







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- Właściwości farmakodynamiczne
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Ostre stany lękowe lub pobudzenie; delirium tremens
Ostre stany spastyczne mięśni; tężec
Ostre stany drgawkowe, w tym stan padaczkowy, stany drgawkowe w przebiegu zatruć, drgawki gorączkowe
Premedykacja przedoperacyjna lub premedykacja przed zabiegami diagnostycznymi (zabiegi stomatologiczne, chirurgiczne, radiologiczne, endoskopowe, cewnikowanie serca, kardiowersja)
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na substancję czynną (lub inne benzodiazepiny) lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1
Myasthenia gravis
Ciężka niewydolność oddechowa, depresja oddechowa
Zespół bezdechu sennego
Ciężka niewydolność wątroby
Fobie lub natręctwa
Przewlekłe psychozy
Nie stosować u noworodków i wcześniaków - patrz punkt 4.4
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
RELANIUM, 5 mg/ml, roztwór do wstrzykiwań
Każdy ml roztworu do wstrzykiwań zawiera 5 mg diazepamu (Diazepamum). Substancje pomocnicze o znanym działaniu:
1 ml roztworu do wstrzykiwań zawiera: 100 mg etanolu 96%, 15 mg alkoholu benzylowego, 48,8 mg sodu benzoesanu, 450 mg glikolu propylenowego.
Produkt zawiera 7,8 mg sodu w 1 ml.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1
Roztwór do wstrzykiwań
Bezbarwny lub żółtozielony, przezroczysty płyn
Dawkowanie
W celu uzyskania optymalnego działania produktu, należy starannie określić dawkowanie indywidualnie dla każdego pacjenta.
Dorośli
Ostre stany lękowe lub pobudzenie: 10 mg we wstrzyknięciu dożylnym lub domięśniowym; wstrzyknięcie można powtórzyć nie wcześniej niż po czterech godzinach.
Delirium tremens: 10 do 20 mg we wstrzyknięciu dożylnym lub domięśniowym. Konieczne może być podanie większych dawek w zależności od nasilenia objawów.
Ostre stany spastyczne mięśni: 10 mg we wstrzyknięciu dożylnym lub domięśniowym; wstrzyknięcie można powtórzyć nie wcześniej niż po czterech godzinach.
Tężec: dawka początkowa podawana dożylnie wynosi od 0,1 mg/kg mc. do 0,3 mg/kg mc., powtarzana co 1 do 4 godzin. Można także podawać w ciągłym wlewie dożylnym trwającym
24 godziny w dawce od 3 mg/kg mc. do 10 mg/kg mc. Wybrana dawka powinna zależeć od nasilenia objawów, w bardzo ciężkich przypadkach można stosować większe dawki.
Stan padaczkowy, drgawki w przebiegu zatrucia: 10 do 20 mg dożylnie lub domięśniowo; dawkę można powtórzyć po 30-60 minutach. Jeśli jest to wskazane, można zastosować powolny wlew dożylny (maksymalna dawka 3 mg/kg mc. w ciągu 24 godzin).
Premedykacja przedoperacyjna lub przed zabiegami diagnostycznymi: 0,2 mg/kg mc. Zazwyczaj stosowana dawka u dorosłych wynosi 10 mg do 20 mg, lecz może być konieczne podawanie
większych dawek w zależności od reakcji klinicznej.
Pacjenci w podeszłym wieku lub osłabieni
Podawane dawki nie powinny być większe niż połowa dawek zazwyczaj zalecanych.
Pacjenci w tej grupie powinni być regularnie monitorowani na początku leczenia w celu minimalizacji podawanych dawek i (lub) częstości ich podawania, aby uniknąć przedawkowania w wyniku kumulacji leku.
Dzieci
Stan padaczkowy, drgawki w przebiegu zatrucia, drgawki gorączkowe: 0,2 mg/kg mc. do 0,3 mg/kg mc. dożylnie lub domięśniowo lub 1 mg na każdy rok życia.
Tężec: dawkowanie jak u dorosłych.
Premedykacja przedoperacyjna lub przed zabiegami diagnostycznymi: 0,2 mg/kg mc.
Leczenie należy ograniczyć do niezbędnego minimum, lek podawać wyłącznie pod ścisłym nadzorem lekarza. Dane dotyczące skuteczności i bezpieczeństwa stosowania benzodiazepin podczas długotrwałego leczenia są ograniczone.
W celu zmniejszenia prawdopodobieństwa wystąpienia działań niepożądanych podczas dożylnej sedacji, należy lek wstrzykiwać powoli (0,5 ml roztworu w ciągu pół minuty) do wystąpienia senności u pacjenta, opadnięcia powiek i niewyraźnej mowy, przy czym pacjent powinien wciąż być zdolny do spełniania poleceń.
Stanowczo zaleca się wykonywanie wstrzyknięć dożylnych produktu do dużej żyły w dole
łokciowym, przy czym przez cały czas trwania zabiegu pacjent powinien być ułożony na plecach.
Przy przestrzeganiu powyższych zaleceń dotyczących podawania dożylnego produktu, ryzyko wystąpienia niedociśnienia tętniczego lub bezdechu jest znacznie zmniejszone.
Z wyjątkiem stanów nagłych, podczas podawania dożylnego leku powinna zawsze być obecna druga osoba; zawsze powinien być też dostępny zestaw do resuscytacji. Zaleca się, aby pacjenci pozostawali pod nadzorem lekarza co najmniej godzinę od podania leku.
W domu pacjentowi powinna zawsze towarzyszyć odpowiedzialna osoba dorosła; należy poinformować pacjenta o zakazie prowadzenia pojazdów i obsługiwania urządzeń mechanicznych przez 24 godziny po podaniu leku.
Zazwyczaj nie należy rozcieńczać roztworu produktu Relanium. Wyjątek stanowi podawanie produktu w powolnym wlewie dożylnym w dużej objętości roztworu NaCl 0,9% lub glukozy w leczeniu tężca
i stanu padaczkowego. Nie należy rozcieńczać więcej niż 40 mg diazepamu (8 ml roztworu) w 500 ml roztworu do wlewu. Roztwór należy przygotowywać bezpośrednio przed podaniem i zużyć w ciągu
6 godzin.
Produktu nie należy mieszać z innymi lekami w roztworze do wlewu lub w jednej strzykawce, ponieważ nie można zagwarantować jego stabilności w przypadku nieprzestrzegania powyższego zalecenia.
Produkt przeznaczony jest do podawania dożylnego lub domięśniowego.
Zazwyczaj nie należy stosować produktu pozajelitowo u pacjentów ze zmianami organicznymi mózgu (szczególnie miażdżycą naczyń) lub z przewlekłą niewydolnością płuc. Jednak w stanach nagłych lub gdy tacy pacjenci są leczeni w warunkach szpitalnych, produkt można podawać pozajelitowo w
mniejszej dawce. W przypadku podawania dożylnego, lek należy wstrzykiwać powoli.
U pacjentów z przewlekłą niewydolnością płuc i pacjentów z przewlekłymi chorobami wątroby, może być konieczne zmniejszenie dawek. W niewydolności nerek okres półtrwania diazepamu jest
niezmieniony, dlatego nie ma konieczności zmniejszenia dawek u pacjentów z zaburzeniami czynności nerek.
Diazepamu nie należy stosować w monoterapii w leczeniu pacjentów z depresją lub stanami lękowymi w przebiegu depresji, ponieważ mogą ujawnić się skłonności samobójcze.
Po kilku godzinach od zastosowania produktu może wystąpić amnezja. Aby zmniejszyć ryzyko jej wystąpienia, pacjenci powinni mieć zapewnione warunki do nieprzerwanego snu przez 7 do 8 godzin.
W przypadkach utraty bliskich osób i żałoby, podczas stosowania benzodiazepin może być hamowane przystosowanie psychologiczne.
Podczas stosowania benzodiazepin, szczególnie u dzieci i pacjentów w podeszłym wieku, opisywano reakcje paradoksalne, takie jak niepokój ruchowy, pobudzenie, drażliwość, agresywność, urojenia, napady złości, koszmary nocne, omamy, psychozy, nieodpowiednie zachowanie i inne zaburzenia
zachowania. W przypadku wystąpienia takich objawów, należy zaprzestać stosowania produktu.
Ryzyko związane z jednoczesnym stosowaniem z opioidami:
Jednoczesne stosowanie produktu Relanium z opioidami może prowadzić do sedacji, depresji oddechowej, śpiączki i zgonu. Z powodu tych zagrożeń przepisywanie leków uspokajających,
np. benzodiazepin lub leków pochodnych, takich jak Relanium, z opioidami powinno ograniczać się tylko do pacjentów, u których zastosowanie alternatywnego leczenia nie jest możliwe. Jeśli zostanie podjęta decyzja o przepisaniu produktu Relanium równocześnie z opioidami, należy stosować możliwie najmniejszą skuteczną dawkę, a czas leczenia powinien być jak najkrótszy (patrz również ogólne zalecenia dotyczące dawkowania w punkcie 4.2).
Pacjentów należy ściśle obserwować w celu wykrycia przedmiotowych i podmiotowych objawów depresji oddechowej oraz sedacji. W związku z tym zdecydowanie zaleca się informowanie pacjentów i ich opiekunów (jeżeli dotyczy) o możliwości wystąpienia tych objawów (patrz punkt 4.5).
Podczas leczenia produktami z grupy benzodiazepin może wystąpić uzależnienie. Ryzyko wystąpienia uzależnienia jest większe u pacjentów leczonych długotrwale i (lub) stosujących duże dawki,
szczególnie u predysponowanych pacjentów nadużywających alkoholu lub leków w wywiadzie. Po wystąpieniu uzależnienia fizycznego od benzodiazepin, przerwanie leczenia może prowadzić do wystąpienia objawów odstawiennych. Należą do nich bóle głowy, bóle mięśni, silny lęk, napięcie,
niepokój ruchowy, stan splątania i drażliwość. W ciężkich przypadkach mogą wystąpić objawy takie jak: utrata poczucia rzeczywistości lub własnej realności, mrowienie i drętwienie kończyn, nadwrażliwość na światło, dźwięk i dotyk, omamy lub napady drgawkowe.
Po długotrwałym podawaniu dożylnym, nagłemu odstawieniu leku mogą towarzyszyć objawy odstawienne, dlatego zaleca się stopniowe zmniejszanie dawek.
Należy zachować szczególną ostrożność stosując diazepam we wstrzyknięciach, szczególnie dożylnych, u pacjentów w podeszłym wieku, w ciężkim stanie oraz u osób o ograniczonej rezerwie sercowej lub płucnej, ze względu na możliwość wystąpienia bezdechu i (lub) zatrzymania czynności serca. Równoczesne stosowanie diazepamu i barbituranów, alkoholu lub innych substancji o działaniu depresyjnym na ośrodkowy układ nerwowy zwiększa ryzyko depresji krążeniowej lub oddechowej
oraz ryzyko bezdechu. Należy zapewnić dostęp do zestawu resuscytacyjnego, włącznie ze sprzętem do wentylacji wspomaganej.
Należy zachować szczególną ostrożność stosując benzodiazepiny u pacjentów nadużywających alkoholu lub leków w wywiadzie.
Produkt leczniczy zawiera 15 mg alkoholu benzylowego w 1 ml roztworu. Alkohol benzylowy może powodować reakcje alergiczne.
Dożylne podawanie alkoholu benzylowego noworodkom wiąże się z ryzykiem ciężkich działań
niepożądanych i śmierci (tzw. „gasping syndrome”). Minimalna ilość alkoholu benzylowego, po której mogą wystąpić objawy toksyczności jest nieznana.
Nie podawać noworodkom (do 4 tygodnia życia).
Z powodu zwiększonego ryzyka kumulacji alkoholu benzylowego u małych dzieci, produktu nie należy podawać dzieciom w wieku poniżej 3 lat dłużej niż przez tydzień.
Duże objętości alkoholu benzylowego należy podawać z ostrożnością i tylko w razie konieczności, zwłaszcza u pacjentów z zaburzeniami czynności nerek lub wątroby oraz u kobiet w ciąży lub
karmiących piersią, ze względu na ryzyko kumulacji i toksyczności (kwasica metaboliczna).
Produkt leczniczy zawiera 12,4% v/v etanolu (alkoholu), tzn. do 200 mg na ampułkę 2 ml, co jest równoważne 4,75 ml piwa lub 1,98 ml wina na ampułkę.
Szkodliwe dla osób z chorobą alkoholową.
Należy wziąć to pod uwagę podczas stosowania u kobiet ciężarnych lub karmiących piersią, dzieci i u osób z grupy dużego ryzyka, takich jak pacjenci z chorobą wątroby lub z padaczką.
Produkt leczniczy zawiera 450 mg glikolu propylenowego w każdym 1 ml. Jednoczesne podawanie z innymi substratami dehydrogenazy alkoholowej, takimi jak etanol, może powodować działania niepożądane u dzieci w wieku poniżej 5 lat.
Chociaż nie wykazano toksycznego wpływu glikolu propylenowego na rozród i rozwój potomstwa, może on przenikać do płodu i do mleka matki. Dlatego też podanie glikolu propylenowego pacjentce w ciąży lub karmiącej piersią należy w każdym przypadku rozważyć indywidualnie.
Pacjenci z zaburzeniami czynności nerek lub wątroby powinni pozostawać pod kontrolą lekarza z powodu różnych działań niepożądanych przypisywanych glikolowi propylenowemu, takich jak zaburzenia czynności nerek (ostra martwica kanalików nerkowych), ostra niewydolność nerek
i zaburzenia czynności wątroby.
Produkt leczniczy zawiera 48,8 mg sodu benzoesanu w 1 ml roztworu. Sodu benzoesan może zwiększać ryzyko żółtaczki u noworodków.
Produkt leczniczy zawiera 15,6 mg sodu na ampułkę 2 ml (7,8 mg sodu na 1 ml), co odpowiada 0,78% zalecanej przez WHO maksymalnej 2 g dobowej dawki sodu u osób dorosłych.
Produkt w może być rozcieńczany w 0,9% roztworze NaCl lub w glukozie. Przy obliczaniu
całkowitej zawartości sodu w przygotowanym rozcieńczeniu produktu należy brać pod uwagę ilość sodu pochodzącego z rozcieńczalnika. W celu uzyskania dokładnej informacji dotyczącej zawartości
sodu w roztworze wykorzystanym do rozcieńczenia produktu, należy zapoznać się z charakterystyką produktu leczniczego stosowanego rozcieńczalnika.
Jeżeli produkt podawany jest jednocześnie z innymi lekami o działaniu ośrodkowym, takimi jak leki przeciwpsychotyczne, przeciwlękowe, uspokajające, przeciwdepresyjne, nasenne, przeciwdrgawkowe, opioidowe leki przeciwbólowe, leki do znieczulenia ogólnego i leki przeciwhistaminowe o działaniu uspokajającym, prawdopodobne jest nasilenie działania uspokajającego. W przypadku opioidowych
leków przeciwbólowych może dojść do nasilenia działania euforyzującego, co może prowadzić do nasilenia uzależnienia psychicznego. Ponadto w przypadku, gdy leki o działaniu depresyjnym na ośrodkowy układ nerwowy podawane są pozajelitowo w skojarzeniu z dożylnie podawanym
diazepamem, może wystąpić ciężka depresja oddechowa i krążeniowa. Pacjenci w podeszłym wieku wymagają specjalnego nadzoru.
Podczas dożylnego podawania produktu Relanium jednocześnie z opioidowymi lekami przeciwbólowymi, np. w stomatologii, zaleca się podawanie diazepamu po podaniu leku przeciwbólowego oraz staranne dostosowanie dawki do indywidualnych potrzeb pacjenta.
Nie zaleca się równoczesnego stosowania produktu i spożywania alkoholu, ze względu na nasilenie działania uspokajającego. Wpływa to na zdolność do prowadzenia pojazdów i obsługiwania maszyn.
Wyniki badań farmakokinetycznych dotyczących potencjalnych interakcji diazepamu z lekami przeciwpadaczkowymi (włącznie z kwasem walproinowym) są sprzeczne. Obserwowano zarówno zmniejszenie jak i zwiększenie, a także brak zmian stężenia leku.
W przypadku równoczesnego stosowania produktu z lekami przeciwpadaczkowymi może dojść do nasilenia działań niepożądanych i toksyczności, szczególnie w przypadku leków z grupy pochodnych hydantoiny lub barbituranów, a także produktów złożonych zawierających te substancje. Dlatego też wymagana jest szczególna ostrożność podczas ustalania dawkowania w początkowym okresie leczenia.
Wykazano, że leki o znanym działaniu hamującym enzymy wątrobowe, np. cymetydyna, fluwoksamina, fluoksetyna i omeprazol, powodują zmniejszenie klirensu benzodiazepin i mogą nasilać ich działanie; leki o znanym działaniu indukującym enzymy wątrobowe, np. ryfampicyna, mogą zwiększać klirens benzodiazepin. Istnieją doniesienia o wpływie diazepamu na eliminację fenytoiny.
Opioidy:
Jednoczesne stosowanie leków uspokajających, np. benzodiazepin, lub leków pochodnych, takich jak Relanium, z opioidami, zwiększa ryzyko wystąpienia sedacji, depresji oddechowej, śpiączki i zgonu ze względu na addytywne działanie depresyjne na ośrodkowy układ nerwowy. Należy ograniczyć dawkę leku i czas trwania leczenia skojarzonego (patrz punkt 4.4).
Ciąża
Nie ma doniesień potwierdzających bezpieczeństwo stosowania leku u kobiet w ciąży. W badaniach na zwierzętach nie uzyskano też dowodów na bezpieczeństwo tego leczenia. Nie należy stosować leku w ciąży, szczególnie w pierwszym i ostatnim trymestrze, o ile okoliczności bezwzględnie tego nie wymagają.
W przypadku przepisywania leku kobietom w wieku rozrodczym, należy poinformować pacjentkę o konieczności skontaktowania się z lekarzem w celu przerwania leczenia w przypadkach, gdy pacjentka planuje ciążę bądź podejrzewa, że jest w ciąży.
Stwierdzono, że stosowanie dużych dawek lub długotrwałe stosowanie niewielkich dawek
benzodiazepin w ostatnim trymestrze ciąży lub podczas porodu powodowało zaburzenia rytmu serca płodu, niedociśnienie tętnicze, zaburzenia ssania, obniżenie temperatury ciała i umiarkowaną depresję oddechową u noworodków. Należy pamiętać, że u noworodków, szczególnie u wcześniaków, układ
enzymatyczny uczestniczący w metabolizmie leku nie jest w pełni rozwinięty. Ponadto noworodki urodzone przez matki, które stosowały długotrwale benzodiazepiny w ostatnim okresie ciąży mogą wykazywać zależność fizyczną i mogą u nich wystąpić objawy odstawienne w okresie pourodzeniowym.
Karmienie piersią
Diazepam przenika do mleka kobiecego, dlatego nie należy stosować diazepamu w okresie karmienia piersią.
Pacjentów należy pouczyć, że podobnie jak w przypadku wszystkich leków z tej grupy, stosowanie diazepamu może prowadzić do zaburzeń zdolności pacjenta do wykonywania złożonych czynności. Uspokojenie, niepamięć, zaburzenia koncentracji i czynności mięśni mogą niekorzystnie wpływać na zdolność do prowadzenia pojazdów lub obsługiwania maszyn. W przypadku niedostatecznej ilości snu, prawdopodobieństwo zaburzeń czujności może być zwiększone. Należy dodatkowo
poinformować pacjentów, że alkohol nasila zaburzenia oraz zalecić unikanie jego spożywania podczas leczenia (patrz punkt 4.5).
Działania niepożądane pogrupowane są według częstości występowania. Rzadko (≥1/10 000, <1/1 000); bardzo rzadko (<1/10 000).
Zaburzenia krwi i układu chłonnego Rzadko: zmiany w obrazie krwi.
Zaburzenia psychiczne
Reakcje paradoksalne, takie jak niepokój ruchowy, pobudzenie, drażliwość, agresywność, urojenia, napady wściekłości, koszmary senne, omamy (niektóre o typie seksualnym), psychozy, niewłaściwe zachowanie oraz inne zaburzenia zachowania (patrz punkt 4.4).
W czasie stosowania leków z grupy benzodiazepin może dojść do ujawnienia istniejącej wcześniej depresji.
Zaburzenia układu nerwowego
Rzadko: splątanie, osłabienie reakcji emocjonalnych, obniżenie poziomu czuwania, niepamięć następcza, ataksja, drżenia, ból głowy, zawroty głowy, zaburzenia mowy lub niewyraźna mowa, senność (występuje najczęściej na początku leczenia i zazwyczaj ustępuje w jego trakcie).
Pacjenci w podeszłym wieku są szczególnie wrażliwi na działanie leków hamujących ośrodkowy układ nerwowy i może u nich wystąpić splątanie, szczególnie w przypadku zmian organicznych w mózgu. Dawka produktu w tej grupie pacjentów nie powinna być większa niż połowa dawki zalecanej innym osobom dorosłym.
Zaburzenia oka
Rzadko: podwójne lub niewyraźne widzenie.
Zaburzenia serca i zaburzenia naczyniowe
Rzadko: niedociśnienie tętnicze, zmiany częstości tętna. Bardzo rzadko: przypadki zatrzymania czynności serca.
Może wystąpić depresja krążeniowa (po szybkim podaniu dożylnym produktu).
Zakrzepowe zapalenie żył i zakrzepica żylna może wystąpić po dożylnym podaniu produktu. W celu
zmniejszenia prawdopodobieństwa wystąpienia takich objawów, należy wstrzykiwać produkt do dużej żyły w dole łokciowym. Nie należy podawać leku do małych żył. W szczególności należy
bezwzględnie unikać podawania dotętniczego oraz wynaczynienia leku. Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia
Rzadko: zaburzenia oddychania, bezdech, depresja oddechowa (po szybkim podaniu dożylnym produktu).
Częstość występowania takich powikłań można zmniejszyć przez ścisłe przestrzeganie zalecanej prędkości podawania produktu. Przez cały czas trwania wstrzyknięcia lub wlewu należy zawsze utrzymywać pacjenta w pozycji leżącej na plecach.
Zaburzenia żołądka i jelit
Rzadko: zaburzenia dotyczące przewodu pokarmowego, nudności, suchość błony śluzowej jamy ustnej lub nadmierne wydzielanie śliny, zwiększenie łaknienia, zaparcia.
Zaburzenia wątroby i dróg żółciowych
Bardzo rzadko: zwiększenie aktywności transaminaz i fosfatazy zasadowej, żółtaczka.
Zaburzenia skóry i tkanki podskórnej Rzadko: reakcje skórne.
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej
Osłabienie mięśni – zależy zazwyczaj od zastosowanej dawki (występuje najczęściej na początku leczenia i zwykle ustępuje w jego trakcie).
Zaburzenia nerek i dróg moczowych:
Rzadko: nietrzymanie lub zatrzymanie moczu.
Zaburzenia układu rozrodczego i piersi
Rzadko: zwiększenie bądź zmniejszenie popędu płciowego.
Zaburzenia ogólne i stany w miejscu podania
Rzadko: zmęczenie (występuje najczęściej na początku leczenia i zazwyczaj ustępuje w trakcie dalszego leczenia).
Ból, a w niektórych przypadkach rumień po podaniu domięśniowym produktu. Stosunkowo często występuje bolesność w miejscu podania.
Osoby w podeszłym wieku oraz pacjenci z zaburzeniami czynności wątroby są szczególnie narażeni na wystąpienie wymienionych powyżej działań niepożądanych. Zaleca się regularne kontrolowanie przebiegu leczenia i odstawienie leku tak szybko, jak to możliwe.
Obserwowano nadużywanie leków z grupy benzodiazepin.
Stosowanie produktu (nawet w dawkach leczniczych) może prowadzić do rozwoju zależności fizycznej i psychicznej (patrz punkt 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań
niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania
Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Przedawkowanie leków z grupy benzodiazepin charakteryzuje się zazwyczaj objawami depresji ośrodkowego układu nerwowego, o nasileniu od senności do śpiączki. W łagodnych przypadkach, objawy mogą obejmować senność, splątanie i letarg. W cięższych przypadkach do objawów należą: ataksja, niedociśnienie tętnicze, depresja oddechowa, śpiączka (rzadko) i zgon (bardzo rzadko).
Nie ustalono dotychczas znaczenia dializy. Należy zwracać szczególną uwagę na czynność układu krążenia i oddechowego w oddziale intensywnej terapii.
Flumazenil jest specyficzną odtrutką podawaną dożylnie w stanach nagłych. Pacjenci wymagający takiego postępowania powinni podlegać ścisłemu monitorowaniu w warunkach szpitalnych. Należy zachować ostrożność podczas podawania flumazenilu pacjentom z padaczką, leczonym produktami z grupy benzodiazepin. W przypadku wystąpienia pobudzenia nie należy stosować barbituranów.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Grupa farmakoterapeutyczna: leki psycholeptyczne; anksjolityki; pochodne benzodiazepiny, kod ATC: N05BA01
Diazepam wykazuje działanie przeciwlękowe, przeciwdrgawkowe, miorelaksacyjne, uspokajające i nasenne. Wykazuje niewielki wpływ na autonomiczny układ nerwowy.
Miejscem działania benzodiazepin jest makromolekularny kompleks obejmujący receptory GABAA, receptory benzodiazepinowe i kanały chlorkowe. Receptory benzodiazepinowe poprzez allosteryczną reakcję z receptorami GABAA powodują otwarcie kanałów chlorkowych i zwiększają przenikanie jonów chlorkowych do wnętrza neuronów. Wynikiem jest nasilenie hamującego działania GABA
w ośrodkowym układzie nerwowym.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Wchłanianie
Po podaniu domięśniowym, wchłanianie jest całkowite, lecz nie zawsze szybsze niż po podaniu doustnym.
Dystrybucja
Diazepam i jego metabolity w dużym stopniu wiążą się z białkami osocza (diazepam w 98%).
Diazepam i jego metabolity przenikają przez barierę krew-mózg i barierę łożyskową; wykrywano je także w mleku kobiecym w stężeniach około dziesięciokrotnie mniejszych niż stężenia w osoczu kobiety. Objętość dystrybucji wynosi 1-2 l/kg.
Metabolizm
Diazepam jest głównie metabolizowany do farmakologicznie czynnych metabolitów, takich jak N-demetylodiazepam, temazepam i oksazepam.
Eliminacja
Zależność stężenia diazepamu w osoczu od czasu po podaniu dożylnym jest dwufazowa,
z początkową szybką i znaczną fazą dystrybucji i przedłużoną fazą końcowej eliminacji (okres półtrwania do 48 godzin). Okres półtrwania w fazie eliminacji czynnego metabolitu
N-demetylodiazepamu wynosi do 100 godzin. Diazepam i jego metabolity wydalane są głównie w moczu, przede wszystkim w postaci sprzężonej. Klirens diazepamu wynosi 20-30 ml/min.
Wielokrotne podawanie dawek prowadzi do kumulacji diazepamu i jego metabolitów. Stan równowagi dynamicznej metabolitów osiągany jest nawet po dwóch tygodniach; metabolity mogą osiągać większe stężenia niż lek macierzysty.
Farmakokinetyka w szczególnych przypadkach klinicznych
Okres półtrwania w fazie eliminacji może ulec wydłużeniu u noworodków, pacjentów w podeszłym wieku i u osób z chorobami wątroby. U pacjentów z niewydolnością nerek okres półtrwania diazepamu nie ulega zmianie.
Podawanie domięśniowe produktu może prowadzić do zwiększenia aktywności fosfokinazy
kreatynowej w surowicy, z maksymalnym stężeniem występującym pomiędzy 12 i 24 godziną po wykonaniu wstrzyknięcia. Należy to uwzględnić podczas diagnostyki różnicowej zawału serca.
Wchłanianie po wstrzyknięciu domięśniowym produktu może być zmienne, szczególnie po wstrzyknięciu do mięśni pośladkowych. Tę drogę podania można stosować wyłącznie w przypadkach, gdy podawanie doustne lub dożylne jest niemożliwe lub niezalecane.
Nie określono.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
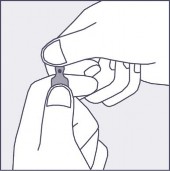
Aby otworzyć ampułkę, należy trzymać ją pionowo w obu dłoniach, kolorową kropką do siebie - patrz rysunek 2. Górną część ampułki należy uchwycić w taki sposób, aby kciuk znajdował się powyżej kolorowej kropki.
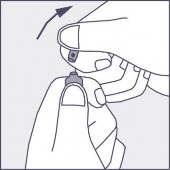
Nacisnąć zgodnie ze strzałką umieszczoną na rysunku 3.
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Glikol propylenowy Etanol 96% Alkohol benzylowy Sodu benzoesan
Kwas octowy lodowaty Kwas octowy 10%
Woda do wstrzykiwań
Produktu nie należy mieszać z innymi lekami w roztworze do wlewu lub w jednej strzykawce. Nie można zapewnić stabilności roztworu w przypadku nieprzestrzegania powyższego zalecenia.
3 lata
Przechowywać w temperaturze poniżej 25C.
Przechowywać ampułki w opakowaniu zewnętrznym w celu ochrony przed światłem. Nie zamrażać.
Ampułki ze szkła oranżowego lub szkła bezbarwnego w tekturowym pudełku 5 ampułek po 2 ml
50 ampułek po 2 ml
Zazwyczaj nie należy rozcieńczać produktu Relanium. Wyjątek stanowi podawanie produktu
w powolnym wlewie dożylnym w dużej objętości roztworu NaCl 0,9% lub glukozy w leczeniu tężca
i stanu padaczkowego. Nie należy rozcieńczać więcej niż 40 mg diazepamu (8 ml roztworu) w 500 ml roztworu do wlewu. Roztwór należy przygotowywać bezpośrednio przed podaniem i zużyć w ciągu
6 godzin.
Podawanie produktu we wstrzyknięciu dożylnym (bolusie) pozwala na dokładniejsze i szybsze dostosowanie dawkowania niż powolny wlew dożylny. Dlatego też jest to sposób podawania preferowany w leczeniu stanów ostrych.
Produktu nie należy mieszać z innymi lekami w roztworze do wlewu lub w jednej strzykawce. Nie można zapewnić stabilności roztworu w przypadku nieprzestrzegania powyższego zalecenia.
Ponad 50% roztworu diazepamu może ulec adsorpcji na ściankach plastikowych pojemników
z roztworem do wlewu; dlatego też nie należy stosować takich pojemników do podawania roztworów diazepamu. Adsorpcja na plastikowych rurkach zestawu do wlewu dożylnego może także powodować początkowe znaczne zmniejszenie stężenia podawanego diazepamu, które następnie powoli zwiększa się w ciągu kilku godzin. Prędkość wlewu należy często dostosowywać do aktualnego stanu pacjenta.
Instrukcja otwierania ampułki
Przed otwarciem ampułki należy upewnić się, że cały roztwór znajduje się w dolnej części ampułki. Można delikatnie potrząsnąć ampułką lub postukać w nią palcem, aby ułatwić spłynięcie roztworu.
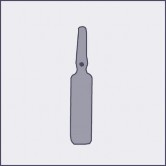
Na każdej ampułce umieszczono kolorową kropkę (patrz rysunek 1.) jako oznaczenie znajdującego się poniżej niej punktu nacięcia.
Ampułki są przeznaczone wyłącznie do jednorazowego użytku, należy je otwierać bezpośrednio przed użyciem. Pozostałą zawartość niezużytego produktu należy zniszczyć zgodnie z obowiązującymi przepisami.
Rysunek 1 Rysunek 2 Rysunek 3
Warszawskie Zakłady Farmaceutyczne Polfa S.A. ul. Karolkowa 22/24
01-207 Warszawa
Pozwolenie nr R/0937
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 10.11.1986 r. Data ostatniego przedłużenia pozwolenia: 11.10.2012 r.








