







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Ezetimibe Alchemia, 10 mg, tabletki
Każda tabletka zawiera 10 mg ezetymibu.
Substancja pomocnicza o znanym działaniu: każda tabletka zawiera 63 mg laktozy jednowodnej. Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka.
Białe lub prawie białe tabletki o kształcie kapsułki, o rozmiarach 8 mm x 4 mm, z wytłoczonym oznakowaniem „713” po jednej stronie i gładkie po drugiej stronie.
Hipercholesterolemia pierwotna
Produkt leczniczy Ezetimibe Alchemia podawany w skojarzeniu z inhibitorem reduktazy HMG-CoA (statyną) jest wskazany jako produkt leczniczy wspomagający wraz z dietą u pacjentów z pierwotną hipercholesterolemią (heterozygotyczną rodzinną lub nierodzinną), u których nie jest możliwe odpowiednie zmniejszenie stężenia lipidów przy zastosowaniu samej statyny.
Produkt leczniczy Ezetimibe Alchemia w monoterapii jest wskazany jako produkt leczniczy wspomagający wraz z dietą u pacjentów z pierwotną hipercholesterolemią (heterozygotyczną rodzinną lub nierodzinną), u których stosowanie statyny jest niewskazane lub ten produkt leczniczy nie jest tolerowany.
Zapobieganie wystąpieniu incydentów sercowo-naczyniowych
Produkt leczniczy Ezetimibe Alchemia, podawany jako lek uzupełniający pacjentom wcześniej leczonym statyną lub włączany do leczenia jednocześnie ze statyną, jest wskazany do stosowania w celu zmniejszenia ryzyka wystąpienia incydentów sercowo-naczyniowych (patrz punkt 5.1) u pacjentów z chorobą wieńcową (ang. CHD, Coronary Heart Disease) i ostrym zespołem wieńcowym w wywiadzie (ang. ACS, Acute Coronary Syndrome).
Homozygotyczna hipercholesterolemia rodzinna
Produkt leczniczy Ezetimibe Alchemia w skojarzeniu ze statyną jest wskazany jako produkt leczniczy wspomagający wraz z dietą u pacjentów z homozygotyczną hipercholesterolemią rodzinną. U pacjentów mogą być również stosowane dodatkowe metody leczenia (np. afereza LDL).
Dawkowanie
Pacjent powinien stosować odpowiednią dietę obniżającą stężenie lipidów przed rozpoczęciem przyjmowania produktu leczniczego Ezetimibe Alchemia oraz kontynuować ją przez cały okres leczenia.
Produkt leczniczy podawany jest doustnie. Zalecana dawka to jedna tabletka produktu leczniczego Ezetimibe Alchemia 10 mg raz na dobę. Produkt leczniczy Ezetimibe Alchemia można przyjmować o dowolnej porze dnia, niezależnie od posiłków.
W przypadku stosowania produktu leczniczego Ezetimibe Alchemia w skojarzeniu ze statyną, należy stosować wskazaną dawkę początkową lub już ustaloną większą dawkę danej statyny. Należy zapoznać się z zaleceniami dotyczącymi stosowania danej statyny.
Stosowanie u pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie
W celu zmniejszenia częstości występowania incydentów sercowo-naczyniowych u pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie produkt leczniczy może być podawany w skojarzeniu ze statyną o udowodnionym korzystnym wpływie na układ
sercowo-naczyniowy.
Jednoczesne stosowanie z produktami leczniczymi wiążącymi kwasy żółciowe
Produkt leczniczy Ezetimibe Alchemia należy przyjmować co najmniej 2 godziny przed lub co najmniej 4 godziny po przyjęciu produktu leczniczego wiążącego kwasy żółciowe.
Osoby w podeszłym wieku
Nie jest wymagane dostosowanie dawki produktu leczniczego dla pacjentów w podeszłym wieku (patrz punkt 5.2).
Dzieci i młodzież
Leczenie należy rozpocząć pod nadzorem lekarza specjalisty.
Dzieci i młodzież w wieku 6 lat i powyżej: Nie ustalono bezpieczeństwa stosowania i skuteczności ezetymibu u dzieci w wieku od 6 do 17 lat. Dostępne obecnie dane opisano w punktach 4.4, 4.8, 5.1 oraz 5.2, jednak na tej podstawie nie można opracować zaleceń dotyczących dawkowania.
Jeśli produkt leczniczy Ezetimibe Alchemia jest podawany w skojarzeniu ze statyną, dawkowanie statyny u młodzieży należy omówić z lekarzem.
Dzieci w wieku poniżej 6 lat: Nie określono bezpieczeństwa stosowania i skuteczności ezetymibu u dzieci w wieku poniżej 6 lat. Brak dostępnych danych.
Zaburzenia czynności wątroby
U pacjentów z łagodnymi zaburzeniami czynności wątroby (5-6 punktów wg skali Child-Pugh) nie jest wymagane dostosowanie dawkowania. Nie zaleca się stosowania produktu leczniczego Ezetimibe Alchemia u pacjentów z umiarkowanymi (7-9 punktów wg skali Child-Pugh) lub ciężkimi zaburzeniami czynności wątroby (>9 punktów wg Child-Pugh) (patrz punkty 4.4 i 5.2).
Zaburzenia czynności nerek
Nie jest konieczne dostosowanie dawkowania u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2).
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
W przypadku stosowania produktu leczniczego Ezetimibe Alchemia w skojarzeniu ze statyną należy zapoznać się z Charakterystyką Produktu Leczniczego (ChPL) danego produktu leczniczego.
Przeciwwskazane jest jednoczesne stosowanie produktu leczniczego Ezetimibe Alchemia i statyny w okresie ciąży i laktacji.
Produkt leczniczy Ezetimibe Alchemia w skojarzeniu ze statyną jest przeciwwskazany u pacjentów z czynną chorobą wątroby lub niewyjaśnionym, utrzymującym się zwiększeniem aktywności aminotransferaz w surowicy.
W przypadku jednoczesnego stosowania ze statyną, należy zapoznać się z ChPL danego produktu leczniczego.
Enzymy wątrobowe
W badaniach z grupą kontrolną, w których pacjenci przyjmowali ezetymib i statynę, obserwowano zwiększenie aktywności aminotransferaz (≥ 3 razy powyżej górnej granicy wartości prawidłowych). W przypadku jednoczesnego stosowania ezetymibu i statyny należy przeprowadzać testy czynnościowe wątroby przed rozpoczęciem leczenia oraz zgodnie z zaleceniami ChPL dotyczącymi danej statyny (patrz punkt 4.8).
W badaniu IMPROVE-IT (ang. IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) 18 144 pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie przydzielano w sposób losowy do grupy otrzymującej ezetymib z symwastatyną w dawce 10 mg + 40 mg na dobę (n=9067) lub symwastatynę w dawce 40 mg na dobę (n=9077). Podczas okresu obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania kolejnych wzrostów poziomu
transaminaz (≥ 3x GGN) w grupie otrzymującej ezetymib z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 2,5% i 2,3% (patrz punkt 4.8).
W kontrolowanym badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało losowo przydzielonych do grupy której raz na dobę podawano produkt leczniczy zawierający 10 mg ezetymibu i 20 mg symwastatyny (n=4650) lub placebo (n=4620) (średni okres obserwacji 4,9 roku), częstość występowania zwiększonej aktywności aminotransferaz (≥ 3 razy powyżej górnej granicy wartości prawidłowych) wynosiła 0,7% dla skojarzenia ezetymibu z symwastatyną oraz 0,6% dla placebo (patrz punkt 4.8).
Mięśnie szkieletowe
Po wprowadzeniu ezetymibu do obrotu zgłaszano przypadki wystąpienia miopatii i rabdomiolizy. Większość pacjentów, u których doszło do rabdomiolizy, przyjmowała statyny jednocześnie z ezetymibem. Jednak bardzo rzadko zgłaszano również występowanie rabdomiolizy w przypadku monoterapii oraz w przypadku jednoczesnego stosowania ezetymibu z innymi produktami leczniczymi, których związek ze zwiększeniem ryzyka rabdomiolizy jest znany. Jeśli podejrzewany jest rozwój miopatii na podstawie objawów ze strony mięśni lub zostanie ona potwierdzona zwiększeniem aktywności kinazy kreatynowej (CK), przekraczającym 10-krotnie górną granicę wartości prawidłowych, należy niezwłocznie przerwać stosowanie ezetymibu, statyn i wszelkich innych wymienionych wcześniej produktów leczniczych, równocześnie przyjmowanych przez pacjenta. Wszystkich pacjentów rozpoczynających leczenie produktem leczniczym Ezetimibe Alchemia należy poinformować o ryzyku wystąpienia miopatii i o konieczności natychmiastowego zgłoszenia wszelkich niewyjaśnionych bólów mięśni, ich tkliwości lub osłabienia (patrz punkt 4.8).
W badaniu IMPROVE-IT 18 144 pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie przydzielano w sposób losowy do grupy otrzymującej ezetymib z symwastatyną w dawce 10 mg + 40 mg na dobę (n=9067) lub symwastatynę w dawce 40 mg na dobę (n=9077). Podczas okresu obserwacji, którego mediana wynosiła 6,0 lat, częstość występowania miopatii w grupie
otrzymującej ezetymib z symwastatyną oraz w grupie przyjmującej symwastatynę wynosiła odpowiednio 0,2% i 0,1%. Miopatię zdefiniowano jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością kinazy kreatynowej (CK) w surowicy wynoszącej ≥ 10x GGN lub dwa kolejne epizody zwiększenia aktywności CK wynoszące ≥ 5 i < 10x GGN. Częstość występowania rabdomiolizy w grupie otrzymującej ezetymib z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,1% i 0,2%. Rabdomiolizę zdefiniowano jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącą ≥ 10x GGN i objawami uszkodzenia nerek, dwa kolejne epizody zwiększenia aktywności CK wynoszącej ≥ 5 i
< 10 x GGN z objawami uszkodzenia nerek lub CK ≥ 10 000 j.m./l bez objawów uszkodzenia nerek (patrz punkt 4.8.).
W badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało losowo przydzielonych do leczenia skojarzeniem 10 mg ezetymibu i 20 mg symwastatyny na dobę (n=4650) lub placebo (n=4620) (średni okres obserwacji 4,9 roku), częstość występowania miopatii i (lub) rabdomiolizy wynosiła 0,2% dla skojarzenia ezetymibu z symwastatyną oraz 0,1% dla placebo (patrz punkt 4.8).
Zaburzenia czynności wątroby
Ze względu na to, że nie są znane skutki zwiększonej ekspozycji na ezetymib u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby, nie zaleca się stosowania produktu leczniczego Ezetimibe Alchemia u tych pacjentów (patrz punkt 5.2).
Dzieci i młodzież
Skuteczność i bezpieczeństwo stosowania ezetymibu u pacjentów w wieku od 6 do 10 lat z heterozygotyczną rodzinną lub nierodzinną hipercholesterolemią oceniono w kontrolowanym placebo badaniu klinicznym trwającym 12 tygodni. W tej grupie wiekowej nie badano działania ezetymibu podczas stosowania powyżej 12 tygodni (patrz punkty 4.2, 4.8, 5.1 i 5.2).
Nie badano stosowania ezetymibu u pacjentów w wieku poniżej 6 lat (patrz punkty 4.2 i 4.8). Bezpieczeństwo i skuteczność stosowania ezetymibu w skojarzeniu z symwastatyną u pacjentów w wieku od 10 do 17 lat z heterozygotyczną hipercholesterolemią rodzinną oceniono w badaniu klinicznym z grupą kontrolną z udziałem dorastających chłopców (w skali Tannera, w fazie II i powyżej) oraz dziewcząt, u których pierwsza miesiączka wystąpiła co najmniej 1 rok wcześniej.
W tym ograniczonym badaniu z grupą kontrolną, ogólnie nie stwierdzono wykrywalnego wpływu na wzrost lub dojrzewanie płciowe u dorastających chłopców lub dziewcząt, ani na długość cyklu miesiączkowego u dziewcząt. Nie przeprowadzono jednak badań dotyczących wpływu stosowania ezetymibu przez okres > 33 tygodni na wzrost i proces dojrzewania płciowego (patrz punkt 4.2 oraz punkt 4.8).
Nie przeprowadzono badań dotyczących oceny bezpieczeństwa i skuteczności stosowania ezetymibu w skojarzeniu z symwastatyną w dawkach przekraczających 40 mg na dobę u dzieci w wieku od 10 do 17 lat.
Nie przeprowadzono badań dotyczących oceny bezpieczeństwa i skuteczności stosowania ezetymibu w skojarzeniu z symwastatyną u dzieci i młodzieży w wieku poniżej 10 lat (patrz punkty 4.2 oraz 4.8.).
Nie przeprowadzono oceny długotrwałej skuteczności stosowania ezetymibu u pacjentów w wieku poniżej 17 lat w zakresie zmniejszenia zachorowalności i śmiertelności w wieku dojrzałym.
Fibraty
Nie ustalono bezpieczeństwa i skuteczności stosowania ezetymibu w skojarzeniu z fibratami. Jeśli u pacjentów przyjmujących produkt leczniczy Ezetimibe Alchemia i fenofibrat podejrzewa się wystąpienie kamicy żółciowej, wskazane są badania pęcherzyka żółciowego, a leczenie to należy przerwać (patrz punkty 4.5 i 4.8).
Cyklosporyna
Należy zachować ostrożność podczas rozpoczynania stosowania produktu leczniczego Ezetimibe Alchemia u pacjentów przyjmujących cyklosporynę. Należy monitorować stężenia cyklosporyny u
pacjentów przyjmujących ją w skojarzeniu z produktem leczniczym Ezetimibe Alchemia (patrz punkt 4.5).
Leki przeciwzakrzepowe
W przypadku rozpoczęcia stosowania produktu leczniczego Ezetimibe Alchemia u pacjentów leczonych warfaryną, inną substancją przeciwzakrzepową z grupy pochodnych kumaryny lub fluindionem, należy odpowiednio monitorować wartości INR (międzynarodowy wskaźnik znormalizowany) (patrz punkt 4.5).
Substancja pomocnicza
Tabletki zawierają laktozę. Produkt leczniczy nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, niedoborem laktazy (typu Lapp) lub z zespołem złego wchłaniania glukozy-galaktozy.
W badaniach przedklinicznych wykazano, że ezetymib nie indukuje metabolizujących produkty lecznicze enzymów z grupy cytochromu P450. Nie obserwowano znamiennych interakcji farmakokinetycznych pomiędzy ezetymibem a produktami leczniczymi, o których wiadomo, że są metabolizowane przez cytochromy P450 1A2, 2D6, 2C8, 2C9 lub 3A4 albo przez
N-acetylotransferazę.
W badaniach klinicznych dotyczących interakcji stwierdzono, że ezetymib nie wpływał na parametry farmakokinetyczne dapsonu, dekstrometorfanu, digoksyny, doustnych środków antykoncepcyjnych (zawierających etynyloestradiol lub lewonorgestrel), glipizydu, tolbutamidu lub midazolamu, podczas jednoczesnego stosowania tych produktów leczniczych. Cymetydyna, podawana jednocześnie z ezetymibem, nie miała wpływu na jego dostępność biologiczną.
Produkty lecznicze zobojętniające
Jednoczesne przyjmowanie produktów leczniczych zobojętniających zmniejszało szybkość wchłaniania ezetymibu, ale nie miało wpływu na jego dostępność biologiczną. Zmniejszenie szybkości wchłaniania nie jest uważane za znamienne klinicznie.
Kolestyramina
Jednoczesne podawanie kolestyraminy zmniejszało średnią wartość pola pod krzywą (AUC) ezetymibu całkowitego (ezetymib + glukuronian ezetymibu) o około 55%. Efekt zwiększonej redukcji stężenia cholesterolu LDL po włączeniu produktu leczniczego Ezetimibe Alchemia do leczenia kolestyraminą może ulec osłabieniu w wyniku tej interakcji (patrz punkt 4.2).
Fibraty
Lekarze powinni wziąć pod uwagę możliwe ryzyko wystąpienia kamicy żółciowej i choroby pęcherzyka żółciowego u pacjentów przyjmujących fenofibrat i produkt leczniczy Ezetimibe Alchemia (patrz punkt 4.4 i 4.8).
Jeśli u pacjentów przyjmujących produkt leczniczy Ezetimibe Alchemia i fenofibrat podejrzewa się wystąpienie kamicy żółciowej, wskazane są badania pęcherzyka żółciowego, a leczenie to należy przerwać (patrz punkt 4.8).
Podczas jednoczesnego przyjmowania fenofibratu lub gemfibrozylu następuje nieznaczne zwiększenie całkowitego stężenia ezetymibu (odpowiednio o 1,5 i 1,7 razy).
Nie przeprowadzono badań dotyczących jednoczesnego stosowania ezetymibu w skojarzeniu z innymi fibratami.
Fibraty mogą zwiększać wydalanie cholesterolu do żółci, co prowadzi do kamicy żółciowej. W badaniu na zwierzętach, ezetymib czasem zwiększał stężenie cholesterolu w żółci zawartej w pęcherzyku żółciowym, ale nie u wszystkich gatunków (patrz punkt 5.3). Ryzyko powstawania kamieni w pęcherzyku żółciowym związane z leczeniem ezetymibem nie może być wykluczone.
Statyny
Nie stwierdzono znamiennych interakcji farmakokinetycznych podczas jednoczesnego stosowania ezetymibu z atorwastatyną, symwastatyną, prawastatyną, lowastatyną, fluwastatyną lub rozuwastatyną.
Cyklosporyna
W badaniu z udziałem 8 pacjentów po przeszczepieniu nerki (klirens kreatyniny > 50 ml/min), którzy przyjmowali ustaloną dawkę cyklosporyny, po podaniu pojedynczej dawki 10 mg ezetymibu stwierdzono 3,4-krotne zwiększenie średniej wartości AUC całkowitego ezetymibu (zakres od 2,3 do 7,9 razy) w porównaniu z grupą osób zdrowych, otrzymujących ezetymib w monoterapii w ramach innego badania (n= 17). W innym badaniu u pacjenta po zabiegu przeszczepienia nerki, u którego rozwinęła się ciężka niewydolność nerek i który przyjmował cyklosporynę i wiele innych produktów leczniczych, wykazano 12-krotne zwiększenie stężenia całkowitego ezetymibu, w porównaniu z osobami z grupy kontrolnej otrzymującymi ezetymib w monoterapii. W badaniu skrzyżowanym złożonym z dwóch okresów, w którym uczestniczyło dwunastu zdrowych ochotników, stosowanie
20 mg ezetymibu na dobę przez 8 dni z podaniem pojedynczej dawki cyklosporyny wielkości 100 mg w dniu 7. badania spowodowało średnie zwiększenie AUC cyklosporyny o 15 % (zakres wynosił od 10% zmniejszenia do 51 % zwiększenia) w porównaniu z podaniem wyłącznie pojedynczej dawki cyklosporyny wielkości 100 mg. Nie przeprowadzono badania kontrolowanego dotyczącego wpływu jednoczesnego stosowania ezetymibu na ekspozycję na cyklosporynę u pacjentów po przeszczepieniu nerki. Należy zachować ostrożność rozpoczynając leczenie produktem leczniczym Ezetimibe Alchemia w trakcie stosowania cyklosporyny. Należy monitorować stężenie cyklosporyny u pacjentów przyjmujących jednocześnie produkt leczniczy Ezetimibe Alchemia i cyklosporynę (patrz punkt 4.4).
Leki przeciwzakrzepowe
Jednoczesne stosowanie ezetymibu (10 mg raz na dobę) nie miało istotnego wpływu na dostępność biologiczną warfaryny i czas protrombinowy w badaniu przeprowadzonym z udziałem dwunastu zdrowych dorosłych mężczyzn. Jednakże istnieją doniesienia z okresu po wprowadzeniu do obrotu dotyczące zwiększenia wartości INR u pacjentów stosujących warfarynę lub fluindion, u których dołączono leczenie ezetymibem. W przypadku stosowania produktu leczniczego Ezetimibe Alchemia jednocześnie z warfaryną, inną substancją przeciwzakrzepową z grupy pochodnych kumaryny lub fluindionem, należy odpowiednio monitorować INR (patrz punkt 4.4).
Dzieci i młodzież
Badania interakcji przeprowadzono jedynie u dorosłych.
Stosowanie produktu leczniczego Ezetimibe Alchemia w skojarzeniu ze statyną jest przeciwwskazane w okresie ciąży i laktacji (patrz punkt 4.3). Należy zapoznać się z ChPL dotyczącą określonej statyny.
Ciąża
Produkt leczniczy Ezetimibe Alchemia wolno podawać kobietom w ciąży jedynie wówczas, gdy jest to bezwzględnie konieczne. Brak danych klinicznych na temat stosowania ezetymibu w okresie ciąży. Badania na zwierzętach dotyczące stosowania ezetymibu w monoterapii nie wykazały żadnego bezpośredniego lub pośredniego szkodliwego oddziaływania na przebieg ciąży, rozwój zarodka i płodu, przebieg porodu lub rozwój noworodka (patrz punkt 5.3).
Karmienie piersią
Produktu leczniczego Ezetimibe Alchemia nie należy stosować w okresie laktacji. Badania na szczurach wykazały, że ezetymib jest wydzielany do mleka tych zwierząt. Nie wiadomo, czy ezetymib przenika do mleka kobiecego.
Płodność
Brak dostępnych danych z badań klinicznych dotyczących wpływu ezetymibu na płodność u ludzi. Ezetymib nie miał wpływu na płodność u samców i samic szczurów (patrz punkt 5.3).
Nie przeprowadzono badań dotyczących wpływu produktu leczniczego na zdolność prowadzenia pojazdów i obsługiwania maszyn. Niemniej jednak, podczas prowadzenia pojazdów i obsługiwania maszyn należy wziąć pod uwagę, że zgłaszano występowanie zawrotów głowy.
Badania kliniczne
W trwających do 112 tygodni badaniach klinicznych ezetymib w dawce 10 mg na dobę podawano 2396 pacjentom w monoterapii, 11308 pacjentom w skojarzeniu ze statyną oraz 185 pacjentom w skojarzeniu z fenofibratem. Działania niepożądane były zwykle łagodne i przemijające. Ogólna częstość występowania działań niepożądanych ezetymibu i placebo była podobna. Również
liczba pacjentów, którzy przerwali leczenie z powodu działań niepożądanych była porównywalna w grupie przyjmującej ezetymib i placebo.
Ezetymib w monoterapii lub w skojarzeniu ze statyną.
Poniżej przedstawiono działania niepożądane obserwowane u pacjentów leczonych ezetymibem w monoterapii (n= 2396), które pojawiły się z większą częstością, niż u pacjentów przyjmujących placebo (n= 1159) lub u pacjentów leczonych ezetymibem w skojarzeniu ze statyną (n= 11308), które pojawiły się z większą częstością niż u pacjentów przyjmujących jedynie statynę (n = 9361). Działania niepożądane po wprowadzeniu do obrotu uzyskano ze zgłoszeń dotyczących ezetymibu podawanego w monoterapii lub w skojarzeniu ze statyną.
Częstości występowania określono w następujący sposób: bardzo często (≥l/10), często (≥l/100 do
<1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥l/10 000 do <1/1 000), bardzo (<1/10 000) oraz nieznana (nie może być określona na podstawie dostępnych danych).
Ezetymib w monoterapii | ||
Klasyfikacja układów i narządów | Działania niepożądane | Częstość |
Zaburzenia metabolizmu i odżywiania | zmniejszone łaknienie | Niezbyt często |
Zaburzenia naczyniowe | nagłe zaczerwienienie twarzy; nadciśnienie tętnicze | Niezbyt często |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | kaszel | Niezbyt często |
Zaburzenia żołądka i jelit | bóle brzucha; biegunka; wzdęcia z oddawaniem wiatrów | Często |
niestrawność; choroba refluksowa przełyku (GERD); nudności | Niezbyt często | |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | bóle stawów; kurcze mięśni; bóle karku | Niezbyt często |
Zaburzenia ogólne i stany w miejscu podania | zmęczenie | Często |
ból w klatce piersiowej; ból | Niezbyt często | |
Badania diagnostyczne | Zwiększenie aktywności AlAT, zwiększenie aktywności AspAT; zwiększenie aktywności CPK we krwi; zwiększenie aktywności gamma-glutamylotransferazy; nieprawidłowe wyniki testów czynnościowych wątroby | Niezbyt często |
Dodatkowe działania niepożądane ezetymibu w skojarzeniu ze statyną | ||
Klasyfikacja układów i narządów | Działania niepożądane | Częstość |
Zaburzenia układu nerwowego | ból głowy | Często |
parestezje | Niezbyt często | |
Zaburzenia żołądka i jelit | suchość w jamie ustnej; zapalenie żołądka | Niezbyt często |
Zaburzenia skóry i tkanki podskórnej | świąd, wysypka, pokrzywka | Niezbyt często |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | bóle mięśni | Często |
bóle pleców; osłabienie mięśni; bóle kończyn | Niezbyt często | |
Zaburzenia ogólne i stany w miejscu podania | osłabienie; obrzęki obwodowe | Niezbyt często |
Badania diagnostyczne | zwiększenie aktywności AlAT i (lub) AspAT | Często |
Doświadczenie po wprowadzeniu do obrotu (w skojarzeniu ze statyną lub w monoterapii) | ||
Klasyfikacja układów i narządów | Działania niepożądane | Częstość |
Zaburzenia krwi i układu chłonnego | trombocytopenia | Nieznana |
Zaburzenia układu immunologicznego | nadwrażliwość, w tym wysypka, pokrzywka, anafilaksja i obrzęk naczynioruchowy | Nieznana |
Zaburzenia psychiczne | depresja | Nieznana |
Zaburzenia układu nerwowego | zawroty głowy; parestezje | Nieznana |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | duszność | Nieznana |
Zaburzenia żołądka i jelit | zapalenie trzustki; zaparcia | Nieznana |
Zaburzenia wątroby | zapalenie wątroby; kamica żółciowa; zapalenie pęcherzyka żółciowego | Nieznana |
Zaburzenia skóry i tkanki podskórnej | rumień wielopostaciowy | Nieznana |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | bóle mięśni; miopatia i (lub) rabdomioliza (patrz punkt 4.4) | Nieznana |
Zaburzenia ogólne i stany w miejscu podania | osłabienie | Nieznana |
Ezetymib w skojarzeniu z fenofibratem
Zaburzenia żołądka i jelit: często - bóle brzucha.
W kontrolowanym placebo klinicznym badaniu wieloośrodkowym, z podwójnie ślepą próbą, uczestniczyli pacjenci z mieszaną hiperlipidemią, spośród których 625 leczonych było przez
12 tygodni, a 576 przez 1 rok. W badaniu tym, 172 pacjentów przyjmujących ezetymib i fenofibrat ukończyło 12-tygodniowe leczenie, a 230 pacjentów przyjmujących ezetymib i fenofibrat (w tym 109
przyjmujących ezetymib w monoterapii przez pierwszych 12 tygodni) ukończyło leczenie trwające 1 rok. Celem badania nie było porównanie leczonych grup pacjentów pod względem występowania rzadkich działań niepożądanych. Wskaźnik występowania (przedział ufności 95%) klinicznie znamiennego zwiększenia (> 3 razy powyżej górnego zakresu wartości prawidłowych, podczas kolejnych oznaczeń) aktywności aminotransferaz w surowicy wynosił odpowiednio 4,5% (1,9; 8,8)
dla fenofibratu stosowanego w monoterapii i 2,7% (1,2; 5,4) dla ezetymibu podawanego w skojarzeniu z fenofibratem, dostosowany do odpowiedzi na leczenie. Natomiast wskaźnik występowania przypadków wycięcia pęcherzyka żółciowego wynosił odpowiednio, dla fenofibratu stosowanego w monoterapii 0,6% (0,0; 3,l) i dla ezetymibu podawanego w skojarzeniu z fenofibratem l,7% (0,6; 4,0) (patrz punkty 4.4 i 4.5).
Dzieci i młodzież (w wieku od 6 do 17 lat)
W badaniu z udziałem dzieci i młodzieży (w wieku od 6 do 10 lat) z heterozygotyczną hipercholesterolemią rodzinną lub nierodzinną (n=138) obserwowano zwiększenie aktywności AlAT i (lub) AspAT (≥ 3 razy powyżej górnego zakresu wartości prawidłowych, podczas kolejnych oznaczeń) u 1,1% pacjentów (1 osoba) w grupie stosującej ezetymib w porównaniu do 0% pacjentów w grupie placebo. Nie odnotowano zwiększenia aktywności CPK (≥ 10 razy powyżej górnego zakresu wartości prawidłowych). Nie zgłaszano przypadków miopatii.
W osobnym badaniu z udziałem dzieci i młodzieży (w wieku od 10 do 17 lat) z heterozygotyczną hipercholesterolemią rodzinną (n = 248), u 3% pacjentów (4 osoby) leczonych ezetymibem w skojarzeniu z symwastatyną obserwowano podwyższenie aktywności AlAT i (lub) AspAT (≥ 3 razy powyżej górnego zakresu wartości prawidłowych, podczas kolejnych oznaczeń) w porównaniu z 2% pacjentów (2 osoby) z grupy stosującej symwastatynę w monoterapii; Zwiększenie aktywności CPK (≥10 razy powyżej górnego zakresu wartości prawidłowych) stwierdzono odpowiednio u 2%
(2 pacjentów) i 0% uczestników badania. Nie zaobserwowano przypadków miopatii.
Te badania nie były dostosowane do oceny porównawczej rzadko występujących działań niepożądanych.
Pacjenci z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie
W badaniu IMPROVE-IT (patrz punkt 5.1) z udziałem 18 144 pacjentów leczonych ezetymibem z symwastatyną w dawce 10 mg + 40 mg (n=9067, przy czym u 6% pacjentów dawkę ezetymibu i symwastatyny zwiększono do 10 mg + 80 mg) lub symwastatyną w dawce 40 mg (n=9077, przy czym u 27% pacjentów dawkę symwastatyny zwiększono do 80 mg) w okresie obserwacji, którego mediana wynosiła 6,0 lat, obserwowano zbliżone profile bezpieczeństwa. Wskaźnik przerwania leczenia z powodu działań niepożądanych wynosił 10,6% u pacjentów leczonych ezetymibem z symwastatyną oraz 10,1% u pacjentów leczonych symwastatyną. Częstość występowania miopatii w grupie otrzymującej ezetymib z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,2% i 0,1%. Miopatię zdefiniowano, jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącą ≥ 10x GGN lub dwa kolejne epizody zwiększenia aktywności CK wynoszące ≥ 5 i < 10x GGN. Częstość występowania rabdomiolizy w grupie otrzymującej ezetymib z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 0,1% i 0,2%. Rabdomiolizę zdefiniowano jako niewyjaśnione osłabienie mięśni lub ból ze zwiększoną aktywnością CK w surowicy wynoszącą ≥ 10x GGN i objawami uszkodzenia nerek, dwa kolejne epizody zwiększenia aktywności CK wynoszącej ≥ 5 i < 10x GGN z objawami uszkodzenia nerek lub CK ≥ 10 000 j.m./l bez objawów uszkodzenia nerek. Częstość występowania kolejnych wzrostów poziomu aminotransferaz (≥ 3x GGN) w grupie otrzymującej ezetymib z symwastatyną oraz w grupie otrzymującej symwastatynę wynosiła odpowiednio 2,5% i 2,3%. (Patrz punkt 4.4.). Działania niepożądane ze strony pęcherzyka żółciowego zgłoszono u 3,1% pacjentów z grupy otrzymującej ezetymib z symwastatyną oraz u 3,5% pacjentów z grupy leczonej symwastatyną. Częstość hospitalizacji z powodu cholecystektomii w obu leczonych grupach wynosiła 1,5%.
Nowotwór (zdefiniowany jako dowolny nowy nowotwór) zdiagnozowano podczas badania u odpowiednio 9,4% i 9,5% pacjentów.
Pacjenci z przewlekłą chorobą nerek
W badaniu SHARP (ang. Study of Heart and Renal Protection; patrz punkt 5.1), obejmującym ponad 9000 pacjentów otrzymujących codziennie produkt złożony zawierający 10 mg ezetymibu i 20 mg symwastatyny (n=4650), albo placebo (n=4620), profile bezpieczeństwa były porównywalne w okresie obserwacji o medianie 4,9 roku. W badaniu tym rejestrowano jedynie ciężkie zdarzenia niepożądane i przypadki przerywania leczenia z powodu zdarzeń niepożądanych. Wskaźniki przerywania leczenia z powodu zdarzeń niepożądanych były porównywalne (10,4% u pacjentów leczonych ezetymibem w skojarzeniu z symwastatyną, 9,8% u pacjentów otrzymujących placebo).
Częstość występowania miopatii i (lub) rabdomiolizy wynosiła 0,2% u pacjentów leczonych ezetymibem w skojarzeniu z symwastatyną i 0,1% u pacjentów otrzymujących placebo. Do zwiększenia aktywności aminotransferaz (>3 x górna granica normy) doszło u 0,7% pacjentów leczonych ezetymibem w skojarzeniu z symwastatyną w porównaniu z 0,6% pacjentów otrzymujących placebo (patrz punkt 4.4). W tym badaniu nie stwierdzono istotnego statystycznie zwiększenia częstości występowania określonych wcześniej zdarzeń niepożądanych, takich jak nowotwory (9,4% w grupie ezetymibu z symwastatyną, 9,5% w grupie placebo), zapalenie wątroby, cholecystektomia lub powikłania kamicy żółciowej czy zapalenie trzustki.
Wpływ na parametry laboratoryjne
W badaniach z grupą kontrolną, w których produkt leczniczy był stosowany w monoterapii, częstość występowania klinicznie znamiennego zwiększenia aktywności aminotransferaz w surowicy (zwiększenie aktywności AlAT i (lub) AspAT 3 razy powyżej górnego zakresu wartości prawidłowych) była podobna w grupie przyjmujących ezetymib (0,5 %) oraz placebo (0,3 %). W badaniach, w których produkty lecznicze stosowano w skojarzeniu, częstość występowania wspomnianego uprzednio zwiększenia aktywności aminotransferaz w surowicy wynosiła 1,3% w grupie pacjentów przyjmujących ezetymib w skojarzeniu ze statyną oraz 0,4 % w grupie pacjentów przyjmujących wyłącznie statynę. Zwiększenie aktywności enzymów przebiegało na ogół bez objawów; nie stwierdzano cech zastoju żółci. Aktywność aminotransferaz powracała do wartości początkowych po zaprzestaniu przyjmowania produktów leczniczych albo podczas dalszego ich stosowania (patrz punkt 4.4).
W badaniach klinicznych zgłaszano zwiększenie aktywności CPK przekraczające 10 razy górną granicę wartości prawidłowych u 4 spośród 1647 (0,2 %) pacjentów stosujących wyłącznie ezetymib w porównaniu z l spośród 786 (0, l%) pacjentów stosujących placebo oraz u l spośród 917 (0, l%) pacjentów stosujących jednocześnie ezetymib i statyny w porównaniu z 4 spośród 929 (0,4%) pacjentów stosujących wyłącznie statyny. Nie stwierdzono większej liczby przypadków miopatii lub rabdomiolizy związanych ze stosowaniem ezetymibu w porównaniu z odpowiednią grupą kontrolną (placebo lub wyłącznie statyny) (patrz punkt 4.4).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych:
Al. Jerozolimskie 181C, 02-222 Warszawa, tel.: + 48 22 49-21-301, fax: +48 22 49-21-309, e-mail: ndl@urpl.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W badaniach klinicznych, w których dawkę dobową 50 mg ezetymibu podawano 15 zdrowym ochotnikom w okresie do 14 dni, bądź też dawkę dobową 40 mg ezetymibu podawano 18 pacjentom z pierwotną hipercholesterolemią w okresie do 56 dni, ezetymib był na ogół dobrze tolerowany. W badaniach przeprowadzonych na zwierzętach nie obserwowano działania toksycznego po przyjęciu
ezetymibu w postaci pojedynczej dawki doustnej 5 000 mg/kg u szczurów i myszy oraz 3 000 mg/kg u psów.
Odnotowano kilka przypadków przedawkowania ezetymibu: większość z nich nie była związana z występowaniem działań niepożądanych. Odnotowane działania niepożądane nie były poważne.
W przypadku przedawkowania produktu leczniczego należy zastosować leczenie objawowe i wspomagające.
Grupa farmakoterapeutyczna: Leki modyfikujące stężenie lipidów; inne leki modyfikujące stężenie lipidów, kod ATC: C10AX09
Mechanizm działania
Ezetymib należy do grupy produktów leczniczych zmniejszających stężenie lipidów, które selektywnie hamują wchłanianie cholesterolu i pokrewnych steroli roślinnych w jelitach. Ezetymib działa po podaniu doustnym, a jego mechanizm działania różni się od innych grup produktów leczniczych zmniejszających stężenie cholesterolu (np. statyn, produktów leczniczych wiążących kwasy żółciowe (żywice), pochodnych kwasu fibrynowego, stanoli roślinnych). Celem działania ezetymibu na poziomie molekularnym jest nośnik steroli, Niemann-Pick C1-Like 1 (NPC1L1), które odgrywa rolę w wychwytywaniu cholesterolu i fitosteroli w jelicie.
Ezetymib wiąże się z rąbkiem szczoteczkowym jelita cienkiego i hamuje wchłanianie cholesterolu, zmniejszając ilość cholesterolu transportowanego z jelit do wątroby. Statyny zmniejszają syntezę cholesterolu w wątrobie, a połączenie tych dwóch mechanizmów działania zapewnia uzupełniające się zmniejszenie stężenia cholesterolu. W trwających 2 tygodnie badaniach klinicznych, przeprowadzonych u 18 pacjentów z hipercholesterolemią, ezetymib hamował wchłanianie cholesterolu z jelit o 54%, w porównaniu z placebo.
Działanie farmakodynamiczne
Przeprowadzono szereg badań przedklinicznych, aby określić czy działanie ezetymibu hamujące wchłanianie cholesterolu jest wybiórcze. Ezetymib hamował wchłanianie cholesterolu znakowanego izotopem węgla 14C, nie miał zaś wpływu na wchłanianie trójglicerydów, kwasów tłuszczowych, kwasów żółciowych, progesteronu, etynyloestradiolu lub rozpuszczalnych w tłuszczach witamin A i D.
Badania epidemiologiczne wykazały, że zachorowalność i umieralność z powodu chorób układu krążenia zmieniają się proporcjonalnie do stężenia cholesterolu całkowitego i cholesterolu LDL, a odwrotnie proporcjonalnie do stężenia cholesterolu HDL.
Podawanie produktu leczniczego Ezetimibe Alchemia ze statyną skutecznie zmniejsza ryzyko wystąpienia incydentów sercowo-naczyniowych u pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym w wywiadzie.
Skuteczność kliniczna i bezpieczeństwo stosowania
W kontrolowanych badaniach klinicznych ezetymib stosowany w monoterapii znacząco zmniejszał stężenie cholesterolu całkowitego, cholesterolu LDL, apolipoproteiny B (Apo B) oraz trójglicerydów, a także zwiększał stężenie cholesterolu HDL u pacjentów z hipercholesterolemią.
Hipercholesterolemia pierwotna
W trwającym 8 tygodni badaniu klinicznym z podwójnie ślepą próbą, kontrolowanym placebo, uczestniczyło 769 pacjentów z hipercholesterolemią, którzy przyjmowali statynę w monoterapii i u
których nie osiągnięto docelowego stężenia cholesterolu LDL (2,6 do 4,1 mmol/l [100 do 160 mg/dl], w zależności od stężenia początkowego) wg National Cholesterol Education Program (NCEP).
Pacjentów losowo dobrano do grup przyjmujących ezetymib w dawce 10 mg lub placebo, w skojarzeniu ze stosowaną już statyną.
Wśród pacjentów leczonych statyną, u których nie osiągnięto docelowego stężenia cholesterolu LDL (ok. 82%), znamiennie więcej pacjentów przyjmujących ezetymib uzyskało docelowe stężenie cholesterolu LDL na zakończenie okresu obserwacji niż pacjentów przydzielonych losowo do grupy placebo, odpowiednio 72 % i 19 %. Znaczne różnice obserwowano również w redukcji stężenia cholesterolu LDL (25% i 4%, odpowiednio u pacjentów przyjmujących ezetymib i placebo). Ponadto dołączenie ezetymibu do leczenia statyną znacząco zmniejszało stężenie cholesterolu całkowitego, apolipoproteiny B i trójglicerydów, a także zwiększało stężenie cholesterolu HDL, w porównaniu z grupą pacjentów otrzymujących placebo. Dołączenie do leczenia statyną ezetymibu lub placebo zmniejszało medianę stężenia białka C-reaktywnego odpowiednio o 10% lub 0%, w porównaniu z wartościami początkowymi.
W dwóch randomizowanych badaniach klinicznych z podwójnie ślepą próbą, kontrolowanych placebo, prowadzonych przez 12 tygodni u 1719 pacjentów z hipercholesterolemią pierwotną, obserwowano znaczące zmniejszenie stężenia cholesterolu całkowitego (o 13%), cholesterolu LDL (o 19%), apolipoproteiny B (o 14%) oraz trójglicerydów (o 8%), a także zwiększenie stężenia cholesterolu HDL (o 3%) po zastosowaniu ezetymibu w dawce 10 mg, w porównaniu z grupą placebo. Ponadto ezetymib nie miał wpływu na osoczowe stężenie rozpuszczalnych w tłuszczach witamin A, D i E oraz na czas protrombinowy, a także, podobnie jak inne produkty lecznicze zmniejszające stężenie lipidów, nie zaburzał wytwarzania hormonów sterydowych kory nadnerczy.
W wieloośrodkowym, podwójnie ślepym, kontrolowanym badaniu klinicznym (ENHANCE) 720 pacjentów z heterozygotyczną hipercholesterolemią rodzinną podzielono losowo do leczenia
ezetymibem 10 mg w skojarzeniu z symwastatyną 80 mg (n = 357) lub samą symwastatyną w dawce 80 mg (n =363) przez 2 lata. Głównym celem tego badania była ocena wpływu terapii skojarzonej ezetymibem/symwastatyną na grubość kompleksu blaszki wewnętrznej i środkowej tętnicy szyjnej (intima-media thickness; IMT) w porównaniu z monoterapią symwastatyną. Znaczenie tego markera zastępczego dla zachorowalności i śmiertelności sercowo-naczyniowej wciąż nie zostało potwierdzone.
Główny punkt końcowy, zmiana średniej IMT wszystkich 6 odcinków tętnicy szyjnej, oceniana na podstawie badania USG w prezentacji B, nie różniła się istotnie (p = 0,29) między dwoma ocenianymi grupami. W czasie dwuletniej obserwacji w grupie ezetymibu 10 mg w skojarzeniu z symwastatyną 80 mg grubość kompleksu blaszki wewnętrznej i środkowej zwiększyła się o 0,0111 mm zaś w grupie samej symwastatyny 80 mg o 0,0058 mm (wyjściowa średnia IMT tętnicy szyjnej wynosiła w tych grupach odpowiednio 0,68 mm i 0,69 mm).
Ezetymib w dawce 10 mg w skojarzeniu z symwastatyną 80 mg zmniejszał stężenie cholesterolu LDL, cholesterolu całkowitego, Apo B i trójglicerydów istotnie silniej niż symwastatyna 80 mg. Procentowy wzrost stężenia cholesterolu HDL był podobny w obu badanych grupach. Działania niepożądane opisywane w przypadku ezetymibu 10 mg w skojarzeniu z symwastatyną 80 mg pokrywały się z dotychczas poznanym profilem bezpieczeństwa tego produktu leczniczego.
Dzieci i młodzież
W wieloośrodkowym, podwójnie ślepym, kontrolowanym placebo badaniu klinicznym 138 pacjentów (59 chłopców i 79 dziewcząt) w wieku od 6 do 10 lat (średni wiek 8,3 lat) z heterozygotyczną hipercholesterolemią rodzinną lub nierodzinną, z wyjściowymi stężeniami cholesterolu LDL między 3,74 a 9,92 mmol/l, przydzielono losowo na okres 12 tygodni do grupy leczonej ezetymibem w dawce 10 mg lub do grupy otrzymującej placebo.
W 12. tygodniu ezetymib istotnie zmniejszył stężenie cholesterolu całkowitego (-21% w porównaniu z 0%), cholesterolu LDL (-28% w porównaniu z -1%), Apo B (-22% w porównaniu z -1%) i cholesterolu nie-HDL (-26% w porównaniu z 0%) w porównaniu z placebo. Wyniki dla TG i cholesterolu HDL były porównywalne w obu grupach badania (odpowiednio -6% w porównaniu z
+8% i +2% w porównaniu z +1%).
W wieloośrodkowym, podwójnie ślepym, kontrolowanym badaniu klinicznym, 142 chłopców (faza II lub wyższa wg Tannera) i 106 dziewczynek po pierwszej miesiączce, w wieku od 10 do 17 lat (średni wiek 14,2 lat), z heterozygotyczną hipercholesterolemią rodzinną (ang. heterozygous familiar hypercholesterolemia; HeFH) i z wyjściowymi stężeniami cholesterolu LDL między 4,1 a 10,4 mmol/l podzielono losowo do leczenia ezetymibem w dawce 10 mg podawanym jednocześnie z symwastatyną (10, 20 lub 40 mg) lub do leczenia samą symwastatyną (10, 20 lub 40 mg) przez okres 6 tygodni, po którym to okresie przez kolejne 27 tygodni stosowano albo ezetymib z 40 mg symwastatyny, albo tylko 40 mg symwastatyny a następnie, przez kolejne 20 tygodni stosowano w ramach próby otwartej ezetymib z symwastatyną (10 mg, 20 mg lub 40 mg).
Po 6 tygodniach ezetymib podawany jednocześnie z symwastatyną (wszystkie dawki) istotnie zmniejszał stężenie cholesterolu całkowitego (38% w porównaniu z 26%), cholesterolu LDL (49% w porównaniu z 34%), Apo B (39% w porównaniu z 27%) i cholesterolu nie-HDL (47% w porównaniu z 33%) w porównaniu z samą symwastatyną (wszystkie dawki). Wyniki dotyczące stężenia trójglicerydów i cholesterolu HDL w obu ocenianych grupach były podobne (odpowiednio -17% w porównaniu z -12% i +7% w porównaniu z +6%). Wyniki w 13. tygodniu były zgodne z wynikami z
6 tygodnia; istotnie więcej pacjentów otrzymujących ezetymib łącznie z 40 mg symwastatyny (62%) osiągnęło idealny cel leczenia według NCEP AAP (<2,8 mmol/l [110 mg/dl]) dla cholesterolu LDL w porównaniu z pacjentami otrzymującymi 40 mg symwastatyny (25%). W tygodniu 53, na koniec badania otwartego stanowiącego przedłużenie podwójnie ślepej próby, wpływ ocenianego leczenia na parametry lipidowe utrzymywał się.
Nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z symwastatyną w dawkach powyżej 40 mg na dobę u dzieci i młodzieży w wieku od 10 do 17 lat. Nie badano bezpieczeństwa i skuteczności ezetymibu podawanego jednocześnie z symwastatyną u pacjentów pediatrycznych w wieku < 10 lat. Nie badano czy długotrwałe leczenie ezetymibem pacjentów poniżej 17 lat wpływa na redukcję zachorowalności i śmiertelności w wieku dorosłym.
Zapobieganie wystąpieniu incydentów sercowo-naczyniowych
Badanie IMPROVE-IT było wieloośrodkowym, randomizowanym badaniem klinicznym, z podwójnie ślepą próbą i z kontrolą aktywną, przeprowadzonym z udziałem 18 144 pacjentów włączonych do badania w ciągu 10 dni od hospitalizacji z powodu wystąpienia ostrego zespołu wieńcowego (OZW, tj. ostrego zawału mięśnia sercowego lub niestabilnej dławicy piersiowej). W dniu przyjęcia do szpitala z powodu OZW u pacjentów, którzy nie stosowali leczenia obniżającego stężenie lipidów, stężenie cholesterolu LDL wynosiło ≤ 125 mg/dl (≤ 3,2 mmol/l), a u pacjentów, którzy stosowali leczenie obniżające stężenie lipidów, wynosiło ≤ 100 mg/dl (≤ 2,6 mmol/l). Wszystkich pacjentów przydzielano w sposób losowy w stosunku 1:1 do grupy otrzymującej ezetymib z symwastatyną w dawce 10 mg+ 40 mg (n=9067) lub symwastatynę w dawce 40 mg (n=9077). Mediana okresu obserwacji wynosiła 6,0 lat.
Średnia wieku pacjentów wynosiła 63,6 lat. 76% pacjentów stanowili mężczyźni, 84% pacjentów było rasy białej, a 27% pacjentów chorowało na cukrzycę. Średnie stężenie cholesterolu LDL w czasie wystąpienia incydentu kwalifikującego do badania u pacjentów stosujących leczenie obniżające stężenie lipidów (n=6390) i u tych, którzy nie stosowali leczenia obniżającego stężenie lipidów (n=11 594) wynosiło odpowiednio 80 mg/dl (2,1 mmol/l) i 101 mg/dl (2,6 mmol/l). Przed hospitalizacją z powodu wystąpienia OZW kwalifikującego do badania 34% pacjentów przyjmowało statynę. Po upływie jednego roku średnie stężenie cholesterolu LDL u pacjentów kontynuujących leczenie wynosiło 53,2 mg/dl (1,4 mmol/l) w grupie otrzymującej ezetymib z symwastatyną i 69,9 mg/dl
(1,8 mmol/l) w grupie przyjmującej symwastatynę w monoterapii. Stężenie lipidów badano na ogół u pacjentów, którzy w dalszym ciągu stosowali badane leczenie.
Na pierwszorzędowy punkt końcowy składał się zgon z przyczyn naczyniowo-sercowych, poważne incydenty wieńcowe (zdefiniowane jako zawał mięśnia sercowego niezakończony zgonem, udokumentowana niestabilna dławica piersiowa wymagająca hospitalizacji lub jakakolwiek procedura rewaskularyzacji wieńcowej przeprowadzona przynajmniej 30 dni po losowym przydzieleniu leczenia) oraz udar mózgu niezakończony zgonem. Badanie wykazało, że leczenie ezetymibem w skojarzeniu z symwastatyną pozwalało uzyskać dodatkowe korzyści w zmniejszeniu pierwszorzędowego złożonego punktu końcowego, tj. zgonu z przyczyn sercowo-naczyniowych, poważnego incydentu wieńcowego i udaru mózgu niezakończonego zgonem, w porównaniu z samą symwastatyną (zmniejszenie ryzyka względnego o 6,4%, p=0,016). Pierwszorzędowy punkt końcowy wystąpił u 2572 z 9067 pacjentów
(7-letni wskaźnik Kaplana-Meiera [KM] wynosił 32,72%) w grupie przyjmującej ezetymib z symwastatyną oraz u 2742 z 9077 pacjentów (7-letni wskaźnik KM wynosił 34,67%) w grupie otrzymującej samą symwastatynę (patrz Rycina 1 i Tabela 1.). Oczekuje się, że w przypadku jednoczesnego podawania innych statyn o udowodnionej skuteczności w zmniejszeniu ryzyka incydentów sercowo-naczyniowych korzyści te będą zbliżone. Całkowita liczba zgonów w tej grupie podwyższonego ryzyka nie uległa zmianie (patrz Tabela 1).
Stwierdzono ogólną korzyść w przypadku wszystkich udarów mózgu; jakkolwiek odnotowano niewielkie, nieistotne zwiększenie liczby udarów krwotocznych w grupie ezetymib-symwastatyna w porównaniu do grupy pacjentów otrzymujących samą symwastatynę (patrz Tabela 1). Ryzyko udaru krwotocznego w przypadku jednoczesnego podawania ezetymibu w skojarzeniu ze statyną o silniejszym działaniu nie zostało ocenione w długoterminowych badaniach.
Efekt leczenia ezetymibem z symwastatyną był na ogół spójny z efektami uzyskiwanymi w szeregu podgrup, tj. zależnie od płci, wieku, rasy, cukrzycy w wywiadzie, wyjściowego stężenia lipidów, wcześniejszego leczenia statyną, przebytego udaru mózgu i nadciśnienia.
Rycina 1: Wpływ leczenia skojarzonego ezetymibem z symwastatyną na pierwszorzędowy złożony punkt końcowy, tj. zgon z przyczyn sercowo-naczyniowych, poważny incydent wieńcowy lub udar mózgu niezakończony zgonem.
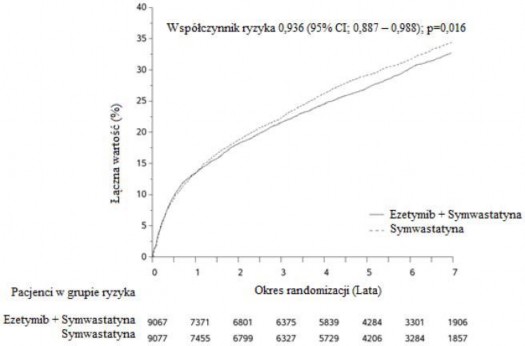
Tabela 1: Poważne incydenty sercowo-naczyniowe u wszystkich przydzielanych w sposób losowy pacjentów w badaniu IMPROVE-IT z podziałem na grupy leczenia
Wynik | Ezetymib +Symwastatyna 10 + 40 mga (N=9067) | Symwastatyna 40 mgb (N=9077) | Współczynnik ryzyka (95% CI) | Wartość p | ||
n | K-M % c | n | K-M % c | |||
Pierwszorzędowy złożony punkt końcowy skuteczności | ||||||
(zgon z przyczyn | 2572 | 32,72% | 2742 | 34,67% | 0,936 (0,887, | 0,016 |
sercowo-naczyniowych, | 0,988) | |||||
poważny incydent | ||||||
wieńcowy i udar mózgu | ||||||
niezakończony | ||||||
zgonem) | ||||||
Drugorzędowe złożone punkty końcowe skuteczności | ||||||
Zgon związany z | 1322 | 17,52% | 1448 | 18,88% | 0,912 (0,847, | 0,016 |
chorobą wieńcową, | 0,983) | |||||
zawał mięśnia | ||||||
sercowego | ||||||
niezakończony zgonem, | ||||||
rewaskularyzacja | ||||||
wieńcowa w trybie | ||||||
pilnym po 30 dniach | ||||||
Poważny incydent | 3089 | 38,65% | 3246 | 40,25% | 0,948 (0,903, | 0,035 |
wieńcowy, udar mózgu | 0,996) | |||||
niezakończony zgonem, | ||||||
zgon (z dowolnej | ||||||
przyczyny) | ||||||
Zgon z przyczyn | 2716 | 34,49% | 2869 | 36,20% | 0,945 (0,897, | 0,035 |
sercowo-naczyniowych, | 0,996) | |||||
zawał mięśnia | ||||||
sercowego | ||||||
niezakończony zgonem, | ||||||
niestabilna dławica | ||||||
piersiowa wymagająca | ||||||
hospitalizacji, | ||||||
jakakolwiek procedura | ||||||
rewaskularyzacji, udar | ||||||
mózgu niezakończony | ||||||
zgonem | ||||||
Składowe pierwszorzędowego złożonego punktu końcowego skuteczności i wybrane punkty końcowe skuteczności (pierwsze wystąpienie określonego zdarzenia w dowolnym momencie) | ||||||
Zgon z przyczyn | 537 | 6,89% | 538 | 6,84% | 1,000 (0,887, | 0,997 |
sercowo-naczyniowych | 1,127) | |||||
Poważny incydent wieńcowy: | ||||||
zawał mięśnia sercowego niezakończony zgonem | 945 | 12,77% | 1083 | 14,41% | 0,871 (0,798, 0,950) | 0,002 |
Niestabilna dławica piersiowa wymagająca hospitalizacji | 156 | 2,06% | 148 | 1,92% | 1,059 (0,846, 1,326) | 0,618 |
Rewaskularyzacja wieńcowa po 30 dniach | 1690 | 21,84% | 1793 | 23,36% | 0,947 (0,886, 1,012) | 0,107 |
Udar mózgu niezakończony zgonem | 245 | 3,49% | 305 | 4,24% | 0,802 (0,678, 0,949) | 0,010 |
Wszystkie przypadki zawału mięśnia sercowego (zakończone i niezakończone zgonem) | 977 | 13,13% | 1118 | 14,82% | 0,872 (0,800, 0,950) | 0,002 |
Wszystkie przypadki udaru (zakończone i niezakończone zgonem) | 296 | 4,16% | 345 | 4,77% | 0,857 (0,734, 1,001) | 0,052 |
Udar niekrwotoczny d | 242 | 3,48% | 305 | 4,23% | 0,793 (0,670, 0,939) | 0,007 |
Udar krwotoczny | 59 | 0,77% | 43 | 0,59% | 1,377 (0,930, 2,040) | 0,110 |
Zgon z dowolnej przyczyny | 1215 | 15,36% | 1231 | 15,28% | 0,989 (0,914, 1,070) | 0,782 |
a u 6% pacjentów dawkę ezetymibu + symwastatyny zwiększono do 10 + 80 mg.
b u 27% pacjentów dawkę symwastatyny zwiększono do 80 mg.
c estymator Kaplana-Meiera po 7 latach.
d w tym udar niedokrwienny mózgu lub udar nieokreślonego rodzaju.
Zapobieganie poważnym zdarzeniom naczyniowym w przewlekłej chorobie nerek
Badanie SHARP było wielonarodowym, randomizowanym badaniem, z grupą kontrolną placebo i podwójnie ślepą próbą, przeprowadzonym z udziałem 9438 pacjentów z przewlekłą chorobą nerek, z których w chwili rozpoczęcia badania 1/3 była poddawana hemodializie. 4650 pacjentów przydzielono do grupy leczonej produktem złożonym zawierającym skojarzenie ezetymibu i symwastatyny w stałej dawce 10/20 mg, a 4620 do grupy placebo. Obserwację prowadzono średnio przez 4,9 roku. Średnia wieku pacjentów wynosiła 62 lata, 63% uczestników stanowili mężczyźni, 72% osób było rasy białej, u 23% stwierdzono cukrzycę, a w przypadku pacjentów niedializowanych średnia szacunkowa szybkość przesączania kłębuszkowego (ang. eGFR, estimated glomerular filtration rate) wynosiła
26,5 ml/min/1,73 m2. W kryteriach przystąpienia do badania nie uwzględniono parametrów lipidowych.
Średnie wyjściowe stężenie frakcji cholesterolu LDL wynosiło 108 mg/dl. Po upływie jednego roku, w tym u pacjentów nie przyjmujących już badanego produktu leczniczego, stężenie cholesterolu LDL zmniejszyło się względem placebo o 26% w grupie leczonej symwastatyną w monoterapii w dawce 20 mg i o 38% w grupie stosującej ezetymib w dawce 10 mg w skojarzeniu z symwastatyną w dawce 20 mg.
Porównaniem pierwotnym określonym w protokole badania SHARP była analiza wyodrębniona zgodnie z zaplanowanym leczeniem „poważnych zdarzeń naczyniowych” (zdefiniowanych jako zawał mięśnia sercowego niezakończony zgonem lub zgon z przyczyn sercowych, udar mózgu lub jakakolwiek procedura rewaskularyzacji) wyłącznie u tych pacjentów przydzielanych w sposób
losowy na początku do grupy leczonej ezetymibem w skojarzeniu z symwastatyną (n=4193) lub do grupy placebo (n=4191). W analizach wtórnych uwzględniono tę samą kombinację zdarzeń, którą analizowano w całej kohorcie pacjentów przydzielanych w sposób losowy (w chwili rozpoczęcia badania lub po upływie 1 roku) do grupy leczonej ezetymibem w skojarzeniu z symwastatyną (n=4650) lub do grupy placebo (n=4620), a także poszczególne składowe tej kombinacji.
Analiza pierwszorzędowego punktu końcowego wykazała, że stosowanie ezetymibu w skojarzeniu z symwastatyną wiązało się z istotnym zmniejszeniem ryzyka wystąpienia poważnych zdarzeń naczyniowych (749 pacjentów ze stwierdzonymi zdarzeniami w grupie placebo, w porównaniu z 639 przypadkami w grupie leczonej ezetymibem w skojarzeniu z symwastatyną), przy czym ryzyko względne zmniejszyło się o 16% (p=0,001).
Jednakże plan tego badania nie pozwala na odrębną ocenę wpływu ezetymibu na skuteczność w zakresie istotnego zmniejszenia ryzyka występowania poważnych zdarzeń naczyniowych u pacjentów z przewlekłą chorobą nerek.
W Tabeli 2 przedstawiono poszczególne zdarzenia opisywane jako poważne incydenty naczyniowe, stwierdzone u pacjentów przydzielanych w sposób losowy. Stosowanie ezetymibu w skojarzeniu z symwastatyną wiązało się z istotnym zmniejszeniem ryzyka wystąpienia udaru mózgu oraz wykonania jakiejkolwiek procedury rewaskularyzacji, przy nieistotnych różnicach liczbowych na korzyść ezetymibu w skojarzeniu z symwastatyną w odniesieniu do zdarzeń w postaci zawału mięśnia sercowego niezakończonego zgonem i zgonu z przyczyn sercowych.
Tabela 2: Poważne zdarzenia naczyniowe stwierdzone u wszystkich przydzielanych w sposób losowy pacjentów w badaniu SHARPa z podziałem na grupy leczenia
Wynik | Ezetymib 10 mg w skojarzeniu z symwastatyną 20 mg (N=4650) | Placebo (N=4620) | Współczynnik ryzyka (95% CI) | Wartość p |
Poważne zdarzenia naczyniowe | 701 (15,1%) | 814 (17,6%) | 0,85 (0,77-0,94) | 0,001 |
Zawał mięśnia sercowego niezakończony zgonem | 134 (2,9%) | 159 (3,4%) | 0,84 (0,66-1,05) | 0,12 |
Zgon z przyczyn sercowych | 253 (5,4%) | 272 (5,9%) | 0,93 (0,78-1,10) | 0,38 |
Jakikolwiek udar | 171 (3,7%) | 210 (4,5%) | 0,81 (0,66-0,99) | 0,038 |
Niekrwotoczny udar mózgu | 131 (2,8%) | 174 (3,8%) | 0,75 (0,60-0,94) | 0,011 |
Krwotoczny udar mózgu | 45 (1,0%) | 37 (0,8%) | 1,21 (0,78-1,86) | 0,40 |
Jakakolwiek procedura rewaskularyzacji | 284 (6,1%) | 352 (7,6%) | 0,79 (0,68-0,93) | 0,004 |
Duże zdarzenia miażdżycowe (ang, MAE, major atherosclerotic events)b | 526 (11,3%) | 619 (13,4%) | 0,83 (0,74-0,94) | 0,002 |
a Analiza wyodrębniona zgodnie z zaplanowanym leczeniem z uwzględnieniem wszystkich uczestników badania SHARP przydzielanych w sposób losowy do grupy leczonej ezetymibem w skojarzeniu z symwastatyną lub do grupy placebo w chwili rozpoczęcia badania lub po upływie 1 roku.
b MAE: zdefiniowane jako złożony punkt końcowy obejmujący zawał mięśnia sercowego niezakończony zgonem, zgon z przyczyn wieńcowych, niekrwotoczny udar mózgu lub jakąkolwiek procedurę rewaskularyzacji.
W przypadku pacjentów, u których wyjściowe stężenie cholesterolu LDL było mniejsze (<
2,5 mmol/l) oraz pacjentów, którzy w chwili rozpoczęcia badania poddawani byli dializie, całkowite zmniejszenie stężenia frakcji cholesterolu LDL uzyskane za pomocą ezetymibu w skojarzeniu z symwastatyną było mniejsze niż w przypadku pozostałych pacjentów i, analogicznie, zmniejszenie ryzyka w obu tych grupach było relatywnie osłabione.
Homozygotyczna hipercholesterolemia rodzinna
W randomizowanym badaniu klinicznym z podwójnie ślepą próbą, trwającym 12 tygodni, wzięło udział 50 pacjentów z rozpoznaną klinicznie i (lub) na podstawie badań genetycznych, homozygotyczną hipercholesterolemią rodzinną, przyjmujących atorwastatynę lub symwastatynę (w dawce 40 mg) w monoterapii lub w połączeniu z aferezą LDL. Jednoczesne stosowanie ezetymibu z atorwastatyną (w dawce 40 lub 80 mg) lub symwastatyną (w dawce 40 lub 80 mg) powodowało znaczące zmniejszenie stężenia cholesterolu LDL (o 15%), w porównaniu z przyjmowaniem zwiększonych dawek atorwastatyny lub symwastatyny (z 40 mg do 80 mg) w monoterapii.
Zwężenie zastawki aorty
Badane Simvastatin and Ezetimibe for the Treatment of Aortic Stenosis (SEAS) było wieloośrodkowym, podwójnie ślepym, kontrolowanym placebo badaniem klinicznym, z medianą czasu trwania 4,4 lat i udziałem 1873 pacjentów z bezobjawowym zwężeniem zastawki aortalnej (ang. Aortic stenosis; AS),udokumentowanym poprzez maksymalną prędkość przepływu w badaniu dopplerowskim między 2,5 a 4,0 m/s. Do badania włączono wyłącznie tych pacjentów, którzy nie wymagali leczenia statyną w celu zmniejszenia ryzyka choroby sercowo-naczyniowej na tle miażdżycy. Pacjentów przydzielano losowo w stosunku 1:1 do grupy otrzymującej placebo lub ezetymib w dawce 10 mg w skojarzeniu z symwastatyną w dawce 40 mg na dobę.
Głównym punktem końcowym były rozpatrywane łącznie poważne zdarzenia sercowo-naczyniowe (major cardiovascular event; MCE), takie jak zgon sercowo-naczyniowy, operacja wymiany zastawki aortalnej (ang. aortic valve replacement; AVR), zastoinowa niewydolność serca (ang. congestive heart failure; CHF) na skutek progresji AS, zawał mięśnia sercowego niezakończony zgonem, operacja pomostowania tętnic wieńcowych (ang. coronary artery bypass grafting; CABG), przezskórna interwencja wieńcowa (ang. percutaneous coronary inrervention; PCI), hospitalizacja z powodu niestabilnej dławicy piersiowej oraz niekrwotoczny udar mózgu. Głównymi dodatkowymi punktami końcowymi były różne zestawienia zdarzeń wymienionych jako kategorie wchodzące w skład głównego punktu końcowego.
W porównaniu z placebo, leczenie ezetymibem/symwastatyną w dawce 10/40 mg nie powodowało istotnego zmniejszenia ryzyka MCE. Główny punkt końcowy wystąpił u 333 pacjentów (35,3%) w grupie ezetymibu/symwastatyny i u 355 pacjentów (38,2%) w grupie placebo (współczynnik ryzyka w grupie ezetymibu/symwastatyny 0,96; 95% przedział ufności 0,83 do 1,12; p = 0,59). Operację wymiany zastawki aortalnej przeprowadzono u 267 pacjentów (28,3%) w grupie ezetymibu/symwastatyny i u 278 pacjentów (29,9%) w grupie placebo (współczynnik ryzyka 1,00; 95% przedział ufności 0,84 do 1,18; p = 0,97). Niedokrwienne zdarzenia sercowo-naczyniowe wystąpiły u mniejszej liczby pacjentów z grupy ezetymibu/symwastatyny (n = 148) niż z grupy placebo (n=187) (współczynnik ryzyka 0,78; 95% przedział ufności 0,63 do 0,97; p = 0,02), głównie z powodu mniejszej liczby pacjentów poddanych operacji pomostowania tętnic wieńcowych.
Nowotwory występowały częściej w grupie ezetymibu/symwastatyny (105 w porównaniu z 70, p=0,01). Kliniczne znaczenie tej obserwacji nie jest pewne, gdyż w większym badaniu SHARP całkowite liczby pacjentów z nowo rozpoznanym nowotworem (438 w grupie ezetymibu/symwastatyny w porównaniu z 439 w grupie placebo) nie różniły się. Ponadto w badaniu IMPROVE-IT całkowita liczba pacjentów z nowo zdiagnozowanym nowotworem złośliwym (853 w grupie ezetymib/symwastatyna w stosunku do 863 w grupie otrzymującej tylko symwastatynę) nie różniła się znacząco, a więc wyniki badania SEAS nie zostały potwierdzone w badaniu SHARP lub IMPROVE-IT.
Wchłanianie
Po podaniu doustnym ezetymib jest szybko wchłaniany i w znacznym stopniu sprzęgany do postaci czynnego farmakologicznie glukuronianu fenolowego (glukuronian ezetymibu). Glukuronian ezetymibu osiąga maksymalne stężenie w osoczu (Cmax) średnio w ciągu 1-2 godzin, natomiast ezetymib w ciągu 4-12 godzin. Bezwzględna dostępność biologiczna ezetymibu nie może być określona, ponieważ jest on praktycznie nierozpuszczalny w wodnych roztworach do wstrzykiwań.
Przyjmowanie produktu leczniczego podczas posiłku (wysokotłuszczowego lub beztłuszczowego) nie miało wpływu na dostępność biologiczną ezetymibu podczas podawania w postaci tabletek o mocy 10 mg. Ezetymib może być przyjmowany z posiłkiem lub na czczo.
Dystrybucja
Ezetymib i glukuronian ezetymibu wiążą się z ludzkimi białkami osocza odpowiednio w 99,7% oraz w 88-92%.
Metabolizm
Ezetymib jest metabolizowany głównie w jelicie cienkim i w wątrobie przez sprzęganie z glukuronianem (reakcja fazy II), a następnie jest wydalany z żółcią. U wszystkich badanych gatunków obserwowano również nieznaczny metabolizm oksydacyjny (reakcja fazy I). Ezetymib i glukuronian ezetymibu są głównymi pochodnymi produktu leczniczego wykrywalnymi w osoczu, stanowiącymi odpowiednio ok. 10-20% i 80-90% całkowitego stężenia produktu leczniczego w osoczu. Obydwie postacie produktu leczniczego są powoli wydalane z osocza przy znaczącym udziale krążenia
jelitowo-wątrobowego. Okres półtrwania ezetymibu i glukuronianu ezetymibu wynosi ok. 22 godziny.
Eliminacja
Po doustnym podaniu ezetymibu znakowanego izotopem węgla 14C (w dawce 20 mg) ludziom, całkowity ezetymib stanowił ok. 93% całkowitej aktywności promieniotwórczej w osoczu. W ciągu dziesięciodniowej zbiórki, około 78% całkowitej ilości izotopu promieniotwórczego zostało wydalone z kałem, zaś 11% z moczem. Po 48 godzinach od podania produktu leczniczego nie stwierdzono wykrywalnego poziomu aktywności promieniotwórczej w osoczu.
Szczególne grupy pacjentów
Dzieci i młodzież
Farmakokinetyka ezetymibu jest podobna u dzieci ≥6 lat i u dorosłych. Brak danych dotyczących farmakokinetyki produktu leczniczego u dzieci w wieku <6 lat. Doświadczenie kliniczne obejmuje dzieci i młodzież z homozygotyczną hipercholesterolemią rodzinną i heterozygotyczną hipercholesterolemią rodzinną.
Osoby w podeszłym wieku
Stężenie całkowitego ezetymibu w osoczu było około dwukrotnie większe u pacjentów w podeszłym wieku (≥ 65 lat), niż u młodszych osób (od 18 do 45 lat). Zmniejszenie stężenia cholesterolu LDL oraz profil bezpieczeństwa były porównywalne po zastosowaniu ezetymibu w obu grupach. W związku z tym nie ma konieczności modyfikacji dawkowania u pacjentów w podeszłym wieku.
Zaburzenia czynności wątroby
Po podaniu ezetymibu w pojedynczej dawce 10 mg u pacjentów z łagodnymi zaburzeniami czynności wątroby (5 lub 6 punktów wg skali Child-Pugh), stwierdzono około 1,7- krotne zwiększenie średniej wartości pola pod krzywą (AUC) dla całkowitego ezetymibu, w porównaniu ze zdrowymi osobami. W 14-dniowym badaniu, w którym podawano dawki wielokrotne produktu leczniczego (10 mg na dobę) u pacjentów z umiarkowanymi zaburzeniami czynności wątroby (od 7 do 9 punktów wg skali Child- Pugh), średnia wartość AUC dla ezetymibu całkowitego zwiększyła się około 4-krotnie między pierwszym, a czternastym dniem badania, w porównaniu ze zdrowymi osobami. U pacjentów z łagodnymi zaburzeniami czynności wątroby nie jest konieczne dostosowanie dawkowania. Ze
względu na brak danych dotyczących wpływu zwiększonej ekspozycji na ezetymib u pacjentów z umiarkowanymi lub ciężkimi (>9 punktów wg skali Child-Pugh) zaburzeniami czynności wątroby, nie zaleca się stosowania ezetymibu w tej grupie pacjentów (patrz punkt 4.4).
Zaburzenia czynności nerek
Po podaniu ezetymibu w pojedynczej dawce 10 mg u pacjentów z ciężkimi zaburzeniami czynności nerek (n=8, średni klirens kreatyniny ≤30 ml/min/1,73 m2), średnia wartość AUC dla ezetymibu całkowitego zwiększyła się około 1,5-krotnie w porównaniu ze zdrowymi osobami (n=9). Różnice te nie są istotne klinicznie i nie ma konieczności dostosowania dawkowania u pacjentów z zaburzeniami czynności nerek.
U jednego pacjenta biorącego udział w tym badaniu (poddanego przeszczepieniu nerki i przyjmującego wiele produktów leczniczych, w tym cyklosporynę) stwierdzono 12-krotne zwiększenie ekspozycji na całkowity ezetymib.
Płeć
Stężenie całkowitego ezetymibu w osoczu jest nieco większe (o około 20%) u kobiet, niż u mężczyzn. Zmniejszenie stężenia cholesterolu LDL i profil bezpieczeństwa produktu leczniczego były porównywalne u kobiet i u mężczyzn leczonych ezetymibem. Nie ma zatem potrzeby dostosowania dawkowania w zależności od płci pacjenta.
Badania na zwierzętach dotyczące przewlekłego działania toksycznego nie wskazały na istnienie narządów narażonych na takie działanie. U psów, którym podawano ezetymib przez cztery tygodnie (≥0,03 mg/kg na dobę) stwierdzono zwiększenie stężenia cholesterolu w żółci znajdującej się w pęcherzyku żółciowym o 2,5 do 3,5 razy. W badaniu trwającym rok, przeprowadzonym u psów otrzymujących ezetymib w dawkach do 300 mg/kg/dobę, nie stwierdzono zwiększenia częstości występowania kamicy żółciowej lub innego oddziaływania na wątrobę i drogi żółciowe. Nie wiadomo, czy wyniki tych badań mają jakieś odniesienie do ludzi. Nie można wykluczyć ryzyka powstawania kamieni żółciowych w przypadku stosowania ezetymibu.
W badaniach, w których ezetymib stosowany był w skojarzeniu ze statynami, działania toksyczne były typowe, jak przy stosowaniu statyn. Niektóre z tych działań były nasilone przy skojarzonym stosowaniu produktów leczniczych w porównaniu ze stosowaniem statyn w monoterapii. Efekt ten ma związek z farmakokinetycznymi i farmakodynamicznymi interakcjami pomiędzy produktami leczniczymi stosowanymi w leczeniu skojarzonym. Nie obserwowano takich interakcji podczas badań klinicznych.
Miopatia występowała u szczurów dopiero po podaniu dawek kilkakrotnie większych od dawki terapeutycznej stosowanej u ludzi (około 20-krotnie większa wartość AUC dla statyn oraz 500-2000 razy większa wartość AUC dla czynnych metabolitów ezetymibu).
W szeregu badań in vivo i in vitro nie stwierdzono działania genotoksycznego ezetymibu stosowanego w monoterapii lub w skojarzeniu ze statynami. Wyniki badań rakotwórczego działania ezetymibu podczas długotrwałego stosowania były negatywne.
Ezetymib nie miał wpływu na płodność samic i samców szczurów, jak również nie wykazywał działania teratogennego na szczury czy króliki, nie miał też wpływu na rozwój zarodków i noworodków. Ezetymib przenikał przez barierę łożyskową u ciężarnych szczurów i królików podczas stosowania wielokrotnych dawek po 1000 mg/kg/dobę. Nie zaobserwowano działania teratogennego u szczurów podczas jednoczesnego stosowania ezetymibu w skojarzeniu ze statyną. U ciężarnych królików odnotowano niewielką liczbę zaburzeń rozwoju kośćca płodu (połączenie kręgów piersiowych i ogonowych, zmniejszenie liczby kręgów ogonowych). Stosowanie ezetymibu w skojarzeniu z lowastatyną wywierało działanie letalne na zarodki.
Kroskarmeloza sodowa Laktoza jednowodna Magnezu stearynian Celuloza mikrokrystaliczna Sodu laurylosiarczan Hypromeloza (3 mPas) Krospowidon
Nie dotyczy.
3 lata
Brak specjalnych środków ostrożności podczas przechowywania.
Przezroczyste blistry typu push-through PVC/Aclar/Aluminium w tekturowym pudełku Przezroczyste blistry typu push-through OPA/Alu/PVC/Aluminium w tekturowym pudełku Przezroczyste blistry zdzieralne PVC/Aclar/Papier/Aluminium w tekturowym pudełku.
Pudełko tekturowe: 10, 20, 28, 30, 50, 98 lub 100 tabletek.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Brak specjalnych wymagań.
Alchemia Limited
5th Floor, 86 Jermyn Street SW1Y 6AW Londyn Wielka Brytania
Pozwolenie nr 23970
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 10.05.2017
15.01.2018








