







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO

Niniejszy produkt leczniczy będzie dodatkowo monitorowany. Umożliwi to szybkie zidentyfikowanie nowych informacji o bezpieczeństwie. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane. Aby dowiedzieć się, jak zgłaszać działania niepożądane - patrz punkt 4.8.
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Jedną fiolkę z proszkiem
Jedną fiolkę z rozpuszczalnikiem z 25 ml jałowej wody do wstrzykiwań
Jedną jałową dwustronną igłę do przenoszenia
Jeden jałowy filtr o średnicy porów 20 mikronów
Rymphysia, 1000 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Proszek: 100 ml fiolka z przezroczystego bezbarwnego szkła typu I z korkiem z gumy butylowej powlekanej ETFE oraz aluminiowym kapslem z polipropylenowym wieczkiem typu „flip-off”.
Rozpuszczalnik: 50 ml fiolka z przezroczystego szkła typu I z korkiem z gumy bromobutylowej pokrytej silikonem oraz aluminiowym kapslem z jasnoniebieskim plastikowym wieczkiem typu „flip-off”.
Każde opakowanie zawiera:
Jedną fiolkę z proszkiem
Jedną fiolkę z rozpuszczalnikiem z 50 ml jałowej wody do wstrzykiwań
Jedną jałową dwustronną igłę do przenoszenia
Jeden jałowy filtr o średnicy porów 20 mikronów
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do
Rymphysia, 500 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji Rymphysia, 1000 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji
Rymphysia, 500 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Jedna fiolka zawiera około 500 mg czynnego inhibitora alfa1-proteinazy wytwarzanego z ludzkiego osocza pochodzącego od dawców.
Roztwór uzyskany po rekonstytucji z użyciem 25 ml jałowej wody do wstrzykiwań zawiera około 20 mg/ml ludzkiego inhibitora alfa1-proteinazy.
Całkowita zawartość białka to 18 – 26 mg/ml na fiolkę.
Rymphysia, 1000 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Jedna fiolka zawiera około 1000 mg czynnego inhibitora alfa1-proteinazy wytwarzanego z ludzkiego osocza pochodzącego od dawców.
Roztwór uzyskany po rekonstytucji z użyciem 50 ml jałowej wody do wstrzykiwań zawiera około 20 mg/ml ludzkiego inhibitora alfa1-proteinazy.
Całkowita zawartość białka to 18 – 26 mg/ml na fiolkę. Substancja pomocnicza o znanym działaniu
Produkt leczniczy Rymphysia zawiera około 108 mg sodu na fiolkę 25 ml (postać 500 mg) oraz 216 mg sodu
na fiolkę 50 ml (postać 1000 mg).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Liofilizowany proszek w kolorze białym lub białawym do lekko żółto-zielonego lub żółtym. Rozpuszczalnik (jałowa woda do wstrzykiwań) jest przezroczysty i bezbarwny.
Po rekonstytucji pH roztworu wynosi od 7,2 do 7,8.
Produkt leczniczy Rymphysia jest wskazany do stosowania w leczeniu podtrzymującym w celu spowolnienia progresji rozedmy płuc u dorosłych z potwierdzonym ciężkim niedoborem inhibitora alfa1-proteinazy (np. z genotypem PiZZ, PiZ (null), Pi (null,null), PiSZ). Pacjenci powinni być objęci optymalnym leczeniem farmakologicznym i niefarmakologicznym i wykazywać oznaki postępującej choroby płuc (np. przewidywana mniejsza natężona objętość wydechowa pierwszosekundowa (FEV1), upośledzona zdolność chodzenia lub zwiększona liczba zaostrzeń) w ocenie fachowego personelu medycznego z doświadczeniem w leczeniu niedoboru inhibitora alfa1-proteinazy.
Pierwsze infuzje produktu leczniczego Rymphysia powinny być podawane pod nadzorem fachowego personelu medycznego z doświadczeniem w leczeniu niedoboru inhibitora alfa1-proteinazy. Kolejne infuzje mogą być podawane przez odpowiednio przeszkolonego pacjenta lub opiekuna, patrz punkt 4.4.
Dawkowanie
Zalecana dawka produktu leczniczego Rymphysia to 60 mg/kg masy ciała (mc.) podawane raz na tydzień. Dzieci i młodzież
Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego Rymphysia u dzieci
i młodzieży (w wieku poniżej 18 lat). Dane nie są dostępne. Pacjenci z zaburzeniami czynności nerek lub wątroby
Nie przeprowadzono specjalnych badań. Nie można zalecić innego schematu dawkowania dla tych pacjentów.
Pacjenci w podeszłym wieku
Nie określono skuteczności produktu leczniczego Rymphysia u pacjentów w podeszłym wieku (w wieku 65 lat i powyżej) w specjalnie zaplanowanych badaniach klinicznych. Jednak ograniczone dane dotyczące bezpieczeństwa stosowania u pacjentów w podeszłym wieku wskazują na to, że profil bezpieczeństwa jest porównywalny do pacjentów w wieku poniżej 65 lat.
Sposób podawania
Produkt leczniczy Rymphysia należy podawać wyłącznie po rekonstytucji w postaci infuzji dożylnej.
Do rozpuszczenia proszku należy używać wyłącznie jałowej wody do wstrzykiwań i podawać przy użyciu zestawu do podawania dożylnego. Zalecana szybkość podawania infuzji wynosi 0,08 ml/kg mc./min.
Szybkość podawania infuzji można dostosować w zależności od odpowiedzi i tolerancji przez pacjenta, jednak nie powinna przekraczać 0,2 ml/kg mc./min.
Instrukcja dotycząca rekonstytucji produktu leczniczego przed podaniem, patrz punkt 6.6.
Produkt leczniczy Rymphysia jest przeciwwskazany u pacjentów z niedoborem immunoglobuliny A (IgA)
z przeciwciałami przeciwko IgA, ze względu na ryzyko ciężkich reakcji nadwrażliwości.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Identyfikowalność
W celu poprawienia identyfikowalności biologicznych produktów leczniczych należy czytelnie zapisać nazwę i numer serii podawanego produktu.
Reakcje nadwrażliwości
Rymphysia może zawierać śladowe ilości immunoglobuliny A (IgA). Pacjenci z potwierdzoną obecnością przeciwciał przeciw IgA, które mogą występować u pacjentów z selektywnym lub ciężkim niedoborem IgA, są narażeni na większe ryzyko wystąpienia ciężkiej nadwrażliwości i reakcji anafilaktycznych. W przypadku wystąpienia objawów ciężkiej nadwrażliwości lub reakcji anafilaktycznych należy przerwać infuzję
i zastosować lecznicze doraźne. Czynniki zakaźne
Standardowe środki ostrożności zapobiegające zakażeniom wynikającym ze stosowania produktów leczniczych przygotowanych z ludzkiej krwi lub osocza obejmują selekcję dawców, badania przesiewowe poszczególnych donacji i pul osocza w kierunku swoistych markerów zakażenia oraz wprowadzenie skutecznych etapów procesu wytwarzania w celu inaktywacji/usunięcia wirusów. Mimo to, w przypadku podawania produktów leczniczych przygotowanych z ludzkiej krwi lub osocza nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych. Dotyczy to również nieznanych lub nowo wykrytych wirusów i innych patogenów.
Podejmowane działania są uważane za skuteczne wobec wirusów otoczkowych, takich jak wirus ludzkiego niedoboru odporności (HIV), wirus zapalenia wątroby typu B (HBV) i wirus zapalenia wątroby typu C (HCV), jak również bezotoczkowego wirusa zapalenia wątroby typu A i parwowirusa B19.
U pacjentów przyjmujących regularnie/wielokrotnie produkty zawierające inhibitory proteinaz pochodzące
z ludzkiego osocza należy rozważyć odpowiednie szczepienia (przeciwko WZW typu A i B). Leczenie w warunkach domowych i (lub) samodzielne podawanie przez pacjenta
Istnieją ograniczone dane dotyczące stosowania produktu leczniczego w warunkach leczenia domowego
i (lub) samodzielnego podawania przez pacjenta.
Potencjalne zagrożenia związane z leczeniem w warunkach domowych i (lub) samodzielnym podawaniem przez pacjenta są związane z przygotowaniem i podawaniem produktu leczniczego, jak również
z postępowaniem w przypadku wystąpienia działań niepożądanych, a zwłaszcza nadwrażliwości. Należy poinformować pacjentów i opiekunów o objawach nadwrażliwości.
Decyzję, czy dany pacjent lub opiekun może stosować leczenie w warunkach domowych i (lub) samodzielne podawanie, podejmuje lekarz prowadzący, który powinien zapewnić właściwe przeszkolenie (np. dotyczące rekonstytucji i podawania, stosowania techniki aseptycznej, prowadzenia dzienniczka leczenia, rozpoznawania działań niepożądanych i środków, jakie należy zastosować w razie wystąpienia takich działań) oraz regularną kontrolę stosowania produktu.
Palenie tytoniu
Palenie tytoniu jest ważnym czynnikiem ryzyka wystąpienia i progresji rozedmy płuc. W związku z tym zdecydowanie zaleca się rzucenie palenia i unikania narażenia na dym tytoniowy.
Zawartość sodu
Rymphysia, 500 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Produkt leczniczy zawiera 108 mg sodu na fiolkę, co odpowiada 5,4% zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych.
Rymphysia, 1000 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Produkt leczniczy zawiera 216 mg sodu na fiolkę, co odpowiada 10,8% zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych.
Nie przeprowadzono badań dotyczących interakcji.
Ciąża
Nie przeprowadzono badań na zwierzętach dotyczących szkodliwego wpływu produktu leczniczego Rymphysia na reprodukcję ani nie określono w kontrolowanych badaniach klinicznych bezpieczeństwa jego stosowania u kobiet w okresie ciąży. Ponieważ inhibitor alfa1-proteinazy jest endogennym ludzkim białkiem, uważa się, że szkodliwe działanie produktu leczniczego Rymphysia na płód podczas podawania w zalecanych dawkach jest mało prawdopodobne. Jednakże produkt leczniczy Rymphysia należy stosować z ostrożnością u kobiet w ciąży.
Karmienie piersią
Nie wiadomo, czy produkt leczniczy Rymphysia i (lub) jego metabolity przenikają do mleka ludzkiego. Nie badano przenikania ludzkiego inhibitora alfa1-proteinazy do mleka zwierząt. Należy podjąć decyzję, czy przerwać/kontynuować karmienie piersią czy przerwać/kontynuować podawanie produktu leczniczego Rymphysia biorąc pod uwagę korzyści z karmienia piersią dla dziecka i korzyści z leczenia ludzkim inhibitorem alfa1-proteinazy dla matki.
Płodność
Nie przeprowadzono badań dotyczących wpływu produktu leczniczego Rymphysia na płodność u zwierząt ani nie określono w kontrolowanych badaniach klinicznych jego wpływu na płodność u ludzi. Ponieważ ludzki inhibitor alfa1-proteinazy jest endogennym ludzkim białkiem, nie oczekuje się jego niepożądanego wpływu na płodność podczas podawania w zalecanych dawkach.
Po podaniu produktu leczniczego Rymphysia mogą wystąpić zawroty głowy. Dlatego produkt leczniczy
Rymphysia może wywierać niewielki wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Podsumowanie profilu bezpieczeństwa
Po podaniu produktu leczniczego Rymphysia zgłaszano występowanie reakcji nadwrażliwości.
Podczas badań klinicznych nie zgłoszono żadnych, czasowo związanych ciężkich działań niepożądanych [tj. ciężkich działań niepożądanych, które w ocenie sponsora mogłyby mieć prawdopodobny związek przyczynowy z produktem leczniczym Rymphysia lub ARALAST (nazwa handlowa produktu leczniczego poprzedzającego produkt leczniczy Rymphysia w Stanach Zjednoczonych), a które rozpoczęły się podczas
infuzji albo w ciągu 72 godzin (lub 3 dni, gdy czas rozpoczęcia działania niepożądanego jest niedostępny) od zakończenia infuzji].
Najczęstszymi, czasowo związanymi działaniami niepożądanymi występującymi u ≥ 5% uczestników badań klinicznych produktu leczniczego Rymphysia lub ARALAST były: zapalenie oskrzeli, kaszel / kaszel
produktywny, biegunka, ból głowy, nudności, ból gardła i krtani, wysypka, zakażenie górnych dróg oddechowych oraz zasinienie w miejscu nakłucia naczynia.
Informacje dotyczące bezpieczeństwa w odniesieniu do czynników zakaźnych, patrz punkt 4.4. Tabelaryczne zestawienie działań niepożądanych
Następujące działania niepożądane wymienione poniżej zaobserwowano u 60 uczestników 4 badań klinicznych (ATC 97-01, 460501, 460502 oraz 460503) oraz w okresie po wprowadzeniu produktu leczniczego Rymphysia lub ARALAST do obrotu.
Poniższa tabela jest zgodna z klasyfikacją układów i narządów MedDRA. Częstość występowania na pacjenta określono zgodnie z następującą konwencją: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1000 do <1/100), rzadko (≥1/10 000 do <1/1000) i bardzo rzadko (<1/10 000). Częstość występowania działań niepożądanych zgłaszanych w okresie po wprowadzeniu produktu leczniczego do obrotu jest określona jako „nieznana” (częstość nie może być określona na podstawie dostępnych danych).
W obrębie każdej grupy o określonej częstości występowania działania niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 1.: Częstość występowania działań niepożądanych w badaniach klinicznych oraz po wprowadzeniu produktu leczniczego Rymphysia lub ARALAST do obrotu
Klasyfikacja układów i narządów | Częstość występowania działań niepożądanycha | ||
Bardzo często (≥1/10) | Często (≥1/100 do <1/10) | Nieznana | |
Zakażenia i zarażenia pasożytnicze | Zapalenie oskrzeli Zakażenie górnych dróg oddechowych | ||
Zaburzenia układu nerwowego | Ból głowy | Zawroty głowy Zaburzenia smaku Dyskomfort w obrębie głowy Letarg Migrena Senność | |
Zaburzenia oka | Zapalenie spojówek Rumień powieki Podrażnienie oczu Zaburzenia widzenia | ||
Zaburzenia naczyniowe | Uderzenia gorąca | ||
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Kaszel Ból gardła i krtani | Duszności Kaszel produktywny Nieżyt nosa Podrażnienie gardła | |
Zaburzenia żołądka i jelit | Wzdęcia Biegunka Owrzodzenie jamy ustnej Nudności Wymioty | ||
Zaburzenia skóry i tkanki podskórnej | Wysypka Pokrzywka Pokrzywka zlokalizowana | ||
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Ból pleców Dyskomfort mięśniowo- szkieletowy Ból mięśniowy Ból kończyny | ||
Zaburzenia ogólne i stany w miejscu podania | Dyskomfort w klatce piersiowej Ból w klatce piersiowej Zmęczenie Choroba grypopodobna Ból w miejscu infuzji Złe samopoczucie Gorączka Ospałość Zasinienie w miejscu nakłucia naczynia | Osłabienie Samopoczucie inne niż normalne Reakcja w miejscu wstrzyknięcia | |
Badania diagnostyczne | Nieprawidłowe skurczowe ciśnienie krwi Zwiększona częstość oddechu |
[a]Działanie niepożądane jest zdefiniowane jako pojawiające się w trakcie leczenia działanie niepożądane, które wystąpiło podczas albo po infuzji produktu leczniczego Rymphysia lub ARALAST i w ocenie sponsora mogłyby mieć prawdopodobny związek przyczynowy z produktem leczniczym Rymphysia lub ARALAST.
Immunogenność
Immunogenność produktu leczniczego Rymphysia oceniono w badaniu popłuczyn oskrzelowo- pęcherzykowych (BAL). U żadnego z leczonych pacjentów nie wystąpiły przeciwciała przeciwko produktowi leczniczemu Rymphysia.
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania ani skuteczności u dzieci i młodzieży. Dane nie są dostępne. Pacjenci w podeszłym wieku
Nie określono skuteczności produktu leczniczego Rymphysia u pacjentów w podeszłym wieku (w wieku 65 lat i powyżej) w specjalnie zaplanowanych badaniach klinicznych. Jednak ograniczone dane dotyczące bezpieczeństwa stosowania u pacjentów w podeszłym wieku wskazują na to, że profil bezpieczeństwa jest porównywalny do pacjentów w wieku poniżej 65 lat.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych
i Produktów Biobójczych Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Następstwa przedawkowania są nieznane.
W przypadku przedawkowania należy uważnie obserwować pacjenta, czy nie występują działania niepożądane, a w razie potrzeby powinny być dostępne odpowiednie środki wspomagające.
Grupa farmakoterapeutyczna: Leki przeciwkrwotoczne, inhibitor proteinazy, kod ATC: B02AB02 Mechanizm działania
Podawanie produktu leczniczego Rymphysia ma na celu inhibicję proteaz serynowych, takich jak elastaza neutrofilowa (NE) w płucach. W płucach zdrowego człowieka NE jest wytwarzana przez aktywowane neutrofile podczas stanu zapalnego (zakażenie dróg oddechowych, palenie tytoniu), a jej ilość jest regulowana przez inhibitor alfa1-proteinazy (alfa1-PI). Zmniejszona inhibicja aktywności NE powoduje zachwianie równowagi proteinaza-inhibitor proteinazy i może uszkadzać ściany pęcherzyków płucnych, prowadząc do schorzeń, takich jak przewlekła obturacyjna choroba płuc lub rozedma płuc u osób
z niedoborem alfa1-PI (nazywanym także niedoborem alfa1-antytrypsyny). Niedobór alfa1-PI to zaburzenie dziedziczone autosomalnie, kodominująco, które charakteryzuje się niskimi stężeniami alfa1-PI w surowicy i płucach. Osoby z ciężkim niedoborem alfa1-PI mają bardzo niewielką ochronę przed zwiększonymi stężeniami NE uwalnianymi podczas stanu zapalnego, co prowadzi do postępującej, umiarkowanej lub ciężkiej przewlekłej obturacyjnej choroby płuc (rozedmy) i ostatecznie manifestuje się objawami klinicznymi w okresie od trzeciej do czwartej dekady życia.
Działanie farmakodynamiczne
Leczenie wspomagające polegające na podawaniu raz na tydzień produktu leczniczego Rymphysia w dawce 60 mg/kg mc. pacjentom z niedoborem alfa1-PI powodowało zwiększenie stężenia alfa1-PI w osoczu oraz w warstwie płynu na powierzchni śródbłonka (ELF), co zostało potwierdzone przy użyciu testu antygenowego. U osób zdrowych stężenia alfa1-PI w osoczu są większe niż 20 µM.
Przeprowadzono otwarte badanie kliniczne w celu oceny wpływu cotygodniowego dożylnego leczenia wspomagającego produktem leczniczym Rymphysia w dawce 60 mg/kg mc. na stężenia alfa1-PI w warstwie płynu na powierzchni śródbłonka (ELF) oraz na aktywność elastazy antyneutrofilowej (ang. antineutrophil elastase capacity, ANEC; czynny alfa1-PI) u uczestników z ciężkim niedoborem alfa1-PI. Łącznie
Leczenie wspomagające produktem leczniczym Rymphysia spowodowało istotne zwiększenie (p<0,0001; n=12) średnich stężeń antygenowego i czynnego alpha1-PI w osoczu oraz ELF. Stężenia antygenowego alfa1-PI w ELF zwiększyły się istotnie (p=0,0195; n=10); jeśli zaś chodzi o stężenia czynnego alfa1-PI
w ELF, to po 8 cotygodniowych infuzjach produktu leczniczego Rymphysia tylko u 2 z 12 uczestników wystąpiły mierzalne stężenia czynnego alfa1-PI w ELF w jednym lub obu płatach płucnych, a różnica w porównaniu do stężenia w punkcie początkowym nie osiągnęła istotności statystycznej.
Tabela 2.: Antygenowy i czynny alfa1-PI mierzony w osoczu i ELF po dożylnym leczeniu wspomagającym produktem leczniczym Rymphysia
Punkt początkowy Średnia ± SD (zakres) (liczba uczestników = 12) | Po leczeniu Średnia ± SD (zakres) (liczba uczestników = 12) | |
Antygenowy alfa1-PI w osoczu (µM) | 4,22 ± 1,08 (od 3,12 do 6,33) | 14,56 ± 2,42 (od 11,10 do 18,10) |
Czynny alfa1-PI w osoczu (µM) | 2,45 ± 0,43 (od 1,62 do 3,05) | 11,91 ± 2,40 (od 7,84 do 16,91) |
Antygenowy alfa1-PI w ELF (µM) | 0,24 ± 0,25 (od 0,02 do 0,81) | 1,00 ± 1,62 (od 0,13 do 5,71) |
Czynny alfa1-PI w ELF (µM) | 2,70 ± 3,55 (od 0,29 do 11,31) | 1,54 ± 0,37 (od 1,16 do 1,90) |
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań produktu leczniczego Rymphysia we wszystkich podgrupach populacji dzieci i młodzieży z niedoborem alfa1-proteinazy w leczeniu rozedmy płuc spowodowanej niedoborem inhibitora alfa1-proteinazy (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Właściwości farmakokinetyczne alfa1-PI oceniono w dwóch badaniach klinicznych po dożylnym podaniu produktu leczniczego Rymphysia lub ARALAST (nazwa handlowa produktu poprzedzającego produkt leczniczy Rymphysia w Stanach Zjednoczonych). Pierwsze badanie kliniczne (ARALAST w porównaniu do produktu referencyjnego) przeprowadzono, aby porównać ARALAST do dostępnego w obrocie produktu zawierającego alfa1-PI (produkt referencyjny) u 28 uczestników z niedoborem alfa1-PI i rozedmą płuc, którzy w ciągu sześciu miesięcy poprzedzających badanie nie otrzymywali terapii wspomagającej alfa1-PI. Uczestników randomizowano do grup otrzymujących ARALAST lub produkt referencyjny w dawce
60 mg/kg mc. dożylnie raz na tydzień, przez 10 tygodni z rzędu. Po pierwszych 10 cotygodniowych infuzjach uczestnicy, którzy otrzymywali produkt referencyjny, zaczęli otrzymywać ARALAST, a ci należący do grupy otrzymującej ARALAST otrzymywali go w dalszym ciągu. Tabela 3. zawiera podsumowanie średnich stężeń minimalnych antygenowego i czynnego alfa1-PI w surowicy przed infuzją w stanie stacjonarnym (tygodnie od 8. do 11.).
Tabela 3.: Minimalne stężenia antygenowego i czynnego alfa1-PI w stanie stacjonarnym w surowicy po dożylnej
terapii wspomagającej produktem leczniczym ARALAST lub produkt referencyjny
ARALAST Średnia ± SD (zakres średnich) (liczba uczestników = 13) | Produkt referencyjny Średnia ± SD (zakres średnich) (liczba uczestników = 13) | |
Antygenowy alfa1-PI (µM) | 15,3 ± 2,5 (od 14,7 do 15,5) | 16,9 ± 2,3 (od 16,2 do 17,2) |
Czynny alfa1-PI (µM) | 15,3 ± 2,4 (od 14,8 do 15,6) | 15,7 ± 2,6 (od 14,4 do 16,4) |
Po cotygodniowym leczeniu wspomagającym polegającym na podawaniu produktu leczniczego ARALAST lub produktu referencyjnego zaobserwowano stopniowe zwiększenie szczytowych i minimalnych stężeń alfa1-PI w surowicy, ze stabilizacją po kilku tygodniach. Okres półtrwania produktu leczniczego ARALAST po podaniu dawki pojedynczej wynosił 5,9 (±1,2) dni. Minimalne stężenia w surowicy czynnego alfa1-PI zwiększyły się znacząco u wszystkich uczestników w tygodniu 2. i 3., a u większości uczestników przekroczyły 11 µM. Z kilkoma wyjątkami stężenia w obu grupach objętych leczeniem utrzymywały się powyżej tego stężenia u poszczególnych uczestników w tygodniach od 3. do 24. Mimo że tylko 5
z 14 uczestników (35,7%) otrzymujących ARALAST miało stężenia w popłuczynach BAL spełniające kryteria akceptacji do analizy zarówno w punkcie początkowym jak i tygodniu 7., zaobserwowano statystycznie istotne zwiększenie stężenia antygenowego alfa1-PI w ELF w popłuczynach BAL. Nie wykryto statystycznie istotnego zwiększenia ANEC w ELF. Ustalono, że podtrzymujące leczenie wspomagające, polegające na podawaniu raz w tygodniu dawki 60 mg/kg mc. dożylnie produktów leczniczych ARALAST
i Produktu referencyjnego miało podobny wpływ na utrzymanie docelowych minimalnych stężeń alfa1-PI w surowicy i zwiększanie stężeń antygenowego alfa1-PI w ELF.
Porównywalność farmakokinetyczną produktów leczniczych Rymphysia i ARALAST zademonstrowano
w randomizowanym, krzyżowym badaniu prowadzonym metodą podwójnie ślepej próby z 25 uczestnikami (mediana wieku: 59 lat; zakres: od 20 do 75 lat) z ciężkim niedoborem alfa1-PI, którzy otrzymali pojedynczą infuzję 60 mg/kg mc. każdego produktu. Rysunek 1. przedstawia średnią ± odchylenie standardowe (SD) stężeń alfa1-PI w osoczu z korekcją według wartości w punkcie początkowym. Tabela 4. przedstawia
podsumowanie kluczowych parametrów farmakokinetycznych produktów leczniczych Rymphysia
i ARALAST. 90% przedział ufności (od 85,8% do 100,2%) dla stosunku średnich geometrycznych AUC0-35d/dawkę dla produktów leczniczych Rymphysia i ARALAST oznaczał, że te dwa produkty są farmakokinetycznie porównywalne.
Rysunek 1.: Profile średnich (± SD) stężeń (w zależności od czasu) alfa1-PI w osoczu po pojedynczej dożylnej infuzji Rymphysia (60 mg/kg) u uczestników z niedoborem alfa1-PI
Czas (dni)
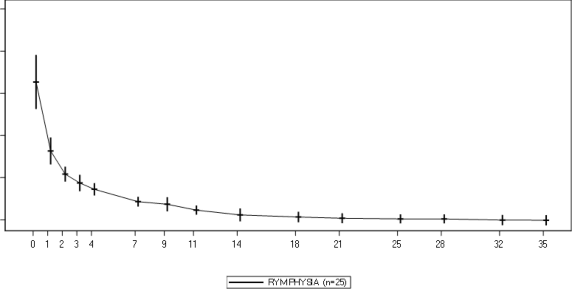
Dostosowane o wartość wyjściową stężenie a1-PI w osoczu (mg/ml)
2,5
2,0
1,5
1,0
0,5
0,0
Uwaga: dostosowane o wartość wyjściową stężenie alfa1-PI (średnia +/- SD) w czasie po leczeniu produktem leczniczym RYMPHYSIA
Tabela 4.: Parametry farmakokinetyczne antygenowego alfa1-PI w osoczu u 25 uczestników po jednorazowej dawce 60 mg/kg mc.
Parametr farmakokinetyczny | Rymphysia Średnia ± SD | ARALAST Średnia ± SD |
Cmaks. mg/ml | 1,6 ± 0,3 | 1,7 ± 0,3 |
AUC0-35d/dawkę, dzień*kg/ml | 0,0837 ± 0,0212 | 0,0897 ± 0,0204 |
t1/2, dni | 4,7 ± 2,7 | 4,8 ± 2,0 |
CL, ml/dzień | 940+275 | 862+206 |
Vss, ml | 5632+2006 | 5618+1618 |
Cmaks. = maksymalne zwiększenie stężenia alfa1-PI w osoczu po infuzji;
AUC0-35d/dawkę = pole pod krzywą od czasu 0 do 35 dni podzielone według dawki;
t1/2 = okres półtrwania;
CL=klirens;
Vss = objętość dystrybucji w stanie stacjonarnym.
Bezpieczeństwo stosowania produktu leczniczego Rymphysia oceniane było w kilku badaniach nieklinicznych prowadzonych na myszach, szczurach, świnkach morskich, królikach i psach. Dane niekliniczne wynikające z badań farmakologicznych dotyczących bezpieczeństwa, badań toksyczności po podaniu pojedynczym oraz po podaniu wielokrotnym do 1 miesiąca, nie ujawniają żadnego szczególnego zagrożenia dla człowieka.
Nie przeprowadzono badań toksyczności przewlekłej, toksyczności rozwojowej ani reprodukcyjnej, jak również badań genotoksyczności ani rakotwórczości. Takie badania nie są traktowane jako istotne z powodu produkcji przeciwciał przeciwko heterologicznym białkom ludzkim u zwierząt. Ponieważ ludzki alfa1-PI jest białkiem i fizjologicznym składnikiem krwi ludzkiej, nie oczekuje się, aby miał efekty rakotwórcze, genotoksyczne ani teratogenne.
Proszek:
Sodu chlorek
Sodu diwodorofosforan jednowodny Sodu wodorotlenek
Rozpuszczalnik:
Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
2 lata
Z mikrobiologicznego punktu widzenia produkt należy zużyć natychmiast. Jeśli produkt nie zostanie użyty natychmiast, za czas i warunki przechowywania przed podaniem odpowiada użytkownik i zwykle nie powinien on przekraczać 3 godzin w temperaturze pokojowej (do 25°C).
Nie przechowywać w temperaturze powyżej 25°C. Nie zamrażać.
Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem. Warunki przechowywania produktu leczniczego po rekonstytucji, patrz punkt 6.3.
Rymphysia, 500 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji.
Proszek: 50 ml fiolka z przezroczystego bezbarwnego szkła typu I z korkiem z gumy butylowej powlekanej ETFE oraz aluminiowym kapslem z polipropylenowym wieczkiem typu „flip-off”.
Rozpuszczalnik: 25 ml fiolka z przezroczystego szkła typu I z korkiem z gumy bromobutylowej pokrytej silikonem oraz aluminiowym kapslem z żółtym plastikowym wieczkiem typu „flip-off”.
Każde opakowanie zawiera:
stosowania
Ogólne instrukcje
Produkt leczniczy Rymphysia jest przeznaczony wyłącznie do stosowania dożylnego.
Rekonstytucję, podawanie i wszelkie czynności z produktem leczniczym Rymphysia należy przeprowadzać z ostrożnością i zachowaniem zasad aseptyki, aby zachować jałowość produktu.
Przed rekonstytucją pozostawić produkt leczniczy Rymphysia i rozpuszczalnik, aby osiągnęły temperaturę pokojową.
Produkt leczniczy Rymphysia po rekonstytucji należy przechowywać w temperaturze pokojowej i podać w ciągu trzech (3) godzin od rekonstytucji. Fiolki zużyte częściowo należy wyrzucić i nie pozostawiać do użycia w przyszłości. Roztwór nie zawiera żadnego środka konserwującego.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami.
Rekonstytucja
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI
OBROTU
Takeda Pharma Sp. z o.o. ul. Prosta 68
00-838 Warszawa
Rymphysia, 500 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji – pozwolenie nr 27196 Rymphysia, 1000 mg, proszek i rozpuszczalnik do sporządzania roztworu do infuzji – pozwolenie nr 27197
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 20 lipca 2022 r.
PRODUKTU LECZNICZEGO
02/2023







