







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY Fiolka 100 mg
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY Fiolka 100 mg
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
zespołami mielodysplastycznymi (ang. myelodysplastic syndromes, MDS) o pośrednim-2 i wysokim ryzyku, zgodnie z Międzynarodowym Punktowym Systemem Prognostycznym (ang. International Prognostic Scoring System, IPSS)
przewlekłą białaczką mielomonocytową (ang. chronic myelomonocytic leukaemia, CMML) z 10-29% blastów w szpiku, bez choroby mieloproliferacyjnej,
ostrą białaczką szpikową (ang. acute myeloid leukaemia, AML) z 20-30% blastów i wieloliniową dysplazją, zgodnie z klasyfikacją Światowej Organizacji Zdrowia (WHO),
AML z >30% blastów w szpiku, zgodnie z klasyfikacją WHO.
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
Azacitidine Glenmark, 25 mg/ml, proszek do sporządzania zawiesiny do wstrzykiwań
Każda fiolka zawiera 100 mg azacytydyny. Po przygotowaniu, każdy ml zawiesiny zawiera 25 mg azacytydyny.
Fiolka 150 mg
Każda fiolka zawiera 150 mg azacytydyny. Po przygotowaniu, każdy ml zawiesiny zawiera 25 mg azacytydyny.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek do sporządzania zawiesiny do wstrzykiwań. Biały liofilizowany proszek.
Produkt leczniczy Azacitidine Glenmark jest wskazany do leczenia pacjentów dorosłych, którzy nie kwalifikują się do przeszczepienia krwiotwórczych komórek macierzystych (ang. haematopoietic stem cell transplantation, HSCT), z:
Leczenie produktem leczniczym Azacitidine Glenmark powinno zostać rozpoczęte, a następnie monitorowane, przez lekarza posiadającego doświadczenie w stosowaniu chemioterapeutyków. Pacjentom należy podać premedykację w postaci leków przeciwwymiotnych, aby zapobiec wystąpieniu nudności i wymiotów.
Dawkowanie
Zalecana dawka początkowa dla pierwszego cyklu leczenia dla wszystkich pacjentów, bez względu na początkowe hematologiczne wartości laboratoryjne, to 75 mg/m2 powierzchni ciała, podawane jako wstrzyknięcie podskórne, codziennie przez 7 dni, po czym następuje okres odpoczynku trwający
21 dni (28-dniowy cykl leczenia).
Zalecane jest leczenie pacjentów przez co najmniej 6 cykli. Leczenie należy kontynuować tak długo, jak pacjent odnosi z niego korzyści, lub do progresji choroby.
Należy monitorować pacjentów w kierunku odpowiedzi/toksyczności hematologicznej oraz toksycznego wpływu na nerki (patrz punkt 4.4); może być konieczne opóźnienie rozpoczęcia następnego cyklu lub zmniejszenie dawki w sposób opisany poniżej.
Produktu Azacitidine Glenmark nie należy podawać zamiennie z azacytydyną podawaną doustnie. Ze względu na różnice w ekspozycji, zalecenia dotyczące dawki i schematu dawkowania azacytydyny podawanej doustnie różnią się od zaleceń dla azacytydyny do wstrzykiwań. Pracownikom opieki zdrowotnej zaleca się sprawdzenie nazwy produktu leczniczego, dawki i drogi podania.
Badania laboratoryjne
Przed rozpoczęciem leczenia i przed każdym cyklem leczenia należy wykonać badania czynności wątroby, oznaczyć stężenie kreatyniny oraz dwuwęglanów w surowicy. Pełną morfologię krwi należy wykonywać przed rozpoczęciem leczenia oraz tak często, jak to jest konieczne, aby monitorować odpowiedź i toksyczność, jednak nie rzadziej niż przed każdym cyklem leczenia.
Dostosowanie dawki z powodu toksyczności hematologicznej
Toksyczność hematologiczna jest zdefiniowana jako najniższa wartość (nadir) osiągnięta w danym cyklu, jeśli liczba płytek ≤50,0 x 109/l i(lub) liczba bezwzględna neutrofili (ang. Absolute Neutrophil Count, ANC) ≤1 x 109/l.
Regeneracja jest zdefiniowana jako zwiększenie linii komórkowej (-ych), dla których zaobserwowano toksyczność hematologiczną, przynajmniej o połowę bezwzględnej różnicy między nadirem a liczbą początkową plus wartości nadiru (np. liczba krwinek w momencie regeneracji ≥ wartość nadiru + (0,5 x [liczba początkowa – wartość nadiru]).
Pacjenci bez obniżonej początkowej liczby krwinek (tzn. liczba krwinek białych (ang. white blood cells,
WBC) ≥ 3,0 x 109/l i ANC ≥ 1,5 x 109/l oraz płytki ≥ 75,0 x 109/l) przed pierwszym leczeniem
W przypadku zaobserwowania toksyczności hematologicznej po leczeniu produktem Azacitidine Glenmark, należy opóźnić następny cykl leczenia do osiągnięcia regeneracji liczby płytek i ANC. Jeśli regeneracja nastąpiła w ciągu 14 dni, dostosowanie dawki nie jest konieczne. Jednak, jeśli regeneracja nie nastąpiła w ciągu 14 dni, należy zmniejszyć dawkę zgodnie z poniższą tabelą. Po zmodyfikowaniu dawki, czas trwania cyklu powinien wynosić ponownie 28 dni.
Wartość nadiru w cyklu | Dawka w następnym cyklu, jeśli regeneracja* nie została osiągnięta w ciągu 14 dni (%) | |
ANC (x 109/l) | Płytki (x 109/l) | |
≤ 1,0 | ≤ 50,0 | 50% |
> 1,0 | > 50,0 | 100% |
*Regeneracja = wartości ≥ wartość nadiru + (0,5 x [liczba początkowa – wartość nadiru])
Pacjenci z obniżoną początkową liczbą krwinek (tzn. WBC < 3,0 x 109/l lub ANC < 1,5 x 109/l lub płytki < 75,0 x 109/l) przed pierwszym leczeniem
Po leczeniu produktem leczniczym Azacitidine Glenmark, jeśli zmniejszenie WBC lub ANC, lub płytek, w porównaniu z wartościami przed leczeniem, wynosi ≤ 50%, lub więcej niż 50 %, ale z poprawą w różnicowaniu którejkolwiek z linii komórkowych, nie należy opóźniać następnego cyklu ani dostosowywać dawki.
Jeśli zmniejszenie WBC lub ANC, lub płytek, w porównaniu z wartościami przed leczeniem, jest większe niż 50%, bez poprawy w różnicowaniu linii komórkowych, należy opóźnić następny cykl leczenia azacytydyną do osiągnięcia regeneracji liczby płytek i ANC. Jeśli regeneracja nastąpiła w ciągu 14 dni, dostosowanie dawki nie jest konieczne. Jednak, jeśli regeneracja nie nastąpiła w ciągu 14 dni, należy oznaczyć komórkowość szpiku kostnego. Jeśli komórkowość szpiku kostnego wynosi
> 50%, nie należy dostosowywać dawki. Jeśli komórkowość szpiku kostnego wynosi ≤ 50%, należy opóźnić leczenie i zmniejszyć dawkę zgodnie z poniższą tabelą:
Komórkowość szpiku kostnego | Dawka w następnym cyklu, jeśli regeneracja nie została osiągnięta w ciągu 14 dni (%) | |
Regeneracja* ≤ 21 dni | Regeneracja* > 21 dni | |
15-50% | 100% | 50% |
< 15% | 100% | 33% |
*Regeneracja = liczby ≥ wartość nadiru + (0,5 x [liczba początkowa – wartość nadiru]) Po modyfikacji dawki czas trwania następnego cyklu powinien wynosić ponownie 28 dni.
Specjalne grupy pacjentów Pacjenci w podeszłym wieku
W przypadku pacjentów w podeszłym wieku nie zaleca się szczególnego dostosowywania dawki. Ponieważ u pacjentów w podeszłym wieku jest bardziej prawdopodobne występowanie zmniejszonej czynności nerek, u tych pacjentów użyteczne może być kontrolowanie czynności nerek.
Pacjenci z niewydolnością nerek
Azacytydyna może być podawana pacjentom z niewydolnością nerek bez dostosowywania dawki początkowej (patrz punkt 5.2). Jeśli wystąpi niewyjaśnione zmniejszenie stężenia dwuwęglanów w surowicy do poziomu poniżej 20 mmol/l, należy zmniejszyć dawkę o 50% w następnym cyklu. Jeśli wystąpi niewyjaśniony wzrost stężenia kreatyniny w surowicy lub azotu mocznikowego we krwi [ang. Blood Urea Nitrogen, BUN] do ≥ 2-krotności wartości początkowych i powyżej górnej granicy normy, należy opóźnić następny cykl, do czasu, gdy wartości powrócą do wartości prawidłowych lub początkowych, oraz należy zmniejszyć dawkę o 50% w następnym cyklu leczenia (patrz punkt 4.4).
Pacjenci z niewydolnością wątroby
Nie przeprowadzono formalnych badań z udziałem pacjentów z niewydolnością wątroby (patrz punkt 4.4). Pacjentów z ciężką niewydolnością narządową wątroby należy uważnie monitorować pod kątem wystąpienia zdarzeń niepożądanych. U pacjentów z niewydolnością wątroby, nie zaleca się szczególnej modyfikacji dawki początkowej przed rozpoczęciem leczenia; późniejsze modyfikacje dawek powinny być oparte na hematologicznych wartościach laboratoryjnych. Azacytydyna jest przeciwwskazana u pacjentów z zaawansowanymi nowotworami złośliwymi wątroby (patrz punkty 4.3 i 4.4).
Dzieci i młodzież
Nie określono dotychczas bezpieczeństwa stosowania ani skuteczności azacytydyny u dzieci w wieku od 0 do 17 lat. Aktualne dane przedstawiono w punktach 4.8, 5.1 i 5.2, ale brak zaleceń dotyczących dawkowania.
Sposób podawania
Przygotowany produkt leczniczy Azacitidine Glenmark należy wstrzykiwać podskórnie w ramię, udo lub brzuch. Należy zmieniać miejsca wstrzyknięcia. Nowe wstrzyknięcia należy podawać przynajmniej 2,5 cm od poprzedniego miejsca i nigdy nie wstrzykiwać w miejsca podrażnione, zasiniaczone, zaczerwienione lub stwardniałe.
Po sporządzeniu, zawiesina nie powinna być filtrowana. Instrukcja dotycząca przygotowania produktu leczniczego przed podaniem, patrz punkt 6.6.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Zaawansowane nowotwory złośliwe wątroby (patrz punkt 4.4). Karmienie piersią (patrz punkt 4.6).
Toksyczność hematologiczna
Leczenie azacytydyną jest związane z występowaniem niedokrwistości, neutropenii i trombocytopenii, szczególnie w czasie pierwszych 2 cykli (patrz punkt 4.8). Należy wykonywać pełną morfologię krwi tak często, jak to jest konieczne, aby monitorować odpowiedź i toksyczność, jednak co najmniej przed każdym cyklem leczenia. Po podaniu zalecanej dawki dla pierwszego cyklu, należy obniżyć dawkę dla następnych cykli lub opóźnić jej podanie w oparciu o wartości nadiru i odpowiedź hematologiczną (patrz punkt 4.2). Należy poinformować pacjenta, by niezwłocznie zgłaszał epizody gorączkowe.
Pacjentów oraz lekarzy należy również poinformować, by zwracali uwagę na objawy przedmiotowe i podmiotowe krwawienia.
Niewydolność wątroby
Nie przeprowadzono formalnych badań z udziałem pacjentów z niewydolnością wątroby. U pacjentów z rozległym obciążeniem nowotworem z powodu przerzutów, zgłaszano występowanie postępującej śpiączki wątrobowej i śmierci podczas leczenia azacytydyną, w szczególności u pacjentów z początkowym stężeniem albuminy w surowicy < 30 g/l. Azacytydyna jest przeciwwskazana u pacjentów z zaawansowanymi nowotworami złośliwymi wątroby (patrz punkt 4.3).
Niewydolność nerek
Zaburzenia czynności nerek, począwszy od podwyższonego stężenia kreatyniny w surowicy aż po niewydolność nerek i śmierć, zgłaszano u pacjentów leczonych dożylnie azacytydyną w skojarzeniu z innymi chemioterapeutykami. Dodatkowo, u 5 pacjentów z przewlekłą białaczką szpikową (ang.
Chronic Myelogenous Leukaemia, CML), leczonych azacytydyną i etopozydem, wystąpiła kwasica kanalikowo-nerkowa, zdefiniowana jako obniżenie stężenia dwuwęglanów w surowicy do
< 20 mmol/l, z moczem zasadowym i hipokaliemią (stężenie potasu w surowicy < 3 mmol/l). W przypadku wystąpienia niewyjaśnionego obniżenia stężenia dwuwęglanów w surowicy (< 20 mmol/l) lub wzrostów stężenia kreatyniny w surowicy lub BUN, należy zmniejszyć dawkę lub opóźnić podanie leku (patrz punkt 4.2).
Należy poinformować pacjentów, aby natychmiast zgłaszali pracownikom służby zdrowia wystąpienie skąpomoczu i bezmoczu.
Chociaż nie zaobserwowano klinicznie istotnych różnic w częstości występowania działań niepożądanych między pacjentami z prawidłową czynnością nerek a pacjentami z niewydolnością nerek, należy ściśle monitorować pacjentów z niewydolnością nerek w celu wykrycia toksyczności, ponieważ azacytydyna i (lub) jej metabolity są wydalane głównie przez nerki (patrz punkt 4.2).
Badania laboratoryjne
Przed rozpoczęciem leczenia i przed każdym cyklem leczenia należy wykonać badania czynności wątroby, oznaczyć stężenie kreatyniny oraz dwuwęglanów w surowicy. Pełną morfologię krwi należy wykonywać przed rozpoczęciem leczenia oraz tak często, jak to jest konieczne, aby monitorować odpowiedź i toksyczność, ale nie rzadziej niż przed każdym cyklem leczenia, patrz punkt 4.8.
Choroby serca i płuc
Pacjenci z ciężką zastoinową niewydolnością serca, klinicznie niestabilną chorobą serca lub chorobą płuc w wywiadzie, byli wyłączeni z głównych badań rejestracyjnych (AZA PH GL 2003 CL 001 oraz AZA-AML-001), i z tego powodu nie ustalono bezpieczeństwa stosowania ani skuteczności azacytydyny u tych pacjentów. Najnowsze dane z badań klinicznych u pacjentów z chorobami serca i płuc w wywiadzie wykazały istotne zwiększenie częstości zdarzeń dotyczących serca związanych ze stosowaniem azacytydyny (patrz punkt 4.8). Dlatego zaleca się zachowanie ostrożności przy przepisywaniu azacytydyny tym pacjentom. Przed oraz w trakcie leczenia należy rozważyć przeprowadzenie oceny wydolności krążeniowo-oddechowej.
Martwicze zapalenie powięzi
U pacjentów leczonych azacytydyną zgłaszano przypadki martwiczego zapalenia powięzi, w tym przypadki zakończone zgonem. U pacjentów, u których wystąpi martwicze zapalenie powięzi należy przerwać podawanie azacytydyny oraz bezzwłocznie wdrożyć właściwe leczenie.
Zespół rozpadu guza
Ryzyko zespołu rozpadu guza dotyczy pacjentów z dużym rozmiarem guza przed zastosowaniem leczenia. Tych pacjentów należy starannie monitorowani i należy zastosować u nich odpowiednie środki ostrożności.
Zespół różnicowania
U pacjentów otrzymujących azacytydyny do wstrzykiwań zgłaszano przypadki zespołu różnicowania (znanego także jako zespół kwasu retinowego). Zespół różnicowania może skutkować zgonem, a objawy kliniczne mogą obejmować niewydolność oddechową, nacieki w płucach, gorączkę, wysypkę, obrzęk płuc, obrzęk obwodowy, szybki przyrost masy ciała, wysięk do opłucnej, wysięk do osierdzia, niedociśnienie i zaburzenia czynności nerek (patrz punkt 4.8). Należy rozważyć leczenie dużymi dawkami dożylnych kortykosteroidów i monitorowanie parametrów hemodynamicznych w razie pierwszego wystąpienia objawów przedmiotowych i podmiotowych, wskazujących na zespół różnicowania. Należy rozważyć czasowe przerwanie stosowania produktu azacytydyny do wstrzykiwań do czasu ustąpienia objawów i w razie ponownego ich wystąpienia należy zachować ostrożność.
W oparciu o dane z badań in vitro, nie wydaje się, by w metabolizmie azacytydyny uczestniczyły izoenzymy cytochromu P450 (CYP), UDP-glukuronozylotransferazy (UGT), sulfotransferazy (SULT) ani transferazy glutationowe (GST). Z tego powodu uznaje się, że interakcje in vivo związane z tymi enzymami metabolizującymi są mało prawdopodobne.
Klinicznie istotne działania hamujące lub indukujące azacytydyny na enzymy cytochromu P450 są mało prawdopodobne (patrz punkt 5.2).
Nie przeprowadzono formalnych badań klinicznych azacytydyny dotyczących interakcji.
Kobiety w wieku rozrodczym/Antykoncepcja u mężczyzn i kobiet
Kobiety w wieku rozrodczym muszą stosować skuteczną metodę antykoncepcji w trakcie leczenia i przez co najmniej 6 miesięcy po okresie leczenia. Należy poinstruować mężczyzn, aby podczas leczenia nie spłodzili dziecka oraz by stosowali skuteczną antykoncepcję w trakcie leczenia i przez co najmniej 3 miesiące po zakończeniu leczenia.
Ciąża
Brak wystarczających danych dotyczących stosowania azacytydyny u kobiet w ciąży. Badania na myszach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3). Możliwe zagrożenie dla człowieka nie jest znane. W oparciu o wyniki badań na zwierzętach i mechanizm działania azacytydyny, nie zaleca się stosowania jej w okresie ciąży, szczególnie w pierwszym trymestrze, chyba że jest to wyraźnie konieczne. W każdym indywidualnym przypadku należy rozważyć stosunek korzyści z leczenia do możliwego ryzyka dla płodu.
Karmienie piersią
Nie wiadomo, czy azacytydyna lub jej metabolity przenikają do mleka ludzkiego. Ze względu na możliwe ciężkie działania niepożądane u karmionego dziecka, karmienie piersią jest przeciwwskazane podczas leczenia azacytydyną.
Płodność
Brak danych dotyczących wpływu azacytydyny na płodność u ludzi. U zwierząt udokumentowano działania niepożądane azacytydyny na płodność u samców (patrz punkt 5.3). Przed rozpoczęciem
leczenia należy doradzić pacjentom płci męskiej, aby zasięgnęli porady na temat przechowywania nasienia.
Azacytydyna wywiera niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Podczas stosowania azacytydyny zgłaszano przypadki zmęczenia. Z tego powodu zaleca się ostrożność podczas prowadzenia pojazdów lub obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Dorośli pacjenci z MDS, CMML oraz AML (20-30% blastów w szpiku)
Działania niepożądane uważane za możliwie lub prawdopodobnie związane z podawaniem azacytydyny wystąpiły u 97% pacjentów.
Najczęściej występujące ciężkie działania niepożądane, zaobserwowane w kluczowym badaniu (AZA PH GL 2003 CL 001) obejmują neutropenię z gorączką (8,0%) oraz niedokrwistość (2,3%), które zgłaszano również w badaniach wspomagających (CALGB 9221 oraz CALGB 8921). Inne ciężkie działania niepożądane w tych 3 badaniach obejmowały zakażenia, takie jak posocznica neutropeniczna (0,8%) i zapalenie płuc (2,5%) (w niektórych przypadkach prowadzące do śmierci), trombocytopenię (3,5%), reakcje nadwrażliwości (0,25%) i zdarzenia krwotoczne (np. krwotok mózgowy [0,5%], krwotok żołądkowo-jelitowy [0,8%] oraz krwotok śródczaszkowy [0,5%]).
Najczęściej zgłaszanymi działaniami niepożądanymi podczas leczenia azacytydyną były reakcje hematologiczne (71,4%), w tym trombocytopenia, neutropenia i leukopenia (zwykle stopnia 3.- 4.), zdarzenia żołądkowo-jelitowe (60,6%), w tym nudności, wymioty (zwykle stopnia 1.-2.) lub reakcje w miejscu podania (77,1%, zwykle stopnia 1.-2.).
Dorośli pacjenci w wieku 65 lat lub starsi z AML z >30% blastów w szpiku
Najczęściej zgłaszane ciężkie działania niepożądane (≥ 10%) w badaniu AZA-AML-001 w grupie azacytydyny obejmowały gorączkę neutropeniczną (25,0%), zapalenie płuc (20,3%) oraz gorączkę (10,6%). Inne, rzadziej występujące, ciężkie działania niepożądane w grupie azacytydyny obejmowały posocznicę (5,1%), niedokrwistość (4,2%), posocznicę neutropeniczną (3,0%), zakażenie dróg
moczowych (3,0%), trombocytopenię (2,5%), neutropenię (2,1%), zapalenie tkanki łącznej (2,1%),
zawroty głowy (2,1%) oraz duszność (2,1%).
Najczęściej zgłaszanymi (≥ 30%) działaniami niepożądanymi podczas leczenia azacytydyną były zdarzenia żołądkowo-jelitowe, w tym zaparcia (41,9%), nudności (39,8%) oraz biegunka (36,9%; zwykle stopnia 1.-2.), zaburzenia ogólne i stany w miejscu podania, w tym gorączka (37,7%; zwykle stopnia 1.-2.), oraz zdarzenia hematologiczne, w tym gorączka neutropeniczna (32,2%) oraz neutropenia (30,1%; zwykle stopnia 3.-4.).
Tabelaryczne zestawienie działań niepożądanych
Tabela 1 poniżej przedstawia działania niepożądane związane z leczeniem azacytydyną, zaobserwowane w głównych badaniach klinicznych dotyczących MDS i AML oraz po wprowadzeniu do obrotu.
Częstości występowania zostały określone jako: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); niezbyt często (≥ 1/1 000 do < 1/100); rzadko (≥ 1/10 000 do < 1/1 000); bardzo rzadko (< 1/10 000); częstość nieznana (nie może być określona na podstawie dostępnych danych). W każdej grupie o określonej częstości występowania, objawy niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem. Działania niepożądane przedstawiono w poniższej tabeli zgodnie z największą częstością występowania obserwowaną w którymkolwiek z głównych badań klinicznych.
Tabela 1: Działania niepożądane zgłaszane u pacjentów z MDS lub AML leczonych azacytydyną (w badaniach klinicznych oraz po wprowadzeniu do obrotu)
Klasyfikacja układów i narządów | Bardzo często | Często | Niezbyt często | Rzadko | Częstość nieznana |
Zakażenia i | zapalenie płuc* (w | posocznica* (w tym | martwi- | ||
zarażenia | tym bakteryjne, | bakteryjna, | cze | ||
pasożytnicze | wirusowe i | wirusowa i | zapalenie | ||
grzybicze), zapalenie | grzybicza), | powięzi* | |||
nosogardzieli | posocznica | ||||
neutropeniczna*, | |||||
zakażenie dróg | |||||
oddechowych (w | |||||
tym górnych dróg | |||||
oddechowych i | |||||
oskrzeli), zakażenie | |||||
dróg moczowych, | |||||
zapalenie tkanki | |||||
łącznej, zapalenie | |||||
uchyłków, grzybica | |||||
jamy ustnej, | |||||
zapalenie zatok, | |||||
zapalenie gardła, | |||||
zapalenie błony | |||||
śluzowej nosa, | |||||
opryszczka | |||||
pospolita, zakażenie | |||||
skóry | |||||
Nowotwory | zespół | ||||
łagodne, złośliwe i nieokreślone (w | różnicowa nia*, a | ||||
tym torbiele i | |||||
polipy) | |||||
Zaburzenia krwi i | gorączka | pancytopenia*, | |||
układu chłonnego | neutropeniczna*, | niewydolność | |||
neutropenia, | szpiku kostnego | ||||
leukopenia, | |||||
trombocytopenia, | |||||
niedokrwistość | |||||
Zaburzenia | reakcje | ||||
układu | nadwrażliwości | ||||
immunologiczne- | |||||
go | |||||
Zaburzenia | anoreksja, | odwodnienie | zespół | ||
metabolizmu i | zmniejszone | rozpaduguza | |||
odżywiania | łaknienie, | ||||
hipokaliemia | |||||
Zaburzenia psychiczne | bezsenność | stan splątania, lęk | |||
Zaburzenia | zawroty głowy, ból | krwotok | |||
układu | głowy | śródczaszkowy*, | |||
nerwowego | omdlenie, senność, | ||||
letarg | |||||
Zaburzenia oka | krwotok oczny, krwotok spojówkowy |
Klasyfikacja układów i narządów | Bardzo często | Często | Niezbyt często | Rzadko | Częstość nieznana |
Zaburzenia serca | wysięk osierdziowy | zapalenie osierdzia | |||
Zaburzenia naczyniowe | niedociśnienie tętnicze*, nadciśnienie tętnicze, niedociśnienie ortostatyczne, krwiaki | ||||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | duszność, krwawieniez nosa | wysięk opłucnowy, duszność wysiłkowa, ból gardła i krtani | choroba śródmiąższo- wa płuc | ||
Zaburzenia żołądka i jelit | biegunka, wymioty, zaparcia, nudności, ból brzucha (w tym dyskomfort nadbrzusza i brzucha) | krwotok żołądkowo- jelitowy* (w tym krwotok z jamy ustnej), krwotok hemoroidalny, zapalenie jamy ustnej, krwawienie dziąseł, dyspepsja | |||
Zaburzenia wątroby i dróg żółciowych | niewydolność wątroby*, postępująca śpiączka wątrobowa | ||||
Zaburzenia skóry i tkanki podskórnej | wybroczyny punktowe, świąd (w tym uogólniony), wysypka, podskórne wylewy krwawe | plamica, łysienie, pokrzywka, rumień, wybroczyny plamkowe | ostra gorączkowa dermatoza neutrofilowa, ropne zgorzelinowe zapalenie skóry (łac. pyoderma gangrenosum) | ||
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | bóle stawów, bóle mięśniowo- szkieletowe (w tym pleców, kości i ból w kończynie) | skurcze mięśni, bóle mięśniowe | |||
Zaburzenia nerek i dróg moczowych | niewydolność nerek*, krwiomocz, podwyższone stężenie kreatyniny w surowicy | nerkowa kwasica kanalikowa | |||
Zaburzenia ogólne i stany w miejscu podania | gorączka*, zmęczenie, osłabienie, bóle klat- ki piersiowej, rumień w miejscu podania, ból w miejscu | siniaki, krwiaki, stwardnienie, wysypka, świąd, stan zapalny, odbarwienie, guzki i krwotok (w | martwica miejsca wstrzyknięcia (w miejscu wstrzyknię- cia) |
Klasyfikacja układów i narządów | Bardzo często | Często | Niezbyt często | Rzadko | Częstość nieznana |
podania, odczyn w miejscu podania (nieokreślony) | miejscu wstrzyknięcia), złe samopoczucie, dreszcze, krwawienie w miejscu wkłucia cewnika | ||||
Badania diagnostyczne | zmniejszenie masy ciała |
* = zgłaszano rzadko przypadki śmiertelne
a = patrz punkt 4.4
Opis wybranych działań niepożądanych
Hematologiczne działania niepożądane
Najczęściej zgłaszane (≥ 10%) hematologiczne działania niepożądane związane z leczeniem azacytydyną obejmują niedokrwistość, trombocytopenię, neutropenię, gorączkę neutropeniczną i leukopenię, które były zazwyczaj stopnia 3. lub 4. Ryzyko wystąpienia tych zdarzeń jest większe podczas pierwszych 2 cykli, po czym u pacjentów z przywróconą czynnością układu krwiotwórczego występują one z mniejszą częstością. W przypadku większości hematologicznych działań niepożądanych, zastosowano rutynowe kontrole pełnej morfologii krwi i opóźnienie podawania azacytydyny w następnym cyklu, zastosowano profilaktyczne podanie antybiotyków i (lub) w miarę potrzeb wspomaganie czynnikami wzrostu (np. G-CSF) w przypadku neutropenii, oraz przetoczenia krwi w niedokrwistości lub trombocytopenii.
Zakażenia
Mielosupresja może prowadzić do neutropenii i zwiększonego ryzyka wystąpienia zakażeń. U pacjentów otrzymujących azacytydynę zgłaszano ciężkie działania niepożądane, takie jak posocznica, w tym posocznica neutropeniczna i zapalenie płuc, w niektórych przypadkach ze skutkiem śmiertelnym. Zakażenia można leczyć stosując leki przeciwzakaźne oraz wspomaganie czynnikami wzrostu (np. G-CSF) w przypadku neutropenii.
Krwawienia
U pacjentów otrzymujących azacytydynę może wystąpić krwawienie. Zgłaszano ciężkie działania niepożądane, takie jak krwotok żołądkowo-jelitowy i krwotok śródczaszkowy. Pacjentów należy monitorować pod kątem objawów przedmiotowych i podmiotowych krwawienia, w szczególności pacjentów z istniejącą wcześniej bądź związaną z leczeniem trombocytopenią.
Nadwrażliwość
U pacjentów otrzymujących azacytydynę zgłaszano ciężkie reakcje nadwrażliwości. W przypadku reakcji rzekomoanafilaktycznych leczenie azacytydyną należy niezwłocznie przerwać i rozpocząć odpowiednie leczenie objawowe.
Działania niepożądane dotyczące skóry i tkanki podskórnej
Większość działań niepożądanych dotyczących skóry i tkanki podskórnej była związana z miejscem podania. W badaniach głównych żadne z tych działań niepożądanych nie prowadziło do przerwania podawania azacytydyny, ani nie było też konieczne zmniejszenie dawki azacytydyny. Większość działań niepożądanych wystąpiła podczas pierwszych 2 cykli leczenia i zwykle zmniejszała się w następnych cyklach. Działania niepożądane dotyczące tkanki podskórnej, takie jak wysypka/stan zapalny/świąd w miejscu podania, wysypka, rumień i zmiany skórne, mogą wymagać jednoczesnego zastosowania produktów leczniczych, takich jak leki przeciwhistaminowe, kortykosteroidy i niesteroidowe leki przeciwzapalne (NLPZ). Tego typu reakcje skórne należy odróżniać od zakażeń tkanek miękkich, występujących niekiedy w miejscu wkłucia. Po wprowadzeniu azacytydyny do obrotu zgłaszano występowanie zakażeń tkanek miękkich, w tym zapalenia tkanki łącznej i
martwiczego zapalenia powięzi, w rzadkich przypadkach prowadzących do śmierci. W celu leczenia działań niepożądanych związanych z zakażeniami, patrz punkt 4.8 Zakażenia.
Działania niepożądane żołądkowo-jelitowe
Najczęściej zgłaszane działania niepożądane dotyczące żołądka i jelit, związane z leczeniem azacytydyną, obejmowały zaparcia, biegunkę, nudności i wymioty. Te działania niepożądane leczono objawowo lekami przeciwwymiotnymi na nudności i wymioty, lekami przeciwbiegunkowymi na biegunkę oraz lekami przeczyszczającymi i (lub) środkami zmiękczającymi stolec na zaparcia.
Działania niepożądane dotyczące nerek
Zaburzenia czynności nerek, począwszy od podwyższonego stężenia kreatyniny w surowicy oraz krwiomoczu, aż po nerkową kwasicę kanalikową, niewydolność nerek i śmierć, zgłaszano u pacjentów leczonych azacytydyną (patrz punkt 4.4).
Działania niepożądane dotyczące wątroby
U pacjentów z rozległym obciążeniem nowotworem z powodu przerzutów, zgłaszano występowanie niewydolności wątroby, postępującej śpiączki wątrobowej i przypadki śmierci podczas leczenia azacytydyną (patrz punkt 4.4).
Zdarzenia dotyczące serca
Dane z badania klinicznego, do którego możliwe było włączenie pacjentów z chorobą układu sercowo-naczyniowego lub chorobą płuc, wykazały zwiększenie częstości zdarzeń dotyczących serca u pacjentów z nowo rozpoznaną ostrą białaczką szpikową (AML) leczonych azacytydyną (patrz punkt 4.4).
Pacjenci w podeszłym wieku
Informacje dotyczące bezpieczeństwa stosowania azacytydyny u pacjentów w wieku ≥ 85 lat są ograniczone (w badaniu AZA-AML-001 leczono 14 [5,9%] pacjentów w wieku ≥ 85 lat).
Dzieci i młodzież
W badaniu AZA-JMML-001 azacytydyną leczono 28 pacjentów z grupy dzieci i młodzieży (w wieku od jednego miesiąca do mniej niż 18 lat) z MDS (n = 10) albo młodzieńczą białaczką mielomonocytową (ang. juvenile myelomonocytic leukaemia, JMML) (n = 18) (patrz punkt 5.1).
U wszystkich 28 pacjentów wystąpiło co najmniej jedno działanie niepożądane, a u 17 (60,7%) wystąpiło co najmniej jedno zdarzenie związane z leczeniem. Najczęściej zgłaszanymi działaniami niepożądanymi w ogólnej populacji dzieci i młodzieży były gorączka, zdarzenia hematologiczne, w tym niedokrwistość, trombocytopenia i gorączka neutropeniczna, oraz zdarzenia dotyczące przewodu pokarmowego, w tym zaparcia i wymioty.
U 3 (trzech) uczestników wystąpiło zdarzenie zaobserwowane w czasie leczenia, prowadzące do przerwania przyjmowania leku (gorączka, progresja choroby i ból brzucha).
W badaniu AZA-AML-004, siedmiu pacjentów z grupy dzieci i młodzieży (w wieku od dwóch do 12 lat) leczono azacytydyną w związku z nawrotem molekularnego AML po pierwszej remisji całkowitej (ang. complete remission, CR1; patrz punkt 5.1).
U wszystkich siedmiu pacjentów wystąpiło przynajmniej jedno działanie niepożądane związane z leczeniem. Najczęściej zgłaszanymi działaniami niepożądanymi były neutropenia, nudności, leukopenia, trombocytopenia, biegunka i zwiększenie aktywności aminotransferazy alaninowej (AlAT). U dwóch pacjentów wystąpiło zdarzenie związane z leczeniem prowadzące do przerwania przyjmowania dawek (gorączka neutropeniczna, neutropenia).
Nie zidentyfikowano żadnych nowych sygnałów dotyczących bezpieczeństwa stosowania wśród ograniczonej liczby pacjentów z gruby dzieci i młodzieży leczonych azacytydyną w badaniu klinicznym. Ogólny profil bezpieczeństwa był zgodny z profilem bezpieczeństwa w populacji osób dorosłych.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301
faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W badaniach klinicznych zgłoszono jeden przypadek przedawkowania azacytydyny. U pacjenta wystąpiła biegunka, nudności i wymioty po otrzymaniu pojedynczej dawki dożylnej wynoszącej około 290 mg/m2, co stanowi prawie 4-krotność zalecanej dawki początkowej.
W razie przedawkowania, pacjenta należy monitorować wykonując odpowiednie badania krwi, oraz w razie potrzeby należy zastosować leczenie wspomagające. Nie jest znane antidotum na przedawkowanie azacytydyny.
Grupa farmakoterapeutyczna: Leki przeciwnowotworowe, analogi pirymidyn; kod ATC: L01BC07. Mechanizm działania
Uważa się, że azacytydyna działa przeciwnowotworowo z wykorzystaniem wielu mechanizmów, w
tym działa cytotoksycznie na nieprawidłowe komórki krwiotwórcze w szpiku kostnym i wpływa na hipometylację DNA. Działania cytotoksyczne azacytydyny mogą wynikać z wielu mechanizmów, w tym zahamowania syntezy DNA, RNA i białek, włączania jej do RNA i DNA oraz aktywacji szlaków odpowiedzi na uszkodzenie DNA. Komórki nieproliferujące są względnie niewrażliwe na azacytydynę. Włączenie azacytydyny do DNA powoduje dezaktywację metylotransferaz DNA, co prowadzi do hipometylacji DNA. Hipometylacja DNA nieprawidłowo metylowanych genów biorących udział w normalnej regulacji cyklu komórkowego, różnicowania i szlakach śmierci komórkowej, może prowadzić do ponownej ekspresji genów oraz przywrócenia komórkom nowotworowym zdolności do supresji nowotworu. Względne znaczenie hipometylacji DNA dla wyników klinicznych, w porównaniu z cytotoksycznością lub innymi aktywnościami azacytydyny, nie zostało ustalone.
Skuteczność kliniczna i bezpieczeństwo stosowania
Dorośli pacjenci (MDS, CMML oraz AML [20-30% blastów w szpiku])
Skuteczność i bezpieczeństwo stosowania azacytydyny badano w międzynarodowym, wieloośrodkowym, kontrolowanym, prowadzonym metodą otwartej próby, randomizowanym badaniu porównawczym fazy III w grupach równoległych (AZA PH GL 2003 CL 001) z udziałem dorosłych pacjentów z: MDS o pośrednim-2 i wysokim ryzyku zgodnie z Międzynarodowym Punktowym Systemem Prognostycznym (IPSS), niedokrwistością oporną na leczenie z nadmiarem blastów (ang. refractory anaemia with excess blasts, RAEB), niedokrwistością oporną na leczenie z nadmiarem blastów w okresie transformacji (ang. refractory anaemia with excess blasts in transformation, RAEB- T) oraz zmodyfikowaną przewlekłą białaczką mielomonocytową (ang. modified chronić
myelomonocytic leukaemia, mCMML) zgodnie z systemem klasyfikacji French American British (FAB). Pacjenci z RAEB-T (21-30% blastów) są obecnie uznawani według aktualnej klasyfikacji WHO za pacjentów z ostrą białaczką szpikową (AML). Azacytydynę i najlepsze leczenie objawowe (ang. best supportive care, BSC) (n = 179) porównywano z konwencjonalnymi metodami leczenia (ang. conventional care regimens, CCR). CCR składało się z samego BSC (n = 105), cytarabiny w małych dawkach z BSC (n = 49) lub standardowej chemioterapii indukcyjnej z BSC (n = 25). Przed randomizacją pacjenci byli wstępnie przydzielani przez lekarza prowadzącego do jednej z 3 metod CCR. Pacjenci, którzy nie zostali przydzieleni losowo do grupy otrzymującej azacytydynę, otrzymywali leczenie wstępnie wybrane przez lekarza prowadzącego. Stan pacjentów w zakresie 0-2 wg. Eastern Cooperative Oncology Group (ECOG) był jednym z kryteriów włączenia. Pacjenci z wtórnym MDS byli wyłączeni z badania. Pierwszorzędowym parametrem końcowym badania był całkowity czas przeżycia. Azacytydyna była podawana podskórnie w dawce 75 mg/m2 na dobę przez 7 dni, po czym następował okres odpoczynku trwający 21 dni (28-dniowy cykl leczenia). Mediana cykli wynosiła 9 (zakres = 1-39) a średnia wynosiła 10,2 cykli. W populacji włączonej do badania (ang. Intent to Treat, ITT) mediana wieku wynosiła 69 lat (zakres 38-88 lat).
W analizie ITT 358 pacjentów (179 otrzymujących azacytydynę i 179 otrzymujących CCR), mediana czasu przeżycia w grupie otrzymującej azacytydynę wynosiła 24,46 miesiąca w porównaniu z 15,02 miesiąca u pacjentów otrzymujących leczenie CCR, czyli różnica wynosiła 9,4 miesiąca, z wartością p wynoszącą 0,0001 w stratyfikowanym teście log-rank. Współczynnik ryzyka (ang. hazard ratio, HR) dla wpływu leczenia wynosił 0,58 (95% CI: 0,43; 0,77). Dwuletni czas przeżycia wynosił 50,8% u pacjentów otrzymujących azacytydynę a 26,2% u pacjentów otrzymujących CCR (p < 0,0001).
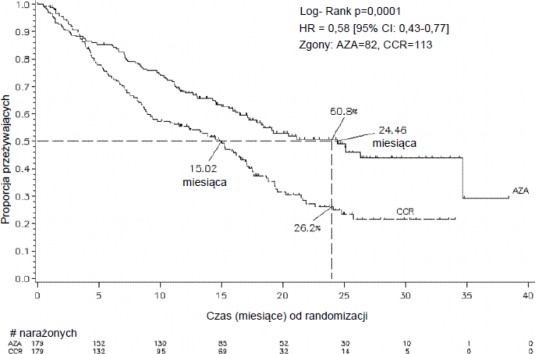
LEGENDA: AZA = azacytydyna; CCR = konwencjonalne metody leczenia; CI (ang. confidence interval) = przedział ufności; HR (ang. hazard ratio) = współczynnik ryzyka
Korzyści dotyczące czasu przeżycia po zastosowaniu azacytydyny były zgodne, niezależnie od opcji leczenia CCR (samo BSC, cytarabina w małej dawce z BSC lub standardowa chemioterapia indukcyjna z BSC) zastosowanego w grupie kontrolnej.
Podczas analizy podgrup cytogenetycznych IPSS zaobserwowano podobne wyniki w odniesieniu do mediany całkowitego czasu przeżycia we wszystkich grupach (dobra, pośrednia, zła cytogenetyka, w tym monosomia 7).
W analizach podgrup wiekowych zaobserwowano zwiększenie mediany całkowitego czasu przeżycia we wszystkich grupach (< 65 lat, ≥ 65 lat i ≥ 75 lat).
Mediana czasu do zgonu lub transformacji w AML po zastosowaniu azacytydyny wynosiła 13,0 miesięcy w porównaniu do 7,6 miesiąca u pacjentów otrzymujących leczenie CCR, poprawa wynosiła 5,4 miesiąca z wartością p wynoszącą 0,0025 w stratyfikowanym teście log-rank.
Leczenie azacytydyną wiązało się również ze zmniejszeniem częstości występowania cytopenii i związanych z nimi objawów. Leczenie azacytydyną prowadziło do obniżenia konieczności przetoczeń czerwonych krwinek (ang. red blood cells, RBC) i płytek krwi. Spośród pacjentów w grupie otrzymującej azacytydynę, zależnych od przetoczeń czerwonych krwinek na początku badania, 45,0% uniezależniło się od przetoczeń czerwonych krwinek podczas okresu leczenia, w porównaniu z 11,4% pacjentów w złożonej grupie leczonej CCR (statystycznie istotna (p < 0,0001) różnica wynosząca 33,6% (95% CI: 22,4; 44,6)). U pacjentów, którzy na początku badania byli zależni od przetoczeń czerwonych krwinek i uniezależnili się od nich, mediana czasu trwania niezależności od przetoczeń czerwonych krwinek, w grupie otrzymującej azacytydynę wynosiła 13 miesięcy.
Odpowiedź była oceniana przez badacza lub niezależną komisję rewizyjną (ang. Independent Review Committee, IRC). Odpowiedź ogółem (remisja całkowita [ang. complete remission, CR] + remisja częściowa [ang. partial remission, PR]) według ustaleń badacza wynosiła 29% w grupie otrzymującej azacytydynę i 12% w złożonej grupie leczonej CCR (p = 0,0001). Odpowiedź ogółem (CR + PR) według ustaleń IRC w badaniu AZA PH GL 2003 CL 001 wynosiła 7% (12/179) w grupie otrzymującej azacytydynę w porównaniu z 1% (2/179) w złożonej grupie leczonej CCR (p = 0,0113). Różnice między oceną odpowiedzi przez IRC i badacza wynikały z kryteriów Międzynarodowej Grupy Roboczej (ang. International Working Group, IWG), według których wymagana jest poprawa morfologii krwi obwodowej i utrzymanie poprawy przez co najmniej 56 dni. Korzyści dotyczące czasu przeżycia wykazano również u pacjentów, którzy nie uzyskali całkowitej/częściowej odpowiedzi po leczeniu azacytydyną. Poprawę hematologiczną (większą lub mniejszą) według ustaleń IRC uzyskano u 49% pacjentów otrzymujących azacytydynę w porównaniu z 29% pacjentów w złożonej grupie leczonej CCR (p < 0,0001).
U pacjentów z jedną lub kilkoma nieprawidłowościami cytogenetycznymi na początku badania, procent pacjentów z większą odpowiedzią cytogenetyczną był podobny w grupie otrzymującej azacytydynę i w złożonej grupie leczonej CCR. Mniejsza odpowiedź cytogenetyczna była statystycznie istotnie (p = 0,0015) wyższa w grupie otrzymującej azacytydynę (34%) w porównaniu ze złożoną grupą leczoną CCR (10%).
Dorośli pacjenci w wieku 65 lat lub powyżej z AML z >30% blastów w szpiku
Przedstawione poniżej wyniki dotyczą populacji ITT badania AZA-AML-001(patrz punkt 4.1, aby zapoznać się z zatwierdzonymi wskazaniami).
Skuteczność oraz bezpieczeństwo stosowania azacytydyny oceniono w międzynarodowym, wieloośrodkowym, kontrolowanym, otwartym badaniu klinicznym fazy III z grupami równoległymi, w grupie pacjentów w wieku 65 lat lub powyżej z nowo rozpoznaną pierwotną lub wtórną AML z
>30% blastów w szpiku zgodnie z klasyfikacją WHO, którzy nie kwalifikowali się do przeszczepienia krwiotwórczych komórek macierzystych. Azacytydynę z BSC (n=241) porównywano z CCR. CCR stanowiło tylko BSC (n=45), cytarabinę w małych dawkach z BSC (n=158) lub standardową intensywną chemioterapią złożoną z cytarabiny i antracykliny z BSC (n=44). Przed randomizacją pacjenci byli wstępnie przydzielani przez lekarza prowadzącego do jednej z 3 metod CCR. Pacjenci, którzy nie zostali przydzieleni losowo do grupy otrzymującej azacytydynę, otrzymywali leczenie wstępnie wybrane przez lekarza prowadzącego. Wśród kryteriów włączenia były wymagania, by stan pacjentów wg ECOG był w zakresie 0-2 oraz, aby ryzyko wynikające z nieprawidłowości cytogenetycznych było umiarkowane lub duże. Pierwszorzędowym punktem końcowym badania był całkowity czas przeżycia.
Azacytydynę podawano podskórnie w dawce 75 mg/m2 pc. na dobę przez 7 dni, po czym następował okres odpoczynku trwający 21 dni (28-dniowy cykl leczenia), mediana leczenia wynosiła 6 cykli
(zakres 1-28), u pacjentów z grupy otrzymujących tylko BSC mediana wynosiła 3 cykle (zakres 1-20), u pacjentów otrzymujących cytarabinę w małych dawkach mediana wynosiła 4 cykle (zakres 1-25) a u pacjentów otrzymujących standardową intensywną chemioterapię mediana wynosiła 2 cykle (zakres 1- 3, cykl indukcyjny plus 1 lub 2 cykle konsolidacyjne).
Indywidualne parametry początkowe były porównywalne między grupą azacytydyny a grupami CCR. Mediana wieku pacjentów wynosiła 75,0 lat (zakres 64 do 91 lat), 75,2% było rasy kaukaskiej, 59,0% stanowili mężczyźni. Na początku badania, u 60,7% pacjentów stwierdzono bliżej nieokreśloną AML, u 32,4% AML ze zmianami mielodysplastycznymi, u 4,1% nowotwory szpiku związane z leczeniem, natomiast u 2,9% AML z nawracającymi nieprawidłowościami genetycznymi, zgodnie z klasyfikacją WHO.
W analizie ITT 488 pacjentów (241 otrzymujących azacytydynę oraz 247 otrzymujących CCR), mediana czasu przeżycia wynosiła 10,4 miesiąca po zastosowaniu leczenia azacytydyną w porównaniu z 6,5 miesiąca u pacjentów otrzymujących CCR, czyli różnica wynosiła 3,8 miesiąca, z wartością p wynoszącą 0,1009 w stratyfikowanym teście log-rank (dwustronnie). Współczynnik ryzyka dla efektu leczenia wynosił 0,85 (95% CI: 0,69; 1,03). Wskaźnik rocznego przeżycia wynosił 46,5% u pacjentów otrzymujących azacytydynę w porównaniu do 34,3% u pacjentów otrzymujących CCR.
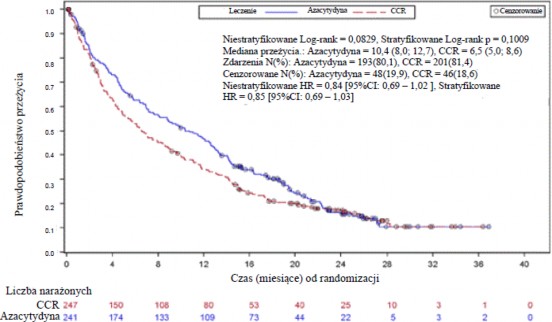
Model hazardu proporcjonalnego Cox’a dostosowywany dla predefiniowanych na wejściu czynników prognostycznych określił wskaźnik ryzyka dla azacytydyny w porównaniu do CCR na poziomie 0,80 (95% CI: 0,66; 0,99; p=0,0355).
Ponadto, pomimo, że badanie nie wykazało statystycznie istotnej różnicy pomiędzy grupą azacytydyny a wybranymi wstępnie grupami CCR, czas przeżycia pacjentów leczonych azacytydyną był dłuższy w porównaniu do wariantów CCR, tylko BSC, cytarabiny w małych dawkach z BSC oraz porównywalny do standardowej, intensywnej chemioterapii z BSC.
We wszystkich określonych przed rozpoczęciem badania podgrupach, ze względu na wiek [<75 lat oraz ≥75 lat], płeć, rasę, wynik w skali ECOG [0, 1 lub 2], wyjściowe ryzyko cytogenetyczne [umiarkowane lub duże], region geograficzny, klasyfikację AML wg WHO [w tym AML ze zmianami mielodysplastycznymi], początkową liczbę białych krwinek [≤5 x 109/l lub >5 x 109/l], początkowy odsetek blastów w szpiku [≤50% lub >50%] i MDS w wywiadzie, obserwowano tendencję większej korzyści w odniesieniu do całkowitego czasu przeżycia w grupie otrzymującej azacytydynę. W kilku określonych przed rozpoczęciem badania podgrupach współczynnik ryzyka całkowitego czasu przeżycia osiągnął wartość istotną statystycznie, w tym u pacjentów z dużym ryzykiem
cytogenetycznym, pacjentów z AML ze zmianami mielodysplastycznymi, pacjentów w wieku
< 75 lat, u kobiet oraz u pacjentów rasy białej.
Wynik odpowiedzi hematologicznej oraz cytogenetycznej były podobnie oceniony przez badacza oraz IRC. Współczynnik odpowiedzi ogółem (remisja całkowita [CR] i remisja całkowita z niepełną regeneracją morfologii krwi [CRi]) określony przez IRC wynosił 27,8% w grupie otrzymującej azacytydynę oraz 25,1% w złożonej grupie CCR (p = 0,5384). U pacjentów, którzy osiągnęli CR lub CRi, mediana trwania remisji wynosiła 10,4 miesiąca (95% CI = 7,2; 15,2) u pacjentów otrzymujących azacytydynę oraz 12,3 miesiąca (95% CI = 9,0; 17,0) u pacjentów otrzymujących CCR. Korzyści dotyczące czasu przeżycia wykazano również u pacjentów, którzy nie uzyskali całkowitej odpowiedzi po leczeniu azacytydyną w porównaniu do CCR.
Leczenie azacytydyną wpływało na poprawę wyników morfologii krwi obwodowej oraz prowadziło do zmniejszenia konieczności przetoczeń czerwonych krwinek i płytek krwi. Pacjenta określano jako zależnego od przetoczeń czerwonych krwinek lub płytek krwi na początku badania, jeśli otrzymał jedno przetoczenie lub więcej przetoczeń czerwonych krwinek lub płytek krwi w ciągu 56 dni (8 tygodni) przed randomizacją. Pacjenta określano jako niezależnego od przetoczeń czerwonych krwinek lub płytek krwi w okresie leczenia, jeśli u pacjenta nie przeprowadzono żadnego przetoczenia czerwonych krwinek lub płytek krwi w okresie 56 dni.
Spośród pacjentów w grupie otrzymującej azacytydynę, zależnych od przetoczeń czerwonych krwinek na początku badania, 38,5% (95% CI = 31,1; 46,2) uniezależniło się od przetoczeń czerwonych krwinek podczas okresu leczenia, w porównaniu z 27,6% (95% CI = 20,9; 35,1) pacjentów w złożonej grupie CCR. U pacjentów, którzy na początku badania byli zależni od przetoczeń czerwonych krwinek i uniezależnili się od nich podczas leczenia, mediana czasu trwania niezależności od przetoczeń czerwonych krwinek w grupie otrzymującej azacytydynę wynosiła 13,9 miesiąca, a nie została osiągnięta w grupie CCR.
Spośród pacjentów w grupie otrzymującej azacytydynę, zależnych od przetoczeń płytek krwi na początku badania, 40,6% (95% CI = 30,9; 50,8) uniezależniło się od przetoczeń płytek krwi podczas okresu leczenia, w porównaniu z 29,3% (95% CI = 19,7; 40,4) pacjentów w złożonej grupie leczonej CCR. U pacjentów, którzy na początku badania byli zależni od przetoczeń płytek krwi i uniezależnili się od nich podczas leczenia, mediana czasu trwania niezależności od przetoczeń płytek krwi, w grupie otrzymującej azacytydynę wynosiła 10,8 miesiąca oraz 19,2 miesiąca w grupie CCR.
Zależną od zdrowia jakość życia (ang. Health- Related Quality of Life, HRQoL) oceniono za pomocą kwestionariusza European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire (EORTC QLQ-C30). Dane dotyczące HRQoL mogły być analizowane dla części lub całej populacji objętej badaniem. Pomimo ograniczeń analizy, z dostępnych danych wynika, że pacjenci nie doświadczają znaczącego pogorszenia jakości życia podczas leczenia azacytydyną.
Dzieci i młodzież
Badanie AZA-JMML-001 było międzynarodowym, wieloośrodkowym badaniem fazy II, prowadzonym metodą otwartej próby, mającym na celu ocenę farmakokinetyki, farmakodynamiki, bezpieczeństwa stosowania oraz aktywności azacytydyny przed przeszczepieniem krwiotwórczych komórek macierzystych u dzieci i młodzieży z nowo rozpoznanym zaawansowanym MDS albo JMML. Głównym celem badania klinicznego była ocena wpływu azacytydyny na wskaźnik odpowiedzi w Dniu 28. cyklu 3.
Pacjentów (z MDS, n = 10; z JMML, n = 18; wiek od trzech miesięcy do 15 lat; 71% płci męskiej) leczono azacytydyną podawaną dożylnie w dawce 75 mg/m² pc. codziennie w dniach 1.–7. w cyklach 28-dniowych przez co najmniej trzy cykle, a maksymalnie sześć cykli.
Włączanie pacjentów do grupy badanej z MDS przerwano po włączeniu 10 pacjentów z powodu braku skuteczności: u tych 10 pacjentów nie odnotowano żadnych potwierdzonych odpowiedzi.
Do grupy badanej z JMML włączono 18 pacjentów (13 z mutacjami somatycznymi w genie PTPN11, trzech w genie NRAS i jednego w genie KRAS oraz jednego z rozpoznaniem klinicznym nerwiakowłókniakowatości typu 1 [ang. neurofibromatosis type 1, NF1]). Szesnastu pacjentów ukończyło trzy cykle leczenia, a pięciu z nich ukończyło sześć cykli leczenia. Łącznie 11 pacjentów z JMML wykazywało odpowiedź kliniczną w dniu 28. cyklu 3, a spośród tych 11 uczestników u
9 (50%) potwierdzono odpowiedź kliniczną (3 uczestników z potwierdzoną remisją całkowitą [ang. confirmed complete remission, cCR] i 6 uczestników z potwierdzoną remisją częściową [ang. confirmed partial remission, cPR]). W kohorcie pacjentów z JMML leczonych azacytydyną, u
7 (43,8%) pacjentów wystąpiła utrzymująca się odpowiedź w zakresie liczby płytek krwi (liczba
≥100 × 109/l), a u 7 (43,8%) konieczne było przetoczenie krwi podczas przeszczepienia krwiotwórczych komórek macierzystych. U 17 z 18 pacjentów przeprowadzono następnie przeszczepienie krwiotwórczych komórek macierzystych.
Ze względu na schemat badania klinicznego (niewielka liczba pacjentów i różne czynniki zakłócające), nie można wnioskować, czy stosowanie azacytydyny przed przeszczepieniem krwiotwórczych komórek macierzystych powoduje poprawę wyników w zakresie przeżycia pacjentów z JMML.
Badanie AZA-AML-004 było wieloośrodkowym, prowadzonym metodą otwartej próby badaniem fazy II oceniającym bezpieczeństwo stosowania, farmakodynamikę i skuteczność azacytydyny w porównaniu z brakiem leczenia przeciwnowotworowego u dzieci i młodych osób dorosłych z nawrotem molekularnym AML po CR1.
Siedmiu pacjentom (mediana wieku 6,7 lat [zakres 2–12 lat]; 71,4% płci męskiej) podawano azacytydynę dożylnie w dawce 100 mg/m2 raz na dobę w dniach 1.–7. każdego 28-dniowego cyklu przez maksymalnie 3 cykle.
U pięciu pacjentów przeprowadzono ocenę w celu wykrycia minimalnej choroby resztkowej w dniu 84., u 4 pacjentów wystąpiła stabilizacja molekularna (n = 3) albo poprawa molekularna (n = 1), a u 1 pacjenta wystąpił nawrót kliniczny. U sześciu z siedmiu pacjentów (90% [95% CI = 0,4, 1,0]) leczonych azacytydyną przeprowadzono przeszczepienie krwiotwórczych komórek macierzystych.
Z powodu małej liczebności próby nie można określić skuteczności azacytydyny w leczeniu AML u dzieci i młodzieży.
Informacje dotyczące bezpieczeństwa znajdują się w punkcie 4.8.
Wchłanianie
Azacytydyna była szybko wchłaniana po pojedynczym podaniu podskórnym dawki 75 mg/m2 pc. z maksymalnymi stężeniami w osoczu wynoszącymi 750 ± 403 ng/ml występującymi 0,5 h po podaniu dawki (pierwszy punkt pobierania próbek). Bezwzględna dostępność biologiczna azacytydyny po podaniu podskórnym w porównaniu do podania dożylnego (pojedyncze dawki 75 mg/m2 pc.) wynosiła około 89% na podstawie pola powierzchni pod krzywą (ang. area under the curve, AUC).
Pole powierzchni pod krzywą oraz maksymalne stężenie w osoczu (Cmax) po podaniu podskórnym azacytydyny były w przybliżeniu proporcjonalne w zakresie dawek od 25 do 100 mg/m2 pc.
Dystrybucja
Po dożylnym podaniu dawki średnia objętość dystrybucji wynosiła 76 ± 26 l, a klirens układowy wynosił 147 ± 47 l/h.
Metabolizm
Na podstawie danych z badań in vitro, wydaje się, że w metabolizmie azacytydyny nie uczestniczyły izoenzymy cytochromu P450 (CYP), UDP-glukuronozylotransferazy (UGT), sulfotransferazy (SULT) ani transferazy glutationowe (GST).
Azacytydyna jest spontanicznie hydrolizowana oraz deaminowana z udziałem deaminazy cytydynowej. W ludzkich frakcjach wątrobowych S9, tworzenie metabolitów było niezależne od NADPH, co wskazuje, że w metabolizmie azacytydyny nie uczestniczą izoenzymy cytochromu P450. Badanie in vitro azacytydyny na hodowlach ludzkich hepatocytów wskazało, że azacytydyna w stężeniach od 1,0 μM do 100 μM (tzn. do około 30-krotnie większych niż stężenia osiągane klinicznie), nie indukuje CYP 1A2, 2C19 ani 3A4 lub 3A5. W badaniach mających na celu ocenę hamowania szeregu izoenzymów P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 i 3A4) azacytydyna w stężeniu do 100 μM nie powodowała hamowania. Dlatego jest mało prawdopodobne, że azacytydyna indukuje lub hamuje enzym CYP w stężeniach klinicznie osiągalnych w osoczu.
Eliminacja
Azacytydyna jest szybko usuwana z osocza ze średnim czasem półtrwania w fazie eliminacji (t½) po podaniu podskórnym wynoszącym 41 ± 8 minut. Po podskórnym podawaniu azacytydyny w dawce 75 mg/m2 raz na dobę przez 7 dni nie występuje akumulacja. Azacytydyna i (lub) jej metabolity są głównie wydalane z moczem. Po podaniu dożylnym i podskórnym znakowanej 14C-azacytydyny, odpowiednio 85 i 50% radioaktywności było wykrywane w moczu, podczas gdy < 1% było wykrywane w kale.
Specjalne grupy pacjentów
Nie przeprowadzono formalnych badań wpływu niewydolności wątroby (patrz punkt 4.2), płci, wieku ani rasy na farmakokinetykę azacytydyny.
Dzieci i młodzież
W badaniu AZA-JMML-001 przeprowadzono analizę farmakokinetyczną w grupie dzieci i młodzieży u 10 pacjentów z MDS i 18 z JMML w dniu 7. cyklu 1 (patrz punkt 5.1). Mediana wieku (zakres) pacjentów z MDS wynosiła 13,3 (1,9–15) lat, a pacjentów z JMML 2,1 (0,2–6,9) lat.
Po podaniu dożylnym w dawce 75 mg/m2 pc. azacytydyna osiągała wartość Cmax szybko, w ciągu 0,083 godziny, zarówno w populacji z MDS, jak i w populacji z JMML. Średnia geometryczna Cmax u pacjentów z MDS i JMML wynosiła odpowiednio 1797,5 i 1066,3 ng/ml, a średnia geometryczna AUC0-∞ wynosiła odpowiednio 606,9 i 240,2 ng∙h/ml. Średnia geometryczna objętości dystrybucji u osób z MDS i JMML wynosiła odpowiednio 103,9 i 61,1 l. Wydaje się, że całkowita ekspozycja osoczowa azacytydyny była wyższa u osób z MDS, jednak stwierdzono umiarkowaną do wysokiej zmienność między pacjentami zarówno w przypadku wartości AUC, jak i Cmax.
Średnia geometryczna t½ w przypadku MDS i JMML wynosiła odpowiednio 0,4 i 0,3 godziny, a średnia geometryczna klirensu wynosiła odpowiednio 166,4 i 148,3 l/h.
Dane farmakokinetyczne pochodzące z badania AZA-JMML-001 połączono i porównano z danymi farmakokinetycznymi pochodzącymi od sześciu osób dorosłych z MDS, którym podawano azacytydynę dożylnie w dawce 75 mg/m2 pc. w ramach badania AZA-2002-BA-002. Średnie wartości Cmax i AUC0-t azacytydyny u pacjentów dorosłych oraz dzieci i młodzieży po podaniu dożylnym były podobne (odpowiednio 2750 ng/ml w porównaniu do 2841 ng/ml oraz 1025 ng∙h/ml w porównaniu do 882,1 ng∙h/ml).
W badaniu AZA-AML-004 przeprowadzono analizę farmakokinetyczną u sześciu z siedmiu pacjentów z grupy dzieci i młodzieży, u których uzyskano przynajmniej jedną mierzalną wartość stężenia leku po podaniu dawki na potrzeby badań farmakokinetycznych (patrz punkt 5.1). Mediana wieku (zakres) pacjentów z AML wynosiła 6,7 (2–12) lat.
Po podaniu dawek wielokrotnych 100 mg/m2 powierzchni ciała, średnie geometryczne dla Cmax i AUC0-tau w dniu 7. cyklu 1. wynosiły odpowiednio 1557 ng/ml i 899,6 ng h/ml, z obserwowaną dużą zmiennością międzyosobniczą (CV% odpowiednio 201,6% i 87,8%). Azacytydyna szybko osiągnęła wartość Cmax, z medianą czasu wynoszącą 0,090 godziny po podaniu dożylnym, a wartość spadała po osiągnięciu średniej geometrycznej t1/2 wynoszącej 0,380 godziny. Średnia geometryczna klirensu i objętości dystrybucji wyniosła odpowiednio 127,2 l/h i 70,2 l.
Ekspozycja farmakokinetyczna (na azacytydynę) obserwowana u dzieci z nawrotem molekularnym AML po CR1 była porównywalna z ekspozycją z danych zbiorczych uzyskanych od 10 dzieci z MDS i 18 dzieci z JMML oraz porównywalna z ekspozycją na azacytydynę u osób dorosłych z MDS.
Niewydolność nerek
Niewydolność nerek nie ma istotnego wpływu na ekspozycję na azacytydynę po pojedynczym ani wielokrotnym podaniu podskórnym. Po podaniu podskórnym pojedynczej dawki 75 mg/m2 pc., średnie wartości ekspozycji (AUC oraz Cmax) u osób z łagodną, umiarkowaną oraz ciężką niewydolnością nerek zwiększyły się o odpowiednio 11-21%, 15-27% oraz 41-66%, w porównaniu do pacjentów z prawidłową czynnością nerek. Jednak ekspozycja mieściła się w zakresie wartości obserwowanych u pacjentów z prawidłową czynnością nerek. Azacytydyna może być podawana pacjentom z niewydolnością nerek bez dostosowania dawki początkowej, pod warunkiem, że pacjenci będą monitorowani w celu wykrycia toksyczności, ponieważ azacytydyna i (lub) jej metabolity są wydalane głównie przez nerki.
Farmakogenomika
Nie przeprowadzono formalnych badań wpływu znanych polimorfizmów deaminazy cytydynowej na metabolizm azacytydyny.
Azacytydyna indukuje mutacje genów i aberracje chromosomowe w układach komórkowych in vitro
bakterii i ssaków. Potencjalne działanie rakotwórcze było oceniane na myszach i szczurach. Azacytydyna indukowała powstawanie nowotworów układu krwiotwórczego u samic myszy po podawaniu dootrzewnowym 3 razy na tydzień przez 52 tygodnie. U myszy, którym podawano azacytydynę dootrzewnowo przez 50 tygodni, obserwowano zwiększoną częstość występowania guzów w układzie chłonno-siateczkowym, płucach, sutkach i skórze. Badanie potencjału rakotwórczego na szczurach wykazało zwiększoną częstość występowania guzów jądra.
Badania wczesnej embriotoksyczności wykazały częstość występowania wewnątrzmacicznej śmierci płodu wynoszącą 44% (zwiększona resorpcja) po pojedynczym wstrzyknięciu dootrzewnowym azacytydyny podczas organogenezy. U myszy, którym podano azacytydynę podczas lub przed zamknięciem podniebienia twardego wykryto nieprawidłowości rozwojowe w mózgu. U szczurów azacytydyna nie powodowała działań niepożądanych, gdy była podawana przed implantacją, ale wyraźnie wykazano działanie embriotoksyczne, gdy była podawana podczas organogenezy.
Nieprawidłowości płodu podczas organogenezy u szczurów obejmowały: nieprawidłowości OUN (egzencefalia/przepuklina mózgowa), anomalie kończyn (mikromelia, stopa zdeformowana, syndaktylia, oligodaktylia) i inne (mikroftalmia, mikrognatia, wytrzewienie, obrzęk i nieprawidłowości żeber).
Podanie azacytydyny samcom myszy przed kopulacją z samicami myszy nienarażonymi na działanie azacytydyny prowadziło do obniżonej płodności i utraty potomstwa podczas późniejszego rozwoju płodowego i pourodzeniowego. Podanie azacytydyny samcom szczurów prowadziło do utraty masy jąder i najądrza, obniżonej liczby plemników, obniżonego współczynnika ciąż, zwiększenia liczby płodów nieprawidłowych i zwiększonej liczby straconych płodów przez pokryte samice (patrz punkt 4.4).
Mannitol
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, oprócz wymienionych w punkcie 6.6.
Nieotwarta fiolka z proszkiem: 3 lata
Po przygotowaniu:
Jeśli do przygotowania produkt leczniczego Azacitidine Glenmark została użyta nieschłodzona woda do wstrzykiwań, wykazano chemiczną i fizyczną stabilność użytkową przygotowanego produktu leczniczego przez 45 minut w temperaturze 25°C i przez 8 godzin w temperaturze 2°C−8°C.
Termin ważności przygotowanego produktu leczniczego może zostać wydłużony poprzez przygotowanie produktu leczniczego z użyciem schłodzonej (2°C-8°C) wody do wstrzykiwań. Jeśli do przygotowania produkt została użyta schłodzona (2°C-8°C) woda do wstrzykiwań, wykazano chemiczną i fizyczną stabilność użytkową przygotowanego produktu leczniczego przez 32 godziny w temperaturze 2°C−8°C.
Z mikrobiologicznego punku widzenia przygotowany produkt należy zużyć natychmiast. Jeśli produkt nie został zużyty natychmiast, użytkownik jest odpowiedzialny za czas i warunki przechowywania przed użyciem. Nie przechowywać produktu dłużej niż 8 godzin w temperaturze 2°C−8°C, jeśli produkt został przygotowany z użyciem nieschłodzonej wody do wstrzykiwań lub nie dłużej niż 32 godziny jeśli produkt został przygotowany z użyciem schłodzonej (2°C-8°C) wody do wstrzykiwań.
Nieotwarte fiolki
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Przygotowana zawiesina
Warunki przechowywania produktu leczniczego po przygotowaniu, patrz punkt 6.3.
Fiolka z bezbarwnego szkła typu I z korkiem z gumy butylowej i aluminiowym uszczelnieniem (biały dla 100 mg i pomarańczowy dla 150 mg) w tekturowym pudełku.
Dostępne wielkości opakowań
1 fiolka zawierająca 100 mg azacytydyny.
1 fiolka zawierająca 150 mg azacytydyny.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Zalecenia dotyczące bezpiecznego postępowania
Produkt leczniczy Azacitidine Glenmark jest cytotoksycznym produktem leczniczym i jak w przypadku innych potencjalnie toksycznych związków, należy zachować ostrożność podczas sporządzania i obchodzenia się z zawiesiną azacytydyny. Należy stosować się do procedur właściwego obchodzenia się i usuwania przeciwnowotworowych produktów leczniczych.
W przypadku kontaktu przygotowanej azacytydyny ze skórą, należy ją natychmiast dokładnie przemyć wodą z mydłem. W przypadku kontaktu z błonami śluzowymi, należy je dokładnie przepłukać wodą.
Kobiety z fachowego personelu medycznego, które są w ciąży nie powinny przygotowywać produktu leczniczego.
Procedura przygotowania
Produkt leczniczy Azacitidine Glenmark powinien zostać przygotowany z zastosowaniem wody do wstrzykiwań. Termin ważności przygotowanego produktu leczniczego można wydłużyć jeśli do przygotowania została użyta schłodzona (2°C-8°C) woda do wstrzykiwań. Szczegóły dotyczące przechowywania przygotowanego produktu zostały podane w punkcie 6.3.
Dawki powyżej 4 ml należy wstrzykiwać w dwa różne miejsca.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Należy zmieniać miejsca wstrzyknięcia. Nowe wstrzyknięcia należy podawać co najmniej 2,5 cm od poprzedniego miejsca i nigdy nie wstrzykiwać w miejsca podrażnione, zasiniaczone, zaczerwienione lub stwardniałe.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Glenmark Pharmaceuticals s.r.o. Hvězdova 1716/2b
140 78 Praga 4 Republika Czeska
Pozwolenie nr: 26708
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 12.11.2021
29.03.2023








