuronid; każdy z nich odpowiada za mniej niż 10% całkowitego stężenia dabigatranu w osoczu. Ślady innych metabolitów były wykrywalne wyłącznie przy użyciu metod analitycznych o wysokiej czułości. Dabigatran ulega wydaleniu przede wszystkim w postaci niezmienionej z moczem, z szybkością około 100 mL/min, odpowiednio do wskaźnika przesączania kłębuszkowego.
Eliminacja
Wartości stężenia dabigatranu w osoczu zmniejszały się dwuwykładniczo, przy czym średni okres półtrwania w fazie eliminacji wynosił u zdrowych ochotników w podeszłym wieku 11 godzin.
Po podaniu wielokrotnym okres półtrwania w fazie eliminacji wynosił od około 12 do 14 godzin. Okres półtrwania nie zależał od dawki. Okres półtrwania jest wydłużony u pacjentów z zaburzeniami czynności nerek, zgodnie z tabelą 16.
Szczególne grupy pacjentów
Niewydolność nerek
W badaniach fazy I całkowity wpływ dabigatranu na organizm (AUC) po doustnym podaniu dabigatranu eteksylanu jest około 2,7 razy większy u dorosłych ochotników z umiarkowaną niewydolnością nerek (CrCL pomiędzy 30 a 50 mL/min) niż u osób bez niewydolności nerek.
U małej liczby dorosłych ochotników z ciężką niewydolnością nerek (CrCL 10-30 mL/min)
całkowity wpływ dabigatranu na organizm (AUC) był około 6 razy większy, a okres półtrwania około 2 razy dłuższy niż w populacji bez niewydolności nerek (patrz punkty 4.2, 4.3 i 4.4).
Tabela 16: Okres półtrwania całkowitego dabigatranu u zdrowych pacjentów oraz pacjentów z niewydolnością nerek.
Wskaźnik przesączania kłębuszkowego (CrCL) [mL/min] | gMean (gCV%; zakres) okres półtrwania [h] |
≥80 | 13,4 (25,7%; 11,0-21,6) |
≥50-<80 | 15,3 (42,7%; 11,7-34,1) |
≥30-<50 | 18,4 (18,5 %; 13,3-23,0) |
<30 | 27,2 (15,3%; 21,6-35,0) |
Ponadto oceniono ekspozycję na dabigatran (w stężeniu minimalnym i maksymalnym)
w prospektywnym, otwartym, randomizowanym badaniu farmakokinetycznym u pacjentów z niezastawkowym migotaniem przedsionków i ciężkimi zaburzeniami czynności nerek (zdefiniowanymi jako klirens kreatyniny [CrCL] 15-30 mL/min), którzy otrzymywali dabigatran eteksylan w dawce 75 mg dwa razy na dobę.
Wynikiem tego schematu była średnia geometryczna minimalnego stężenia, mierzonego bezpośrednio przed podaniem kolejnej dawki, wynosząca 155 ng/mL (gCV 76,9%) oraz średnia geometryczna maksymalnego stężenia, mierzonego dwie godziny po podaniu ostatniej dawki, wynosząca 202 ng/mL (gCV 70,6%).
Klirens dabigatranu w wyniku hemodializy badano u 7 dorosłych pacjentów ze schyłkową
niewydolnością nerek bez migotania przedsionków. Dializa trwała cztery godziny, tempo przepływu dializatu wynosiło 700 mL/min, a tempo przepływu krwi wynosiło 200 mL/min lub 350-390 mL/min. Usunięto odpowiednio od 50% do 60% stężenia dabigatranu. Ilość substancji usunięta podczas dializy jest proporcjonalna do tempa przepływu krwi aż do wartości 300 mL/min.
Działanie przeciwzakrzepowe dabigatranu uległo zmniejszeniu wraz ze spadającym stężeniem dabigatranu w osoczu. Dializa nie miała wpływu na stosunek PK/PD.
Pacjenci w podeszłym wieku
Specjalne badania farmakokinetyczne fazy I przeprowadzone z udziałem pacjentów w podeszłym wieku wykazały zwiększenie AUC o 40-60% i zwiększenie Cmax o ponad 25% w porównaniu do młodych pacjentów.
Wpływ wieku na ekspozycję na dabigatran potwierdzono w badaniu RE-LY, w którym obserwowano wyższe o około 31% stężenia minimalne u pacjentów w wieku ≥75 lat oraz o około 22% niższe stężenia minimalne u pacjentów w wieku <65 lat w porównaniu do pacjentów w wieku pomiędzy 65 i 75 lat (patrz punkty 4.2 i 4.4).
Niewydolność wątroby
U 12 dorosłych pacjentów z umiarkowaną niewydolnością wątroby (stopnia B wg klasyfikacji Childa- Pugha) nie stwierdzono zmian całkowitego wpływu dabigatranu na organizm w porównaniu do
12 pacjentów z grupy kontrolnej (patrz punkty 4.2 i 4.4).
Masa ciała
Minimalne stężenia dabigatranu były o około 20% niższe u dorosłych pacjentów o masie ciała
> 100 kg w porównaniu do pacjentów o masie ciała 50-100 kg. Większość pacjentów (80,8%) mieściła się w kategorii wagowej ≥50 kg i <100 kg bez wyraźnej różnicy (patrz punkty 4.2 i 4.4). Dane kliniczne u dorosłych pacjentów o masie ciała <50 kg są ograniczone.
Płeć
U pacjentek płci żeńskiej w badaniach prewencji pierwotnej żylnej choroby zakrzepowo-zatorowej całkowity wpływ substancji czynnej na organizm był około 40% do 50% większy, w związku z czym nie zaleca się modyfikacji dawkowania.
Pochodzenie etniczne
Nie stwierdzono klinicznie znaczących różnic pomiędzy pacjentami rasy białej, czarnej, pochodzenia latynoamerykańskiego, rasy żółtej pod względem właściwości farmakokinetycznych i farmakodynamicznych dabigatranu.
Dzieci i młodzież
Doustne podawanie dabigatranu eteksylanu zgodnie ze zdefiniowanym w protokole algorytmem dawkowania prowadziło do ekspozycji w zakresie obserwowanym u dorosłych z ZŻG / ZP. W oparciu o zbiorczą analizę danych farmakokinetycznych z badań DIVERSITY i 1160.108 obserwowana średnia geometryczna najmniejszej ekspozycji wynosiła 53,9 ng/mL, 63,0 ng/mL i 99,1 ng/mL odpowiednio u dzieci i młodzieży z ŻChZZ w wieku od 0 do <2 lat, od 2 do <12 lat oraz od 12 do
<18 lat.
Interakcje farmakokinetyczne
Badania nad interakcjami in vitro nie wykazały zahamowania ani indukcji głównych izoenzymów cytochromu P450. Wynik ten potwierdziły badania przeprowadzone in vivo z udziałem zdrowych ochotników, u których nie stwierdzono jakichkolwiek interakcji pomiędzy omawianym produktem leczniczym a następującymi substancjami czynnymi: atorwastatyną (CYP3A4), digoksyną (interakcja z białkiem transportowym P-gp) i diklofenakiem (CYP2C9).
Dane niekliniczne, wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, badań toksyczności po podaniu wielokrotnym i genotoksyczności nie ujawniają szczególnego zagrożenia dla człowieka.
Skutki stosowania produktu leczniczego obserwowane w badaniach toksyczności po podaniu wielokrotnym wynikały z nasilonego działania farmakodynamicznego dabigatranu.
Obserwowano wpływ produktu leczniczego na płodność samic w postaci zmniejszenia liczby zagnieżdżeń zapłodnionego jaja i zwiększenia częstości utraty zapłodnionego jaja przed
zagnieżdżeniem po dawce 70 mg/kg (5-krotnie większej od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów). Po dawkach toksycznych dla matek (od 5- do 10-krotnie większych od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów) u szczurów i królików stwierdzono zmniejszenie masy ciała i
przeżywalności
płodów, łącznie ze zwiększeniem liczby wad rozwojowych płodów. W badaniach pre- i postnatalnych zaobserwowano zwiększenie umieralności płodów po dawkach toksycznych dla samic (4-krotnie większych od całkowitego wpływu produktu leczniczego zawartego w osoczu na organizm u pacjentów).
W badaniu toksyczności u młodych szczurów Han Wistar umieralność była związana z incydentami krwawienia przy ekspozycji podobnej do tej, przy której krwawienie obserwowano u dorosłych zwierząt. Uważa się, że zarówno u dorosłych, jak i młodych szczurów śmiertelność jest związana
z nadmierną aktywnością farmakologiczną dabigatranu w połączeniu z siłą mechaniczną wywieraną podczas podawania produktu leczniczego. Dane z badania toksyczności u młodych nie wykazały zwiększonej wrażliwości na toksyczność ani specyficznej dla młodych zwierząt toksyczności.
W badaniach toksykologicznych w całym okresie życia u szczurów i myszy nie stwierdzono dowodów na potencjalne działanie guzotwórcze dabigatranu po podaniu maksymalnych dawek do 200 mg/kg.
Dabigatran, czynna cząstka dabigatranu eteksylanu (w postaci mezylanu) nie ulega rozpadowi w
środowisku.
Zawartość kapsułki
Nie dotyczy.
2 lata
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego
Produkt leczniczy Wasedoc, 75 mg jest dostępny w opakowaniach zawierających 10, 30 lub 60 kapsułek twardych w perforowanych blistrach Aluminium/OPA/Aluminium/PE z odrywaną folią ze środkiem pochłaniającym wilgoć, w tekturowym pudełku.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Podczas wyjmowania kapsułek produktu leczniczego Wasedoc z blistra należy postępować zgodnie z poniższymi zaleceniami:
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Ranbaxy (Poland) Sp. z o.o. ul. Kubickiego 11
02-954 Warszawa
Pozwolenie nr
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:







