







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
ogólny stan pacjenta,
reakcje po ostatnim wstrzyknięciu,
przedział czasowy między wstrzyknięciami.
Konwencjonalne zwiększanie dawki
Jedno wstrzyknięcie co 3 – 7 dzień (patrz Tabela 1.).
Podczas tego sposobu podwyższania dawki, można – zamiast tego produktu – rozważyć zastosowanie postaci depot (wolno uwalnianej), Alutard SQ produktu ALK 802 Jad osy lub ALK 801 Jad pszczoły.
Jeśli nastąpi przekroczenie 7-dniowego odstępu między wstrzyknięciami w fazie zwiększania dawki, należy odpowiednio zmniejszyć dawkę:
Odstęp do 2 tygodni:
Dawka bez zmian.
Odstęp do 3 tygodni:
Zmniejszenie dawki do połowy ostatnio podanej dawki.
Odstęp do 4 tygodni:
Zmniejszenie dawki do 1/10 dawki ostatnio podanej.
Odstęp dłuższy niż 4 tygodnie:
Ponowne rozpoczęcie dawkowania od stężenia początkowego.
Wstrzyknięcia wykonuje się ambulatoryjnie w odstępach 1-tygodniowych. Dawkę ustala się Indywidualnie, w zależności od wrażliwości pacjenta. Tabela 1. może być wykorzystana jako orientacyjna propozycja dawkowania.
Tabela 1. Orientacyjna instrukcja dawkowania podczas konwencjonalnej immunoterapii.
Numer tygodnia
Numer dnia
Stężenie mikrogram/ml
Objętość ml
Mikrogramy jadu owada/iniekcję
1
1
0,1
0,1
0,01●
2
8
1
0,1
0,1
3
15
10
0,1
1
4
22
10
0,5
5●●
5
29
100
0,1
10●●
6
36
100
0,2
20●●
7
43
100
0,3
30●●
8
50
100
0,4
40●●
9
57
100
0,5
50●●
10
64
100
0,6
60●●
11
71
100
0,8
80●●
12
78
100
1,0
100●●
Jeśli konieczne, dawka może być mniejsza, zależnie od wrażliwości pacjenta.
●● Aby uniknąć wtórnych odczynów zaleca się, by każda dawka (5-100 μg) została podzielona na połowę, tzn. te dawki należy wstrzykiwać zachowując 30 minutową przerwę.
Immunoterapia przyspieszona zmodyfikowana („clustered”).
Od dwóch do czterech wstrzyknięć co 30 minut raz w tygodniu (Tabela 2.). Jeśli to konieczne, odstępy między sesjami można zwiększyć maksymalnie do 2 tygodni. Pacjent powinien pozostawać
w placówce medycznej przez przynajmniej 30 minut po ostatnim wstrzyknięciu. W przypadku przekroczenia przerwy dwóch tygodni, należy zastosować zmniejszenie dawki zgodnie ze schematem zmniejszania dawki w schemacie konwencjonalnym (patrz punkt a).
Dawkę zmniejsza się indywidualnie. Generalnie, maksymalnie 4 wstrzyknięcia co 30 minut raz w tygodniu aż do osiągnięcia dawki podtrzymującej. Jeśli to konieczne, odstępy między sesjami można zwiększyć maksymalnie do 2 tygodni (patrz Tabela 2.).
Pacjenci powinni pozostawać w placówce medycznej przez przynajmniej 30 minut po ostatnim wstrzyknięciu.
Tabela 2. Schemat dawkowania w zmodyfikowanej immunoterapii przyspieszonej (clustered)
Numer tygodnia
Numer dnia
Stężenie mikrogram/ml
Objętość ml
Mikrogramy jadu owada/iniekcję
1
1
0,1
0,1
0,01●
1
0,1
0,1
1
1
1
10
0,3
3
2
8
10
0,25
2,5
10
0,25
2,5
3
15
100
0,05
5
100
0,05
5
4
22
100
100
0,1
0,1
10
10
5
29
100
100
0,2
0,2
20
20
6
36
100
100
0,3
0,3
30
30
7
43
100
100
0,5
0,5
50
50
Jeśli konieczne, dawka może być mniejsza, zależnie od wrażliwości pacjenta.
Immunoterapia przyspieszona („rush”).
Jeśli konieczne dawka może być mniejsza, w zależności od wrażliwości pacjenta
Przeciwwskazania
Nadwrażliwość na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1;
Choroby immunologiczne (np. choroby kompleksów immunologicznych i niedobory odporności);
Ciężka, chroniczna lub sezonowa astma oskrzelowa (FEV1 stale poniżej 70% wartości przewidywanej po odpowiednim leczeniu farmakologicznym);
Choroby lub stany uniemożliwiające leczenie potencjalnych reakcji anafilaktycznych (w tym wstrząsu anafilaktycznego) za pomocą adrenaliny, np. przewlekłe choroby serca, ciężkie nadciśnienie tętnicze, przewlekłe choroby płuc lub leczenie lekami blokującymi receptory beta- adrenergiczne (w tym także krople do oczu);
Leczenie inhibitorami ACE (patrz punkt 4.5);
Nowotwory złośliwe;
Ostre i chroniczne postacie chorób infekcyjnych;
Poważne wypryski lub zapalenie skóry;
U pacjentów leczonych trójcyklicznymi lekami przeciwdepresyjnymi lub inhibitorami monoaminooksydazy (MAO) działanie adrenaliny (w przypadku wstrząsu anafilaktycznego) może być zwiększone, co w konsekwencji może prowadzić do zgonu. Należy mieć to na uwadze przed rozpoczęciem swoistej immunoterapii.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Należy przedsięwziąć odpowiednie środki ostrożności na wypadek wystąpienia reakcji anafilaktycznych;
Przed każdym wstrzyknięciem należy u pacjenta wykonać badanie czynnościowe płuc (patrz punkt 4.3);
Przed każdym wstrzyknięciem należy dwukrotnie sprawdzić alergen, stężenie, objętość oraz poprzednią datę wstrzyknięcia (odstęp czasu między dawkami);
Nie wolno podawać donaczyniowo;
Nie należy wstrzykiwać w pobliżu nerwów obwodowych oraz dużych nerwów;
U pacjentów ze zwiększonym stężeniem tryptazy w surowicy krwi i (lub) mastocytozą, ryzyko ogólnoustrojowych reakcji alergicznych i stopień ich nasilenia może być większy.
Środki ostrożności związane ze stanem ogólnym pacjenta
Pacjent powinien unikać wysiłku fizycznego, gorących kąpieli oraz spożywania alkoholu w dniu wstrzyknięcia produktu;
Należy dokumentować wszelkie reakcje alergiczne (zarówno miejscowe, jak i ogólnoustrojowe) które wystąpiły po poprzednich wstrzyknięciach i na tej podstawie ustalić odpowiednie dawkowanie produktu (patrz punkt 4.2);
Tolerancja produktu przez pacjenta może się zmieniać, jeżeli zostanie zmieniona dawka innych leków przeciwalergicznych (patrz punkt 4.5);
W przypadku wszelkich zmian w przyjmowaniu leków od czasu ostatniego wstrzyknięcia należy również ocenić stan ogólny i alergiczny pacjenta;
Wstrzyknięcie należy przełożyć jeśli pacjent:
ma gorączkę lub objawy ostrego lub przewlekłego zakażenia,
miał objawy alergiczne w ciągu ostatnich 3–4 dni przed wstrzyknięciem,
ma istotne pogorszenie czynności płuc (przepływ szczytowy FEV1 ≤ 70% wartości prawidłowej u danego pacjenta),
ma zaostrzenie atopowego zapalenia skóry.
U pacjentów z mastocytozą można spodziewać się zmniejszenia skuteczności działania produktu w porównaniu z ogólną populacją uczuloną na jady owadów.
Środki ostrożności po każdym wstrzyknięciu.
Należy odnotować wszelkie reakcje alergiczne (zarówno miejscowe, jak i ogólnoustrojowe) przed wypisem pacjenta ze szpitala;
Należy zmierzyć przepływ szczytowy płuc;
Należy pouczyć pacjenta o konieczności obserwacji wszystkich reakcji miejscowych lub ogólnoustrojowych, które mogą wystąpić z opóźnieniem i poinformowania o nich lekarza w czasie następnej wizyty;
Należy poinformować pacjenta o konieczności natychmiastowego skontaktowania się z lekarzem lub oddziałem ratunkowym szpitala, jeśli wystąpią poważne, uogólnione, późne rekacje.
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
10 mikrogramów/ml (1:10): 0,55 ml roztworu o stężeniu 100 mikrogramów/ml jadu + 5 ml rozcieńczalnika z albuminą.
1 mikrogram/ml (1:100): 0,55 ml roztworu o stężeniu 10 mikrogramów/ml jadu + 5 ml rozcieńczalnika z albuminą.
0,1 mikrograma/ml (1:1000): 0,55 ml roztworu o stężeniu 1 mikrogram/ml jadu + 5 ml rozcieńczalnika z albuminą.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Pharmalgen Hymenoptera venoms
Wyciągi alergenowe jadów owadów błonkoskrzydłych (osy, pszczoły) Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań
Substancja czynna:
Wyciągi alergenowe jadów owadów błonkoskrzydłych (osy, pszczoły) - 120 mikrogramów. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania roztworu do wstrzykiwań.
Proszek w kolorze białawym po rozpuszczeniu rozpuszczalnikiem staje się przejrzysty.
Diagnostyka i leczenie (immunoterapia) alergii IgE zależnej na jad osy lub pszczoły.
Immunoterapia powinna być stosowana wyłącznie po swoistej diagnozie, a dawka powinna być zawsze ustalona indywidualnie dla pacjenta.
Leczenie powinno być prowadzone wyłącznie przez lekarzy mających doświadczenie w swoistej immunoterapii.
Należy dysponować wyposażeniem niezbędnym do leczenia wstrząsu anafilaktycznego.
Pacjent powinien pozostać pod obserwacją przez przynajmniej 30 minut po każdym wstrzyknięciu.
Diagnostyka
Przed rozpoczęciem immunoterapii za pomocą jadu pszczoły lub osy, powinna być wykonana szczegółowa diagnoza na podstawie wywiadu na temat objawów oraz diagnostyki in vivo (test skórny) i (lub) diagnostyki in vitro.
Diagnostyka in vivo
Testy skórne:
Test punktowy (prick test)
Do wykonania testu skórnego punktowego zaleca się zastosowanie jadu owadów błonkoskrzydłych o stężeniu 100 mikrogramów/ml. W przypadku bardzo wrażliwych pacjentów zaleca się wykonanie testu za pomocą mniejszych stężeń (1 lub 10 mikrogramów/ml).
Alternatywnie, do ustalenia indywidualnych bezpiecznych stężeń początkowych w swoistej
immunoterapii można zastosować oznaczenie punktu końcowego. Oznaczanie punktu końcowego przeprowadza się z użyciem stężenia jadu od 0,01 mikrograma/ml do 100 mikrogramów/ml. Instrukcja dotycząca rekonstytucji i rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6.
W przypadku wystąpienia dodatniego odczynu po podaniu stężenia 0,01 mikrogramaw/ml należy przeprowadzić ponowne badania za pomocą mniejszego stężenia. Jeżeli nie obserwuje się dodatniego odczynu po podaniu stężenia 0,01 mikrograma/ml, można stosować kolejne stężenia.
Najmniejsze stężenie, powodujące dodatnią reakcję, jest uznawane za punkt końcowy.
Odczyny odczytuje się po 15-20 minutach. Reakcję uważa się za dodatnią, gdy wystąpi bąbel o średnicy większej niż 3 mm.
Do rozcieńczania i jako kontrolę należy stosować rozcieńczalnik z albuminą. Test śródskórny:
Test śródskórny jest 100-1000 razy bardziej czuły niż test punktowy.
Test śródskórny powinien być wykonywany wyłącznie jako określenie punktu końcowego. Zaleca się, aby rozpocząć test od stężenia 0,0001 mikrograma/ml i objętości 0,02-0,05 ml. Instrukcja dotycząca rekonstytucji i rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6. Jeśli po zastosowaniu stężeniu 0,0001 mikrograma/ml występuje znacząca reakcja, należy ustalić punkt końcowy za pomocą mniejszych stężeń. Jeśli po 15-20 min nie wystąpi reakcja, oznaczanie należy kontynuować za pomocą stężeń 0,001-0,01-0,1 mikrograma/ml. Stężenia te można stosować wyłącznie u pacjentów, którzy tolerowali dobrze test skórny punktowy o stężeniu 100 mikrogramów/ml.
Do testu śródskórnego nie należy stosować stężeń 1 mikrograma/ml, gdyż mogą one powodować reakcje nieswoiste.
Odczyny odczytuje się po 15-20 minutach. Reakcję uważa się za dodatnią, gdy wystąpi bąbel o średnicy większej niż 5 mm i rumień.
Do rozcieńczania i jako kontrolę należy stosować rozcieńczalnik z albuminą. Diagnostyka in vitro
Ustalenie gatunku powodującego objawy można przeprowadzić za pomocą testu na swoistość IgE,
który pozwala potwierdzić, że występuje alergia wzbudzana za pośrednictwem IgE.
Leczenie
Leczenie przebiega w dwóch fazach: faza podstawowa (zwiększanie dawki) i faza podtrzymująca (stała dawka).
Leczenie za pomocą produktu Pharmalgen Hymenoptera venoms jest szczególnie korzystne, gdy pożądane jest szybkie zwiększenie dawki. Zwiększenie dawki może przebiegać stopniowo (według schematu „clustered”) w warunkach ambulatoryjnych lub jako szybkie zwiększanie dawki w warunkach pobytu szpitalnego (według schematu „rush”). Podczas konwencjonalnego zwiększania dawki zaleca się stosowanie Alutard SQ odpowiednio ALK 802 Jad osy lub ALK 801 Jad pszczoły.
Sposób podawania
Zaleca się stosowanie 1 ml strzykawki tuberkulinowej i igły 27 G 0,4 x 19.
Ewentualnie podczas zwiększenia dawki można stosować 0,5 ml strzykawkę insulinową. Pobrać właściwą ilość.
Wstrzyknięcie wykonuje się głęboko podskórnie bocznie (lateralnie) w ramię lub grzbietowo (dorsalnie) w przedramię.
Tkankę podskórną unosi się dwoma palcami i wprowadza igłę ok. 1 cm w głąb do tkanki podskórnej, pod katem 300 -600.
Przed rozpoczęciem wstrzykiwania aspiruje się lekko strzykawkę, aby uniknąć wstrzyknięcia donaczyniowo. Powtarza się to po każdej dawce 0,1 ml.
Wstrzyknięcie wykonuje się powoli, tak aby 1 ml podać w ciągu 60 sekund.
Dawkowanie
Zwiększanie dawki
Doświadczenia z zastosowaniem Pharmalgen Hymenoptera venoms wykazały, iż dawka początkowa 0,01 mikrograma jest tolerowana przez 90-95% pacjentów. Dlatego zalecana dawka początkowa to 0,1 ml stężenia 0,1 mikrograma/ml. W przypadku bardzo wrażliwych pacjentów, zaleca się dziesięciokrotne zmniejszenie dawki tzn. 0,1 ml o stężeniu 0,01 mikrograma/ml. Instrukcja dotycząca rekonstytucji i rozcieńczania produktu leczniczego przed podaniem, patrz punkt 6.6.
Jeśli do ustalenia indywidualnych stężeń początkowych stosuje się oznaczenie punktu końcowego, dawka początkowa wynosi 0,1 ml roztworu o stężeniu, które jest 1000 razy mniejsze, niż stężenie punktu końcowego w teście skórnym punktowym i 10 razy mniejsze niż stężenie punktu końcowego w teście śródskórnym.
W kolejnych wstrzyknięciach dawka będzie zwiększana dziesięciokrotnie, aż do stężenia 0,01 mikrograma/ml. Dalsze wstrzyknięcia dokonywane są według zaleceń Tabel 1, 2 lub 3.
Podczas każdego wstrzyknięcia zwiększa się dawkę aż do osiągnięcia dawki maksymalnej (tolerowanej przez pacjenta). W niektórych przypadkach obserwuje się niższy próg tolerancji
z powodu miejscowych lub uogólnionych reakcji. Po trzykrotnej konieczności zmniejszania dawki, kontynuuje się podawanie tym pacjentom największej osiągniętej dawki.
Zwiększoną dawkę powinno się podawać wyłącznie po dokonaniu oceny miejscowych i uogólnionych reakcji opisanych poniżej.
Wszystkie zalecenia mają charakter ogólnych wytycznych. Zawsze należy dokonywać indywidualnej oceny zwiększania dawki w oparciu o:
Każda dawka musi być dostosowana do reaktywności pacjenta i może być zwiększona jedynie wtedy, gdy pacjent tolerował poprzednie dawki. Za pomocą sukcesywnie wzrastających dawek stymuluje się system immunologiczny bez wywoływania reakcji.
Istnieją trzy schematy leczenie początkowego:
Wstrzyknięcia w odstępach 2 godzinnych podczas pobytu szpitalnego. Maksymalnie 4 wstrzyknięcia dziennie (patrz Tabela 3.).
Wstrzyknięcia wykonuje się w odstępach 2-godzinnych podczas pobytu pacjenta w szpitalu. Na początku, wstrzyknięcia mogą być wykonywane co 30 minut.
Tabela 3. może być wykorzystana jako orientacyjna propozycja dawkowania.
Pacjenta należy poddawać obserwacji przez przynajmniej 30 minut po ostatnim wstrzyknięciu. Pierwsza dawka w dniu następnym zawsze powinna być taka jak ostatnia dawka w poprzednim dniu.
Tabela 3. Schemat dawkowania w immunoterapii przyspieszonej („rush”).
Stężenie mikrogram/ml | Dawka ml | Dawka mikrogramy |
0,1 | 0,1 | 0,01● |
0,1 | 0,2 | 0,02 |
0,1 | 0,4 | 0,04 |
1 | 0,05 | 0,05 |
1 | 0,1 | 0,1 |
1 | 0,2 | 0,2 |
1 | 0,4 | 0,4 |
10 | 0,05 | 0,5 |
10 | 0,1 | 1 |
10 | 0,2 | 2 |
10 | 0,4 | 4 |
100 | 0,05 | 5 |
100 | 0,1 | 10 |
100 | 0,2 | 20 |
100 | 0,3 | 30 |
100 | 0,4 | 40 |
100 | 0,5 | 50 |
100 | 0,6 | 60 |
100 | 0,8 | 80 |
100 | 0,9 | 90 |
100 | 1 | 100 |
Maksymalnie 4 wstrzyknięcia dziennie
Leczenie podtrzymujące
Zalecana dawka podtrzymująca wynosi 1 ml 100 mikrogramów/ml produktu Pharmalgen Hymenoptera venoms.
W niektórych przypadkach próg tolerancji może być niższy z powodu miejscowych lub uogólnionych reakcji. Po trzykrotnej konieczności zmniejszania dawki, kontynuuje się podawanie największej osiągniętej dawki.
Po osiągnięciu dawki podtrzymującej, zaleca się zmianę tego produktu na produkt typu depot (powoli uwalnianej postaci leku), zgodnie z wytycznymi ujętymi w punkcie „Zmiana na produkt typu depot”.
Jeśli po osiągnięciu dawki podtrzymującej nie jest podjęta decyzja o zmianie na produkt typu depot, zwiększa się odstęp do 1, 2 i czterech tygodni.
W fazie podtrzymującej, Pharmalgen Hymenoptera venoms podaje się co 4 tygodnie. Jeśli
u pacjentów otrzymujących w fazie podtrzymującej dawkę 100 mikrogramów obserwuje się reakcje alergiczne po ukłuciu owadów, można powoli zwiększyć dawkę do 200 mikrogramów.
Immunoterapia powinna być kontynuowana przez co najmniej trzy lata (3-5 lat).
Zmniejszenie dawki w przypadku przekroczenia odstępu czasu pomiędzy dawkami w fazie podtrzymującej
Odstęp dłuższy niż 4 ale krótszy niż 6 tygodni:
Zmniejszenie dawki do poziomu 3/4 ostatnio podanej dawki.
Odstęp dłuższy niż 6 ale krótszy niż 8 tygodni:
Zmniejszenie dawki do poziomu 1/2 ostatnio podanej dawki.
Odstęp dłuższy niż 8 ale krótszy niż 10 tygodni:
Zmniejszenie dawki do poziomu ¼ ostatnio podanej dawki.
Odstęp dłuższy niż 10 tygodni:
Ponowne rozpoczęcie dawkowania od stężenia początkowego.
W przypadku wątpliwości, w jakim stopniu zmniejszyć dawkę, należy albo wybrać najmniejszą
z rozważanych alternatyw albo podzielić dawkę na dwie równe części i po każdym wstrzyknięciu obserwować pacjenta przez 30 minut.
Zmiana na produkt typu depot.
Po osiągnięciu dawki podtrzymującej (największej dawki tolerowanej), zaleca się kontynuację terapii za pomocą produktu typu depot Alutard SQ (ALK 801 Jad pszczoły lub ALK 802 Jad osy). Po zmianie na Alutard SQ ALK 801 Jad pszczoły lub Alutard SQ 802 Jad osy, dawkę podtrzymującą podaje się w dwóch porcjach.
Najpierw podaje się 1/3 dawki, a po 30 minutach obserwacji pacjenta, podaje się pozostałą część. Po drugim wstrzyknięciu, pacjenta obserwuje się przez 30 minut.
Następnie zaleca się zwiększenie odstępów czasowych między podaniem ALK 801 Jad pszczoły lub ALK 802 Jad osy (Alutard SQ) o 2, 4 i 6 tygodni.
Gdy kontynuuje się terapię za pomocą ALK 801 Jad pszczoły lub ALK 802 Jad osy (Alutard SQ), dawkę podtrzymującą podaje się co 6 tygodni ± 2 tygodnie.
Jeśli zmiana jest w fazie zwiększania dawki lub w przypadkach, kiedy nie jest możliwe osiągnięcie dawki podtrzymującej na poziomie 100 μg/ml, podaje się takie stężenie ALK 801 Jad pszczoły lub ALK 802 Jad osy (Alutard SQ) jakie odpowiada dawce podtrzymującej.
0,01mikrograma odpowiada 10 SQ-U 0,1 mikrograma odpowiada 100 SQ-U
1 mikrogram odpowiada 1 000 SQ –U
10 mikrogramów odpowiada 10 000 SQ – U
100 mikrogramów odpowiada 100 000 SQ – U
ALK 801 Jad pszczoły lub ALK 802 Jad osy (Alutard SQ) do terapii podtrzymującej powinno się zamówić jednocześnie z zestawem Pharmalgen Hymenoptera venoms.
Równoczesne leczenie więcej niż jednym alergenem
Produkty Pharmalgen Hymenoptera venoms nie mogą być mieszane. Pacjenci uczuleni na jad pszczoły jak i jad osy powinni być leczeni najpierw jednym produktem (zawierającym jad osy lub jad pszczoły) Gdy osiągnie się dawkę podtrzymującą, można rozpocząć leczenie drugim produktem. Dawki podtrzymujące obu produktów należy podawać z zachowaniem przerwy 2-3 dni.
Zmiana dawkowania po reakcji w miejscu wstrzyknięcia lub ogólnoustrojowej.
Pacjenta obserwuje się przez 30 minut po każdym wstrzyknięciu i odnotowuje wszystkie reakcje alergiczne. Reakcje opóźnione mogą powstać w ciągu 24 godzin lub później i powinny być także odnotowane. Jeśli u pacjenta wystąpią reakcje, których nie akceptuje, należy zmniejszyć dawkę zgodnie z poniższymi zaleceniami.
Natychmiastowa reakcja w miejscu podania
Obrzęk, zaczerwienienie i swędzenie wokół miejsca wstrzyknięcia. Mierzy się maksymalną średnicę obrzęku (nie zaczerwienienia). W przypadku wystąpienia natychmiastowych reakcji w miejscu podania, nie wykonuje się żadnych kolejnych wstrzyknięć tego samego dnia.
Średnica obrzęku:
Maksymalna średnica obrzęku | ||
Dzieci | Dorośli | Zalecane zmniejszanie dawki. |
< 5 cm | < 8 cm | Kontynuować zwiększanie dawki według schematu dawkowania. |
5–7 cm | 8–12 cm | Powtórzyć ostatnio podaną dawkę. |
7–12 cm | 12–20 cm | Zmniejszyć dawkę do dawki podanej wcześniej niż ostatnia. |
12-17 cm | > 20 cm | Zmniejszyć dawkę do odpowiedniej dawki podanej dwa okresy wcześniej niż ostatnia. |
>17 cm | - | Zmniejszyć dawkę do odpowiedniej dawki podanej trzy okresy wcześniej niż ostatnia. |
Po zmniejszeniu dawki, należy podjąć stopniowe zwiększanie dawki.
Stopień zmniejszenia dawki, zależy od ciężkości reakcji alergicznej i przerwy między wstrzyknięciami itd. Nadrzędną wartością jest tu bezpieczeństwo pacjenta i w razie wątpliwości zalecane jest zmniejszenie dawki.
Opóźniona reakcja w miejscu wstrzyknięcia (może wystąpić w ciągu 24-48 godzin po wstrzyknięciu). Jeśli reakcja, która wystąpiła w fazie zwiększania dawki, była dla pacjenta uciążliwa, należy powtórzyć poprzednią dawkę. Dotyczy to także przypadków, w których średnica obrzęku była większa niż 8 cm. Jeśli uciążliwa dla pacjenta reakcja wystąpiła podczas terapii podtrzymującej, należy zmniejszyć dawkę. W przypadku nieuciążliwych dla pacjenta reakcji, nie ma powodu do wprowadzania zmian, ponieważ takie reakcje nie powodują reakcji ogólnoustrojowych.
Reakcje o charakterze ogólnoustrojowym:
Łagodne reakcje ogólnoustrojowe
Na przykład: zapalenie spojówki, nieżyt nosa. Jeśli łagodne reakcje uogólnione wystąpią w ciągu 24 godzin po wstrzyknięciu, zmniejsza się kolejną dawkę o 2-3 stopnie wstecz, ewentualnie kontynuuje się terapię z wolniejszym zwiększaniem dawki.
Umiarkowane reakcje ogólnoustrojowe
Na przykład: pokrzywka i astma, które łatwo poddają się leczeniu. Jeśli umiarkowane reakcje uogólnione wystąpią do 24 godzin po iniekcji, należy zmniejszyć kolejną dawkę o 3-4 stopnie wstecz i kontynuować terapię z wolniejszym zwiększaniem dawki.
Ciężkie reakcje ogólnoustrojowe
Na przykład: złe ogólne samopoczucie, kilka jednoczesnych objawów lub słaba odpowiedź na leczenie objawowe. Jeśli ciężkie reakcje uogólnione wystąpią do 24 godzin po iniekcji, należy ponownie rozważyć stosowność terapii. Jeśli leczenie będzie kontynuowane, należy zredukować kolejną dawkę do 1/10 ostatnio podanej a następnie zwiększać dawkę.
Wstrząs anafilaktyczny
Szybko rozwijająca się reakcja ze świądem skóry, poczuciem gorąca, rumieniem, skurczem oskrzeli itd. wymaga zaawansowanego leczenia. W przypadku wystąpienia wstrząsu anafilaktycznego, należy ponownie ocenić bezpieczeństwo terapii. Jeśli podejmie się kontynuację terapii, zaleca się rozpoczęcie dawkowania od początku.
Leczenie produktem Pharmalgen Hymenoptera venoms powinno być prowadzone wyłącznie przez lekarzy doświadczonych w stosowaniu swoistej immunoterapii.
Przed zastosowaniem należy dokładnie przeczytać punkt 4.2 Dawkowanie i sposób podawania.
Przed zastosowaniem tego produktu leczniczego u dzieci poniżej 5 lat należy szczególnie rozważyć stosunek ryzyka do korzyści. Dane dotyczące skuteczności klinicznej u dzieci w wieku 5 lat
i starszych są ograniczone, natomiast dane dotyczące bezpieczeństwa nie wskazują na wyższe ryzyko niż u dorosłych.
Ze względu na ryzyko ciężkich reakcji anafilaktycznych, należy zapewnić bezpośredni dostęp do pełnego wyposażenia do resucytacji krażeniowo-oddechowej oraz odpowiednich leków, w tym adrenaliny do wstrzyknięć oraz obecność personelu wyszkolonego w ich stosowaniu.
Pacjent powinien znajdować się pod obserwacją przez 30 minut po wstrzyknięciu.
Jeśli wystąpią jakiekolwiek objawy reakcji ogólnoustrojowej, takie jak pokrzywka, obrzęk naczynioruchowy lub ciężka astma, należy niezwłocznie rozpocząć leczenie objawowe.
Środki ostrożności związane z leczeniem
Inne szczepienia
Nie należy wykonywać innych szczepień jeden tydzień przed i jeden tydzień od podania produktu Pharmalgen Hymenoptera venoms.
Substancje pomocnicze
Pharmalgen Hymenoptera venoms zawiera mniej niż 1 mmol (22 mg) sodu w każdym ml.
Jednoczesne leczenie lekami przeciwalergicznymi np. lekami przeciwhistaminowymi do podawania ogólnego, lekami rozszerzającymi oskrzela, kortykosteroidami i stabilizatorami mastocytów może zwiększyć poziom tolerancji alergenu przez pacjenta.
U pacjentów leczonych inhibitorami ACE, poddawanych równoczesnemu odczulaniu produktem Pharmalgen Hymenoptera venoms bardzo rzadko zgłaszano występowanie zagrażających życiu reakcji anafilaktycznych. Można temu zapobiec, przerywając na pewien czas leczenie inhibitorem ACE (w oparciu o okres półtrwania inhibitora ACE). Jednakże, u każdego pacjenta należy rozważyć dokładnie ryzyko przerwania leczenia inhibitorem ACE w stosunku do korzyści wynikających z immunoterapii (patrz punkt 4.3). Nie należy jednocześnie prowadzić immunoterapii i leczenia lekami immunosupresyjnymi.
Ciąża
Pharmalgen Hymenoptera venoms można stosować w okresie ciąży wyłącznie wtedy, gdy jest to absolutnie konieczne.
Nie ma danych klinicznych na temat stosowania produktu podczas ciąży. Nie należy rozpoczynać immunoterapii w okresie ciąży. Jeżeli zajście w ciążę nastąpi w trakcie leczenia, terapię można kontynuować po starannej ocenie stanu ogólnego pacjentki i jej reakcji na uprzednie wstrzyknięcia produktu Pharmalgen Hymenoptera venoms.
Należy skrupulatnie rozważyć ryzyko ciężkich reakcji alergicznych u matki i płodu.
Karmienie piersią
Nie ma danych klinicznych dotyczących stosowania produktu podczas karmienia piersią.
Wpływ na płodność
Nie ma danych klinicznych dotyczących wpływu produktu Pharmalgen Hymenoptera venoms na płodność.
Ze względu na działania niepożądane (zmęczenie) produkt leczniczy Pharmalgen Hymenoptera venoms może wywierać niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Zazwyczaj reakcje, związane ze stosowaniem produktu leczniczego Pharmalgen Hymenoptera venoms spowodowane są reakcjami immunologicznymi (w miejscu podania i (lub) ogólnoustrojowymi) na dany alergen).
Objawy wczesnej reakcji pojawiają się w ciągu pierwszych 30 minut po wstrzyknięciu. Objawy późnej reakcji pojawiają się zazwyczaj w ciągu 24 godzin od wstrzyknięcia.
Bardzo często zgłaszanymi reakcjami niepożądanymi u pacjentów leczonych Pharmalgen Hymenoptera venoms były reakcje w miejscu iniekcji.
Działania niepożądane uszeregowano według układów narządowych, zgodnie z częstością występowania wg. konwencji MedDRA:
bardzo często (≥1/10), często (≥1/100, <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1 000), bardzo rzadko (≥1/10 000). Częstość występowania oparta jest o wyniki z badań klinicznych dotyczących immunoterapii.
Określenie „częstość nieznana” oznacza, że nie można jej ocenić na podstawie dostępnych danych z badań klinicznych a jest oparta na podstawie danych po wprowadzeniu do obrotu.
Klasyfikacja układów i narządów | Częstość występowania | Działania niepożądane |
Zaburzenia układu immunologicznego | Niezbyt często Rzadko | Reakcja anafilaktyczna Wstrząs anafilaktyczny |
Zaburzenia układu nerwowego | Bardzo często Nieznana | Ból głowy Zawroty głowy, parestezje |
Zaburzenia oka | Często Nieznana | Zapalenie spojówek Obrzęk powieki |
Zaburzenia ucha i błędnika | Nieznana | Zawroty głowy |
Zaburzenia serca | Nieznana | Kołatanie, tachykardia, sinica |
Zaburzenia naczyniowe | Często Nieznana | Uderzenia gorąca Niedociśnienie tętnicze, bladość |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | Często Nieznana | Świsty, kaszel, duszność Astma, nieżyt nosa, alergiczny nieżyt nosa, kichanie, skurcz oskrzeli, podrażnienie gardła, uczucie ucisku w gardle |
Zaburzenia żołądka i jelit | Często Nieznana | Biegunka, wymioty, nudności, niestrawność Ból brzucha |
Zaburzenia skóry i tkanki podskórnej | Często Nieznana | Pokrzywka, świąd, wysypka Obrzęk naczynioruchowy, rumień |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | Niezbyt często Nieznana | Ból pleców Obrzęk stawów, ból stawów |
Zaburzenia ogólne i stany w miejscu podania | Bardzo często Często Nieznana | Obrzęk w miejscu iniekcji Świąd w miejscu iniekcji, pokrzywka w miejscu iniekcji, uczucie dyskomfortu, uczucie zmęczenia Świąd, uczucie dyskomfortu w klatce piersiowej, dreszcze, rumień w miejscu iniekcji, ból w miejscu iniekcji, uczucie obcego ciała |
Miejscowe reakcje występują w miejscu iniekcji i obejmują obrzęk, zaczerwienienie, ból, swędzenie, odbarwienie i krwiaki w miejscu iniekcji.
Reakcje układowe są to wszystkie objawy pochodzące z narządów odległych od miejsca iniekcji. Reakcje układowe mogą mieć postać od alergicznego nieżytu nosa aż do wstrząsu anafilaktycznego. Leczenie ciężkich reakcji układowych należy wdrożyć niezwłocznie.
W przypadku nasilonych reakcji miejscowych i reakcji układowych należy ocenić możliwość kontynuowania leczenia (patrz punkt 4.2).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych.
Al. Jerozolimskie 181 C, 02-222 Warszawa
Tel.: +48 22 49 21 301
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Objawy
W przypadku wstrzyknięcia większej dawki, niż to zamierzono, wzrasta ryzyko rozwoju ciężkich reakcji alergicznych.
Leczenie
Pacjent powinien znajdować się pod obserwacją i w przypadku wystąpienia reakcji należy wszcząć leczenie objawowe.
Grupa farmakoterapeutyczna: wyciągi alergenowe jadów owadów błonkoskrzydłych. Kod ATC: V01AA07
Reakcja biologiczna na wyciągi alergenowe jest głównie wynikiem odpowiedzi immunologicznej ustroju.
Ponieważ mogą wystąpić reakcje alergiczne I typu, właściwe postępowanie lecznicze obejmuje podanie leków, które przeciwdziałają odpowiedzi ustrojowej oraz leków o właściwościach sympatykomimetycznych.
Ekstrakty składają się z substancji wielkocząsteczkowych i stosowane dawki są małe (µg). Podaje się je podskórnie, przy czym działaniem objęty jest układ immunologiczny. Nie przeprowadzono badań w celu wyjaśnienia drogi metabolizowania poszczególnych składników. Większa część ekstraktów to polipeptydy i białka i przypuszcza się, że są one metabolizowane do aminokwasów i mniejszych polipeptydów.
Celem badań przedklinicznych jest uzyskanie informacji odnośnie ryzyka związanego ze stosowaniem produktu u ludzi. Doświadczenia na zwierzętach wykazały, że jady osy i pszczoły (Hymenoptera
Venoms) mogą prowadzić do degeneracji peryferyjnych nerwów. Zaobserwowano to także u pacjentów. Dlatego nie należy podawać iniekcji w bliskości nerwów obwodowych.
Nie prowadzono żadnych badań, aby to potwierdzić.
Proszek:
Mannitol Albumina ludzka
Rozcieńczalnik z albuminą:
Sodu chlorek Albumina ludzka Fenol
Woda do wstrzykiwań
Kwas solny (do ustalenia pH)
Nie dotyczy.
Proszek: 5 lat
Rozcieńczalnik z albuminą: 3 lata
Okres ważności po rekonstytucji i rozcieńczeniu rozcieńczalnikiem z albuminą: 100 mikrogramów/ml – 6 miesięcy.
0,1 mikrograma/ml – 1 tydzień
1 mikrogram/ml – 1 tydzień,
10 mikrogramów/ml – 2 tygodnie
Po rozcieńczeniu do stężenia poniżej 0,1 mikrograma/ml – należy wykorzystać tego samego dnia. Po rekonstytucji i rozcieńczeniu, każdą fiolkę produktu Pharmalgen Hymenoptera venoms należy oznaczyć terminem przydatności. Termin przydatności od chwili rekonstytucji i rozcieńczenia
produktu nie może przekraczać terminu ważności liofilizowanego Pharmalgen Hymenoptera venoms lub rozcieńczalnika z albuminą. Do rekonstytucji i rozcieńczania powinno się stosować wyłącznie rozcieńczalnik z albuminą.
Przechowywać w lodówce (20C – 80C). Nie zamrażać.
Przechowywać fiolki w oryginalnym opakowaniu, w celu ochrony przed światłem.
Fiolki ze szkła typu I z korkiem z gumy chlorobutylowej i aluminiowym wieczkiem. Zestaw do leczenia podstawowego i podtrzymującego:
4 fiolki z proszkiem po 120 mikrogramów + 4 fiolki z rozcieńczalnikiem z albuminą po 5 ml.
Zestaw do rozcieńczania:
10 fiolek po 5 ml rozcieńczalnika
W analogiczny sposób można wykonać kolejne rozcieńczenie (patrz rycina poniżej).









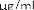






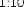





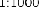

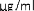
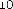





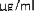




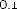
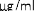



UWAGA: nie należy używać tej samej jałowej strzykawki do przenoszenia roztworu o różnych stężeniach. Do każdego stężenia należy użyć nowej jałowej strzykawki jednorazowej. Aby nie pomylić stężeń, należy je uprzednio opisać na etykietach fiolek [minimum informacji: alergen, stężenie, data wykonania rozcieńczenia i data ważności (patrz punkt 6.3)].
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami
ALK-Abelló A/S Bøge Allé 6-8
DK-2970 Hørsholm Dania
R/0104
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 05.02.1999 Data ostatniego przedłużenia pozwolenia: 20.11.2013







