







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER(-Y) POZWOLENIA(Ń) NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
Lipegis, 10 mg, tabletki
Każda tabletka zawiera 10 mg ezetymibu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka.
Białe lub prawie białe, okrągłe, płaskie ze ściętymi krawędziami tabletki, ze stylizowanym E po jednej stronie i kodem „612” po drugiej stronie.
Pierwotna hipercholesterolemia
Lipegis, stosowany jednocześnie z inhibitorem reduktazy HMG-CoA (statyną), jest wskazany jako terapia wspomagająca dietę u chorych z pierwotną (heterozygotyczną rodzinną lub nierodzinną) hipercholesterolemią, u których sama statyna nie zapewnia odpowiedniej kontroli stężenia cholesterolu.
Lipegis stosowany w monoterapii zaleca się jako środek wspomagający dietę u pacjentów z pierwotną hipercholesterolemią (heterozygotyczną rodzinną lub nierodzinną), u których stosowanie statyny jest niewskazane bądź lek ten nie jest tolerowany.
Zapobieganie zdarzeniom sercowo-naczyniowym
Lipegis, dołączany do prowadzonej terapii statyną lub włączany jednocześnie ze statyną, jest wskazany w celu zmniejszania ryzyka zdarzeń sercowo-naczyniowych (patrz punkt 5.1) u pacjentów z chorobą wieńcową i z wywiadem ostrego zespołu wieńcowego.
Homozygotyczna hipercholesterolemia rodzinna (Homozygous Familial Hypercholesterolaemia; HoFH)
Lipegis stosowany jednocześnie ze statyną, jest wskazany jako terapia wspomagająca dietę u chorych z HoFH. U pacjentów można stosować również inne metody leczenia (np aferezę LDL).
U pacjentów należy zastosować odpowiednią dietę zmniejszającą stężenie lipidów we krwi i kontynuować ją podczas leczenia produktem Lipegis.
Dawkowanie
Zalecana dawka to jedna tabletka 10 mg leku Lipegis na dobę.
W przypadku stosowania produktu Lipegis w skojarzeniu ze statyną należy stosować zalecaną dawkę początkową lub kontynuować leczenie już ustaloną wyższą dawką danej statyny. Należy zapoznać się z zaleceniami dotyczącymi stosowania odpowiedniej statyny.
Stosowanie u pacjentów z chorobą wieńcową i wywiadem ostrego zespołu wieńcowego
W celu dodatkowego zmniejszenia występowania zdarzeń sercowo-naczyniowych u pacjentów z chorobą wieńcową i wywiadem ostrego zespołu wieńcowego Lipegis 10 mg można stosować łącznie ze statyną o udowodnionym korzystnym wpływie na zdarzenia sercowo-naczyniowe.
Jednoczesne stosowanie leków wiążących kwasy żółciowe
Lipegis należy przyjmować co najmniej 2 godziny przed lub co najmniej 4 godziny po podaniu leku wiążącego kwasy żółciowe.
Pacjenci w podeszłym wieku
Nie ma konieczności modyfikacji dawki u pacjentów w podeszłym wieku (patrz punkt 5.2).
Dzieci i młodzież
Rozpoczynanie leczenia musi odbywać się pod nadzorem specjalisty.
Dzieci i młodzież ≥6. roku życia: Nie ustalono bezpieczeństwa i skuteczności leku Lipegis u dzieci od
6. do 17. roku życia. Obecnie dostępne dane przedstawiono w punktach 4.4, 4.8, 5.1 i 5.2, ale sformułowanie zaleceń dotyczących dawkowania nie jest możliwe.
W przypadku stosowania leku Lipegis jednocześnie ze statyną należy zapoznać się z informacją dotyczącą dawkowania tej statyny u dzieci.
Dzieci w wieku <6 lat: Nie ustalono bezpieczeństwa i skuteczności ezetymibu u dzieci poniżej 6. roku życia. Żadne dane nie są dostępne.
Zaburzenia czynności wątroby
Nie ma konieczności modyfikacji dawki leku u pacjentów z łagodnym upośledzeniem czynności wątroby (od 5 do 6 punktów wg skali Child-Pugh). Nie zaleca się stosowania produktu Lipegis u pacjentów z umiarkowaną (od 7 do 9 punktów wg skali Child-Pugh) lub ciężką (>9 punktów wg skali Child-Pugh) niewydolnością wątroby (patrz punkty 4.4 i 5.2).
Zaburzenia czynności nerek
Nie ma konieczności modyfikacji dawki u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2).
Sposób podawania
Preparat jest przeznaczony do stosowania doustnego. Lipegis może być stosowany o dowolnej porze dnia, z posiłkiem lub bez.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
W przypadku stosowania produktu Lipegis jednocześnie ze statyną należy zapoznać się z Charakterystyką Produktu Leczniczego danego leku.
Leczenie produktem Lipegis w połączeniu ze statyną jest przeciwwskazane u kobiet w ciąży i karmiących piersią.
Stosowanie produktu Lipegis jednocześnie ze statyną jest przeciwwskazane u pacjentów z czynną chorobą wątroby lub niewyjaśnionym trwałym zwiększeniem stężenia aminotransferaz w surowicy.
W przypadku stosowania produktu Lipegis w połączeniu ze statyną należy zapoznać się z Charakterystyką Produktu Leczniczego danego leku.
Enzymy wątrobowe
W kontrolowanych badaniach klinicznych przeprowadzonych u pacjentów przyjmujących ezetymib w skojarzeniu ze statyną, obserwowano zwiększone stężenie aminotransferaz (co najmniej 3-krotnie przekroczona górna granica normy). W przypadku stosowania leku Lipegis w skojarzeniu ze statyną zaleca się przeprowadzenie testów czynnościowych wątroby na początku leczenia, w sposób zgodny z zaleceniami dotyczącymi stosowania statyny (patrz punkt 4.8).
W badaniu IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) 18144 pacjentów z chorobą wieńcową, po przebytym ostrym zespole wieńcowym, zostało losowo rozdzielonych do grupy otrzymującej ezetymib/symwastatynę 10/40 mg dziennie (n = 9067) lub do grupy otrzymującej symwastatynę 40 mg dziennie (n = 9077). W okresie obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania zwiększonej aktywności transaminaz (≥ 3x górna granica normy) po włączeniu leczenia wyniosła 2,5% w grupie ezetymibu/symwastatyny i 2,3% w grupie symwastatyny (patrz punkt 4.8).
W kontrolowanym badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało losowo rozdzielonych albo do grupy leczonej ezetymibem 10 mg w połączeniu z symwastatyną 20 mg dziennie (n = 4650), albo do grupy otrzymującej placebo (n = 4620) (mediana okresu obserwacji 4,9 lat) częstość występowania nowych przypadków zwiększenia aktywności transaminaz (> 3 razy ponad górną granicę normy) wynosiła 0,7% w grupie leczenia skojarzonego ezetymibem i symwastatyną w porównaniu z 0,6% w grupie placebo (patrz punkt 4.8).
Mięśnie szkieletowe
Po wprowadzeniu ezetymibu do obrotu odnotowano przypadki wystąpienia miopatii oraz rabdomiolizy. Większość pacjentów, u których stwierdzono rabdomiolizę, przyjmowało statynę w skojarzeniu z ezetymibem. Rabdomiolizę odnotowywano jednak bardzo rzadko w przypadku stosowania ezetymibu w monoterapii, a także podczas stosowania ezetymibu jednocześnie z innymi środkami o znanym działaniu zwiększającym ryzyko jej wystąpienia. Jeżeli istnieje podejrzenie miopatii oparte na objawach ze strony mięśni, bądź jest ona potwierdzona przez wynik badania stężenia fosfokinazy kreatynowej (CPK, ang. creatine phosphokinase), w którym górna granica normy przekroczona jest ponad 10-krotnie, należy natychmiast przerwać leczenie ezetymibem, jakąkolwiek statyną oraz innymi środkami stosowanymi jednocześnie z tymi lekami. Podczas rozpoczynania terapii z zastosowaniem ezetymibu należy poinformować pacjentów o ryzyku wystąpienia miopatii oraz konieczności natychmiastowego zgłaszania jakiegokolwiek niewyjaśnionego bólu mięśni, ich tkliwości lub osłabienia (patrz punkt 4.8).
W badaniu IMPROVE-IT 18 144 pacjentów z chorobą wieńcową, po przebytym ostrym zespole wieńcowym, zostało losowo rozdzielonych do grupy otrzymującej ezetymib/symwastatynę 10 mg/40 mg dziennie (n = 9067) lub do grupy otrzymującej symwastatynę 40 mg dziennie (n = 9077).
W okresie obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania miopatii wyniosła 0,2% w grupie ezetymibu/symwastatyny i 0,1% w grupie symwastatyny, przy czym miopatię zdefiniowano jako niewyjaśnione osłabienie lub ból mięśni ze zwiększeniem stężenia CK w surowicy
≥10 razy ponad górną granicę normy lub jako stwierdzenie w 2 kolejnych pomiarach zwiększenia stężenia CK ≥ 5 i <10 razy ponad górną granicę normy. Częstość występowania rabdomiolizy wyniosła 0,1% w grupie ezetymibu/symwastatyny i 0,2% w grupie symwastatyny, przy czym rabdomiolizę zdefiniowano jako niewyjaśnione osłabienie lub ból mięśni ze zwiększeniem stężenia CK ≥10 razy ponad górną granicę normy i cechami uszkodzenia nerek, zwiększenie stężenia CK w 2 kolejnych pomiarach ≥ 5 razy i < 10 razy ponad górną granicę normy z cechami uszkodzenia nerek lub zwiększenie stężenia CK ≥ 10 000 j.m./l bez cech uszkodzenia nerek (patrz punkt 4.8).
W badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało losowo rozdzielonych albo do grupy leczonej ezetymibem 10 mg w połączeniu z symwastatyną 20 mg dziennie (n = 4650), albo do grupy otrzymującej placebo (n = 4620) (mediana okresu obserwacji 4,9 lat) częstość występowania nowych przypadków miopatii/rabdomiolizy wynosiła 0,2% w grupie leczenia skojarzonego ezetymibem i symwastatyną w porównaniu z 0,1% w grupie placebo (patrz punkt 4.8).
Zaburzenia czynności wątroby
Brak danych dotyczących skutków zwiększonej ekspozycji na ezetymib u osób z umiarkowanym lub ciężkim upośledzeniem czynności wątroby, nie zaleca się stosowania produktu leczniczego Lipegis w tej grupie pacjentów (patrz punkt 5.2).
Dzieci i młodzież
Skuteczność i bezpieczeństwo ezetymibu u pacjentów w wieku od 6 do 10 lat z heterozygotyczną hipercholesterolemią rodzinną i nierodzinną oceniono w 12-tygodniowym kontrolowanym placebo badaniu klinicznym. W tej grupie wiekowej nie badano efektów stosowania ezetymibu w okresach dłuższych niż 12 tygodni (patrz punkty 4.2, 4.8, 5.1 i 5.2).
Nie badano ezetymibu u pacjentów poniżej 6. roku życia (patrz punkty 4.2 i 4.8).
Skuteczność i bezpieczeństwo ezetymibu podawanego jednocześnie z symwastatyną u pacjentów z heterozygotyczną hipercholesterolemią rodzinną w wieku od 10 do 17 lat oceniano w kontrolowanym badaniu klinicznym obejmującym dojrzewających chłopców (faza II lub wyższa wg Tannera) i dziewczynki będące co najmniej rok po pierwszej miesiączce.
W tym ograniczonym kontrolowanym badaniu klinicznym nie stwierdzono wykrywalnego
wpływu leku na wzrost lub dojrzewanie płciowe u dojrzewających chłopców i dziewcząt ani żadnego wpływu na długość cyklu menstruacyjnego u dziewcząt. Nie badano jednak wpływu ezetymibu na wzrost i dojrzewanie płciowe w okresie leczenia > 33 tygodni (patrz punkty 4.2 i 4.8).
Nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z symwastatyną w dawkach powyżej 40 mg dziennie u młodzieży w wieku od 10 do 17 lat.
Nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z symwastatyną u pacjentów pediatrycznych w wieku <10 lat (patrz punkty 4.2 i 4.8).
Nie badano czy długotrwałe leczenie ezetymibem pacjentów poniżej 17 roku życia wpływa na redukcję chorobowości i śmiertelności w wieku dorosłym.
Fibraty
Nie określono bezpieczeństwa i skuteczności działania ezetymibu stosowanego jednocześnie z fibratami.
Jeżeli u pacjentów przyjmujących Lipegis i fenofibrat istnieje podejrzenie kamicy żółciowej, zalecane jest przeprowadzenie badań pęcherzyka żółciowego oraz przerwanie leczenia (patrz punkty 4.5 i 4.8).
Cyklosporyna
Należy zachować ostrożność na początku stosowania preparatu Lipegis u pacjentów przyjmujących cyklosporynę. Należy monitorować stężenie cyklosporyny u pacjentów przyjmujących Lipegis oraz cyklosporynę (patrz punkt 4.5).
Leki przeciwzakrzepowe
W przypadku stosowania produktu Lipegis jednocześnie z warfaryną, innym lekiem przeciwzakrzepowym z grupy pochodnych kumaryny lub fluindionem, zaleca się monitorowanie wartości Międzynarodowego Współczynnika Znormalizowanego (INR) (patrz punkt 4.5).
Sód
Lek zawiera mniej niż 1 mmol (23 mg) sodu na tabletkę, to znaczy lek uznaje się za „wolny od sodu”.
W badaniach przedklinicznych wykazano, iż ezetymib nie indukuje metabolizujących leki enzymów cytochromu 450. Nie obserwowano klinicznie istotnych interakcji farmakokinetycznych pomiędzy ezetymibem oraz lekami metabolizowanymi przez enzymy cytochromu P450 1A2, 2D6, 2C8, 2C9 i 3A4 lub N-acetylotransferazę.
W badaniach dotyczących interakcji klinicznych stwierdzono brak wpływu ezetymibu na farmakokinetykę dapsonu, dekstrometorfanu, digoksyny, doustnych środków antykoncepcyjnych (etynyloestradiol i lewonogestrel), glipizydu, tolbutamidu lub midazolamu podczas jednoczesnego stosowania tych leków. Cymetydyna podawana jednocześnie z ezetymibem nie miała wpływu na jego biodostępność.
Leki zobojętniające kwas solny
Jednoczesne stosowanie leków zobojętniających kwas solny zmniejsza szybkość wchłaniania ezetymibu, jednak nie ma wpływu na jego biodostępność. Zmniejszona szybkość wchłaniania nie jest uważana za klinicznie istotną.
Cholestyramina
Jednoczesne stosowanie cholestyraminy zmniejsza średnią wartość pola pod krzywą (AUC) dla całkowitego ezetymibu (substancja macierzysta + glukuronian ezetymibu) o ok. 55 %. W wyniku tej interakcji redukcja stężenia cholesterolu LDL po jednoczesnym zastosowaniu ezetymibu i cholestyraminy może ulec osłabieniu (patrz punkt 4.2).
Fibraty
Należy wziąć pod uwagę ryzyko wystąpienia kamicy żółciowej lub choroby pęcherzyka żółciowego u pacjentów przyjmujących fenofibrat i Lipegis (patrz punkty 4.4 i 4.8).
Jeżeli u pacjentów przyjmujących Lipegis i fenofibrat istnieje podejrzenie kamicy żółciowej, zalecane jest przeprowadzenie badań pęcherzyka żółciowego oraz przerwanie leczenia (patrz punkt 4.8).
Jednoczesne stosowanie fenofibratu lub gemfibrozylu nieznacznie zwiększa całkowite stężenie ezetymibu (odpowiednio ok. 1,5 i 1,7 razy).
Nie przeprowadzono badań dotyczących jednoczesnego stosowania ezetymibu z innymi fibratami. Fibraty mogą zwiększać wydzielanie cholesterolu do żółci, co prowadzi do kamicy żółciowej.
W badaniach przeprowadzonych na zwierzętach ezetymib zwiększał stężenie cholesterolu w żółci
zawartej w pęcherzyku żółciowym, jednak nie u wszystkich gatunków zwierząt (patrz punkt 5.3). Nie można wykluczyć ryzyka litogennego działania ezetymibu.
Statyny
Nie obserwowano klinicznie istotnych interakcji farmakokinetycznych podczas jednoczesnego stosowania ezetymibu z atorwastatyną, symwastatyną, prawastatyną, lowastatyną, fluwastatyną lub rozuwastatyną.
Cyklosporyna
W badaniu przeprowadzonym u ośmiu pacjentów po przeszczepie nerki z klirensem kreatyniny powyżej 50 ml/min, otrzymujących stałą dawkę cyklosporyny, po podaniu pojedynczej dawki 10 mg ezetymibu stwierdzono 3,4-krotne (w zakresie 2,3 - 7,9 razy) zwiększenie średniej wartości AUC całkowitego ezetymibu w porównaniu z kontrolną populacją zdrowych pacjentów z innego badania (n = 17), otrzymujących ezetymib w monoterapii. W kolejnym badaniu, u pacjenta po przeszczepie nerki, z ciężkim upośledzeniem czynności nerek, otrzymującego cyklosporynę i wiele innych leków, stwierdzono 12-krotnie większą ekspozycję na całkowity ezetymib w porównaniu z pacjentami z grupy kontrolnej, otrzymującymi ezetymib w monoterapii. W dwuokresowym badaniu
skrzyżowanym, przeprowadzonym z udziałem 12 zdrowych ochotników, stosowanie dawki dobowej 20 mg ezetymibu przez 8 dni oraz pojedynczej dawki 100 mg cyklosporyny w 7. dniu leczenia powodowało zwiększenie wartości AUC dla cyklosporyny średnio o 15% (zakres od 10% zmniejszenia do 51% zwiększenia), w porównaniu z podaniem wyłącznie pojedynczej dawki 100 mg cyklosporyny. Nie przeprowadzono kontrolowanego badania dotyczącego skutków jednoczesnego stosowania ezetymibu i cyklosporyny u pacjentów poddanych przeszczepowi nerki. Należy zachować ostrożność na początku stosowania produktu Lipegis u pacjentów przyjmujących cyklosporynę.
Należy monitorować stężenie cyklosporyny u pacjentów przyjmujących Lipegis oraz cyklosporynę (patrz punkt 4.4).
Leki przeciwzakrzepowe
Jednoczesne stosowanie ezetymibu (w dawce 10 mg raz na dobę) nie wykazało istotnego wpływu na biodostępność warfaryny oraz czas protrombinowy w badaniu przeprowadzonym z udziałem 12 zdrowych mężczyzn. Odnotowano jednak zwiększenie Międzynarodowego Współczynnika Znormalizowanego (INR) u pacjentów przyjmujących ezetymib jednocześnie z warfaryną lub fluindionem. W przypadku stosowania produktu Lipegis w połączeniu z warfaryną, innym kumarynowym środkiem przeciwzakrzepowym lub fluindionem zaleca się monitorowanie wartości INR (patrz punkt 4.4).
Dzieci i młodzież
Badania dotyczące interakcji przeprowadzono wyłącznie u dorosłych pacjentów.
Stosowanie leku Lipegis w połączeniu ze statyną jest przeciwwskazane w okresie ciąży i karmienia piersią (patrz punkt 4.3). Należy zapoznać się z Charakterystyką Produktu Leczniczego odpowiedniej statyny.
Ciąża
Lipegis może być podawany kobietom w ciąży jedynie w przypadku bezwzględnej konieczności. Brak danych klinicznych dotyczących stosowania ezetymibu w czasie ciąży. Badania przeprowadzone na zwierzętach nie wykazały bezpośredniego bądź pośredniego szkodliwego wpływu ezetymibu stosowanego w monoterapii na przebieg ciąży, rozwój zarodka i (lub) płodu, przebieg porodu lub rozwój noworodka (patrz punkt 5.3).
Karmienie piersią
Nie należy stosować leku Lipegis w okresie karmienia piersią. Stwierdzono, iż ezetymib jest wydzielany do mleka karmiących szczurów. Nie wiadomo, czy ezetymib jest wydzielany do mleka kobiecego.
Płodność
Nie ma dostępnych żadnych danych z badań klinicznych dotyczących wpływu ezetymibu na płodność u ludzi. Ezetymib nie miał wpływu na płodność samców i samic szczura (patrz punkt 5.3).
Nie przeprowadzono badań dotyczących wpływu preparatu na zdolność prowadzenia pojazdów i obsługiwania maszyn. Odnotowano jednak przypadki wystąpienia zawrotów głowy w trakcie stosowania leku, co należy wziąć pod uwagę podczas prowadzenia pojazdów lub obsługiwania urządzeń mechanicznych.
Tabelaryczne zestawienie reakcji niepożądanych (badania kliniczne i doświadczenie po wprowadzeniu leku do obrotu)
W badaniach klinicznych trwających do 112 tygodni, 2396 pacjentom podawano ezetymib w dawce 10 mg na dobę w monoterapii, 11 308 pacjentom w połączeniu ze statyną oraz 185 pacjentom w połączeniu z fenofibratem. Działania niepożądane były zwykle łagodne i przemijające. Całkowita częstość występowania działań niepożądanych była podobna w grupach pacjentów przyjmujących ezetymib i w grupach przyjmujących placebo. Podobnie, liczba pacjentów, którzy przerwali leczenie z powodu działań niepożądanych była porównywalna w grupie przyjmującej ezetymib i placebo.
Ezetymib stosowany w monoterapii lub w połączeniu ze statyną
Niżej wymienione działania niepożądane obserwowano u pacjentów leczonych ezetymibem (n = 2396) z częstością większą niż podczas stosowania placebo (n = 1159) jak również
u pacjentów leczonych ezetymibem w połączeniu ze statyną (n = 11308) z częstością większą
niż podczas stosowania samej statyny (n = 9361). Po wprowadzeniu do obrotu informacje na temat reakcji niepożądanych uzyskiwano z doniesień dotyczących stosowania samego ezetymibu lub ezetymibu w połączeniu ze statyną. Działania niepożądane obserwowane w badaniach klinicznych ezetymibu (w monoterapii lub w skojarzeniu ze statyną) lub zgłoszone po wprowadzeniu ezetymibu do obrotu, w monoterapii lub w skojarzeniu ze statyną wymieniono w Tabeli 1. Objawy te przedstawiono według klasyfikacji układów i narządów oraz częstości występowania.
Częstość występowania działań niepożądanych określono następująco: bardzo często (>1/10), często (>1/100 do <1/10), niezbyt często (>1/1000 do <1/100), rzadko (>1/10 000 do <1/1000), bardzo rzadko (<1/10 000) i częstość nieznana (nie może być ustalona na podstawie dostępnych danych).
Tabela 1 Działania niepożądane
Klasyfikacja narządów i układów Częstość występowania | Działanie niepożądane |
Zaburzenia krwi i układu chłonnego | |
Częstość nieznana | małopłytkowość |
Zaburzenia układu immunologicznego | |
Częstość nieznana | nadwrażliwość, w tym wysypka; pokrzywka; wstrząs anafilaktyczny i obrzęk naczynioruchowy |
Zaburzenia metabolizmu i odżywiania | |
Niezbyt często | zmniejszony apetyt |
Zaburzenia psychiczne | |
Częstość nieznana | Depresja |
Zaburzenia układu nerwowego | |
Często | ból głowy |
Niezbyt często | Parestezje |
Częstość nieznana | zawroty głowy |
Zaburzenia naczyniowe | |
Niezbyt często | uderzenia gorąca; nadciśnienie |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Niezbyt często | Kaszel |
Częstość nieznana | Duszność |
Zaburzenia żołądka i jelit | |
Często | ból brzucha; biegunka; wzdęcia |
Niezbyt często | dyspepsja; refluks żołądkowo-przełykowy; nudności; suchość w jamie ustnej; zapalenie błony śluzowej żołądka |
Częstość nieznana | zapalenie trzustki; zaparcia |
Zaburzenia wątroby i dróg żółciowych | |
Częstość nieznana | zapalenie wątroby; kamica żółciowa; zapalenie pęcherzyka żółciowego |
Zaburzenia skóry i tkanki podskórnej | |
Niezbyt często | świąd; wysypka; pokrzywka |
Częstość nieznana | rumień wielopostaciowy |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często | ból mięśni |
Niezbyt często | ból stawów; skurcze mięśni; ból szyi; ból pleców, osłabienie mięśni; ból kończyn |
Częstość nieznana | miopatia/rabdomioliza (patrz punkt 4.4) |
Zaburzenia ogólne i stany w miejscu podania | |
Często | Zmęczenie |
Niezbyt często | ból w klatce piersiowej; ból; osłabienie; obrzęk obwodowy |
Badania diagnostyczne | |
Często | zwiększenie aktywności ALAT i/lub ASPAT |
Niezbyt często | zwiększenie aktywności CPK we krwi; zwiększenie aktywności gammaglutamylotransferazy; nieprawidłowe wyniki testów czynności wątroby |
Stosowanie ezetymibu w połączeniu z fenofibratem:
Zaburzenia żołądka i jelit: ból brzucha (często).
W wieloośrodkowym, podwójnie zaślepionym, kontrolowanym placebo badaniu klinicznym, przeprowadzonym u pacjentów z mieszaną hiperlipidemią, 625 pacjentów leczonych było przez okres do 12 tygodni, a 576 pacjentów do 1 roku. Spośród pacjentów otrzymujących ezetymib w połączeniu z fenofibratem, 172 ukończyło leczenie trwające 12 tygodni, zaś 230 (w tym 109 otrzymujących ezetymib w monoterapii w ciągu pierwszych 12 tygodni) ukończyło leczenie trwające 1 rok.
Badanie nie miało na celu porównania badanych grup pod względem występowania rzadkich działań niepożądanych. Wskaźniki występowania (przedział ufności 95%) klinicznie istotnego zwiększenia (> 3 razy powyżej górnego zakresu wartości prawidłowych, kolejno) aminotransferaz w surowicy wynosiły 4,5% (1,9; 8,8) oraz 2,7% (1,2; 5,4), odpowiednio dla fenofibratu w monoterapii oraz ezetymibu w skojarzeniu z fenofibratem, w dawkach dostosowanych do odpowiedzi na leczenie.
Częstość cholecystektomii wynosiła 0,6% (0,0; 3,1) oraz 1,7% (0,6; 4,0), odpowiednio dla fenofibratu w monoterapii oraz ezetymibu w połączeniu z fenofibratem (patrz punkty 4.4 i 4.5).
Dzieci i młodzież (od 6 do 17 lat)
W badaniu obejmującym pacjentów pediatrycznych (w wieku od 6 do 10 lat) z heterozygotyczną hipercholesterolemią rodzinną i nierodzinną (n = 138) zwiększenie aktywności ALAT i/lub ASPAT (≥ 3 x górna granica normy) obserwowano u 1,1% (1 pacjent) pacjentów leczonych ezetymibem w porównaniu z 0% w grupie placebo. Nie stwierdzano zwiększenia aktywności CPK (≥ 10 x górna granica normy). Nie opisywano przypadków miopatii.
W oddzielnym badaniu obejmującym młodocianych pacjentów (od 10 do 17 lat) z heterozygotyczną hipercholesterolemią rodzinną (n = 248) wzrost ALAT i/lub ASPAT (≥3 x górna granica normy) obserwowano u 3% (4 pacjentów) w grupie ezetymibu/symwastatyny w porównaniu z 2% (2 pacjentów) w grupie monoterapii symwastatyną; w odniesieniu do wzrostu CPK
(≥ 10 x górna granica normy) analogiczne odsetki wynosiły odpowiednio 2% i 0%. Nie odnotowano żadnego przypadku miopatii.
Badania te nie były zaplanowane do porównania rzadkich reakcji niepożądanych. Pacjenci z chorobą wieńcową i wywiadem ostrego zespołu wieńcowego
W badaniu IMPROVE-IT (patrz punkt 5.1) z udziałem 18144 pacjentów leczonych albo ezetymibem/symwastatyną 10 mg/40 mg (n = 9067; u 6% z nich dawkę ezetymibu/symwastatyny zwiększono do 10 mg/80 mg), albo symwastatyną 40 mg (n = 9077; u 27% dawkę symwastatyny
zwiększono do 80 mg) profile bezpieczeństwa leków w okresie obserwacji, którego mediana wynosiła 6,0 lat, były podobne. Częstość odstawiania leku z powodu zdarzeń niepożądanych wyniosła 10,6% w grupie leczonej ezetymibem/symwastatyną i 10,1% w grupie leczonej symwastatyną. Częstość występowania miopatii wyniosła 0,2% w przypadku ezetymibu/symwastatyny i 0,1% w przypadku symwastatyny, przy czym miopatię zdefiniowano jako niewyjaśnione osłabienie lub ból mięśni ze zwiększeniem stężenia CK w surowicy ≥10 razy ponad górną granicę normy lub jako stwierdzenie w 2 kolejnych pomiarach zwiększenia stężenia CK ≥ 5 i <10 razy ponad górną granicę normy. Częstość występowania rabdomiolizy wyniosła 0,1% w grupie ezetymibu/symwastatyny i 0,2% w grupie symwastatyny, przy czym rabdomiolizę zdefiniowano jako niewyjaśnione osłabienie lub ból mięśni ze zwiększeniem stężenia CK ≥10 razy ponad górną granicę normy i cechami uszkodzenia nerek, zwiększenie stężenia CK w 2 kolejnych pomiarach ≥5 razy i <10 razy ponad górną granicę normy z cechami uszkodzenia nerek lub zwiększenie stężenia CK ≥10 000 j.m./l bez cech uszkodzenia nerek. Częstość występowania zwiększonej aktywności transaminaz po włączeniu leczenia (≥3 razy ponad górną granicę normy) wyniosła 2,5% w grupie ezetymibu/symwastatyny i 2,3% w grupie symwastatyny (patrz punkt 4.4). Objawy niepożądane związane z pęcherzykiem żółciowym
zgłaszało 3,1% pacjentów przydzielonych do leczenia ezetymibem/symwastatyną i 3,5% pacjentów przydzielonych do leczenia symwastatyną. Częstość hospitalizacji związanej z cholecystektomią wyniosła 1,5% w obu grupach badania. Nowotwór (zdefiniowany jako każdy nowy przypadek nowotworu złośliwego) zdiagnozowano w trakcie badania odpowiednio u 9,4% i 9,5% pacjentów.
Pacjenci z przewlekłą chorobą nerek
W badaniu Study of Heart and Renal Protection (SHARP) (patrz punkt 5.1) obejmującym ponad 9000 pacjentów otrzymujących codziennie albo preparat złożony zawierający 10 mg ezetymibu i 20 mg symwastatyny (n = 4650), albo placebo (n = 4620) profile bezpieczeństwa były porównywalne w okresie obserwacji o medianie 4,9 lat. W badaniu tym rejestrowano jedynie ciężkie zdarzenia niepożądane i przypadki przerywania leczenia z powodu jakichkolwiek zdarzeń niepożądanych.
Wskaźniki przerywania leczenia z powodu zdarzeń niepożądanych były porównywalne (10,4% u pacjentów leczonych ezetymibem w skojarzeniu z symwastatyną, 9,8% u pacjentów otrzymujących placebo). Częstość występowania miopatii/rabdomiolizy wynosiła 0,2% u pacjentów leczonych ezetymibem w skojarzeniu z symwastatyną i 0,1% u pacjentów otrzymujących placebo. Do zwiększenia aktywności transaminaz (>3 x górna granica normy) doszło u 0,7% pacjentów leczonych ezetymibem w połączeniu z symwastatyną w porównaniu z 0,6% pacjentów otrzymujących placebo (patrz punkt 4.4). W tym badaniu nie stwierdzono istotnego statystycznie zwiększenia częstości występowania określonych wcześniej zdarzeń niepożądanych, takich jak nowotwory (9,4% w grupie ezetyminu z symwastatyną, 9,5% w grupie placebo), zapalenie wątroby, cholecystektomia lub powikłania kamicy żółciowej czy zapalenie trzustki.
Wpływ na wyniki badań diagnostycznych
W kontrolowanych badaniach klinicznych, w których ezetymib stosowano w monoterapii, częstość występowania klinicznie istotnego zwiększenia aminotransferaz w surowicy (AlAT i (lub) AspAT
≥3 razy powyżej górnego zakresu wartości prawidłowych, kolejno) była zbliżona w grupach pacjentów przyjmujących ezetymib (0,5%) oraz placebo (0,3%). W badaniach, w których produkty lecznicze stosowano w skojarzeniu, wzrost aminotransferaz w surowicy wystąpił u 1,3% pacjentów
w grupie przyjmującej ezetymib w skojarzeniu ze statyną oraz 0,4% pacjentów w grupie przyjmującej wyłącznie statynę. Zwiększenia aktywności enzymów przebiegało zwykle bez objawów, nie było związane z cholestazą i powracało do początkowych wartości po zakończeniu leczenia lub w trakcie jego trwania (patrz punkt 4.4).
W badaniach klinicznych obserwowano ponad 10-krotne zwiększenie stężenia fosfokinazy kreatynowej (CPK) w stosunku do górnej granicy wartości prawidłowych u 4 z 1 674 (0,2%) pacjentów przyjmujących ezetymib w monoterapii w porównaniu z 1 z 786 (0,1%) pacjentów przyjmujących placebo, a także u 1 z 917 (0,1%) pacjentów przyjmujących ezetymib w połączeniu ze statyną w porównaniu z 4 z 929 (0,4%) pacjentów przyjmujących statynę w monoterapii. Nie stwierdzono zwiększenia liczby przypadków miopatii lub rabdomiolizy związanych ze stosowaniem ezetymibu w porównaniu z grupą kontrolną (pacjenci przyjmujący placebo lub statynę w monoterapii) (patrz punkt 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem:
Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych
Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C
02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W badaniach klinicznych, w których dawkę dobową 50 mg ezetymibu podawano 15 zdrowym ochotnikom w okresie do 14 dni, bądź też dawkę dobową 40 mg ezetymibu podawano 18 pacjentom z pierwotną hipercholesterolemią w okresie do 56 dni, ezetymib był zazwyczaj dobrze tolerowany.
W badaniach przeprowadzonych na zwierzętach nie obserwowano działania toksycznego po przyjęciu ezetymibu w postaci pojedynczej dawki doustnej 5 000 mg/kg u szczurów i myszy oraz 3 000 mg/kg u psów.
Odnotowano kilka przypadków przedawkowania ezetymibu: większość z nich nie była związana z występowaniem działań niepożądanych. Odnotowane działania niepożądane nie były poważne.
W przypadku przedawkowania leku należy zastosować leczenie objawowe i wspomagające.
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Grupa farmakoterapeutyczna: Inne środki modyfikujące stężenie lipidów, kod ATC: C10A X09 Mechanizm działania
Ezetymib należy do nowej grupy leków zmniejszających stężenie lipidów, które selektywnie hamują
wchłanianie cholesterolu i pokrewnych steroli roślinnych w jelitach. Ezetymib działa po podaniu doustnym. Mechanizm jego działania różni się od innych grup leków zmniejszających stężenie cholesterolu (np. statyn, leków wiążących kwasy żółciowe (żywice), pochodnych kwasu fibrynowego, stanoli roślinnych). Celem działania ezetymibu na poziomie molekularnym jest nośnik steroli, białko Niemann-Pick C1-Like 1 (NPC1L1), które odgrywa rolę w wychwytywaniu cholesterolu i fitosteroli w jelicie.
Ezetymib wiąże się z rąbkiem szczoteczkowym jelita cienkiego i hamuje wchłanianie cholesterolu, zmniejszając ilość cholesterolu transportowanego z jelit do wątroby. W trwających 2 tygodnie badaniach klinicznych, przeprowadzonych u 18 pacjentów z hipercholesterolemią, ezetymib hamował wchłanianie cholesterolu z jelit o 54% w porównaniu z placebo.
Właściwości farmakodynamiczne
Przeprowadzono szereg badań przedklinicznych, aby określić czy działanie ezetymibu hamujące wchłanianie cholesterolu jest wybiórcze. Ezetymib hamował wchłanianie cholesterolu znakowanego izotopem węgla C14, nie miał za to wpływu na wchłanianie trójglicerydów, kwasów tłuszczowych, kwasów żółciowych, progesteronu, etynyloestradiolu lub rozpuszczalnych w tłuszczach
witamin A i D.
Badania epidemiologiczne wykazały, że zapadalność i umieralność z powodu chorób układu krążenia zmieniają się proporcjonalnie do stężenia cholesterolu całkowitego i cholesterolu LDL, a odwrotnie proporcjonalnie do stężenia cholesterolu HDL.
Podawanie ezetymibu razem ze statyną skutecznie zmniejsza ryzyko zdarzeń sercowo-naczyniowych u pacjentów z chorobą wieńcową i wywiadem ostrego zespołu wieńcowego.
Kliniczna skuteczność i bezpieczeństwo
W kontrolowanych badaniach klinicznych ezetymib stosowany w monoterapii znacząco zmniejszał stężenie cholesterolu całkowitego, cholesterolu LDL, apolipoproteiny B (Apo B) oraz trójglicerydów, a także zwiększał stężenie cholesterolu HDL u pacjentów z hipercholesterolemią.
Hipercholesterolemia pierwotna
W trwającym 8 tygodni badaniu klinicznym z podwójnie ślepą próbą, kontrolowanym placebo, wzięło udział 769 pacjentów z hipercholesterolemią, którzy przyjmowali statynę w monoterapii i u których nie osiągnięto docelowego stężenia cholesterolu LDL (2,6 do 4,1 mmol/l [100 do 160 mg/dl], w zależności od stężenia początkowego) wg National Cholesterol Education Program (NCEP).
Pacjentów losowo dobrano do grup przyjmujących ezetymib w dawce 10 mg lub placebo, w połączeniu ze stosowaną już statyną.
Wśród pacjentów leczonych statyną, u których nie osiągnięto docelowego stężenia cholesterolu LDL (ok. 82%), znamiennie więcej pacjentów przyjmujących ezetymib uzyskało docelowe stężenie cholesterolu LDL na zakończenie okresu obserwacji niż pacjentów przydzielonych losowo do grupy placebo, odpowiednio 72 % i 19 %. Znaczne różnice obserwowano również w redukcji stężenia cholesterolu LDL (25% i 4%, odpowiednio u pacjentów przyjmujących ezetymib i placebo). Ponadto dołączenie ezetymibu do leczenia statyną znacząco zmniejszało stężenie cholesterolu całkowitego, apolipoproteiny B i trójglicerydów, a także zwiększało stężenie cholesterolu HDL, w porównaniu z grupą pacjentów otrzymujących placebo. Dołączenie do leczenia statyną ezetymibu lub placebo zmniejszało medianę stężenia białka C-reaktywnego odpowiednio o 10% lub 0%, w porównaniu z wartościami początkowymi.
W dwóch randomizowanych badaniach klinicznych z podwójnie ślepą próbą, kontrolowanych placebo, prowadzonych przez 12 tygodni u 1 719 pacjentów z hipercholesterolemią pierwotną, obserwowano znaczące zmniejszenie stężenia cholesterolu całkowitego (o 13%), cholesterolu LDL (o 19%), apolipoproteiny B (o 14%) oraz trójglicerydów (o 8%), a także zwiększenie stężenia cholesterolu HDL (o 3%) po zastosowaniu ezetymibu w dawce 10 mg/dobę, w porównaniu z grupą placebo. Ponadto ezetymib nie miał wpływu na osoczowe stężenie rozpuszczalnych w tłuszczach witamin A, D i E oraz na czas protrombinowy, a także, podobnie jak inne środki zmniejszające stężenie lipidów, nie zaburzał wytwarzania hormonów sterydowych kory nadnerczy.
W wieloośrodkowym, podwójnie ślepym, kontrolowanym badaniu klinicznym (ENHANCE) 720 pacjentów z heterozygotyczną hipercholesterolemią rodzinną podzielono losowo do leczenia ezetymibem 10 mg w połączeniu z symwastatyną 80 mg (n = 357) lub samą symwastatyną 80 mg (n = 363) przez 2 lata. Głównym celem tego badania była ocena wpływu terapii skojarzonej ezetymibem/symwastatyną na grubość kompleksu blaszki wewnętrznej i środkowej tętnicy szyjnej (intima-media thickness; IMT) w porównaniu z monoterapią symwastatyną. Znaczenie tego markera zastępczego dla chorobowości i śmiertelności sercowo-naczyniowej wciąż nie zostało potwierdzone.
Główny punkt końcowy, zmiana średniej IMT wszystkich 6 segmentów tętnicy szyjnej, oceniana na podstawie badania USG w prezentacji B, nie różniła się istotnie (p = 0,29) między dwoma ocenianymi grupami. W czasie dwuletniej obserwacji w grupie ezetymibu 10 mg w połączeniu z symwastatyną 80 mg grubość kompleksu blaszki wewnętrznej i środkowej zwiększyła się o 0,0111 mm zaś w grupie samej symwastatyny 80 mg o 0,0058 mm (wyjściowa średnia IMT tętnicy szyjnej wynosiła w tych grupach odpowiednio 0,68 mm i 0,69 mm).
Ezetymib 10 mg w połączeniu z symwastatyną 80 mg zmniejszał stężenie cholesterolu LDL, cholesterolu całkowitego, Apo B i trójglicerydów istotnie silniej niż symwastatyna 80 mg. Procentowy wzrost stężenia cholesterolu HDL był podobny w obu badanych grupach. Działania niepożądane opisywane w przypadku ezetymibu 10 mg w połączeniu z symwastatyną 80 mg pokrywały się z dotychczas poznanym profilem bezpieczeństwa tego leku.
Dzieci i młodzież
W wieloośrodkowym, podwójnie ślepym, kontrolowanym badaniu klinicznym 138 pacjentów (59 chłopców i 79 dziewcząt) w wieku od 6 do 10 lat (średni wiek 8,3 lat) z heterozygotyczną hipercholesterolemią rodzinną lub nierodzinną, z wyjściowymi stężeniami cholesterolu LDL między 3,74 a 9,92 mmol/l, rozdzielono losowo na okres 12 tygodni do grupy leczonej ezetymibem 10 mg lub do grupy otrzymującej placebo.
W tygodniu 12. ezetymib istotnie zmniejszył stężenie cholesterolu całkowitego (-21% w porównaniu z 0%), cholesterolu LDL (-28% w porównaniu z -1%), Apo B (-22% w porównaniu z -1%) i cholesterolu nie-HDL (-26% w porównaniu z 0%) w porównaniu z placebo. Wyniki dla TG i cholesterolu HDL były porównywalne w obu grupach badania (odpowiednio -6% w porównaniu z
+8% i +2% w porównaniu z +1%).
W wieloośrodkowym, podwójnie ślepym, kontrolowanym badaniu klinicznym, 142 chłopców (faza II lub wyższa wg Tannera) i 106 dziewczynek po pierwszej miesiączce, w wieku od 10 do 17 lat (średni wiek 14,2 lat), z heterozygotyczną hipercholesterolemią rodzinną (heterozygous familiar hypercholesterolemia; HeFH) i z wyjściowymi stężeniami cholesterolu LDL między 4,1 a 10,4 mmol/l podzielono losowo do leczenia ezetymibem 10 mg podawanym jednocześnie z simwastatyną (10 mg, 20 mg lub 40 mg) lub do leczenia samą simwastatyną (10 mg, 20 mg lub 40 mg) przez okres 6 tygodni, po którym to okresie przez kolejne 27 tygodni stosowano albo ezetymib z 40 mg symwastatyny, albo tylko 40 mg symwastatyny a następnie, przez kolejne 20 tygodni stosowano w ramach próby otwartej ezetymib z symwastatyną (10 mg, 20 mg lub 40 mg).
Po 6 tygodniach ezetymib podawany łącznie z symwastatyną (wszystkie dawki) istotnie zmniejszał stężenie cholesterolu całkowitego (38% w porównaniu z 26%), cholesterolu LDL (49% w porównaniu z 34%), Apo B (39% w porównaniu z 27%) i cholesterolu nie-HDL (47% w porównaniu z 33%) w porównaniu z samą symwastatyną (wszystkie dawki). Wyniki dotyczące stężenia trójglicerydów i cholesterolu HDL w obu ocenianych grupach były podobne (odpowiednio -17% w porównaniu z
-12% i +7% w porównaniu z +6%). Wyniki w 13 tygodniu. były zgodne z wynikami z 6 tygodnia; istotnie więcej pacjentów otrzymujących ezetymib łącznie z 40 mg symwastatyny (62%) osiągnęło idealny cel leczenia według NCEP AAP (<2,8 mmol/l [110 mg/dl]) dla cholesterolu LDL w porównaniu z pacjentami otrzymującymi 40 mg symwastatyny (25%). W tygodniu 53, na koniec badania otwartego stanowiącego przedłużenie podwójnie ślepej próby, wpływ ocenianego leczenia na parametry lipidowe utrzymywał się.
U dzieci i młodzieży w wieku od 10 do 17 lat nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z symwastanyną w dawkach powyżej 40 mg na dobę. Nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z simwastatyną u pacjentów pediatrycznych w wieku <10 lat. Nie badano czy długotrwałe leczenie ezetymibem pacjentów poniżej 17 roku życia wpływa
na redukcję chorobowości i śmiertelności w wieku dorosłym.
Zapobieganie zdarzeniom sercowo-naczyniowym
Badanie IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) było to wieloośrodkowe, podwójnie zaślepione, kontrolowane aktywnym lekiem badanie z randomizacją, które objęło 18144 pacjentów włączanych do badania w ciągu 10 dni od przyjęcia do szpitala z powodu ostrego zespołu wieńcowego (ostry zawał serca lub niestabilna dławica piersiowa).
W momencie zgłoszenia się z ostrym zespołem wieńcowym pacjenci mieli stężenie cholesterolu LDL
≤125 mg/dl (≤3,2 mmol/l), jeśli nie przyjmowali terapii zmniejszającej stężenie lipidów lub ≤100 mg/dl (≤2,6 mmol/l), jeśli przyjmowali wcześniej terapię zmniejszającą stężenie lipidów. Wszystkich
pacjentów rozdzielono losowo w stosunku 1:1 albo do grupy otrzymującej ezetymib/symwastatynę
10 mg/40 mg (n = 9067), albo do grupy otrzymującej symwastatynę 40 mg (n = 9077), i obserwowano przez okres (mediana) 6,0 lat.
Średni wiek pacjentów wynosił 63,6 lat; 76% badanych stanowili mężczyźni, 84% osoby razy białej; 27% pacjentów miało cukrzycę. Przeciętne stężenie cholesterolu LDL w momencie zdarzenia decydującego o włączeniu pacjenta do badania wynosiło 80 mg/dl (2,1 mmol/l) u osób stosujących terapię hipolipemizującą (n = 6390) i 101 mg/dl (2,6 mmol/l) u osób niestosujących wcześniej terapii hipolipemizującej (n = 11594). Przed hospitalizacją z powodu ostrego zespołu wieńcowego kwalifikującego pacjenta do badania 34% pacjentów stosowało statynę, Po roku badania przeciętne stężenie cholesterolu LDL u pacjentów kontynuujących leczenie wyniosło 53,2 mg/dl (1,4 mmol/l) w grupie ezetymibu/symwastatyny i 69,9 mg/dl (1,8 mmol/l) w grupie monoterapii symwastatyną.
Generalnie stężenia lipidów uzyskano od pacjentów, którzy kontynuowali ocenianą w badaniu terapię.
Głównym punktem końcowym był złożony punkt końcowy obejmujący zgon sercowo-naczyniowy, poważne zdarzenia wieńcowe (zdefiniowane jako niezakończony zgonem zawał serca, udokumentowana niestabilna dławica piersiowa wymagająca hospitalizacji lub zabieg rewaskularyzacji naczyń wieńcowych po co najmniej 30 dniach od losowego przydziału leczenia) i udar mózgu niezakończony zgonem. Badanie wykazało, że leczenie ezetymibem dołączanym do symwastatyny zapewniało dodatkową korzyść w aspekcie redukcji głównego złożonego punktu końcowego obejmującego zgon sercowo-naczyniowe, poważne zdarzenia wieńcowe i niezakończony zgonem udar mózgu w porównaniu z samą symwastatyną (względna redukcja ryzyka 6,4%,
p = 0,016).
Główny punkt końcowy wystąpił u 2572 spośród 9067 pacjentów (7-letni wskaźnik Kaplana-Meiera [KM] 32,72%) w grupie ezetymibu/symwastatyny i u 2742 spośród 9077 pacjentów (7-letni wskaźnik KM 34,67%) w grupie samej symwastatyny. (Patrz Rycina 1 i Tabela 2) Przypuszcza
się, że ta dodatkowa korzyść będzie podobna w przypadku jednoczesnego podawania innych statyn o udowodnionej skuteczności w redukowaniu ryzyka zdarzeń sercowo-naczyniowych. Śmiertelność całkowita w tej grupie wysokiego ryzyka nie zmieniła się (patrz Tabela 2).
Stwierdzono ogólny korzystny wpływ na występowanie udarów; zaobserwowano jednak niewielkie, nieistotne zwiększenie występowania udarów krwotocznych w grupie ezetymibu/symwastatyny w porównaniu z grupą samej symwastatyny (patrz Tabela 2). Nie badano ryzyka udaru krwotocznego dla ezetymibu podawanego jednocześnie z silniejszymi statynami w badaniach oceniających odległe wyniki leczenia.
Efekt leczenia ezetymibem/symwastatyną był generalnie podobny w wielu różnych podgrupach wyróżnionych ze względu na płeć, wiek, rasę, wywiad cukrzycy, wyjściowe stężenia lipidów, wcześniejszą terapię statynami, przebyty udar mózgu i nadciśnienie.
Rycina 1: Wpływ ezetymibu/symwastatyny na główny złożony punkt końcowy obejmujący zgon sercowo-naczyniowy, poważne zdarzenia wieńcowe i niezakończony zgonem udar mózgu.
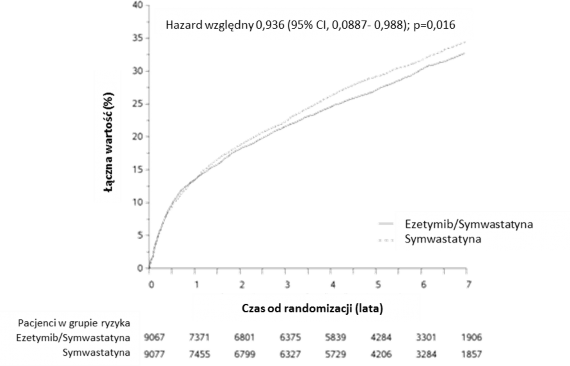
Tabela 2
Poważne zdarzenia sercowo-naczyniowe z rozdziałem na poszczególne grupy badania u wszystkich pacjentów objętych randomizacją w badaniu IMPROVE-IT
Punkt końcowy | Ezetymib/symwastatyna 10/40 mga (n = 9067) | Symwastatyna 40 mgb (n = 9077) | Hazard względny (95% CI) | Wartość p | ||
n | K-M %c | n | K-M %c | |||
Główny złożony punkt końcowy w ocenie skuteczności | ||||||
(Zgon sercowo- | 2572 | 32,72% | 2742 | 34,67% | 0,936 | 0,016 |
naczyniowy, | (0,887; | |||||
poważne | 0,988) | |||||
zdarzenia | ||||||
wieńcowe i | ||||||
niezakończony | ||||||
zgonem udar | ||||||
mózgu) | ||||||
Dodatkowe złożone punkty końcowe w ocenie skuteczności | ||||||
Zgon z powodu | 1322 | 17,52% | 1448 | 18,88% | 0,912 | 0,016 |
choroby | (0,847; | |||||
wieńcowej, | 0,983) | |||||
niezakończony | ||||||
zgonem zawał | ||||||
serca, pilna | ||||||
rewaskularyzacja | ||||||
wieńcowa po 20 | ||||||
dniach | ||||||
Poważne | 3089 | 38,65% | 3246 | 40,25% | 0,948 | 0,035 |
zdarzenia | (0,903; | |||||
wieńcowe, | 0,996) | |||||
niezakończony | ||||||
zgonem udar | ||||||
mózgu, zgon | ||||||
(wszystkie przyczyny) | ||||||
Zgon sercowo- naczyniowy, niezakończony zgonem zawał serca, niestabilna dławica piersiowa wymagająca hospitalizacji, rewaskularyzacja, niezakończony zgonem udar mózgu | 2716 | 34,49% | 2869 | 36,20% | 0,945 (0,897; 0,996) | 0,035 |
Składowe głównego złożonego punktu końcowego w ocenie skuteczności i wybrane punkty końcowe w ocenie skuteczności (wystąpienie w dowolnym momencie określonego zdarzenia po raz pierwszy) | ||||||
Zgon sercowo- naczyniowy | 537 | 6,89% | 538 | 6,84% | 1,000 (0,887; 1,127) | 0,997 |
Poważne zdarzenie wieńcowe: | ||||||
Niezakończony zgonem zawał serca | 945 | 12,77% | 1083 | 14,41% | 0,871 (0,798; 0,950) | 0,002 |
Niestabilna dławica wymagająca hospitalizacji | 156 | 2,06% | 148 | 1,92% | 1,059 (0,846; 1,326) | 0,618 |
Rewaskularyzacja wieńcowa po 30 dniach | 1690 | 21,84% | 1793 | 23,36% | 0,947 (0,886; 1,012) | 0,107 |
Niezakończony zgonem udar mózgu | 245 | 3,49% | 305 | 4,24% | 0,802 (0,678; 0,949) | 0,010 |
Wszystkie zawały serca (zakończone i niezakończone zgonem) | 977 | 13,13% | 1118 | 14,82% | 0,872 (0,800; 0,950) | 0,002 |
Wszystkie udary mózgu (zakończone i niezakończone zgonem) | 296 | 4,16% | 345 | 4,77% | 0,857 (0,734; 1,001) | 0,052 |
Udar niekrwotocznyd | 242 | 3,48% | 305 | 4,23% | 0,793 (0,670; 0,939) | 0,007 |
Udar krwotoczny | 59 | 0,77% | 43 | 0,59% | 1,377 (0,930; 2,040) | 0,110 |
Zgon z dowolnej przyczyny | 1215 | 15,36% | 1231 | 15,28% | 0,089 (0,914; 1,070) | 0,782 |
aU 6% dawkę ezetymibu/symwastatyny zwiększono do 10/80 mg.
bU 27% dawkę symwastatyny zwiększono do 80 mg.
cEstymata Kaplana.Meiera po 7 latach.
dW tym udar niedokrwienny lub udar nieokreślony.
Zapobieganie poważnym zdarzeniom naczyniowym w przewlekłej chorobie nerek
Badanie Study of Heart and Renal Protection (SHARP) było to międzynarodowe, randomizowane, kontrolowane placebo, podwójnie ślepe badanie kliniczne obejmujące 9438 pacjentów z przewlekłą chorobą nerek, z których 1/3 od początku badania była leczona dializami. W sumie 4650 zostało przydzielonych do grupy leczonej preparatem złożonym zawierającym 10 mg ezetymibu i 20 mg symwastatyny a 4620 pacjentów do grupy otrzymującej placebo. Pacjentów obserwowano przez okres czasu o medianie 4,9 lat. Średni wiek pacjentów wynosił 62 lata, 63% badanych stanowili mężczyźni, 72% były to osoby rasy białej, 23% chorzy na cukrzycę. U pacjentów, którzy nie byli dializowani średni szacowany współczynnik przesączania kłębuszkowego (estimated glomerular filtration rate; eGFR) wynosił 26,5 ml/min/1,73 m2. Nie było ustalonych lipidowych kryteriów włączenia. Średnie wyjściowe stężenie cholesterolu LDL wynosiło 108 mg/dl. Po roku, uwzględniając pacjentów, którzy przerwali przyjmowanie badanego leku, stężenie cholesterolu LDL zmniejszyło się o 26% względem placebo przy stosowaniu samej simwastatyny 20 mg i o 38% przy stosowaniu ezetymibu 10 mg w połączeniu z symwastatyną 20 mg.
Głównym porównaniem określonym w protokole badania SHARP była analiza „poważnych zdarzeń naczyniowych” (major vascular event; MVE) (zdefiniowanych jako niezakończony zgonem zawał serca, zgon sercowy, udar mózgu lub zabieg rewaskularyzacji) w populacji wyodrębnionej zgodnie z zamiarem leczenia, wyłącznie u chorych randomizowanych początkowo do grupy terapii skojarzonej ezetymibem i symwastatyną (n = 4193) lub do grupy placebo (n = 4191). Analizy dodatkowe obejmowały ten sam złożony punkt końcowy analizowany dla całej kohorty randomizowanej (na początku badania lub po roku) do terapii skojarzonej ezetymibem i symwastatyną (n = 4650) lub placebo (n = 4620) oraz poszczególne elementy tego złożonego punktu końcowego. Analiza głównego punktu końcowego wykazała, że ezetymib w połączeniu z symwastatyną istotnie zmniejszał ryzyko poważnych zdarzeń naczyniowych (749 pacjentów z takimi zdarzeniami w grupie placebo w porównaniu z 639 pacjentami w grupie terapii skojarzonej ezetymibem i symwastatyną), z redukcją ryzyka względnego 16% (p = 0,001).
Konstrukcja badania nie pozwoliła jednak na określenie udziału samego ezetymibu w skuteczności zmniejszania ryzyka poważnych zdarzeń naczyniowych u pacjentów z przewlekłą chorobą nerek.
Poszczególne elementy MVE u wszystkich pacjentów objętych randomizacją przedstawiono w Tabeli
3. Ezetymib w skojarzeniu z symwastatyną istotnie zmniejszał ryzyko udaru mózgu i konieczności rewaskularyzacji, przy nieistotnych liczbowych różnicach na korzyść terapii skojarzonej ezetymibem i symwastatyną dla niezakończonego zgonem zawału serca i zgonu sercowego.
Tabela 3
Poważne zdarzenia naczyniowe z rozdziałem na poszczególne grupy badania u wszystkich pacjentów objętych randomizacją w badaniu SHARPa
Punkt końcowy | Ezetymib 10 mg w połączeniu z symwastatyną 20 mg (n = 4650) | Placebo (n = 4620) | Współczynnik ryzyk (95% CI) | Wartość P |
Poważne zdarzenia naczyniowe | 701 (15,1%) | 814 (17,6%) | 0,85 (0,77-0,94) | 0,001 |
Niezakończony zgonem zawał serca | 134 (2,9%) | 159 (3,4%) | 0,84 (0,66-1,05) | 0,12 |
Zgon sercowy | 253 (5,4%) | 272 (5,9%) | 0,93 (0,78-1,10) | 0,38 |
Udar mózgu | 171 (3,7%) | 210 (4,5%) | 0,81 (0,66-0,99) | 0,038 |
Udar niekrwotoczny | 131 (2,8%) | 174 (3,8%) | 0,75 (0,60-0,94) | 0,011 |
Udar krwotoczny | 45 (1,0%) | 37 (0,8%) | 1,21 (0,78-1,86) | 0,40 |
Zabieg rewaskularyzacji | 284 (6,1%) | 352 (7,6%) | 0,79 (0,68-0,93) | 0,004 |
Poważne zdarzenia na tle miażdżycy b | 526 (11,3%) | 619 (13,4%) | 0,83 (0,74-0,94) | 0,002 |
a Analiza w populacji wyodrębnionej zgodnie z zamiarem leczenia obejmująca wszystkich pacjentów z badania SHARP randomizowanych do ezetymibu w połączeniu z symwastatyną lub do placebo, czy to na początku badania, czy po roku.
b Poważne zdarzenia na tle miażdżycy; zdefiniowane jako złożony punkt końcowy obejmujący niezakończony zgonem zawał serca, zgon wieńcowy, niekrwotoczny udar mózgu lub zabieg rewaskularyzacji.
Bezwzględne zmniejszenie stężenia cholesterolu LDL uzyskane podczas stosowania ezetymibu w połączeniu z simwastatyną było mniejsze u pacjentów z mniejszym wyjściowym stężeniem cholesterolu LDL (<2,5 mmol/l) i u pacjentów dializowanych w momencie rozpoczynania badania w porównaniu z pozostałymi pacjentami a redukcje ryzyka w tych 2 grupach były odpowiednio mniejsze.
Homozygotyczna hipercholesterolemia rodzinna
W randomizowanym badaniu klinicznym z podwójnie ślepą próbą, trwającym 12 tygodni, wzięło udział 50 pacjentów z rozpoznaną klinicznie i (lub) na podstawie badań genetycznych, homozygotyczną hipercholesterolemią rodzinną, przyjmujących atorwastatynę lub symwastatynę (w dawce 40 mg) w monoterapii lub w połączeniu z aferezą LDL. Jednoczesne stosowanie ezetymibu z atorwastatyną (w dawce 40 mg lub 80 mg) lub symwastatyną (w dawce 40 mg lub 80 mg) powodowało znaczące zmniejszenie stężenia cholesterolu LDL( o 15%), w porównaniu z przyjmowaniem zwiększonych dawek atorwastatyny lub symwastatyny (z 40 mg do 80 mg) w monoterapii.
Zwężenie zastawki aorty
Badane Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis (SEAS) było wieloośrodkowym, podwójnie ślepym, kontrolowanym placebo badaniem klinicznym, z medianą czasu trwania 4,4 lat i udziałem 1873 pacjentów z bezobjawowym zwężeniem zastawki
aortalnej (aortic stenosis; AS),udokumentowanym poprzez maksymalną prędkość przepływu w badaniu dopplerowskim między 2,5 a 4,0 m/s. Do badania włączono wyłącznie tych pacjentów, którzy nie wymagali leczenia statyną w celu zmniejszenia ryzyka choroby sercowo-naczyniowej na tle miażdżycy. Pacjentów podzielano losowo w stosunku 1:1 do grupy otrzymującej placebo lub ezetymib 10 mg w połączeniu z symwastatyną 40 mg dziennie.
Głównym punktem końcowym były rozpatrywane łącznie poważne zdarzenia sercowo-naczyniowe (major cardiovascular event; MCE), takie jak zgon sercowo-naczyniowy, operacja wymiany zastawki aortalnej (aortic valve replacement; AVR), zastoinowa niewydolność serca (congestive heart failure; CHF) na skutek progresji AS, zawał serca niezakończony zgonem, operacja pomostowania tętnic wieńcowych (coronary artery bypass grafting; CABG), przezskórna interwencja wieńcowa (percutaneous coronary inrervention; PCI), hospitalizacja z powodu niestabilnej dławicy piersiowej oraz niekrwotoczny udar mózgu. Głównymi dodatkowymi punktami końcowymi były różne zestawienia zdarzeń wymienionych jako kategorie wchodzące w skład głównego punktu końcowego.
W porównaniu z placebo leczenie ezetymibem/symwastatyną 10/40 mg nie powodowało istotnego zmniejszenia ryzyka MCE. Główny punkt końcowy wystąpił u 333 pacjentów (35,3%) w grupie ezetymibu/symwastatyny i u 355 pacjentów (38,2%) w grupie placebo (hazard względny w grupie ezetymibu / symwastatyny 0,96; 95% przedział ufności 0,83 do 1,12; p = 0,59). Operację wymiany zastawki aortalnej przeprowadzono u 267 pacjentów (28,3%) w grupie ezetymibu / symwastatyny i u 278 pacjentów (29,9%) w grupie placebo (hazard względny 1,00; 95% przedział ufności 0,84 do 1,18; p = 0,97). Niedokrwienne zdarzenia sercowo-naczyniowe wystąpiły u mniejszej liczby pacjentów z grupy ezetymibu / symwastatyny (n = 148) niż z grupy placebo (n = 187) (hazard względny 0,78; 95%
przedział ufności 0,63 do 0,97; p = 0,02), głównie z powodu mniejszej liczby pacjentów poddanych operacji pomostowania tętnic wieńcowych.
Nowotwory występowały częściej w grupie ezetymibu / symwastatyny (105 w porównaniu z 70, p = 0,01). Kliniczne znaczenie tej obserwacji nie jest pewne, gdyż w większym badaniu SHARP całkowite liczby pacjentów z nowo rozpoznanym nowotworem (438 w grupie
ezetymibu/symwastatyny w porównaniu z 439 w grupie placebo) nie różniły się. Również w badaniu IMPROVE-IT całkowite liczba pacjentów z nowym nowotworem złośliwym (853 w grupie ezetymibu/symwastatyny w porównaniu z 863 w grupie symwastatyny) nie różniły się, a więc wyniki badania SEAS nie zostały potwierdzone w badaniu SHARP ani w badaniu IMPROVE-IT.
Wchłanianie
Po podaniu doustnym ezetymib jest szybko wchłaniany i w znacznym stopniu sprzęgany do postaci czynnego farmakologicznie glukuronianu fenolowego (glukuronian ezetymibu). Glukuronian ezetymibu osiąga maksymalne stężenie w osoczu (Cmax) średnio w ciągu 1-2 godzin, zaś ezetymib w ciągu 4-12 godzin. Bezwzględna biodostępność ezetymibu nie może być określona, ponieważ lek jest właściwie nierozpuszczalny w wodnych roztworach do wstrzykiwań.
Przyjmowanie leku podczas posiłku (wysokotłuszczowego lub beztłuszczowego) nie miało wpływu na biodostępność ezetymibu. Ezetymib może być przyjmowany z posiłkiem lub na czczo.
Dystrybucja
Ezetymib i glukuronian ezetymibu wiążą się z ludzkimi białkami osocza odpowiednio w 99,7% oraz w 88-92%.
Metabolizm
Ezetymib jest metabolizowany głównie w jelicie cienkim i w wątrobie przez sprzęganie z glukuronianem (reakcja fazy II), a następnie jest wydalany z żółcią. U wszystkich badanych gatunków obserwowano również nieznaczny metabolizm oksydacyjny (reakcja fazy I). Ezetymib i glukuronian ezetymibu są głównymi pochodnymi leku wykrywalnymi w osoczu, stanowiącymi odpowiednio ok.
10-20% i 80-90% całkowitego stężenia leku w osoczu. Obydwie postacie leku są powoli eliminowane z osocza przy znaczącym udziale krążenia jelitowo-wątrobowego. Okres półtrwania ezetymibu i glukuronianu ezetymibu wynosi ok. 22 godziny.
Eliminacja
Po doustnym podaniu ezetymibu znakowanego izotopem węgla C14 (w dawce 20 mg) ludziom, całkowity ezetymib stanowił ok. 93% całkowitej aktywności promieniotwórczej w osoczu.
W ciągu dziesięciodniowej zbiórki, około 78% całkowitej ilości izotopu promieniotwórczego zostało wydalone z kałem, zaś 11% z moczem,. Po 48 godzinach od podania leku nie stwierdzono wykrywalnego poziomu aktywności promieniotwórczej w osoczu.
Szczególne grupy pacjentów
Dzieci i młodzież
Farmakokinetyka ezetymibu jest podobna u dzieci ≥6. roku życia i u dorosłych.
Brak danych dotyczących farmakokinetyki leku u dzieci w wieku <6 lat. Doświadczenie kliniczne obejmuje dzieci i młodzież z homozygotyczną hipercholesterolemią rodzinną i heterozygotyczną hipercholesterolemią rodzinną.
Pacjenci w podeszłym wieku
Stężenie całkowitego ezetymibu w osoczu było ok. dwukrotnie większe u pacjentów w podeszłym wieku (> 65 lat), niż u młodszych osób (od 18 do 45 lat). Zmniejszenie stężenia cholesterolu LDL oraz profil bezpieczeństwa leku były porównywalne po zastosowaniu ezetymibu w obu grupach. W związku z tym nie ma konieczności modyfikacji dawki leku u pacjentów w podeszłym wieku.
Zaburzenia czynności wątroby
Po podaniu ezetymibu w pojedynczej dawce 10 mg u pacjentów z łagodnym upośledzeniem czynności wątroby (5 lub 6 punktów wg skali Child-Pugh), stwierdzono około 1,7- krotne zwiększenie średniej wartości pola pod krzywą (AUC) dla całkowitego ezetymibu, w porównaniu ze zdrowymi osobami. W 14 dniowym badaniu, w którym podawano dawki wielokrotne leku (10 mg na dobę) u pacjentów z umiarkowanym upośledzeniem czynności wątroby (od 7 do 9 punktów wg skali Child-Pugh), średnia wartość AUC dla ezetymibu całkowitego zwiększyła się około 4-krotnie między pierwszym a czternastym dniem badania, w porównaniu ze zdrowymi osobami. U pacjentów z łagodnym upośledzeniem czynności wątroby nie jest konieczna modyfikacja dawki . Ze względu na brak danych dotyczących wpływu zwiększonej ekspozycji na ezetymib u pacjentów z umiarkowanym lub ciężkim (>9 punktów wg skali Child-Pugh) upośledzeniem czynności wątroby, nie zaleca się stosowania produktu Lipegis w tej grupie pacjentów (patrz punkt 4.4).
Zaburzenia czynności nerek
Po podaniu ezetymibu w pojedynczej dawce 10 mg u pacjentów z ciężką chorobą nerek (n = 8, średni klirens kreatyniny ≤30 ml/min./1,73 m2), średnia wartość AUC dla ezetymibu całkowitego zwiększyła się około 1,5-krotnie w porównaniu ze zdrowymi osobami (n = 9). Różnice te nie są istotne klinicznie i nie ma konieczności modyfikacji dawki u pacjentów z upośledzoną czynnością nerek.
U jednego pacjenta biorącego udział w tym badaniu (poddanego przeszczepieniu nerki i przyjmującego wiele leków, w tym cyklosporynę) stwierdzono 12-krotne zwiększenie ekspozycji na całkowity ezetymib.
Płeć
Stężenie całkowitego ezetymibu w osoczu jest nieco większe (o ok. 20%) u kobiet, niż u mężczyzn. Zmniejszenie stężenia cholesterolu LDL i profil bezpieczeństwa leku były porównywalne u kobiet i u mężczyzn leczonych ezetymibem. Nie ma więc potrzeby modyfikacji dawki leku w zależności od płci pacjenta.
Badania na zwierzętach dotyczące przewlekłego działania toksycznego nie wskazały na istnienie narządu narażonego na takie działanie. U psów, którym podawano ezetymib przez cztery tygodnie (≥0,03 mg/kg na dobę) stwierdzono zwiększenie stężenia cholesterolu w żółci znajdującej się
w pęcherzyku żółciowym o 2,5 do 3,5 razy. W badaniu trwającym rok, przeprowadzonym u psów otrzymujących lek w dawkach do 300 mg/kg/dobę, nie stwierdzono zwiększenia częstości występowania kamicy żółciowej lub innego oddziaływania na wątrobę i drogi żółciowe. Nie wiadomo, czy wyniki tych badań mają jakieś odniesienie do ludzi. Nie można wykluczyć ryzyka powstawania kamieni żółciowych w przypadku stosowania produktu Lipegis.
W badaniach, w których ezetymib stosowany był w skojarzeniu ze statynami, działania toksyczne były typowe, jak przy stosowaniu statyn. Niektóre z tych działań były nasilone przy skojarzonym stosowaniu leków w porównaniu ze stosowaniem statyn w monoterapii. Efekt ten ma związek z farmakokinetycznymi i farmakodynamicznymi interakcjami pomiędzy lekami stosowanymi w leczeniu skojarzonym. Nie obserwowano takich interakcji podczas badań klinicznych. Miopatia występowała u szczurów dopiero po podaniu dawek kilkakrotnie większych od dawki terapeutycznej stosowanej u ludzi (około 20-krotnie większa wartość AUC dla statyn oraz 500-2000 razy większa wartość AUC dla czynnych metabolitów ezetymibu).
W szeregu badań in vivo i in vitro nie stwierdzono działania genotoksycznego ezetymibu stosowanego w monoterapii lub w połączeniu ze statynami. Wyniki badań rakotwórczego działania ezetymibu podczas długotrwałego stosowania były ujemne.
Ezetymib nie ma wpływu na płodność samic i samców szczurów, jak również nie działa teratogennie na szczury czy króliki, nie ma też wpływu na rozwój zarodków i noworodków. Ezetymib przenikał przez barierę łożyskową u ciężarnych szczurów i królików podczas stosowania wielokrotnych dawek
po 1000 mg/kg/dobę. Nie zaobserwowano działania teratogennego u szczurów podczas jednoczesnego stosowania ezetymibu w skojarzeniu ze statyną,. U ciężarnych królików odnotowano niewielką liczbę zaburzeń rozwoju kośćca płodu (połączenie kręgów piersiowych i ogonowych, zmniejszenie liczby kręgów ogonowych). Stosowanie ezetymibu w skojarzeniu z lowastatyną wywierało działanie letalne na zarodki.
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER(-Y) POZWOLENIA(Ń) NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Celuloza mikrokrystaliczna Mannitol
Kroskarmeloza sodowa Hydroksypropyloceluloza niskopodstawiona Powidon
Sodu laurylosiarczan Magnezu stearynian
Nie dotyczy.
3 lata.
Produkt leczniczy nie wymaga żadnych specjalnych warunków przechowywania dotyczących temperatury.
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Blistry OPA/Aluminium/PVC/Aluminium zawierające 30 x (3x10), 60x (6x10), 80x (8x10), 90x (9 x10) lub 100x (10x10) tabletek w tekturowym pudełku.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Brak szczególnych wymagań dotyczących usuwania.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Egis Pharmaceuticals PLC
H-1106 Budapest, Keresztúri út 30-38. Węgry
Pozwolenie nr 20385
/ DATA PRZEDŁUŻENIA POZWOLENIA
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 11.07.2012
14.01.2021








