







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
dorosłych oraz dzieci i młodzieży z nowo rozpoznaną przewlekłą białaczką szpikową (ang. chronic myeloid leukaemia, CML) z chromosomem Philadelphia (bcr-abl, Ph+), którzy nie kwalifikują się do zabiegu transplantacji szpiku jako leczenia pierwszego rzutu;
dorosłych oraz dzieci i młodzieży z CML Ph+ w fazie przewlekłej, gdy leczenie interferonem alfa okazało się nieskuteczne lub w fazie akceleracji choroby albo w przebiegu przełomu blastycznego;
dorosłych pacjentów oraz dzieci i młodzieży z nowo rozpoznaną ostrą białaczką limfoblastyczną (ang. acute lymphoblastic leukaemia, ALL) z chromosomem Philadelphia (Ph+) w skojarzeniu
z chemioterapią;
dorosłych pacjentów z nawracającą lub oporną na leczenie Ph+ ALL w monoterapii;
dorosłych pacjentów z zespołami mielodysplastycznymi/mieloproliferacyjnymi (ang. myelodysplastic/myeloproliferate, MDS/MPD) związanymi z rearanżacją genu receptora płytkopochodnego czynnika wzrostu (ang. platelet-derived growth factor receptor, PDGFR);
dorosłych pacjentów z zaawansowanym zespołem hipereozynofilowym (ang. hypereosinophilic syndrome, HES) i (lub) przewlekłą białaczką eozynofilową (ang. chronic eosinophilic leukemia, CEL) z rearanżacją FIP1L1-PDGFRα.
Nie oceniano wpływu imatynibu na wynik przeszczepienia szpiku.
Produkt leczniczy Imatinib Sandoz jest wskazany w:
leczeniu dorosłych pacjentów ze złośliwymi, nieoperacyjnymi i (lub) z przerzutami, Kit (CD 117) - dodatnimi nowotworami podścieliskowymi przewodu pokarmowego (ang. Gastrointestinal Stromal Tumors - GIST).
leczeniu adjuwantowym dorosłych pacjentów z istotnym ryzykiem nawrotu po zabiegu usunięcia Kit (CD 117) - dodatnich nowotworów podścieliskowych przewodu pokarmowego (GIST). Pacjenci z małym lub bardzo małym ryzykiem nawrotu nie powinni otrzymywać leczenia adjuwantowego.
leczeniu dorosłych pacjentów z nieoperacyjnymi guzowatymi włókniakomięsakami skóry (ang. dermatofibrosarcoma protuberans - DFSP) oraz dorosłych pacjentów z nawracającymi i (lub) z przerzutami DFSP, którzy nie kwalifikują się do zabiegu chirurgicznego.
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Zapalenie płuc zgłaszano najczęściej u pacjentów z CML po transformacji oraz u pacjentów
z GIST.
Ból głowy występował najczęściej u pacjentów z GIST.
W analizie pacjentolat działania niepożądane dotyczące serca, w tym zastoinową niewydolność serca, częściej obserwowano u pacjentów z CML po transformacji niż u pacjentów z przewlekłą CML.
Zaczerwienienie skóry (głównie twarzy) występowało najczęściej u pacjentów z GIST,
a krwawienie (krwiak, krwotok) u pacjentów z GIST i pacjentów z CML po transformacji (CML- AP i CML-BC).
Wysięk opłucnowy zgłaszano częściej u pacjentów z GIST i u pacjentów z CML po transformacji
Zgłoszono kilka przypadków niewydolności wątroby i martwicy wątroby zakończonych zgonem.
Bóle mięśniowo-szkieletowe i podobne działania występowały częściej u pacjentów z CML
w porównaniu z pacjentami z GIST.
Przypadki zgonów były zgłaszane u pacjentów z zaawansowaną chorobą, silnymi zakażeniami, ciężką neutropenią i innymi poważnymi chorobami współistniejącymi.
Opisywano reaktywację wirusowego zapalenia wątroby typu B powiązaną ze stosowaniem inhibitorów kinazy tyrozynowej BCR-ABL. Niektóre przypadki prowadziły do ostrej niewydolności wątroby lub piorunującego zapalenia wątroby, a w konsekwencji do przeszczepienia wątroby lub zgonu pacjenta (patrz punkt 4.4).
Bóle mięśniowo-szkieletowe w trakcie leczenia imatynibem lub po jego zakończeniu
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu leczniczego do
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Imatinib Sandoz, 100 mg, tabletki powlekane Imatinib Sandoz, 400 mg, tabletki powlekane
Imatinib Sandoz, 100 mg
Każda tabletka powlekana zawiera 100 mg imatynibu (Imatinibum) w postaci imatynibu mezylanu.
Imatinib Sandoz, 400 mg
Każda tabletka powlekana zawiera 400 mg imatynimu (Imatinibum) w postaci imatynibu mezylanu.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana. Imatinib Sandoz, 100 mg
Ciemnożółta do brązowo-pomarańczowej, okrągła, obustronnie wypukła, ze skośnie ściętymi
krawędziami o średnicy około 9,2 mm, z wytłoczonym napisem „NVR” na jednej stronie i linią ułatwiającą podział między literami „SA” na drugiej stronie.
Tabletkę powlekaną można podzielić na równe dawki. Imatinib Sandoz, 400 mg
Ciemnożółta do brązowo-pomarańczowej, owalna, obustronnie wypukła, ze skośnie ściętymi
krawędziami o przybliżonej długości 19,2 mm i szerokości 7,7 mm, z wytłoczonym napisem „400” na jednej stronie i linią ułatwiającą podział między literami „SL” na drugiej stronie.
Tabletkę powlekaną można podzielić na równe dawki.
Produkt leczniczy Imatinib Sandoz jest wskazany w leczeniu:
U dorosłych pacjentów oraz dzieci i młodzieży, skuteczność imatynibu została oceniona na podstawie współczynnika ogólnej odpowiedzi hematologicznej i cytogenetycznej oraz okresu przeżycia wolnego od progresji choroby w CML, współczynnika odpowiedzi hematologicznej i cytogenetycznej w Ph+ ALL, MDS/MPD, współczynnika odpowiedzi hematologicznej w HES/CEL oraz na podstawie obiektywnego współczynnika odpowiedzi u dorosłych pacjentów z nieoperacyjnymi i (lub)
z przerzutami GIST i DFSP oraz na podstawie okresu przeżycia bez wznowy w leczeniu adjuwantowym GIST. Doświadczenie ze stosowaniem imatynibu u pacjentów z MDS/MPD związanymi z rearanżacją genu PDGFR jest bardzo ograniczone (patrz punkt 5.1). Z wyjątkiem nowo rozpoznanej przewlekłej białaczki szpikowej, brak kontrolowanych badań klinicznych wykazujących korzyść kliniczną lub zwiększone przeżycie w tych wskazaniach.
Leczenie powinien rozpoczynać lekarz z doświadczeniem w leczeniu pacjentów z hematologicznymi nowotworami złośliwymi i mięsakami złośliwymi.
Imatinib Sandoz, 100 mg
W celu podania dawki 400 mg i większej (patrz niżej zalecenia dotyczące dawkowania) można zastosować podzielną tabletkę o mocy 400 mg.
Imatinib Sandoz, 400 mg
W celu podania dawek innych niż 400 mg, 600 mg i 800 mg (patrz niżej zalecenia dotyczące dawkowania) można zastosować podzielną tabletkę o mocy 100 mg.
Zalecaną dawkę należy przyjmować doustnie podczas posiłku, popijając dużą ilością wody w celu zminimalizowania ryzyka podrażnienia przewodu pokarmowego. Dawki 400 mg lub 600 mg należy podawać raz na dobę, zaś dobową dawkę 800 mg należy podawać w dwóch dawkach po 400 mg, rano i wieczorem.
Dla pacjentów, którzy nie są w stanie połknąć tabletek powlekanych, tabletkę można wymieszać ze szklanką wody niegazowanej lub soku jabłkowego. Zaleconą ilość tabletek należy wsypać do napoju w odpowiedniej objętości (około 50 ml dla tabletki 100 mg i 200 ml dla tabletki 400 mg) i wymieszać łyżeczką. Uzyskaną zawiesinę należy podać pacjentowi natychmiast po całkowitym rozpadnięciu się tabletki (tabletek).
Dawkowanie w CML u dorosłych
Zalecaną dawką imatynibu u dorosłych pacjentów z przewlekłą białaczką szpikową w fazie przewlekłej jest 400 mg/dobę. Faza przewlekła jest definiowana, jako stan spełniający wszystkie następujące kryteria: liczba blastów we krwi i w szpiku <15%, liczba bazofilów we krwi obwodowej
<20%, liczba płytek krwi >100 x 109/l.
Zalecaną dawką imatynibu u dorosłych pacjentów z przewlekłą białaczką szpikową w fazie przyspieszonej (akceleracji) jest 600 mg/dobę. Faza akceleracji jest definiowana jako stan, który spełnia którekolwiek z podanych kryteriów: odsetek blastów we krwi lub szpiku ≥15%, ale <30%, liczba blastów i promielocytów we krwi lub szpiku ≥30% (pod warunkiem, że liczba blastów <30%), liczba bazofilów we krwi obwodowej ≥20%, liczba płytek <100 x 109/l i nie jest związana
z leczeniem.
Zalecaną dawką imatynibu u dorosłych pacjentów w przebiegu przełomu blastycznego jest
600 mg/dobę. Przełom blastyczny jest definiowany jako stan, w którym liczba blastów we krwi lub szpiku wynosi ≥30% lub jako obecność ognisk pozaszpikowych choroby innych niż w wątrobie
i śledzionie.
Czas trwania leczenia
W badaniach klinicznych leczenie imatynibem kontynuowano do czasu progresji choroby. Nie badano
wpływu zaprzestania leczenia po osiągnięciu pełnej odpowiedzi cytogenetycznej.
Zwiększenie dawki od 400 mg do 600 mg lub 800 mg u pacjentów z chorobą w fazie przewlekłej lub od 600 mg do maksymalnie 800 mg (podawanych, jako 400 mg dwa razy na dobę) u pacjentów
z chorobą w fazie przyspieszonej lub w przebiegu przełomu blastycznego, u których nie występują poważne działania niepożądane ani niezwiązana z białaczką poważna neutropenia lub trombocytopenia, można rozważyć w następujących przypadkach: postęp choroby (na każdym jej etapie); brak zadowalającej odpowiedzi hematologicznej po co najmniej 3 miesiącach leczenia; brak odpowiedzi cytogenetycznej po 12 miesiącach leczenia lub utrata uzyskanej uprzednio odpowiedzi hematologicznej i (lub) cytogenetycznej. Ze względu na ryzyko większej częstości działań niepożądanych, po zwiększeniu dawki należy uważnie obserwować stan pacjentów.
Dawkowanie w CML u dzieci
Dawkowanie u dzieci należy ustalać na podstawie powierzchni ciała (mg/m2 pc.). U dzieci w fazie przewlekłej CML i fazie zaawansowanej CML zaleca się dawkę 340 mg/m2 pc. na dobę (nie należy przekraczać całkowitej dawki 800 mg). Imatynib można podawać w jednej dawce na dobę lub dawkę dobową można podzielić na dwie części - jedną podawaną rano i drugą wieczorem. Zalecenia dotyczące dawkowania są oparte na danych uzyskanych u małej liczby dzieci i młodzieży (patrz punkty 5.1 i 5.2). Brak doświadczenia w leczeniu dzieci w wieku poniżej 2 lat.
U dzieci, u których nie występują poważne działania niepożądane ani niezwiązana z białaczką poważna neutropenia lub trombocytopenia, można rozważyć zwiększenie dawki z 340 mg/m2 pc. do 570 mg/m2 pc. na dobę (nie należy przekraczać całkowitej dawki 800 mg) w następujących przypadkach: postęp choroby (na każdym jej etapie); brak zadowalającej odpowiedzi hematologicznej po co najmniej 3 miesiącach leczenia; brak odpowiedzi cytogenetycznej po 12 miesiącach leczenia lub utrata osiągniętej uprzednio odpowiedzi hematologicznej i (lub) cytogenetycznej. Ze względu na ryzyko większej częstości działań niepożądanych, po zwiększeniu dawki należy uważnie obserwować stan pacjentów.
Dawkowanie w Ph+ ALL u dorosłych
Zalecana dawka imatynibu u dorosłych pacjentów z Ph+ ALL wynosi 600 mg/dobę. Wszystkie fazy leczenia powinni nadzorować hematolodzy z doświadczeniem w postępowaniu w tej chorobie.
Schemat dawkowania
Na podstawie aktualnych danych wykazano skuteczność i bezpieczeństwo stosowania imatynibu w dawce 600 mg na dobę w skojarzeniu z chemioterapią w fazie indukcji, konsolidacji i leczenia
podtrzymującego (patrz punkt 5.1) u dorosłych pacjentów z nowo rozpoznaną Ph+ ALL. Czas trwania leczenia imatynibem może różnić się w zależności od wybranego programu leczenia, ale na ogół dłuższa ekspozycja na imatynib dawała lepsze wyniki.
Dla dorosłych pacjentów z nawracającą lub oporną na leczenie Ph+ ALL monoterapia imatynibem w dawce 600 mg na dobę jest bezpieczna, skuteczna i może być stosowana do czasu wystąpienia
progresji choroby.
Dawkowanie w Ph+ ALL u dzieci
Dawkowanie u dzieci należy ustalać na podstawie powierzchni ciała (mg/m2 pc.). U dzieci z Ph+ ALL zaleca się dawkę dobową 340 mg/m2 pc. (nie należy przekraczać całkowitej dawki 600 mg).
Dawkowanie w MDS/MPD
Zalecana dawka imatynibu u dorosłych pacjentów z MDS/MPD wynosi 400 mg/dobę.
Czas trwania leczenia
W jedynym przeprowadzonym do tej pory badaniu klinicznym leczenie imatynibem kontynuowano do czasu wystąpienia progresji choroby (patrz punkt 5.1). W momencie przeprowadzania analizy mediana czasu leczenia wynosiła 47 miesięcy (24 dni - 60 miesięcy).
Dawkowanie w HES/CEL
Zalecana dawka imatynibu u dorosłych pacjentów z HES/CEL wynosi 100 mg na dobę.
Jeśli badania wykażą niewystarczającą odpowiedź na leczenie i u pacjenta nie występują działania niepożądane leku, można rozważyć zwiększenie dawki ze 100 mg do 400 mg.
Leczenie należy kontynuować tak długo, jak długo jest ono korzystne dla pacjenta. Dawkowanie w GIST
Zalecana dawka imatynibu u dorosłych pacjentów z postaciami GIST nieoperacyjnymi i (lub) z przerzutami, wynosi 400 mg/dobę.
Dane dotyczące zwiększenia dawki leku z 400 mg do 600 mg lub 800 mg u pacjentów, u których wystąpiła progresja choroby w czasie stosowania mniejszej dawki, są ograniczone (patrz punkt 5.1).
Czas leczenia: w badaniach klinicznych prowadzonych u pacjentów z GIST imatynib był podawany aż do wystąpienia progresji choroby. W momencie analizy danych mediana czasu leczenia wynosiła 7 miesięcy (od 7 dni do 13 miesięcy). Skutek zaprzestania leczenia po osiągnięciu odpowiedzi na leczenie nie został zbadany.
Zalecana dawka imatynibu w leczeniu adjuwantowym dorosłych pacjentów po resekcji GIST wynosi 400 mg na dobę. Optymalny czas trwania leczenia nie został jeszcze ustalony.
Długość leczenia tego wskazania w badaniu klinicznym wynosiła 36 miesięcy (patrz punkt 5.1).
Dawkowanie w DFSP
U dorosłych pacjentów z DFSP zalecana dawka imatynibu wynosi 800 mg na dobę.
Zmiana dawki ze względu na działania niepożądane
Pozahematologiczne działania niepożądane
Jeśli podczas stosowania imatynibu wystąpią u pacjenta poważne pozahematologiczne działania niepożądane, leczenie należy przerwać do czasu ich ustąpienia. Następnie, w zależności od początkowego nasilenia działania niepożądanego, można wznowić właściwe leczenie.
Jeśli stężenie bilirubiny przekroczy 3-krotnie górną granicę normy (GGN) lub aktywność aminotransferaz wątrobowych przekroczy 5-krotnie GGN, należy wstrzymać podawanie imatynibu do czasu, gdy stężenie bilirubiny będzie mniejsze niż 1,5-krotna wartość GGN, a aktywność aminotransferaz będzie mniejsza niż 2,5-krotna wartość GGN. Leczenie imatynibem można kontynuować, stosując zmniejszoną dawkę dobową. U dorosłych dawkę należy zmniejszyć z 400 do 300 mg lub z 600 do 400 mg albo z 800 do 600 mg, a u dzieci z 340 do 260 mg/m2 pc. na dobę.
Hematologiczne działania niepożądane
Zaleca się zmniejszenie dawki lub przerwanie leczenia w przypadku ciężkiej neutropenii lub trombocytopenii, zgodnie ze wskazówkami podanymi w poniższej tabeli.
Modyfikacja dawki u pacjentów z neutropenią i małopłytkowością:
HES/CEL (dawka początkowa 100 mg) | ANC <1,0 x 109/l i (lub) płytki krwi <50 x 109/l | ≥75 x 109/l. poprzednio stosowanej (tzn. przed wystąpieniem ciężkiego działania niepożądanego). |
CML w fazie przewlekłej, MDS/MPD i GIST (dawka początkowa 400 mg) HES/CEL (po dawce 400 mg) | ANC <1,0 x 109/l i (lub) płytki krwi <50 x 109/l | uzyskania ANC ≥1,5 x 109/l i płytek krwi ≥75 x 109/l. <1,0 x 109/l i (lub) płytek krwi <50 x 109/l, powtórzyć postępowanie z punktu 1, a następnie wrócić do podawania imatynibu w zmniejszonej dawce 300 mg. |
CML w fazie przewlekłej u dzieci i młodzieży (po dawce 340 mg/m2 pc.) | ANC <1,0 x 109/l i (lub) płytki krwi <50 x 109/l | ≥75 x 109/l. <1,0 x 109/l i (lub) płytek krwi <50 x 109/l, powtórzyć postępowanie z punktu 1, a następnie wrócić do podawania imatynibu w zmniejszonej dawce 260 mg/m2 pc. |
CML w fazie akceleracji i w fazie przełomu blastycznego i Ph+ ALL (dawka początkowa 600 mg) | aANC <0,5 x 109/l i (lub) płytki krwi <10 x 109/l | dawkę zmniejszyć do 300 mg. leczenie dawką 300 mg. |
CML w fazie akceleracji i przełomu blastycznego u dzieci i młodzieży 340 mg/m2 pc.) | aANC <0,5 x 109/l i (lub) płytki krwi <10 x 109/l | leczenie dawką 200 mg/m2 pc. |
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną
w punkcie 6.1.
Podczas stosowania imatynibu z innymi produktami leczniczymi istnieje prawdopodobieństwo wystąpienia interakcji. Należy zachować ostrożność podczas stosowania imatynibu z inhibitorami proteazy, azolowymi lekami przeciwgrzybiczymi, niektórymi antybiotykami makrolidowymi (patrz punkt 4.5), substratami CYP3A4 o wąskim indeksie terapeutycznym (tj. cyklosporyna, pimozyd, takrolimus, syrolimus, ergotamina, diergotamina, fentanyl, alfentanyl, terfenadyna, bortezomib, docetaksel, chynidyna) lub warfaryną i innymi pochodnymi kumaryny (patrz punkt 4.5).
Jednoczesne podawanie imatynibu i innych produktów leczniczych, które indukują CYP3A4 (tj. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital lub ziele dziurawca [Hypericum perforatum]) może istotnie zmniejszać ekspozycję na imatynib, z możliwością zwiększenia ryzyka niepowodzenia terapeutycznego. Dlatego należy unikać jednoczesnego stosowania silnych induktorów CYP3A4 i imatynibu (patrz punkt 4.5).
Niedoczynność tarczycy
Istnieją doniesienia o klinicznych przypadkach niedoczynności tarczycy u leczonych imatynibem pacjentów po usunięciu gruczołu tarczowego, którym zastępczo podawano lewotyroksynę (patrz punkt 4.5). U takich pacjentów należy ściśle kontrolować stężenie hormonu tyreotropowego (TSH).
Toksyczność dla wątroby
Metabolizm imatynibu zachodzi głównie w wątrobie, a tylko 13% jest wydalane przez nerki.
U pacjentów z zaburzeniami czynności wątroby (lekkimi, umiarkowanymi lub ciężkimi) należy uważnie kontrolować obraz krwi obwodowej oraz aktywność enzymów wątrobowych (patrz punkty 4.2, 4.8 i 5.2). Należy zauważyć, że pacjenci z nowotworami podścieliskowymi przewodu pokarmowego (ang. gastrointestinal stromal tumors, GIST) mogą mieć przerzuty do wątroby, które mogą spowodować zaburzenia jej czynności.
Podczas stosowania imatynibu obserwowano przypadki uszkodzenia wątroby, w tym niewydolność wątroby i martwicę wątroby. Podczas leczenia skojarzonego imatynibem i chemioterapią w dużych dawkach odnotowano zwiększenie częstości ciężkich działań niepożądanych dotyczących wątroby. Jeśli imatynib stosowany jest razem ze schematami chemioterapii, które mogą powodować zaburzenia czynności wątroby, należy dokładnie kontrolować czynność wątroby (patrz punkt 4.5 i 4.8).
Zatrzymanie płynów
U około 2,5% otrzymujących imatynib pacjentów z nowo rozpoznaną CML występowało znacznego stopnia zatrzymanie płynów (wysięk opłucnowy, obrzęki, obrzęk płuc, wodobrzusze, powierzchowny obrzęk). Dlatego bardzo zaleca się regularne kontrolowanie masy ciała tych pacjentów.
Nieoczekiwane, szybkie zwiększenie masy ciała należy dokładnie przeanalizować i, w razie konieczności, wdrożyć odpowiednie postępowanie wspomagające i lecznicze. W badaniach klinicznych stwierdzono zwiększenie liczby takich przypadków u osób w podeszłym wieku oraz pacjentów z chorobą serca w wywiadzie. Dlatego należy zachować ostrożność u pacjentów
z zaburzeniem czynności serca.
Pacjenci z chorobą serca
Należy uważnie monitorować pacjentów z chorobami serca, czynnikami ryzyka niewydolności serca lub niewydolnością nerek w wywiadzie, a wszyscy pacjenci z przedmiotowymi lub podmiotowymi objawami odpowiadającymi niewydolności serca lub nerek wymagają oceny lekarskiej i leczenia.
U pacjentów z zespołem hipereozynofilowym (HES) z utajonym naciekaniem komórek zespołu hipereozynofilowego w obrębie mięśnia sercowego, pojedyncze przypadki wstrząsu kardiogennego
i (lub) zaburzeń funkcji lewej komory związały się z degranulacją komórek HES przed rozpoczęciem leczenia imatynibem. Donoszono, że stan ten jest odwracalny po podaniu steroidów o działaniu ogólnym, zastosowaniu środków podtrzymujących krążenie i czasowym odstawieniu imatynibu.
Ponieważ po zastosowaniu imatynibu niezbyt często zgłaszano działania niepożądane dotyczące serca, przed rozpoczęciem leczenia należy dokonać uważnej oceny stosunku korzyści do ryzyka związanego
z leczeniem imatynibem w populacji z HES/CEL.
Zespoły mielodysplastyczne/mieloproliferacyjne (MDS/MPD) z rearanżacją genu PDGFR mogą być związane z dużą liczbą eozynofilów. Dlatego u pacjentów z HES/CEL i u pacjentów z MDS/MPD związanymi z dużą liczbą eozynofilów przed rozpoczęciem leczenia należy rozważyć konsultację
z kardiologiem, wykonanie echokardiogramu i oznaczenie stężenia troponiny w surowicy. Jeśli którykolwiek z wyników tych badań okaże się nieprawidłowy, należy rozważyć dalszą obserwację kardiologiczną i profilaktyczne zastosowanie steroidów układowych (1-2 mg/kg mc.) przez jeden lub dwa tygodnie na początku leczenia, jednocześnie z podawaniem imatynibu.
Krwawienie z przewodu pokarmowego
W badaniu z udziałem pacjentów z GIST nieoperacyjnymi i (lub) z przerzutami stwierdzono zarówno krwawienia z przewodu pokarmowego, jak i krwawienia wewnątrz guza (patrz punkt 4.8). Na podstawie dostępnych danych nie określono czynników predysponujących (tj. wielkość guza, umiejscowienie guza, zaburzenia krzepnięcia), które mogłyby identyfikować pacjentów z GIST do grupy zwiększonego ryzyka jednego z tych rodzajów krwawienia. Ponieważ zwiększenie unaczynienia i skłonność do krwawień jest cechą i naturalnym obrazem klinicznym GIST, należy zastosować standardowe postępowanie i procedury w celu monitorowania i leczenia krwawienia
u wszystkich pacjentów.
Ponadto, po wprowadzeniu imatynibu do obrotu zgłaszano u pacjentów z CML, ALL i innymi chorobami występowanie poszerzenia naczyń okolicy przedodźwiernikowej żołądka, tzw. żołądka arbuzowatego(ang. gastric antral vascular ectasia, GAVE), rzadkiej przyczyny krwawienia z przewodu pokarmowego (patrz punkt 4.8). W razie konieczności można rozważyć przerwanie leczenia imatynibem.
Zespół rozpadu guza
Ze względu na możliwość wystąpienia zespołu rozpadu guza (ang. tumour lysis syndrome, TLS), przed rozpoczęciem leczenia imatynibem zaleca się wyrównanie klinicznie istotnego odwodnienia oraz leczenie w celu zmniejszenia podwyższonego stężenia kwasu moczowego (patrz punkt 4.8).
Reaktywacja wirusowego zapalenia wątroby typu B
U pacjentów będących przewlekłymi nosicielami wirusa zapalenia wątroby typu B dochodziło do reaktywacji zapalenia wątroby po otrzymaniu przez nich inhibitorów kinazy tyrozynowej BCR-ABL. Niektóre przypadki prowadziły do ostrej niewydolności wątroby lub piorunującego zapalenia wątroby, a w konsekwencji do przeszczepienia wątroby lub zgonu pacjenta.
U pacjentów należy wykonać badania pod kątem zakażenia HBV przed rozpoczęciem leczenia produktem leczniczym Imatinib Sandoz. Przed rozpoczęciem leczenia u pacjentów z dodatnim wynikiem badania serologicznego w kierunku wirusowego zapalenia wątroby typu B (w tym
u pacjentów z aktywną chorobą) i w przypadku pacjentów z dodatnim wynikiem badania w kierunku zakażenia HBV w trakcie leczenia, należy skonsultować się z ekspertami ds. chorób wątroby
i leczenia wirusowego zapalenia wątroby typu B. Nosiciele wirusa HBV, którzy wymagają leczenia produktem leczniczym Imatinib Sandoz, powinni być poddawani ścisłej obserwacji pod kątem objawów podmiotowych i przedmiotowych aktywnego zakażenia HBV w trakcie całego okresu leczenia i przez kilka miesięcy po jego zakończeniu (patrz punkt 4.8).
Fototoksyczność
Należy unikać bezpośredniej ekspozycji lub zminimalizować bezpośrednią ekspozycję na światło słoneczne ze względu na ryzyko fototoksyczności związanej z leczeniem imatynibem. Pacjentów należy poinformować o konieczności stosowania środków zapobiegawczych, takich jak odzież ochronna oraz preparaty z filtrem o wysokim wskaźniku ochrony przeciwsłonecznej (SPF).
Mikroangiopatia zakrzepowa
Stosowanie inhibitorów kinazy tyrozynowej (TKI) BCR-ABL jest związane z występowaniem mikroangiopatii zakrzepowej (ang. thrombotic microangiopathy, TMA), co obejmuje zgłoszenia pojedynczych przypadków po zastosowaniu imatynibu (patrz punkt 4.8). Jeśli u pacjenta
otrzymującego produkt Imatinib Sandoz wystąpią laboratoryjne lub kliniczne cechy TMA, leczenie należy przerwać i przeprowadzić gruntowną ocenę w celu wykrycia TMA, obejmującą aktywność ADAMTS13 i oznaczenie miana przeciwciał przeciwko ADAMTS13. Jeśli miano przeciwciał przeciwko ADAMTS13 jest podwyższone z jednocześnie występującą małą aktywnością ADAMTS13, leczenia produktem Imatinib Sandoz nie należy wznawiać.
Badania laboratoryjne
Podczas leczenia imatynibem należy regularnie wykonywać pełne badanie krwi. Leczenie imatynibem pacjentów z CML wiązało się z wystąpieniem neutropenii lub małopłytkowości. Jednak wystąpienie tych cytopenii zależy prawdopodobnie od stopnia zaawansowania choroby i jest częstsze u pacjentów w fazie akceleracji CML lub w przełomie blastycznym niż u pacjentów w fazie przewlekłej choroby.
W takich przypadkach można przerwać leczenie imatynibem lub zmniejszyć jego dawkę, zgodnie
z zaleceniami w punkcie 4.2.
U pacjentów otrzymujących imatynib należy regularnie oceniać czynność wątroby (aminotransferazy, bilirubina, fosfataza zasadowa).
Wydaje się, że u pacjentów z zaburzeniami czynności nerek stężenie imatynibu w osoczu jest większe niż u pacjentów z prawidłową czynnością nerek, prawdopodobnie na skutek zwiększonego stężenia
u nich kwaśnej alfa-glikoproteiny (ang. alpha-acid glycoprotein, AGP), białka wiążącego imatynib.
U pacjentów z zaburzeniami czynności nerek należy stosować minimalną dawkę początkową.
U pacjentów z ciężkimi zaburzeniami czynności nerek należy zachować ostrożność podczas leczenia.
Jeśli dawka nie jest tolerowana, można ją zmniejszyć (patrz punkty 4.2 i 5.2).
Długotrwałe leczenie imatynibem może wiązać się z klinicznie istotnym pogorszeniem czynności nerek. Dlatego czynność nerek należy ocenić przed rozpoczęciem leczenia imatynibem i ściśle kontrolować w trakcie terapii, ze zwróceniem szczególnej uwagi na pacjentów z czynnikami ryzyka dysfunkcji nerek. W razie stwierdzenia zaburzeń czynności nerek, należy wdrożyć odpowiednie postępowanie i leczenie, zgodnie z wytycznymi dla standardowej terapii.
Dzieci i młodzież
Istnieją doniesienia o przypadkach opóźnienia wzrostu u otrzymujących imatynib dzieci i młodzieży przed okresem dojrzewania. W obserwacyjnym badaniu w populacji dzieci i młodzieży z CML,
w dwóch małych podgrupach zgłoszono statystycznie istotne (ale o niepewnym znaczeniu klinicznym) zmniejszenie mediany odchylenia standardowego wzrostu po 12 i 24 miesiącach leczenia, niezależnie od dojrzewania płciowego lub płci pacjentów. Zaleca się ścisłe kontrolowanie wzrostu u dzieci
w czasie leczenia imatynibem (patrz punkt 4.8).
Substancje czynne, które mogą zwiększać stężenie imatynibu w osoczu
Substancje hamujące aktywność izoenzymu CYP3A4 cytochromu P450 (np. inhibitory proteazy, takie jak indynawir, lopinawir z rytonawirem, rytonawir, sakwinawir, telaprewir, nelfinawir, boceprewir; azolowe leki przeciwgrzybicze, w tym ketokonazol, itrakonazol, pozakonazol, worykonazol; niektóre antybiotyki makrolidowe, takie jak erytromycyna, klarytromycyna i telitromycyna) mogą spowalniać metabolizm imatynibu i zwiększać jego stężenia. U zdrowych ochotników stwierdzono znaczące zwiększenie ekspozycji na imatynib (średnie wartości Cmax i AUC imatynibu zwiększyły się odpowiednio o 26% i 40%) po jednoczesnym podaniu jednorazowo ketokonazolu (inhibitor CYP3A4). Należy zachować ostrożność podczas jednoczesnego stosowania imatynibu z inhibitorami enzymów rodziny CYP3A4.
Substancje czynne, które mogą zmniejszać stężenie imatynibu w osoczu
Substancje będące induktorami CYP3A4 (np. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital, fosfenytoina, prymidon lub ziele dziurawca [Hypericum perforatum]) mogą znacząco zmniejszyć ekspozycję na imatynib, potencjalnie zwiększając ryzyko niepowodzenia terapeutycznego. Wielokrotne podawanie ryfampicyny w dawce 600 mg, po którym nastąpiło podanie jednej dawki imatynibu 400 mg, spowodowało zmniejszenie wartości Cmax i AUC(0-∞) odpowiednio o co najmniej
54% i 74% wobec wartości uzyskanych bez uprzedniego podawania ryfampicyny. Podobne wyniki obserwowano u pacjentów z glejakami złośliwymi, otrzymujących imatynib podczas stosowania przeciwpadaczkowych leków pobudzających enzymy (ang. enzyme-inducing anti-epileptic drugs, EIAED), takich jak karbamazepina, okskarbazepina i fenytoina. Wartość AUC imatynibu w osoczu zmniejszyła się o 73% w porównaniu z pacjentami nieotrzymującymi EIAED. Należy unikać jednoczesnego stosowania ryfampicyny lub innych silnych induktorów CYP3A4 i imatynibu.
Substancje czynne, których stężenie w osoczu może zostać zmienione przez imatynib
Imatynib zwiększa średnie wartości Cmax i AUC symwastatyny (substratu CYP3A4) odpowiednio
2- oraz 3,5-krotnie, co wskazuje na hamowanie aktywności CYP3A4 przez imatynib. Dlatego zaleca się ostrożność podczas podawania imatynibu z substratami CYP3A4 z wąskim indeksem terapeutycznym (tj. cyklosporyna, pimozyd, takrolimus, syrolimus, ergotamina, diergotamina, fentanyl, alfentanyl, terfenadyna, bortezomib, docetaksel i chynidyna). Imatynib może zwiększać stężenie w osoczu innych produktów leczniczych metabolizowanych przez CYP3A4 (np. triazolobenzodiazepin, antagonistów kanału wapniowego z grupy dihydropirydyny, niektórych inhibitorów reduktazy HMG-CoA takich jak statyny itd.).
Ze względu na znane ryzyko zwiększenia krwawienia (np. krwotok) związane z zastosowaniem imatynibu, pacjentom, u których konieczne jest stosowanie leków przeciwzakrzepowych, należy podawać niskocząsteczkową lub standardową heparynę zamiast pochodnych kumaryny, takich jak warfaryna.
W warunkach in vitro imatynib hamuje aktywność izoenzymu CYP2D6 cytochromu P450
w stężeniach podobnych do tych, które mają wpływ na aktywność CYP3A4. Imatynib w dawce
400 mg dwa razy na dobę hamował metabolizm metoprololu z udziałem CYP2D6, przy czym wartości Cmax i AUC metoprololu zwiększało się o około 23% (90% CI [1,16-1,30]). Wydaje się, że podczas jednoczesnego stosowania imatynibu z substratami CYP2D6 modyfikacja dawki nie jest konieczna, ale zaleca się ostrożność, jeśli stosowane są substraty CYP2D6 o wąskim indeksie terapeutycznym, takie jak metoprolol. U pacjentów leczonych metoprololem należy rozważyć monitorowanie kliniczne.
Imatynib hamuje in vitro O-glukuronidację paracetamolu przy wartości Ki 58,5 mikromol/l. Takiego hamowania nie obserwowano in vivo po podaniu 400 mg imatynibu i 1000 mg paracetamolu. Nie badano jednoczesnego stosowania większych dawek imatynibu i paracetamolu.
Należy zachować ostrożność podczas jednoczesnego stosowania dużych dawek imatynibu i paracetamolu.
U otrzymujących lewotyroksynę pacjentów po usunięciu tarczycy stężenie w osoczu lewotyroksyny może zmniejszyć się podczas jednoczesnego stosowania imatynibu (patrz punkt 4.4). Z tego względu zaleca się ostrożność. Mechanizm obserwowanej interakcji jest obecnie nieznany.
Istnieje kliniczne doświadczenie dotyczące jednoczesnego stosowania imatynibu i chemioterapii
u pacjentów z Ph+ ALL (patrz punkt 5.1), ale nie określono dokładnie interakcji typu lek-lek między imatynibem a chemioterapeutykami. Możliwe jest nasilenie działań niepożądanych imatynibu, takich jak hepatotoksyczność, mielosupresja lub inne. Istnieją doniesienia, że jednoczesne stosowanie
z L-asparaginazą mogło wiązać się ze zwiększeniem toksycznego działania na wątrobę (patrz punkt 4.8). Dlatego stosowanie imatynibu razem z innymi produktami leczniczymi wymaga szczególnej ostrożności.
Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznych metod antykoncepcji w trakcie
leczenia i przez co najmniej 15 dni po zakończeniu leczenia imatynibem.
Ciąża
Istnieją ograniczone dane dotyczące stosowania imatynibu u kobiet w ciąży. Po wprowadzeniu do
obrotu zgłaszano występowanie samoistnych poronień i wad wrodzonych u dzieci matek, które przyjmowały imatynib. Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3), a potencjalne zagrożenie dla płodu nie jest znane. Imatynibu nie należy stosować w okresie ciąży, jeśli nie jest to bezwzględnie konieczne. Jeśli imatynib jest stosowany w okresie ciąży, pacjentka musi być poinformowana o możliwym ryzyku dla płodu.
Karmienie piersią
Informacje dotyczące przenikania imatynibu do mleka kobiecego są ograniczone. Badania z udziałem dwóch kobiet karmiących piersią wykazały, że zarówno imatynib, jak i jego czynny metabolit mogą przenikać do mleka. Oceniana u jednej z pacjentek proporcja stężenia w mleku do stężenia w osoczu wynosiła 0,5 dla imatynibu i 0,9 dla metabolitu, co wskazuje na większe przenikanie metabolitu do mleka. Biorąc pod uwagę połączone stężenie imatynibu i metabolitu oraz maksymalne dzienne spożycie mleka przez niemowlęta, całkowite przewidywane narażenie powinno być małe (~10% dawki leczniczej). Jednak ze względu na to, że nieznane są skutki narażenia niemowlęcia na małe dawki imatynibu, kobiety nie powinny karmić piersią w trakcie leczenia imatynibem i przez co najmniej 15 dni po jego zakończeniu.
Płodność
W badaniach nieklinicznych wykazano brak wpływu na płodność samców i samic szczura, jednak obserwowano wpływ na parametry reprodukcyjne (patrz punkt 5.3). Nie przeprowadzono badań wpływu na płodność i gametogenezę z udziałem pacjentów otrzymujących imatynib. Pacjenci, którzy są zaniepokojeni wpływem imatynibu na płodność, powinni skonsultować się z lekarzem.
Pacjentów należy poinformować o możliwości wystąpienia podczas leczenia imatynibem działań niepożądanych, takich jak zawroty głowy, niewyraźne widzenie lub senność. Dlatego należy zalecić zachowanie ostrożności podczas prowadzenia pojazdów lub obsługiwania maszyn.
Pacjenci w zaawansowanym stadium nowotworów złośliwych mogą mieć szereg współistniejących zaburzeń, które utrudniają ustalenie związku przyczynowego z działaniami niepożądanymi, ze względu na różnorodność objawów związanych z chorobą podstawową, jej postęp i jednoczesne przyjmowanie licznych produktów leczniczych.
W badaniach klinicznych u pacjentów z CML przerwanie leczenia ze względu na wystąpienie działań niepożądanych odnotowano u 2,4% pacjentów z nowo rozpoznaną chorobą, u 4% pacjentów
w późnym okresie fazy przewlekłej po niepowodzeniu terapii interferonem i 4% pacjentów w fazie akceleracji choroby po niepowodzeniu terapii interferonem oraz u 5% pacjentów z przełomem blastycznym po niepowodzeniu terapii interferonem. W badaniach klinicznych dotyczących GIST leczenie przerwano u 4% pacjentów z powodu wystąpienia działań niepożądanych związanych
z lekiem.
Poza dwoma wyjątkami, działania niepożądane były podobne we wszystkich wskazaniach.
U pacjentów z CML obserwowano więcej przypadków mielosupresji niż u pacjentów z GIST, co jest prawdopodobnie związane z chorobą podstawową. W badaniu z udziałem pacjentów
z nieoperacyjnymi i (lub) z przerzutami GIST u 7 pacjentów (5%) wystąpiły krwawienia z przewodu pokarmowego stopnia 3. lub 4. wg CTC (ang. Common Toxicity Criteria) (3 pacjentów), krwawienia w obrębie guza (3 pacjentów) lub krwawienia obu rodzajów (1 pacjent). Umiejscowienie guza
w przewodzie pokarmowym może być przyczyną krwawienia z przewodu pokarmowego (patrz punkt 4.4). Krwawienia z przewodu pokarmowego i krwawienia wewnątrz guza mogą być poważne
i czasami zakończone zgonem. Najczęściej zgłaszanymi (≥10%) działaniami niepożądanymi związanymi z lekiem były w obu wskazaniach lekkie nudności, wymioty, biegunka, ból brzucha, zmęczenie, ból mięśni, kurcze mięśni i wysypka. We wszystkich badaniach często obserwowano powierzchowne obrzęki, opisywane głównie jako obrzęki wokół oczu i obrzęk kończyn dolnych. Jednak rzadko były one ciężkie i ustępowały po podaniu leków moczopędnych, innych środków
wspomagających lub po zmniejszeniu dawki imatynibu.
Gdy imatynib był stosowany razem z chemioterapią w dużych dawkach u pacjentów z Ph+ ALL, obserwowano przemijające działanie uszkadzające wątrobę w postaci zwiększenia aktywności aminotransferaz i hiperbilirubinemii. Biorąc pod uwagę ograniczoną wielkość bazy danych
o bezpieczeństwie, działania niepożądane zgłaszane do tej pory u dzieci są zgodne ze znanym profilem bezpieczeństwa u dorosłych pacjentów z Ph+ ALL. Ilość danych dotyczących bezpieczeństwa u dzieci z Ph+ ALL jest bardzo ograniczona, ale nie odnotowano żadnych nowych problemów związanych z bezpieczeństwem.
Różnorodne działania niepożądane, takie jak wysięk opłucnowy, wodobrzusze, obrzęk płuc i szybkie
zwiększenie masy ciała z obrzękami powierzchniowymi lub bez nich można ogólnie opisać, jako
„zatrzymanie płynów”. Działania te można najczęściej opanować wstrzymując czasowo stosowanie imatynibu, podając leki moczopędne i stosując inne odpowiednie metody podtrzymujące. Jednak niektóre z tych działań niepożądanych mogą być poważne lub zagrażające życiu, a opisano kilka przypadków zgonów pacjentów w przełomie blastycznym, ze złożoną historią kliniczną (wysięk opłucnowy, zastoinowa niewydolność serca i niewydolność nerek). W badaniach klinicznych u dzieci i młodzieży nie stwierdzono szczególnych działań niepożądanych.
Działania niepożądane
Niżej wymieniono działania niepożądane zgłaszane częściej niż jeden raz, zgodnie z klasyfikacją układów i narządów i częstością. Częstości zdefiniowano następująco: bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1000 do <1/100), rzadko (≥1/10 000 do <1000), bardzo rzadko (<1/10 000), częstość nieznana (nie może być określona na podstawie dostępnych danych).
W obrębie każdej grupy o określonej częstości objawy niepożądane przedstawiono, zaczynając od najczęstszych.
Działania niepożądane i ich częstość przedstawione w tabeli 1.
Tabela 1 Tabelaryczne zestawienie działań niepożądanych
Zakażenia i zarażenia pasożytnicze | |
Niezbyt często: | Ospa, opryszczka, zapalenie nosa i gardła, zapalenie płuc1, zapalenie zatok, zapalenie tkanki łącznej, zakażenie górnych dróg oddechowych, grypa, zakażenie dróg moczowych, zapalenie błony śluzowej żołądka i jelit, posocznica |
Rzadko: | Zakażenie grzybicze |
Częstość nieznana: | Reaktywacja wirusowego zapalenia wątroby typu B11 |
Nowotwory łagodne, złośliwe i nieokreślone (w tym torbiele i polipy) | |
Rzadko: | Zespół rozpadu guza |
Częstość nieznana: | Krwawienie z guza/martwica guza* |
Zaburzenia układu immunologicznego | |
Częstość nieznana: | Wstrząs anafilaktyczny* |
Zaburzenia krwi u układu chłonnego | |
Bardzo często: | Neutropenia, małopłytkowość, niedokrwistość |
Często: | Pancytopenia, gorączka neutropeniczna |
Niezbyt często: | Trombocytoza, limfopenia, zahamowanie czynności szpiku kostnego, eozynofilia, powiększenie węzłów chłonnych |
Rzadko: | Niedokrwistość hemolityczna, mikroangiopatia zakrzepowa |
Zaburzenia metabolizmu i odżywiania | |
Często: | Jadłowstręt |
Niezbyt często: | Hipokaliemia, zwiększenie apetytu, hipofosfatemia, zmniejszenie apetytu, odwodnienie, dna, hiperurykemia, hiperkalcemia, hiperglikemia, hiponatremia |
Rzadko: | Hiperkaliemia, hipomagnezemia |
Zaburzenia psychiczne | |
Często: | Bezsenność |
Niezbyt często: | Depresja, osłabienie libido, lęk |
Rzadko: | Stan splątania |
Zaburzenia układu nerwowego | |
Bardzo często: | Ból głowy2 |
Często: | Zawroty głowy pochodzenia ośrodkowego, parestezje, zaburzenia smaku, niedoczulica |
Niezbyt często: | Migrena, senność, omdlenie, neuropatia obwodowa, zaburzenia pamięci, rwa kulszowa, zespół niespokojnych nóg, drżenie, krwawienie mózgowe |
Rzadko: | Zwiększenie ciśnienia śródczaszkowego, drgawki, zapalenie nerwu wzrokowego |
Częstość nieznana: | Obrzęk mózgu* |
Zaburzenia oka | |
Często: | Obrzęk powiek, nasilone łzawienie, krwawienie w obrębie spojówek, zapalenie spojówek, suchość oka, niewyraźne widzenie |
Niezbyt często: | Podrażnienie oka, ból oka, obrzęk oczodołu, krwotok twardówkowy, krwawienie do siatkówki, zapalenie powiek, obrzęk plamki |
Rzadko: | Zaćma, jaskra, tarcza zastoinowa |
Częstość nieznana: | Krwotok do ciała szklistego* |
Zaburzenia ucha i błędnika | |
Niezbyt często: | Zawroty głowy pochodzenia obwodowego, szumy uszne, utrata słuchu |
Zaburzenia serca | |
Niezbyt często: | Kołatanie serca, tachykardia, zastoinowa niewydolność serca3, obrzęk płuc |
Rzadko: | Zaburzenia rytmu serca, migotanie przedsionków, zatrzymanie akcji serca, zawał mięśnia sercowego, dławica piersiowa, wysięk osierdziowy |
Częstość nieznana: | Zapalenie osierdzia*, tamponada serca* |
Zaburzenia naczyniowe4 | |
Często: | Zaczerwienienie skóry, krwotok |
Niezbyt często: | Nadciśnienie tętnicze, krwiak, krwiak podtwardówkowy, ziębnięcie rąk i stóp, niedociśnienie tętnicze, objaw Raynauda |
Częstość nieznana: | Zakrzepica/zator* |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Często: | Duszność, krwawienie z nosa, kaszel |
Niezbyt często: | Wysięk opłucnowy5, ból gardła i krtani, zapalenie gardła |
Rzadko: | Ból związany z zapaleniem opłucnej, zwłóknienie płuc, nadciśnienie płucne, krwotok płucny |
Częstość nieznana: | Ostra niewydolność oddechowa10*, śródmiąższowa choroba płuc* |
Zaburzenia żołądka i jelit | |
Bardzo często: | Nudności, biegunka, wymioty, niestrawność, ból brzucha6 |
Często: | Wzdęcia, rozdęcie brzucha, refluks żołądkowo-przełykowy, zaparcie, suchość w jamie ustnej, zapalenie błony śluzowej żołądka |
Niezbyt często: | Zapalenie jamy ustnej, owrzodzenie jamy ustnej, krwotok z przewodu pokarmowego7, odbijanie się, smoliste stolce, zapalenie przełyku, wodobrzusze, wrzód żołądka, krwawe wymioty, zapalenie czerwieni wargowej, dysfagia, zapalenie trzustki |
Rzadko: | Zapalenie jelita grubego, ileus, zapalna choroba jelit |
Częstość nieznana: | Ileus/niedrożność jelit*, perforacja żołądka i jelit*, zapalenie uchyłka*, poszerzenie naczyń okolicy przedodźwiernikowej żołądka* |
Zaburzenia wątroby i dróg żółciowych | |
Często: | Zwiększenie aktywności enzymów wątrobowych |
Niezbyt często: | Hiperbilirubinemia, zapalenie wątroby, żółtaczka |
Rzadko: | Niewydolność wątroby8, martwica wątroby |
Zaburzenia skóry i tkanki podskórnej | |
Bardzo często: | Obrzęk okołooczodołowy, zapalenie skóry/wyprysk/wysypka |
Często: | Świąd, obrzęk twarzy, suchość skóry, rumień, łysienie, nocne poty, reakcja nadwrażliwości na światło |
Niezbyt często: | Wysypka krostkowa, siniaki, nasilone pocenie, pokrzywka, wylew krwawy podskórny, wzmożona tendencja do występowania siniaków, skąpe owłosienie, odbarwienie skóry, złuszczające zapalenie skóry, łamliwość paznokci, zapalenie mieszków włosowych, wybroczyny, łuszczyca, plamica, nadmierna pigmentacja skóry, wysypki pęcherzowe, zapalenie tkanki tłuszczowej (w tym rumień guzowaty). |
Rzadko: | Ostra dermatoza z gorączką i neutrofilią (zespół Sweeta), przebarwienie paznokci, obrzęk naczynioruchowy, wysypka pęcherzykowa, rumień wielopostaciowy, leukoklastyczne zapalenie naczyń, zespół Stevensa- Johnsona, ostra uogólniona osutka krostkowa (AGEP), pęcherzyca* |
Częstość nieznana: | Zespół ręka-stopa*, rogowacenie liszajowate*, liszaj płaski*, toksyczne martwicze oddzielanie się naskórka*, osutka polekowa z eozynofilią i objawami układowymi (DRESS)*, pseudoporfiria* |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Bardzo często: | Skurcze i kurcze mięśni, bóle mięśniowo-szkieletowe, w tym ból mięśni12, ból stawów, ból kości9 |
Często: | Obrzęk stawów |
Niezbyt często: | Sztywność mięśni i stawów, martwica kości* |
Rzadko: | Osłabienie mięśni, zapalenie stawów, rabdomioliza/miopatia |
Częstość nieznana: | Opóźnienie wzrastania u dzieci* |
Zaburzenia nerek i dróg moczowych | |
Niezbyt często: | Ból nerki, krwiomocz, ostra niewydolność nerek, zwiększona częstość oddawania moczu |
Częstość nieznana: | Przewlekła niewydolność nerek |
Zaburzenia układu rozrodczego i piersi | |
Niezbyt często: | Ginekomastia, zaburzenia wzwodu, krwotok miesiączkowy, nieregularne miesiączkowanie, zaburzenia funkcji seksualnych, ból sutków, powiększenie piersi, obrzęk moszny |
Rzadko: | Krwotoczne ciałko żółte/krwotoczna torbiel jajnika |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często: | Zatrzymanie płynów i obrzęk, zmęczenie |
Często: | Osłabienie, gorączka, obrzęk tkanki podskórnej, dreszcze, zesztywnienie mięśni |
Niezbyt często: | Ból w klatce piersiowej, złe samopoczucie |
Badania diagnostyczne | |
Bardzo często: | Zwiększenie masy ciała |
Często: | Zmniejszenie masy ciała |
Niezbyt często: | Zwiększenie stężenia kreatyniny, zwiększenie aktywności kinazy kreatynowej we krwi, zwiększenie aktywności dehydrogenazy mleczanowej, zwiększenie aktywności fosfatazy zasadowej |
Rzadko: | Zwiększenie aktywności amylazy we krwi |
* Ten rodzaj działań niepożądanych zgłaszano głównie w okresie po wprowadzeniu imatynibu do
obrotu. Obejmuję one doniesienia spontaniczne, a także poważne działania niepożądane
z trwających badań, programów rozszerzonego dostępu, badań farmakologii klinicznej i badań eksploracyjnych w niezarejestrowanych wskazaniach. Ponieważ działania te notowano
w populacji o nieokreślonej liczebności, oszacowanie ich częstości lub ustalenie związku przyczynowego z ekspozycją na imatynib nie zawsze jest możliwe.
(CML-AP i CML-BC) niż u pacjentów z CML w fazie przewlekłej.
6+7 Ból brzucha i krwotok z przewodu pokarmowego były najczęściej obserwowane u pacjentów
z GIST.
obserwowano w okresie po wprowadzeniu imatynibu do obrotu.
Zmiany wyników badań laboratoryjnych
Hematologia
We wszystkich badaniach u pacjentów z CML obserwowano niedobór krwinek, zwłaszcza neutropenię i małopłytkowość, sugerując ich większą częstość po zastosowaniu imatynibu w dużych dawkach ≥750 mg (badanie I fazy). Jednak niedobór krwinek miał także ścisły związek ze stopniem zaawansowania choroby, a częstość neutropenii 3. lub 4. stopnia (ANC <1,0 x 109/l)
i małopłytkowości (liczba płytek krwi < 50 x 109/l) była 4 do 6 razy większa u pacjentów w przełomie
blastycznym i fazie akceleracji choroby (odpowiednio 59-64% i 44-63% dla neutropenii
i małopłytkowości) w porównaniu z pacjentami z nowo rozpoznaną CML w fazie przewlekłej (16,7% neutropenia i 8,9% małopłytkowość). U pacjentów z nowo rozpoznaną CML w przewlekłej fazie choroby neutropenię 4. stopnia (ANC <0,5 x 109/l) i małopłytkowość (liczba płytek krwi
<10 x 109/l) obserwowano odpowiednio u 3,6% i <1% pacjentów. Mediana czasu trwania neutropenii wynosiła od 2 do 3 tygodni, zaś małopłytkowości od 3 do 4 tygodni. Zdarzenia te można było zazwyczaj opanować zmniejszając dawkę lub przerywając stosowanie imatynibu, ale w rzadkich przypadkach konieczne było trwałe przerwanie leczenia. U dzieci i młodzieży z CML najczęściej obserwowanymi zaburzeniami były niedobory krwinek 3. lub 4. stopnia obejmujące neutropenię, małopłytkowość i niedokrwistość. Występowały one na ogół w ciągu kilku pierwszych miesięcy leczenia.
W badaniu z udziałem pacjentów z nieoperacyjnymi i (lub) przerzutowymi GIST zgłaszano niedokrwistość 3. i 4. stopnia występującą odpowiednio u 5,4% i 0,7% pacjentów. Mogła się ona wiązać z krwawieniami z przewodu pokarmowego oraz krwawieniami wewnątrz guza, przynajmniej u niektórych pacjentów. Neutropenię stopnia 3. i 4. stwierdzono odpowiednio u 7,5% i 2,7% pacjentów, a małopłytkowość stopnia 3. u 0,7% pacjentów. U żadnego z pacjentów nie stwierdzono trombocytopenii 4. stopnia. Zmniejszenie liczby białych krwinek oraz neutrofilów występowało głównie w ciągu pierwszych 6 tygodni leczenia i pozostawało względnie stałe w trakcie dalszego leczenia.
Biochemia
U pacjentów z CML obserwowano znaczne zwiększenie aktywności aminotransferaz (<5%) lub stężenia bilirubiny (<1%). Zmiany te zazwyczaj ustępowały po zmniejszeniu dawki lub przerwaniu leczenia (mediana czasu trwania tych incydentów wynosił około jednego tygodnia). Leczenie trwale przerywano z powodu nieprawidłowych parametrów laboratoryjnych wątroby u mniej niż 1% pacjentów z CML. U pacjentów z GIST (badanie B2222) obserwowano zwiększenie aktywności AlAT (aminotransferazy alaninowej) stopnia 3. lub 4. u 6,8% pacjentów, a zwiększenie aktywności AspAT (aminotransferazy asparaginianowej) stopnia 3. lub 4. u 4,8% pacjentów. Zwiększenie stężenia bilirubiny nie przekraczało 3%.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301/faks: + 48 22 49 21 309/ strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Doświadczenie dotyczące skutków zastosowania dawek większych niż zalecana dawka lecznicza jest ograniczone. Pojedyncze przypadki przedawkowania imatynibu zgłaszano spontanicznie i opisywano w piśmiennictwie. W wypadku przedawkowania należy obserwować pacjenta i zastosować odpowiednie leczenie objawowe. Na ogół zgłaszanym w tych przypadkach wynikiem było
„polepszenie” lub „wyzdrowienie”. Zdarzenia notowane po zastosowaniu różnych zakresów dawek
były następujące:
Dorośli
1200 do 1600 mg (czas trwania w zakresie od 1 do 10 dni): nudności, wymioty, biegunka, wysypka, rumień, obrzęk, obrzmienie, zmęczenie, kurcze mięśni, małopłytkowość, pancytopenia, ból brzucha, ból głowy, zmniejszenie apetytu.
1800 do 3200 mg (aż do 3200 mg/dobę przez 6 dni): osłabienie, ból mięśni, zwiększenie aktywności
kinazy kreatynowej, zwiększenie stężenia bilirubiny, ból żołądkowa i jelit.
6400 mg (dawka pojedyncza, jedno doniesienie literaturowe dotyczące jednego pacjenta): nudności, wymioty, ból brzucha, gorączka, obrzęk twarzy, zmniejszenie liczby neutrofilów, zwiększenie aktywności aminotransferaz.
8 do 10 g (dawka pojedyncza): wymioty i ból żołądka i jelit.
Dzieci i młodzież
U jednego 3-letniego chłopca zastosowanie pojedynczej dawki 400 mg wywołało wymioty, biegunkę i jadłowstręt, u innego 3-letniego chłopca po podaniu pojedynczej dawki 980 mg wystąpiło zmniejszenie liczby białych krwinek i biegunka.
W razie przedawkowania pacjenta należy obserwować oraz zastosować odpowiednie leczenie
wspomagające.
Grupa farmakoterapeutyczna: inhibitor kinazy białkowo-tyrozynowej, kod ATC: L01EA01
Imatynib jest inhibitorem kinazy białkowo-tyrozynowej o małej cząsteczce, która silnie hamuje aktywność kinazy tyrozynowej (KT) Bcr-Abl oraz kilku receptorów kinaz tyrozynowych: Kit, receptora czynnika wzrostu komórek macierzystych (SCF) kodowanego przez protoonkogen c-Kit, receptory domeny dyskoidynowej (DDR1 i DDR2), receptory czynnika stymulującego kolonie (CSF- 1R) oraz receptory alfa i beta płytkopochodnego czynnika wzrostu (PDGFR-alfa i PDGFR-beta).
Imatynib może również hamować procesy komórkowe, w aktywacji których pośredniczą wymienione receptory kinaz.
Działania farmakodynamiczne
Imatynib jest inhibitorem kinazy białkowo-tyrozynowej, który silnie hamuje kinazę tyrozynową Bcr-Abl in vitro, w komórce i in vivo. Związek ten wybiórczo hamuje proliferację i wywołuje apoptozę w komórkach linii Bcr-Abl dodatnich, a także w komórkach białaczkowych świeżo pobranych od pacjentów z CML z dodatnim chromosomem Philadelphia i od pacjentów z ostrą białaczką limfoblastyczną (ALL) z dodatnim chromosomem Philadelphia.
W badaniach in vivo na modelach zwierzęcych z użyciem Bcr-Abl dodatnich komórek nowotworowych imatynib sam wykazuje działanie przeciwnowotworowe.
Imatynib jest również inhibitorem receptorów kinaz tyrozynowych płytkopochodnego czynnika aktywacji (ang. platelet-derived growth factor, PDGF), PDGF-R i czynnika komórek pnia (ang. stem cell factor, SCF), c-Kit i hamuje procesy komórkowe aktywowane przez PDGF i SCF. In vitro imatynib hamuje proliferację i indukuje apoptozę komórek nowotworów podścieliskowych przewodu pokarmowego (GIST), w których stwierdzono ekspresję mutacji kit. Istotna aktywacja receptora PDGF lub kinaz białkowo-tyrozynowych Abl w wyniku połączenia się różnych odpowiadających sobie białek lub istotne wytwarzanie PDGF są wpisane w patogenezę MDS/MPD, HES/CEL i DFSP. Imatynib hamuje przekazywanie sygnałów i proliferację komórek zależną od zaburzonej regulacji aktywności PDGFR i kinazy Abl.
Badania kliniczne w przewlekłej białaczce szpikowej
Skuteczność imatynibu jest oceniana na podstawie stopnia całkowitej odpowiedzi hematologicznej
i cytogenetycznej oraz czasu przeżycia wolnego od progresji choroby. Z wyjątkiem nowo rozpoznanej przewlekłej białaczki szpikowej w fazie przewlekłej brak kontrolowanych badań klinicznych, które wykazywałyby takie korzyści kliniczne, jak zmniejszenie objawów związanych z chorobą lub zwiększone przeżycie.
Trzy duże, międzynarodowe, otwarte, niekontrolowane badania II fazy przeprowadzono u pacjentów z CML z dodatnim chromosomem Philadelphia (Ph+) w fazie zaawansowanej, w przełomie blastycznym lub fazie akceleracji, z innymi białaczkami (Ph+) lub z CML w fazie przewlekłej, po niepowodzeniu wcześniejszej terapii interferonem-alfa (IFN). Jedno duże, otwarte, wieloośrodkowe, międzynarodowe, randomizowane badanie III fazy przeprowadzono u pacjentów z nowo rozpoznaną CML z dodatnim chromosomem Philadelphia (Ph+). Ponadto dzieci leczono w ramach dwóch badań I fazy i jednego badania II fazy.
We wszystkich badaniach klinicznych 38-40% pacjentów było w wieku ≥60 lat, a 10-12% pacjentów było w wieku ≥70 lat.
Faza przewlekła, nowo rozpoznana
W badaniu III fazy u dorosłych pacjentów porównywano leczenie imatynibem w monoterapii z leczeniem skojarzonym interferonem alfa (IFN) z cytarabiną (Ara-C). Pacjenci, u których nie uzyskano odpowiedzi na leczenie [brak całkowitej odpowiedzi hematologicznej (ang. complete
haematological response, CHR) w 6. miesiącu, zwiększenie liczby krwinek białych, brak większej odpowiedzi cytogenetycznej (ang. major cytogenetic response, MCyR) w 24. miesiącu), utrata odpowiedzi (utrata CHR lub MCuR) lub ciężka nietolerancja leczenia, mogli być przeniesieni do innego ramienia badania. W grupie imatynibu pacjenci otrzymywali dawkę 400 mg/dobę. W ramieniu IFN pacjenci otrzymywali podskórnie dawkę docelową IFN wynoszącą 5 mln j.m./m2/dobę
w połączeniu z Ara-C podawaną podskórnie w dawce 20 mg/m2/dobę przez 10 dni w miesiącu.
Łącznie 1106 pacjentów poddano randomizacji (po 553 w każdym z ramion badania). Charakterystyka pacjentów obu ramion przed rozpoczęciem badania była zbliżona. Mediana wieku wynosiła 51 lat
(w zakresie od 18 do 70), a 21,9% pacjentów miało co najmniej 60 lat; 59% stanowili mężczyźni, a 41% kobiety; 89,9% było rasy kaukaskiej, a 4,7% rasy czarnej. Po siedmiu latach od włączenia ostatniego pacjenta mediana czasu trwania leczenia pierwszego rzutu wyniosła 82 miesiące
w ramieniu imatynibu oraz 8 miesięcy w ramieniu IFN. Mediana czasu trwania leczenia drugiego rzutu imatynibem wyniosła 64 miesiące. U pacjentów otrzymujących imatynib w leczeniu pierwszego rzutu średnia dawka dobowa wyniosła 406 ± 76 mg. Głównym parametrem skuteczności był okres
przeżycia bez progresji choroby. Postęp choroby definiowano, jako jedno z następujących zdarzeń: progresja do fazy akceleracji lub przełom blastyczny, zgon, utrata CHR lub MCyR, a u pacjentów, którzy nie osiągnęli CHR, zwiększenie liczby krwinek białych mino odpowiedniego leczenia. Większa odpowiedź cytogenetyczna, odpowiedź hematologiczna, odpowiedź molekularna (ocena choroby resztkowej), czas do wystąpienia fazy akceleracji lub przełomu blastycznego i czas przeżycia były głównymi parametrami drugorzędowymi. Dane dotyczące odpowiedzi przedstawiono w Tabeli 2.
Tabela 2 Odpowiedź na leczenie w badaniu (dane po 84 miesiącach) u pacjentów z nowo rozpoznaną CML
(Wskaźniki najlepszej odpowiedzi) | Imatynib n=553 | IFN+Ara-C n=553 |
Odpowiedź hematologiczna Wskaźnik CHR n (%) [95% CI] | 534 (96,6%)* [94,7%, 97,9%] | 313 (56,6%)* [52,4%, 60,8%] |
Odpowiedź cytogenetyczna | ||
Większa odpowiedź n (%) | 490 (88,6%)* | 129 (23,3%)* |
[95% CI] | [85,7%, 91,1%] | [19,9%, 27,1%] |
Całkowita CyR n (%) | 456 (82,5%)* | 64 (11,6%)* |
Częściowa CyR n (%) | 34 (6,1%) | 65 (11,8%) |
Odpowiedź molekularna** | ||
Większa odpowiedź w 12. miesiącu (%) | 153/305=50,2% | 8/83=9,6% |
Większa odpowiedź w 24. miesiącu (%) | 73/104=70,2% | 3/12=25% |
Większa odpowiedź w 84. miesiącu (%) | 02/116=87,9% | 3/4=75% |
* p<0.001, test Fischera ** odsetki odpowiedzi molekularnej na podstawie oceny dostępnych próbek Kryteria odpowiedzi hematologicznej (wszystkie odpowiedzi należy potwierdzić po ≥4 tygodniach): leukocyty <10 x 109/l, płytki krwi <450 x 109/l, mielocyty+metamielocyty <5% we krwi, brak blastów i promielocytów we krwi, bazofile <20%, brak pozaszpikowych ognisk białaczki Kryteria odpowiedzi cytogenetycznej: całkowita (0% Ph+ metafaz), częściowa (1–35%), mniejsza (36–65%) lub minimalna (66–95%). Większa odpowiedź (0–35%) stanowi połączenie odpowiedzi całkowitej i częściowej. Kryteria większej odpowiedzi molekularnej: zmniejszenie ilości transkryptu Bcr-Abl we krwi obwodowej ≥3 logarytmy (mierzone ilościową metodą RT-PCR w czasie rzeczywistym) w stosunku do wystandaryzowanych wartości wyjściowych. | ||
Wskaźniki całkowitej odpowiedzi hematologicznej, większej odpowiedzi cytogenetycznej oraz całkowitej odpowiedzi cytogenetycznej w leczeniu pierwszego rzutu szacowano stosując metodę Kaplana-Meiera, w której obserwacje przypadków bez uzyskanej odpowiedzi były cenzorowane datą ostatniego badania. Uzyskane z zastosowaniem tej metody szacowane skumulowane wskaźniki odpowiedzi u pacjentów leczonych w pierwszym rzucie imatynibem poprawiły się po 12 i 84 miesiącach leczenia, odpowiednio: CHR z 96,4% do 98,4% i CCyR z 69,5% do 87,2%.
W ciągu 7 lat obserwacji odnotowano 93 (16,8%) przypadki progresji choroby u pacjentów otrzymujących imatynib: 37 (6,7%) przypadki progresji do fazy akceleracji / przełomu blastycznego, 31 (5,6%) przypadków utraty MCyR, 15 (2,7%) utraty CHR lub zwiększenia liczby krwinek białych oraz 10 (1,8%) przypadków zgonu bez związku z CML. Natomiast w ramieniu IFN+Ara-C odnotowano 165 (29,8%) zdarzeń, z czego 130 wystąpiło podczas stosowania IFN+Ara-C jako leczenia pierwszego rzutu.
Szacowany odsetek pacjentów bez progresji do fazy akceleracji lub przełomu blastycznego po
84 miesiącach był istotnie większy w grupie pacjentów otrzymujących imatynib niż u pacjentów leczonych IFN (92,5% w porównaniu z 85,1%, p<0,001). Roczny współczynnik progresji do fazy
akceleracji lub przełomu blastycznego zmniejszał się z czasem trwania leczenia i był mniejszy niż 1% rocznie w czwartym i piątym roku. Szacowany współczynnik przeżycia bez postępu choroby po
84 miesiącach wyniósł 81,2% w ramieniu imatynibu i 60,6% w ramieniu kontrolnym (p<0,001). Roczne współczynniki progresji jakiegokolwiek typu dla imatynibu także zmniejszały się w czasie.
Łącznie zmarło 71 (12,8%) pacjentów z grupy leczonej imatynibem i 85 (15,4%) pacjentów z grupy otrzymującej IFN+Ara-C. Po 84 miesiącach szacowane przeżycie całkowite wynosiło 86,4% (83, 90) w porównaniu z 83,3% (80, 87) odpowiednio dla pacjentów zakwalifikowanych do grupy leczonej imatynibem oraz do grupy otrzymującej IFN+Ara-C (p=0,073, log-rank test). Na ten punkt końcowy, czas do wystąpienia zdarzenia, znaczny wpływ miał duży odsetek pacjentów, u których zmieniono leczenie z IFN+Ara-C na imatynib. Wpływ leczenia imatynibem na przeżycie pacjentów z nowo rozpoznaną CML w fazie przewlekłej był dalej badany w retrospektywnej analizie powyższych danych dotyczących imatynibu z uwzględnieniem danych z innego badania III fazy z zastosowaniem IFN+Ara-C (n=325) o identycznym schemacie dawkowania. W tej retrospektywnej analizie wykazano przewagę imatynibu nad IFN+Ara-C w odniesieniu do wpływu na czas całkowitego przeżycia (p<0,001); w ciągu 42 miesięcy zmarło 47 (8,5%) pacjentów leczonych imatynibem i 63 (19,4%) pacjentów leczonych wg schematu IFN+Ara-C.
Stopień odpowiedzi cytogenetycznej i odpowiedzi molekularnej miał wyraźny wpływ na długoterminowe wyniki leczenia pacjentów otrzymujących imatynib. Podczas gdy szacunkowo 96% (93%) pacjentów z CCyR (PCyR) w 12. miesiącu było wolnych od progresji do fazy akceleracji / przełomu blastycznego po 84 miesiącach, jedynie u 81% pacjentów bez MCyR w 12. miesiącu nie doszło do progresji do zaawansowanej fazy CML po 84 miesiącach badania (całkowite p<0,001, p=0,25 między CCyR a PCyR). U pacjentów ze zmniejszeniem ilości transkrypcji Bcr-Abl nie mniejszej niż 3 logarytmy po 12 miesiącach, prawdopodobieństwo pozostawania w grupie bez progresji do fazy akceleracji/przełomu blastycznego wynosiło 99% po 84 miesiącach. Podobne obserwacje zebrano w oparciu o analizę badania po 18 miesiącach.
W tym badaniu możliwe było zwiększenie dawki z 400 mg na dobę do 600 mg na dobę, a następnie z 600 mg na dobę do 800 mg na dobę. Po 42 miesiącach obserwacji u 11 pacjentów nastąpiła potwierdzona utrata (w ciągu 4 tygodni) odpowiedzi cytogenetycznej. Spośród tych 11 pacjentów, u 4 dawkę leku zwiększono do 800 mg na dobę, przy czym u 2 z nich odpowiedź cytogenetyczną odzyskano (u 1 pacjenta częściową i u 1 pacjenta całkowitą, a w tym ostatnim przypadku uzyskano
również odpowiedź molekularną), podczas gdy z pozostałych 7 pacjentów, u których nie zwiększano dawki leku, całkowitą odpowiedź cytogenetyczną odzyskano tylko u 1 pacjenta. Odsetek pewnych działań niepożądanych był większy u 40 pacjentów, którym dawkę leku zwiększono do 800 mg na dobę, w porównaniu z populacją pacjentów przed zwiększeniem dawki (n=551). Do częściej występujących działań niepożądanych należały krwotoki z przewodu pokarmowego, zapalenie spojówek, podwyższona aktywność aminotransferaz lub stężenia bilirubiny. Inne działania niepożądane występowały z mniejszą lub równą częstością.
Faza przewlekła, niepowodzenie leczenia interferonem
532 dorosłych pacjentów leczono dawką początkową 400 mg. Pacjentów podzielono na trzy główne grupy: niepowodzenie w zakresie parametrów hematologicznych (29%), niepowodzenie w zakresie parametrów cytogenetycznych (35%) oraz nietolerancja interferonu (36%). Uprzednio pacjenci byli leczeni IFN przez średnio (mediana) 14 miesięcy w dawkach ≥25 x 106 j.m./tydzień. Wszyscy pacjenci byli w późnej fazie przewlekłej, a mediana czasu od chwili rozpoznania choroby wynosiła 32 miesiące. Głównym parametrem skuteczności w tym badaniu był wskaźnik większej odpowiedzi cytogenetycznej (odpowiedź całkowita + odpowiedź częściowa, 0 do 35% metafaz Ph+ w szpiku kostnym).
W badaniu u 65% pacjentów osiągnięto większą odpowiedź cytogenetyczną, w tym u 53% była to odpowiedź całkowita (potwierdzona u 43%) (Tabela 3). Całkowitą odpowiedź hematologiczną uzyskano u 95% pacjentów.
Faza akceleracji
Do badania włączono 235 dorosłych pacjentów w fazie akceleracji. U pierwszych 77 pacjentów
rozpoczęto leczenie dawką 400 mg, a po wprowadzeniu poprawek do protokołu umożliwiających podawanie większych dawek leku, kolejnych 158 pacjentów rozpoczynało leczenie od dawki 600 mg.
Głównym parametrem skuteczności był wskaźnik odpowiedzi hematologicznej, określany jako całkowita odpowiedź hematologiczna, brak objawów białaczki (tzn. brak blastów w szpiku i krwi, ale bez pełnej normalizacji obrazu krwi obwodowej, jak w przypadku całkowitej odpowiedzi hematologicznej) lub powrót do fazy przewlekłej CML. Potwierdzoną odpowiedź hematologiczną uzyskano u 71,5% pacjentów (Tabela 3). Co ważne, 27,7% pacjentów osiągnęło także większą odpowiedź cytogenetyczną, która u 20,4% pacjentów była całkowitą odpowiedzią cytogenetyczną (potwierdzoną u 16%). Dla pacjentów leczonych dawką 600 mg aktualna szacunkowa mediana przeżycia bez progresji choroby i przeżycie całkowite wynosiły odpowiednio 22,9 i 42,5 miesiąca.
Mieloidalny przełom blastyczny
Do badania włączono 260 pacjentów z mieloidalnym przełomem blastycznym. 95 pacjentów (37%) otrzymywało uprzednio chemioterapię z powodu fazy akceleracji lub przełomu blastycznego („pacjenci uprzednio leczeni”), zaś 165 pacjentów (63%) nie otrzymywało chemioterapii („pacjenci uprzednio nieleczeni”). U pierwszych 37 pacjentów rozpoczęto leczenie dawką 400 mg, a następnie, po wprowadzeniu do protokołu badania poprawek umożliwiających podawanie większych dawek, kolejnych 223 pacjentów rozpoczynało leczenie od dawki 600 mg.
Głównym parametrem skuteczności był wskaźnik odpowiedzi hematologicznej, określany, jako całkowita odpowiedź hematologiczna, brak objawów białaczki lub powrót do fazy przewlekłej CML. Przyjęto takie same kryteria oceny, jak w badaniu u pacjentów w fazie akceleracji. W tym badaniu odpowiedź hematologiczną uzyskano u 31% pacjentów (36% w grupie pacjentów uprzednio nieleczonych i 22% w grupie pacjentów uprzednio leczonych). Wskaźnik odpowiedzi hematologicznej był również większy wśród pacjentów leczonych dawką 600 mg (33%) niż u pacjentów leczonych dawką 400 mg (16%, p=0,0220). Szacowana mediana przeżycia w grupie pacjentów uprzednio nieleczonych oraz pacjentów uprzednio leczonych wynosi odpowiednio 7,7 i 4,7 miesiąca.
Limfoidalny przełom blastyczny
Do badań I fazy została włączona ograniczona liczba pacjentów (n=10). Wskaźnik odpowiedzi hematologicznej wynosił 70% i utrzymywał się 2-3 miesiące.
Tabela 3 Odpowiedź na leczenie u dorosłych pacjentów z CML
Badanie 0110 Dane po 37 miesiącach Faza przewlekła, niepowodzenie leczenia IFN (n=532) | Badanie 0109 Dane po 40,5 miesiąca Faza akceleracji (n=235) | Badanie 0102 Dane po 38 miesiącach Mieloidalny przełom blastyczny (n=260) | |
% pacjentów (CI95%) | |||
Odpowiedź hematologiczna1 | 95% (92,3-96,3) | 71% (65,3-77,2) | 31% (25,2-36,8) |
Całkowita odpowiedź | 95% | 42% | 8% |
hematologiczna (CHR) | |||
Brak objawów białaczki (NEL) | Nie dotyczy | 12% | 5% |
Powrót do fazy przewlekłej (RTC) | Nie dotyczy | 17% | 18% |
Większa odpowiedź cytogenetyczna2 | 65% (61,2-69,5) | 28% (22,0-33,9) | 15% (11,2-20,4) |
Całkowita | 53% | 20% | 7% |
(Potwierdzona3) [95% CI] | (43%) [38,6-47,2] | (16%) [11,3-21,20] | (2%) [0,6-4,4] |
Częściowa | 12% | 7% | 8% |
1 Kryteria odpowiedzi hematologicznej (wszystkie odpowiedzi powinny być potwierdzone po ≥4 tygodniach): CHR: badanie 0110 [leukocyty <10 x 109/l, płytki krwi <450 x 109/l, mielocyty+metamielocyty <5% we krwi, brak blastów i promielocytów we krwi, bazofile <20%, brak pozaszpikowych ognisk białaczki], w badaniach 0102 oraz 0109 ANC ≥1,5 x 109/l, płytki krwi ≥100 x 109/l, brak blastów we | |||
krwi, blasty w szpiku <5%
i brak pozaszpikowych ognisk białaczki
NEL: Te same kryteria, co dla CHR, ale ANC ≥1 x 109/l i płytki krwi ≥20 x 109/l
RTC: <15% blastów w szpiku i krwi obwodowej, <30% blastów + promielocytów w szpiku i krwi obwodowej, <20% bazofilów w szpiku, brak pozaszpikowych ognisk białaczki z wyjątkiem śledziony i wątroby (tylko badania 0102 i 0109)
2 Kryteria odpowiedzi cytogenetycznej:
Większa odpowiedź cytogenetyczna łączy odpowiedź całkowitą i odpowiedzi częściowe: odpowiedź całkowita (0% metafaz Ph+), odpowiedź częściowa (1-35%)
3 Całkowita odpowiedź cytogenetyczna potwierdzona drugim badaniem cytogenetycznym szpiku
wykonanym po co najmniej jednym miesiącu od badania wstępnego.
Dzieci i młodzież
Do badania I fazy dotyczącego ustalenia dawki włączono 26 pacjentów w wieku <18 lat z fazą przewlekłą CML (n=11) lub z CML w przełomie blastycznym lub z ostrymi białaczkami Ph+ (n=15). Pacjenci ci byli uprzednio bardzo intensywnie leczeni: 46% poddano przeszczepieniu szpiku (BMT), a 73% otrzymywało chemioterapię wielolekową. Pacjenci otrzymywali imatynib w dawkach dobowych 260 mg/m2 pc. (n=5), 340 mg/m2 pc. (n=9), 440 mg/m2 pc. (n=7) i 570 mg/m2 pc. (n=5). Spośród 9 pacjentów w fazie przewlekłej CML, dla których dostępne były badania cytogenetyczne,
u 4 (44%) i 3 (33%) uzyskano, odpowiednio, całkowitą i częściową odpowiedź cytogetyczną,
z odsetkiem MCyR wynoszącym 77%.
Łącznie 51 pacjentów z grupy dzieci i młodzieży z nowo rozpoznaną i nieleczoną CML w fazie przewlekłej włączono do otwartego, wieloośrodkowego, jednoramiennego badania II fazy. Pacjenci byli leczeni imatynibem w dawce 340 mg/m2 pc. na dobę bez przerw przy braku toksyczności zależnej od dawki. Imatynib indukował szybką całkowitą odpowiedź hematologiczną (CHR) u 78% dzieci
i młodzieży z nowo zdiagnozowaną CML po 8 tygodniach leczenia. Wysokiemu współczynnikowi CHR towarzyszyła u 65% pacjentów całkowita odpowiedź cytogenetyczna (ang. complete cytogenetic response, CCyR), co jest porównywalne do wyników obserwowanych u dorosłych. Ponadto obserwowano częściową odpowiedź cytogenetyczną (ang. partial cytogenetic response, PCyR) u 16% pacjentów, a większą odpowiedź cytogenetyczną (ang. major cytogenetic response, MCyR) u 81% pacjentów. U większości pacjentów, u których wystąpiła CCyR, miało to miejsce między 3.
a 10. miesiącem, z medianą czasu do uzyskania odpowiedzi na podstawie analizy Kaplana-Meiera, wynoszącą 5,6 miesiąca.
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań z zastosowaniem referencyjnego produktu leczniczego zawierającego imatynib we wszystkich podgrupach populacji dzieci i młodzieży z nowo rozpoznaną przewlekłą białaczką szpikową z chromosomem Philadelphia (bcr-abl translokacja) (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Badania kliniczne w Ph+ ALL
Nowo rozpoznana Ph+ ALL
W kontrolowanym badaniu (ADE10) porównującym imatynib z chemioterapią indukcyjną
u 55 pacjentów w wieku co najmniej 55 lat z nowo rozpoznaną chorobą, po zastosowaniu imatynibu w monoterapii uzyskano istotnie większy wskaźnik całkowitej odpowiedzi hematologicznej niż po chemioterapii (96,3% vs. 50%; p=0,0001). Gdy u pacjentów bez odpowiedzi lub ze słabą odpowiedzią na chemioterapię zastosowano ratunkowo imatynib, całkowitą odpowiedź hematologiczną uzyskano
u 9 z 11 pacjentów (81,8%). Ten efekt kliniczny związany był z większą redukcją ilości transkryptów bcr-abl po 2 tygodniach leczenia wśród pacjentów leczonych imatynibem w porównaniu z pacjentami leczonymi chemioterapią (p=0,02). Po indukcji wszyscy pacjenci otrzymywali imatynib oraz chemioterapię konsolidacyjną (patrz tabela 3) i po 8 tygodniach ilość transkryptów bcr-abl była identyczna w obu ramionach badania. Jak można się było spodziewać na podstawie projektu badania, nie obserwowano różnic w czasie trwania remisji, okresie przeżycia bez choroby lub całkowitym czasie przeżycia, chociaż pacjenci, u których stwierdzono całkowitą odpowiedź molekularną
i minimalną chorobę resztkową osiągnęli lepsze wyniki zarówno w odniesieniu do czasu trwania
remisji (p=0,01), jak i czasu przeżycia bez choroby (p=0,02).
Wyniki uzyskane w grupie 211 pacjentów z nowo rozpoznaną Ph+ ALL, uczestniczących w czterech niekontrolowanych badaniach klinicznych (AAU02, ADE04, AJP01 i AUS01) są zgodne z wynikami opisanymi wyżej. Imatynib w połączeniu z chemioterapią indukcyjną (patrz tabela 4) wywołał całkowitą odpowiedź hematologiczną u 93% pacjentów (u 147 z 158 pacjentów podlegających ocenie), a wskaźnik większej odpowiedzi cytogenetycznej u 90% pacjentów (19 z 21 pacjentów podlegających ocenie). Wskaźnik całkowitej odpowiedzi molekularnej wyniósł 48% (49 ze 102 pacjentów podlegających ocenie). Okres przeżycia bez choroby (ang. diseaese-free survival, DFS) oraz całkowity czas przeżycia (ang. overall survival, OS) stale przekraczały 1 rok i w dwóch badaniach (AJP01 i AUS01) były zwiększone w porównaniu z historyczną grupą kontrolną (DFS p<0,001; OS p<0,0001).
Tabela 4 Chemioterapia stosowana w połączeniu z imatynibem
Badanie ADE10 | |
Faza wstępna | DEX 10 mg/m2 doustnie, dni 1-5; CP 200 mg/m2 iv., dni 3, 4, 5; MTX 12 mg dooponowo, dzień 1 |
Indukcja remisji | DEX 10 mg/m2 doustnie, dni 6-7, 13-16; VCR 1 mg iv., dni 7, 14; IDA 8 mg/m2 iv. (0,5 h), dni 7, 8, 14, 15; CP 500 mg/m2 iv. (1 h) dzień 1; Ara-C 60 mg/m2 iv., dni 22-25, 29-32 |
Leczenie konsolidacyjne I, III, V | MTX 500 mg/m2 iv. (24 h), dni 1, 15; 6-MP 25 mg/m2 doustnie, dni 1-20 |
Leczenie konsolidacyjne II, IV | Ara-C 75 mg/m2 iv. (1 h), dni 1-5; VM26 60 mg/m2 iv. (1 h), dni 1-5 |
Badanie AAU02 | |
Leczenie indukcyjne (de novo Ph+ ALL) | Daunorubicyna 30 mg/m2 iv., dni 1-3, 15-16; VCR 2 mg całkowita dawka iv., dni 1, 8, 15, 22; CP 750 mg/m2 iv., dni 1, 8; Prednizon 60 mg/m2 doustnie, dni 1-7, 15-21; IDA 9 mg/m2 doustnie, dni 1-28; MTX 15 mg dooponowo, dni 1, 8, 15, 22; Ara-C 40 mg dooponowo, dni 1, 8, 15, 22; Metyloprednizolon 40 mg dooponowo, dni 1, 8, 15, 22 |
Konsolidacja (de novo Ph+ ALL) | Ara-C 1000 mg/m2/12 h iv. (3 h), dni 1-4; Mitoksantron 10 mg/m2 iv. dni 3-5; MTX 15 mg dooponowo, dzień 1; Metyloprednizolon 40 mg dooponowo, dzień 1 |
Badanie ADE04 | |
Faza wstępna | DEX 10 mg/m2 doustnie, dni 1-5; CP 200 mg/m2 iv., dni 3-5; MTX 15 mg dooponowo, dzień 1 |
Leczenie indukcyjne I | DEX 10 mg/m2 doustnie, dni 1-5; VCR 2 mg iv., dni 6, 13, 20; Daunorubicyna 45 mg/m2 iv., dni 6-7, 13-14 |
Leczenie indukcyjne II | CP 1 g/m2 iv. (1 h), dni 26, 46; Ara-C 75 mg/m2 iv. (1 h), dni 28-31, 35-38, 42-45; 6-MP 60 mg/m2 doustnie, dni 26-46 |
Leczenie konsolidacyjne | DEX 10 mg/m2 doustnie, dni 1-5; Windezyna 3 mg/m2 iv., dzień 1; MTX 1,5 g/m2 iv. (24 h), dzień 1; Etopozyd 250 mg/m2 iv. (1 h) dni 4-5; Ara-C 2x 2 g/m2 iv. (3 h, co 12 h), dzień 5 |
Badanie AJP01 | |
Leczenie indukcyjne | CP 1,2 g/m2 iv. (3 h), dzień 1; |
Daunorubicyna 60 mg/m2 iv. (1 h), dni 1-3; Winkrystyna 1,3 mg/m2 iv., dni 1, 8, 15, 21; Prednizolon 60 mg/m2/dobę doustnie | |
Leczenie konsolidacyjne | Naprzemienna chemioterapia: duża dawka chemioterapii MTX 1 g/m2 iv. (24 h), dzień 1 i Ara-C 2 g/m2 iv. (co 12 h), dni 2-3, przez 4 cykle |
Podtrzymanie remisji | VCR 1,3 g/m2 iv., dzień 1; Prednizolon 60 mg/m2 doustnie, dni 1-5 |
Badanie AUS01 | |
Leczenie indukcyjno- konsolidacyjne | Schemat Hyper-CVAD: CP 300 mg/m2 iv. (3 h co 12 h), dni 1-3; Winkrystyna 2 mg iv., dni 4, 11; Doksorubicyna 50 mg/m2 iv. (24 h), dzień 4; DEX 40 mg/dobę w dniach 1-4 i 11-14, naprzemiennie z MTX 1 g/m2 iv. (24 h), dzień 1, Ara-C 1 g/m2 iv. (2 h co 12 h), dni 2-3 (w sumie 8 kursów) |
Podtrzymanie remisji | VCR 2 mg iv. Co miesiąc przez 13 miesięcy; Prednizolon 200 mg doustnie, 5 dni w miesiącu przez 13 miesięcy |
Wszystkie schematy leczenia obejmują podawanie steroidów w ramach profilaktyki dla OUN. | |
Ara-C: arabinozyd cytozyny; CP: cyklofosfamid; DEX: deksametazon; MTX: metotreksat; 6-MP: 6-merkaptopuryna VM26: tenipozyd; VCR: winkrystyna; IDA: idarubicyna; iv.: dożylnie | |
Dzieci i młodzież
Do otwartego, wieloośrodkowego, nierandomizowanego sekwencyjnego, kohortowego badania III fazy I2301 włączono w sumie 93 dzieci, młodzieży i młodych dorosłych (w wieku od 1 do 22 lat) z Ph+ ALL, którym podawano imatynib (340 mg/m2 pc./dobę) w skojarzeniu z intensywną
chemioterapią po leczeniu indukcyjnym. Imatynib podawano z przerwami kohortom 1-5, wydłużając czas trwania i przyspieszając rozpoczęcie leczenia imatynibem w poszczególnych kohortach; przy czym kohorta 1 otrzymywała leczenie imatynibem o najmniejszej intensywności, zaś kohorta
5 otrzymywała leczenie o największej intensywności (najdłuższy czas trwania liczony w dniach
z ciągłym, codziennym podawaniem imatynibu w pierwszych cyklach chemioterapii). Nieprzerwana, codzienna ekspozycja na imatynib na wczesnym etapie leczenia w skojarzeniu z chemioterapią
w kohorcie 5 (n=50) spowodowała poprawę 4-letniego przeżycia bez zdarzeń (ang. event-free survival, EFS) w porównaniu z historyczną grupą kontrolną (n=120), która otrzymywała standardową chemioterapię bez imatynibu (odpowiednio 69,6% vs. 31,6%). Szacowane 4-letnie przeżycie całkowite (OS) w kohorcie 5 wyniosło 83,6% w porównaniu z 44,8% w historycznej grupie kontrolnej. U 20 z 50 (40%) pacjentów z kohorty 5 dokonano przeszczepienia hematopoetycznych komórek macierzystych.
Tabela 5 Schemat chemioterapii stosowany w skojarzeniu z imatynibem w badaniu I2301
Konsolidacja blok 1 (3 tygodnie) | VP-16 (100 mg/m2/dobę, iv.): dni 1-5 Ifosfamid (1,8 g/m2/dobę, iv.): dni 1-5 MESNA (360 mg/m2/dawkę co 3 h, x 8 dawek/dobę, iv.): dni 1-5 G-CSF (5 µg/kg, sc.): dni 6-15 lub do ANC >1500 po osiągnieciu nadiru Metotreksat it.(dostosowany do wieku): TYLKO dzień 1 Potrójna terapia it. (dostosowana do wieku): dzień 8, 15 |
Konsolidacja blok 2 (3 tygodnie) | Metotreksat (5 g/m2 przez 24 godziny, iv.): dzień 1 Leukoworyna (75 mg/m2 w godzinie 36, iv.; 15 mg/m2 iv. lub po. co 6 h x 6 dawek)iii: dni 2 i 3 Potrójna terapia it. (dostosowana do wieku): dzień 1 ARA-C (3 g/m2/dawkę co 12 h x 4, iv.): dni 2 i 3 G-CSF (5 µg/kg, sc.): dni 4-13 lub do ANC >1500 po osiągnieciu nadiru |
Reindukcja blok 1 (3 tygodnie) | VCR (1,5 mg/m2/dobę, iv.): dni 1, 8, i 15 DAUN (45 mg/m2/dobę bolus, iv.): dni 1 i 2 CPM (250 mg/m2/dawkę co 12 h x 4 dawki, iv.): dni 3 i 4 PEG-ASP (2500 j.m./m2, im.): dzień 4 G-CSF (5 µg/kg, sc.): dni 5-14 lub do ANC >1500 po osiągnieciu nadiru Potrójna terapia it. (dostosowana do wieku): dni 1 i 15 DEX (6 mg/m2/dobę, po.): dni 1-7 i 15-21 |
Intensyfikacja blok 1 (9 tygodni) | Metotreksat (5 g/m2 przez 24 h, iv.): dni 1 i 15 Leukoworyna (75 mg/m2 w godzinie 36, iv.; 15 mg/m2 iv. lub po. co 6 h x 6 dawek)iii: dni 2, 3, 16 i 17 Potrójna terapia it. (dostosowana do wieku): dni 1 i 22 VP-16 (100 mg/m2/dobę, iv.): dni 22-26 CPM (300 mg/m2/dobę, iv.): dni 22-26 MESNA (150 mg/m2/dobę, iv.): dni 22-26 G-CSF (5 µg/kg, sc.): dni 27-36 lub do ANC >1500 po osiągnieciu nadiru ARA-C (3 g/m2, co 12 h, iv.): dni 43, 44 L-ASP (6000 j.m./m2, im.): dzień 44 |
Reindukcja blok 2 (3 tygodnie) | VCR (1,5 mg/m2/dobę, iv.): dni 1, 8 i 15 DAUN (45 mg/m2/dobę bolus, iv.): dni 1 i 2 CPM (250 mg/m2/dawkę co 12 h x 4 dawki, iv.): dni 3 i 4 PEG-ASP (2500 j.m./m2, im.): dzień 4 G-CSF (5 µg/kg, sc.): dni 5-14 lub do ANC >1500 po osiągnieciu nadiru Potrójna terapia it. (dostosowana do wieku): dni 1 i 15 DEX (6 mg/m2/dobę, po.): dni 1-7 i 15-21 |
Intensyfikacja blok 2 (9 tygodni) | Metotreksat (5 g/m2 przez 24 h, iv.): dni 1 i 15 Leukoworyna (75 mg/m2 w godzinie 36, iv.; 15 mg/m2 iv. lub po. co 6 h x 6 dawek)iii: dni 2, 3, 16 i 17 Potrójna terapia it. (dostosowana do wieku): dni 1 i 22 VP-16 (100 mg/m2/dobę, iv.): dni 22-26 CPM (300 mg/m2/dobę, iv.): dni 22-26 MESNA (150 mg/m2/dobę iv.): dni 22-26 G-CSF (5 μg/kg, sc.): dni 27-36 lub do ANC >1500 po osiągnieciu nadiru ARA-C (3 g/m2, co 12 h, iv.): dni 43, 44 L-ASP (6000 j.m./m2, im.): dzień 44 |
Leczenie podtrzymujące (cykle 8-tygodniowe) Cykle 1-4 | MTX (5 g/m2 przez 24 h, iv.): dzień 1 Leukoworyna (75 mg/m2 w godzinie 36, iv.; 15 mg//m2 iv. lub po. co 6 h x 6 dawek)iii: dni 2 i 3 Potrójna terapia IT (dostosowana do wieku): dni 1, 29 VCR (1,5 mg/m2, iv.): dni 1, 29 DEX (6 mg/m2/dobę po.): dni 1-5; 29-33 6-MP (75 mg/m2/dobę, po.): dni 8-28 Metotreksat (20 mg/m2/tydzień, po.): dni 8, 15, 22 VP-16 (100 mg/m2, iv.): dni 29-33 CPM (300 mg/m2, iv.): dni 29-33 MESNA iv. dni 29-33 G-CSF (5 µg/kg, sc.): dni 34-43 |
Leczenie podtrzymujące (cykle 8-tygodniowe) Cykl 5 | Napromienianie czaszki (Tylko blok 5) 12 Gy w 8 frakcjach u wszystkich pacjentów zakwalifikowanych w chwili rozpoznania jako CNS1 i CNS2 18 Gy w 10 frakcjach u pacjentów zakwalifikowanych w chwili rozpoznania jako CNS3 VCR (1.5 mg/m2/dobę, iv.): dni 1, 29 DEX (6 mg/m2/dobę, po.): dni 1-5; 29-33 6-MP (75 mg/m2/dobę, po.): dni 11-56 (wstrzymanie 6-MP podczas 6-10 dni napromieniania czaszki, poczynając od dnia 1. cyklu 5. Rozpoczęcie podawania 6-MP w 1. dniu po zakończeniu napromieniania czaszki) Metotreksat (20 mg/m2/tydzień, po.): dni 8, 15, 22, 29, 36, 43, 50 |
Leczenie podtrzymujące (cykle 8-tygodniowe) Cykle 6-12 | VCR (1.5 mg/m2/ dobę, iv.): dni 1, 29 DEX (6 mg/m2/ dobę, po.): dni 1-5; 29-33 6-MP (75 mg/m2/ dobę, po.): dni 1-56 Metotreksat (20 mg/m2/tydzień, po.): dni 1, 8, 15, 22, 29, 36, 43, 50 |
G-CSF=czynnik stymulujący kolonie granulocytarne, VP-16=etopozyd, iv.=dożylnie, sc.=podskórnie,
it.=dooponowo, po.=doustnie, im.=domięśniowo, ARA-C=cytarabina, CPM=cyklofosfamid, VCR=winkrystyna, DEX=deksametazon, DAUN=daunorubicyna, 6-MP=6-merkaptopuryna, E.Coli
L-ASP=L-Asparaginaza, PEG-ASP=PEG-Asparaginaza, MESNA=2-merkaptoetanosulfonian sodowy,
iii= lub do czasu, gdy stężenie MTX wyniesie <0,1 μM, Gy=Gray
AIT07 było wieloośrodkowym, otwartym, randomizowanym badaniem II/III fazy, w którym
uczestniczyło 128 pacjentów (w wieku 1 do <18 lat) leczonych imatynibem w skojarzeniu
z chemioterapią. Dane z tego badania dotyczące bezpieczeństwa wydają się być zgodne z profilem bezpieczeństwa imatynibu u pacjentów z Ph+ ALL.
Nawracająca/oporna na leczenie Ph+ ALL
Po podaniu imatynibu w monoterapii pacjentom z nawracającą/oporną na leczenie Ph+ ALL, u 53
z 411 pacjentów, u których odpowiedź była możliwa do oceny, wskaźnik odpowiedzi hematologicznej wyniósł 30% (9% odpowiedzi całkowitej), a wskaźnik większej odpowiedzi cytogenetycznej wyniósł 23%. (Co istotne, 353 z 411 pacjentów otrzymywało leczenie według rozszerzonego programu dostępu, bez zebrania danych dotyczących pierwszej odpowiedzi). Mediana czasu do progresji w całej populacji 411 pacjentów z nawracającą/oporną na leczenie Ph+ ALL wynosiła od 2,6 do 3,1 miesiąca, a mediana całkowitego przeżycia u 401 pacjentów podlegających ocenie od 4,9 do 9 miesięcy.
Podobne dane uzyskano po powtórnej analizie, do której włączono tylko pacjentów w wieku 55 lat lub
starszych.
Badania kliniczne w MDS/MPD
Doświadczenie z zastosowaniem imatynibu w tym wskazaniu jest bardzo ograniczone i opiera się na wskaźnikach odpowiedzi hematologicznej i cytogenetycznej. Nie ma kontrolowanych badań klinicznych wykazujących korzyść kliniczną lub wydłużone przeżycie. Przeprowadzono jedno otwarte, wieloośrodkowe badanie kliniczne II fazy (badanie B2225), oceniające imatynib w różnych populacjach pacjentów z zagrażającymi życiu chorobami związanymi z kinazą białkowo-tyrozynową Abl, Kit lub PDGFR. W badaniu tym uczestniczyło 7 pacjentów z MDS/MPD leczonych imatynibem w dawce 400 mg na dobę. U 3 pacjentów wystąpiła całkowita odpowiedź hematologiczna (CHR),
a u jednego pacjenta odpowiedź częściowa (PHR). W czasie pierwszej analizy u trzech z czterech
pacjentów, u których wykryto rearanżacje genu PDGFR, wystąpiła odpowiedź hematologiczna
(2 CHR i 1 PHR). Wiek tych pacjentów sięgał od 20 do 72 lat.
Prowadzono obserwacyjny rejestr (badanie L2401) w celu zgromadzenia danych o długotrwałym bezpieczeństwie stosowania i skuteczności imatynibu u pacjentów z nowotworami mieloproliferacyjnymi z rearanżacją PDGFR-β. Dwudziestu trzech pacjentów włączonych do tego rejestru otrzymywało imatynib w dawce dobowej o medianie 264 mg (zakres od 100 do 400 mg) przez średnio (mediana) 7,2 roku (zakres od 0,1 do 12,7 roku). Ze względu na obserwacyjny charakter tego rejestru, dane z oceny hematologicznej, cytogenetycznej i molekularnej były dostępne odpowiednio od 22,9 i 17 z 23 włączonych do rejestru pacjentów. Przyjmując zachowawcze założenie, że pacjentami
z brakującymi danymi byli pacjenci bez odpowiedzi, CHR obserwowano u 20/23 (87%) pacjentów, CCyR u 9/23 (39,1%) pacjentów, a MR u 11/23 (47,8%) pacjentów. Gdy wskaźnik odpowiedzi obliczano na podstawie danych pochodzących od pacjentów z co najmniej jedną ważną oceną, wskaźnik odpowiedzi CHR, CCyR i MR wyniósł odpowiednio 20/22 (90,9%), 9/9 (100%) i 11/17 (64,7%).
Ponadto, w 13 publikacjach opisano kolejnych 24 pacjentów z MDS/MPD, z których 21 otrzymywało imatynib w dawce 400 mg na dobę, a kolejnych 3 było leczonych mniejszymi dawkami. U 11 pacjentów wykryto rearanżacje genu PDGFR, u 9 pacjentów uzyskano całkowitą odpowiedź hematologiczną (CHR), a u 1 pacjenta odpowiedź częściową (PHR). Wiek tych pacjentów wynosił od 2 do 79 lat. W ostatniej publikacji uaktualnione dane dotyczące 6 z tych 11 pacjentów wskazują, że wszyscy oni pacjenci pozostawali w fazie remisji cytogenetycznej (zakres 32-38 miesięcy). W tej samej publikacji opisywano dane z długotrwałej obserwacji 12 pacjentów z MDS/MPD
i rearanżacjami genu PDGFR (5 pacjentów z badania B2225). Pacjenci ci otrzymywali imatynib przez średnio (mediana) 47 miesięcy (od 24 dni do 60 miesięcy). U 6 z tych pacjentów czas obserwacji przekracza obecnie 4 lata. U 11 pacjentów całkowita odpowiedź cytogenetyczna (CHR) wystąpiła szybko; u dziesięciu pacjentów anomalie cytogenetyczne ustąpiły całkowicie i obserwowano również
zmniejszenie się lub zanik liczby transkryptów fuzyjnych mierzonych za pomocą RT-PCR. Odpowiedź hematologiczna i cytogenetyczna utrzymywała się odpowiednio przez średnio (mediana) 49 miesięcy (zakres 19-60) i 47 miesięcy (zakres 16-59). Całkowite przeżycie od chwili diagnozy wynosi 65 miesięcy (zakres od 25 do 234). Podawanie imatynibu pacjentom bez translokacji genów zazwyczaj nie daje poprawy.
Nie ma kontrolowanych badań z udziałem dzieci i młodzieży z MDS/MPD. W 4 publikacjach opisano 5 pacjentów z MDS/MPD i rearanżacjami genu PDGFR. Wiek tych pacjentów mieścił się w zakresie od 3 miesięcy do 4 lat, a imatynib podawano w dawce 50 mg na dobę lub w dawkach od 92,5 do
340 mg/m2 pc. na dobę. U wszystkich pacjentów uzyskano całkowitą odpowiedź hematologiczną, cytogenetyczną i (lub) kliniczną.
Badania kliniczne w HES/CEL
Przeprowadzono jedno otwarte, wieloośrodkowe badanie kliniczne II fazy (badanie B2225), oceniające imatynib w różnych populacjach pacjentów z zagrażającymi życiu chorobami związanymi z kinazą białkowo-tyrozynową Abl, Kit lub PDGFR. W badaniu tym 14 pacjentów z HES/CEL otrzymywało imatynib w dawce od 100 mg do 1000 mg na dobę, kolejnych 162 pacjentów
z HES/CEL opisywanych w 35 opublikowanych opisach przypadków i seriach przypadków otrzymywało imatynib w dawce od 75 do 800 mg na dobę. Anomalie cytogenetyczne oceniano u 117 ze wszystkich 176 pacjentów. U 61 z tych 117 pacjentów zidentyfikowano kinazę fuzyjną FIP1L1- PDGFRα. W 3 innych publikacjach opisano dodatkowo czterech pacjentów z HES i dodatnim wynikiem na obecność kinazy fuzyjnej FIP1L1-PDGFRα. U wszystkich 65 pacjentów z obecnością kinazy fuzyjnej FIP1L1-PDGFRα uzyskano całkowitą odpowiedź hematologiczną utrzymującą się przez wiele miesięcy (zakres od 1+ do 44+ miesięcy do czasu publikacji). Jak donoszono w ostatnio opublikowanej pracy, 21 ze wspomnianych 65 pacjentów również uzyskało całkowitą remisję molekularną, przy medianie czasu trwania obserwacji wynoszącej 28 miesięcy (zakres 13 do
67 miesięcy). Wiek tych pacjentów wynosił od 25 do 72 lat. Ponadto, w kartach obserwacji klinicznej badacze donosili o poprawie w zakresie objawów i innych zaburzeń funkcji narządów. Poprawa dotyczyła serca, układu nerwowego, skóry/tkanki podskórnej, układu oddechowego/klatki piersiowej/śródpiersia, układu mięśniowo-szkieletowego/tkanki łącznej/naczyń i przewodu pokarmowego.
Nie ma kontrolowanych badań z udziałem dzieci i młodzieży z HES/CEL. W 3 publikacjach opisano 3 pacjentów z HES/CEL i rearanżacjami genu PDGFR. Wiek tych pacjentów mieścił się w zakresie od 2 do 16 lat, a imatynib podawano w dawce 300 mg/m2 pc. na dobę lub w dawkach od 200 do 400 mg na dobę. U wszystkich pacjentów uzyskano całkowitą odpowiedź hematologiczną, cytogenetyczną
i (lub) molekularną.
Badania kliniczne w nieoperacyjnych i (lub) z przerzutami GIST
Przeprowadzono jedno, międzynarodowe, randomizowane, niekontrolowane, otwarte badanie drugiej fazy z udziałem pacjentów ze złośliwymi, nieoperacyjnymi lub przerzutowymi nowotworami podścieliskowymi przewodu pokarmowego (GIST). Do badania włączono i randomizowano
147 pacjentów, którzy otrzymywali doustnie 400 lub 600 mg imatynibu jeden raz na dobę przez okres do 36 miesięcy. Pacjenci byli w wieku od 18 do 83 lat z rozpoznaną Kit pozytywną, złośliwą, nieoperacyjną i (lub) z przerzutami postacią GIST. Przeprowadzono rutynowe badanie immunohistochemiczne z przeciwciałem Kit (A-4502, królicza poliklonalna surowica odpornościowa, 1:100; DAKO Corporation, Carpinteria, CA) zgodnie z analizą prowadzoną metodą z kompleksem awidyna-biotyna-peroksydaza po odzyskaniu antygenu.
Pierwszorzędowe kryterium skuteczności ustalono na podstawie obiektywnego odsetka odpowiedzi. Guzy musiały być mierzalne przynajmniej w jednym ognisku choroby, a charakterystyka odpowiedzi opierała się na kryteriach SWOG (ang. Southwestern Oncology Group). Wyniki przedstawiono w Tabeli 6.
Tabela 6 Najlepsza odpowiedź na leczenie w badaniu STIB2222 (GIST)
Najlepsza odpowiedź | Wszystkie dawki (n=147) 400 mg (n=73) 600 mg (n=74) n (%) |
Odpowiedź całkowita | 1(0,7) |
Odpowiedź częściowa | 98 (66,7) |
Stabilizacja choroby | 23 (15,6) |
Progresja choroby | 18 (12,2) |
Nie podlegające ocenie | 5 (3,4) |
Nieznane | 2 (1,4) |
Nie było różnicy w stopniu odpowiedzi między obu grupami. Znaczna liczba pacjentów ze stabilizacją choroby w czasie analizy tymczasowej uzyskała częściową odpowiedź w wyniku dłuższego leczenia (mediana czasu obserwacji 31 miesięcy). Mediana czasu do uzyskania odpowiedzi wynosiła 13 tygodni (95% CI, 12–23). Mediana czasu do niepowodzenia leczenia u osób z odpowiedzią wynosiła 122 tygodnie (95% CI, 106–147), natomiast w całej populacji badania, były to 84 tygodnie (95% CI, 71–109). Nie uzyskano mediany całkowitego przeżycia. Wynik analizy przeżywalności przeprowadzonej metodą Kaplana-Meiera dla przeżycia po 36 miesiącach obserwacji wynosi 68%.
W dwóch badaniach klinicznych (badanie B2222 i badanie międzygrupowe S0033) dobową dawkę imatynibu zwiększono do 800 mg u pacjentów z progresją po najmniejszych dawkach dobowych w wysokości 400 mg lub 600 mg. Dawkę dobową zwiększono do 800 mg u 103 pacjentów; u 6 pacjentów uzyskano odpowiedź częściową, a u 21 pacjentów – stabilizację choroby po zwiększeniu dawki, w sumie uzyskano poprawę kliniczną u 26%. Jak wynika z dostępnych danych o bezpieczeństwie, zwiększenie dawki do 800 mg na dobę u pacjentów z progresją po mniejszych dawkach leku w wysokości 400 mg i 600 mg na dobę wydaje się nie mieć wpływu na profil bezpieczeństwa imatynibu.
Badania kliniczne z leczeniem adjuwantowym w GIST
W leczeniu adjuwantowym stosowanie imatynibu badano w wieloośrodkowym, podwójnie zaślepionym, długoterminowym badaniu III fazy z grupą kontrolną otrzymującą placebo
(Z9001) z udziałem 773 pacjentów. Wiek pacjentów wahał się od 18 do 91 lat. Do badania włączono pacjentów z histologicznym rozpoznaniem pierwotnego GIST z ekspresją białka Kit w badaniu immunochemicznym oraz guzem wielkości ≥ 3 cm w największym wymiarze, z całkowitą resekcją pierwotnego GIST w okresie 14-70 dni przed rejestracją. Po resekcji pierwotnego guza GIST pacjentów zrandomizowano do jednego z dwóch ramion badania: z imatynibem w dawce 400 mg na dobę lub odpowiadającym mu placebo. Leczenie było prowadzone przez rok.
Pierwszorzędowym punktem końcowym był czas przeżycia wolnego od nawrotu (ang. recurrence-free survival - RFS), określany jako czas od daty randomizacji do daty wystąpienia nawrotu choroby lub zgonu z jakiejkolwiek przyczyny.
Imatynib znamiennie wydłużał RFS. W grupie pacjentów otrzymujących imatynib 75% było wolnych od nawrotu po 38 miesiącach w porównaniu z 20 miesiącami w grupie otrzymującej placebo (95% CI odpowiednio [30 – nimożliwe do oceny]); [14 – niemożliwe do oceny]); (współczynnik ryzyka = 0,398 [0,259-0,610], p<0,0001). Po jednym roku całkowity wskaźnik RFS był znamiennie lepszy dla grupy otrzymującej imatynib (97,7%) w porównaniu z grupą otrzymującą placebo
(82,3%), (p<0,0001). Ryzyko wznowy w porównaniu z grupą otrzymującą placebo było zatem zmniejszone o około 89% (współczynnik ryzyka = 0,113 [0,049-0,264]).
Ryzyko nawrotu u pacjentów po operacji pierwotnego nowotworu GIST było oceniane retrospektywnie na podstawie następujących czynników prognostycznych: wielkość guza, indeks mitotyczny, umiejscowienie guza pierwotnego. Dane dotyczące indeksu mitotycznego uzyskano od 556 z 713 pacjentów z populacji ITT. W Tabeli 7 przedstawiono wyniki analizy podgrup pacjentów zgodnie z klasyfikacją ryzyka wg Narodowego Instytutu Zdrowia Stanów Zjednoczonych (NIH) oraz
Instytutu Patologii Sił Zbrojnych (AFIP). Nie zaobserwowano korzyści w grupach małego i bardzo małego ryzyka. Nie obserwowano poprawy przeżycia całkowitego.
Tabela 7 Wyniki analizy RFS badania Z9001 w klasyfikacji ryzyka wg NIH i AFIP
Kryterium | Stopień | % | Liczba zdarzeń / | Całkowity | Wskaźnik RFS (%) | |
ryzyka | ryzyka | pacjen -tów | Liczba pacjentów | współczynnik ryzyka (95%CI)* | 12 miesięcy | 24 miesiące |
imatynib vs. placebo | imatynib vs. placebo | imatynib vs. placebo | ||||
NIH | Małe | 29,5 | 0/86 vs. 2/90 | N.E. | 100 vs. 98,7 | 100 vs. 95,5 |
Średnie | 25,7 | 4/75 vs. 6/78 | 0,59 (0,17; 2,10) | 100 vs. 94,8 | 97,8 vs. 89,5 | |
Duże | 44,8 | 21/140 vs. 51/127 | 0,29 (0,18; 0,49) | 94,8 vs. 64,0 | 80,7 vs. 46,6 | |
AFIP | Bardzo małe | 20,7 | 0/52 vs. 2/63 | N.E. | 100 vs. 98,1 | 100 vs. 93,0 |
Małe | 25,0 | 2/70 vs. 0/69 | N.E. | 100 vs. 100 | 97,8 vs. 100 | |
Umiarko- wane | 24,6 | 2/70 vs. 11/67 | 0,16 (0,03; 0,70) | 97,9 vs. 90,8 | 97,9 vs. 73,3 | |
Wysokie | 29,7 | 16/84 vs. 39/81 | 0,27 (0,15; 0,48) | 98,7 vs. 56,1 | 79,9 vs. 41,5 | |
* Cały okres obserwacji (follow-up); NE – niemożliwe do oceny
Drugie, wieloośrodkowe otwarte badanie III fazy (SSG XVIII/AIO), w którym porównano 12- miesięczne leczenie imatynibem w dawce 400 mg na dobę z 36-miesięcznym leczeniem u pacjentów po chirurgicznej resekcji GIST, spełniających jeden z wymienionych warunków: średnica guza > 5 cm i indeks mitotyczny > 5/50 pól widzenia w dużym powiększeniu; lub średnica guza > 10 cm i każda wartość wskaźnika mitotycznego lub każdy wymiar guza z odsetkiem mitoz > 10/50 pól widzenia w dużym powiększeniu lub guzy, które ulegają pęknięciu do jamy otrzewnej. Łącznie 397 pacjentów wyraziło zgodę na udział w badaniu i zostali oni randomizowani do grup badawczych (199 pacjentów do grupy leczonej przez 12 miesięcy i 198 pacjentów do grupy leczonej przez 36 miesięcy). Mediana wieku wyniosła 61 lat (zakres 22-84 lata). Mediana okresu obserwacji pacjentów wyniosła 54 miesiące (od daty randomizacji do momentu zakończenia zbierania danych). Od randomizacji pierwszego pacjenta do daty zakończenia upłynęły 83 miesiące.
Pierwszorzędowym punktem końcowym badania był czas przeżycia bez nawrotu (RFS), określany jako okres od randomizacji do daty nawrotu lub śmierci z jakiegokolwiek powodu.
Leczenie imatynibem przez 36 miesięcy znacząco wydłużało czas przeżycia bez nawrotu w porównaniu z 12-miesięczną terapią imatynibem (całościowy współczynnik ryzyka (HR) = 0,46 [0,32; 0,65], p<0,0001) (Tabela 8, Rysunek 1).
Ponadto, 36-miesięczne leczenie imatynibem znacząco wydłużało całkowity czas przeżycia w porównaniu z 12-miesięcznym leczeniem imatynibem (HR = 0,45 [0,22; 0,89], p=0,0187) (Tabela 8,
Rysunek 2)
Dłuższe leczenie (>36 miesięcy) może opóźnić wystąpienie kolejnych nawrotów; jednak wpływ tego wyniku na całkowite przeżycie pacjentów pozostaje nieznany.
Całkowita liczba zgonów wyniosła 25 w grupie leczonej przez 12 miesięcy i 12 w grupie leczonej
przez 36 miesięcy.
Analiza populacji ITT, tzn. całej populacji badania, wykazała, że leczenie imatynibem przez 36 miesięcy było lepsze od leczenia trwającego 12 miesięcy. W planowanej analizie podgrup wyodrębnionych na podstawie typu mutacji, HR dla RFS w przypadku leczenia trwającego 36 miesięcy u pacjentów z mutacją w eksonie 11 wyniósł 0,35 [95% CI: 0,22; 0,56]. Z uwagi na małą liczbę obserwowanych zdarzeń nie można wyciągnąć wniosków dla podgrup z innymi, rzadszymi mutacjami.
Tabela 8 12-miesięczne i 36-miesięczne leczenie imatynibem (Badanie SSGXVIII/AIO)
RFS | 12-miesięczne leczenie % (przedział ufności) | 36-miesięczne leczenie % (przedział ufności) |
12 miesięcy | 93,7 (89,2-96,4) | 95,9 (91,9-97,9) |
24 miesięcy | 75,4 (68,6-81,0) | 90,7 (85,6-94,0) |
36 miesięcy | 60,1 (52,5-66,9) | 86,6 (80,8-90,8) |
48 miesięcy | 52,3 (44,0-59,8) | 78,3 (70,8-84,1) |
60 miesięcy | 47,9 (39,0-56,3) | 65,6 (56,1-73,4) |
Przeżycie | ||
36 miesięcy | 94,0 (89,5-96,7) | 96,3 (92,4-98,2) |
48 miesięcy | 87,9 (81,1-92,3) | 95,6 (91,2-97,8) |
60 miesięcy | 81,7 (73,0-87,8) | 92,0 (85,3-95,7) |
Rysunek 1 Estymator Kaplana-Meiera dla pierwszorzędowego punktu końcowego - czasu
przeżycia bez nawrotu (populacja ITT)
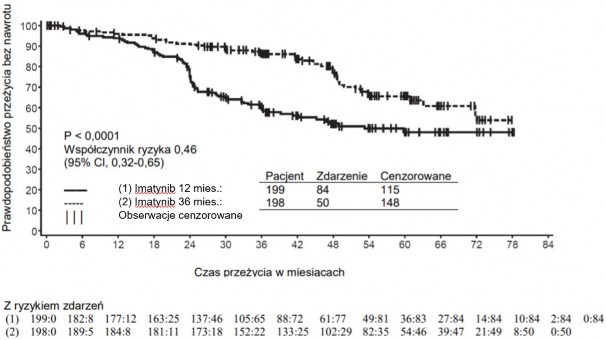
Rysunek 2 Estymator Kaplana-Meiera dla całkowitego czasu przeżycia (populacja ITT)
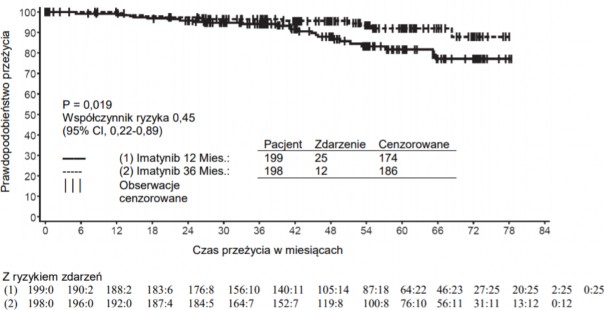
Nie ma kontrolowanych badań z udziałem dzieci i młodzieży z c-Kit dodatnim GIST. W 7 publikacjach opisano przypadki 17 pacjentów z GIST (z lub bez obecności Kit oraz mutacjami PDGFR). Wiek tych pacjentów wahał się od 8 do 18 lat, a imatynib podawano zarówno w leczeniu adjuwantowym, jak i w leczeniu przerzutów w dawkach wynoszących od 300 do 800 mg na dobę. U większości dzieci i młodzieży leczonych z powodu GIST brak jest danych potwierdzających występowanie c-kit lub mutacji PDGFR, co mogłoby prowadzić do mieszanych rezultatów klinicznych.
Badania kliniczne w DFSP
Przeprowadzono jedno otwarte, wieloośrodkowe badanie kliniczne II fazy (badanie B2225)
z udziałem 12 pacjentów z DFSP leczonych imatynibem w dawce 800 mg na dobę. Wiek pacjentów z DFSP wynosił od 23 do 75 lat. Byli to pacjenci z DFSP z przerzutami i miejscową wznową po wstępnej resekcji, którzy w chwili włączenia do badania zostali uznani jako niekwalifikujący się do ponownej resekcji. Podstawowe dowody skuteczności leku uzyskano na podstawie obiektywnych wskaźników odpowiedzi. Spośród 12 pacjentów włączonych do badania, u 9 uzyskano całkowitą odpowiedź, a u 8 odpowiedź częściową. Trzech spośród pacjentów z odpowiedzią częściową zostało
następnie wyleczonych za pomocą zabiegu chirurgicznego. Mediana czasu trwania leczenia w badaniu B2225 wynosiła 6,2 miesiąca, maksymalnie 24,3 miesiąca. Kolejnych 6 pacjentów z DFSP (w wieku od 18 miesięcy do 49 lat) leczonych imatynibem opisano w 5 opublikowanych opisach przypadków.
Dorosłych pacjentów opisywanych w literaturze leczono imatynibem w dawce dobowej 400 mg
(4 przypadki) lub 800 mg (1 przypadek). Odpowiedź uzyskano u pięciu (5) pacjentów: u 3 całkowitą, a u 2 częściową. Mediana czasu trwania leczenia opisywanego w literaturze wynosiła od 4 tygodni do ponad 20 miesięcy. Translokacja (17:22)[(q22:q13)] lub jej produkt genowy były obecne u prawie wszystkich pacjentów odpowiadających na leczenie imatynibem.
Nie ma kontrolowanych badań z udziałem dzieci i młodzieży z DFSP. W 3 publikacjach opisano
5 pacjentów z DFSP i rearanżacjami genu PDGFR. Byli to pacjenci w wieku od noworodka do 14 lat, a imatynib podawano im w dawce 50 mg na dobę lub od 400 do 520 mg/m2 pc. na dobę. U wszystkich pacjentów uzyskano częściową i (lub) całkowitą odpowiedź.
Farmakokinetyka imatynibu
Właściwości farmakokinetyczne imatynibu oceniano w zakresie dawek od 25 do 1000 mg. Profile farmakokinetyczne w osoczu analizowano w 1. dniu oraz w 7. lub 28. dniu, kiedy stężenie imatynibu w osoczu osiągnęło stan stacjonarny.
Wchłanianie
Średnia bezwzględna biodostępność imatynibu wynosi 98%. Po podaniu doustnym stwierdzono dużą międzyosobniczą zmienność wartości AUC imatynibu w osoczu. Po podaniu imatynibu
z wysokotłuszczowym posiłkiem stopień wchłaniania był w porównaniu z przyjmowaniem leku na czczo minimalnie zmniejszony (zmniejszenie wartości Cmax o 11% i wydłużenie tmax o 1,5 godziny), z niewielkim zmniejszeniem AUC (7,4%). Nie badano wpływu uprzedniej operacji w obrębie przewodu pokarmowego na wchłanianie leku.
Dystrybucja
Badania in vitro wykazały, że imatynib w stężeniach istotnych klinicznie wiąże się w 95% z białkami
osocza, głównie z albuminą i kwaśną alfa-glikoproteiną oraz w niewielkim stopniu z lipoproteiną.
Metabolizm
Głównym metabolitem imatynibu we krwi człowieka jest N-demetylowa pochodna piperazyny, która in vitro charakteryzuje się podobną siłą działania, co związek macierzysty. Wykazano, że wartość AUC tego metabolitu w osoczu stanowi zaledwie 16% AUC imatynibu. Wiązanie N-demetylowanego metabolitu i wiązanie związku macierzystego z białkami osocza jest podobne.
Imatynib i jego N-demetylowy metabolit stanowią łącznie około 65% radioaktywności we krwi (AUC(0-48h)). Pozostała część radioaktywności we krwi jest związana z obecnymi w mniejszej ilości metabolitami.
Badania in vitro wykazały, że u ludzi CYP3A4 jest głównym enzymem cytochromu P450 katalizującym biotransformację imatynibu. Spośród leków, które można by stosować jednocześnie z imatynibem (acetaminofen, acyklowir, allopurynol, amfoterycyna, cytarabina, erytromycyna, flukonazol, hydroksymocznik, norfloksacyna, penicylina V), tylko erytromycyna (IC50=50 μmol/l) i flukonazol (IC50=118 μmol/l) hamowały metabolizm imatynibu w stopniu, który może mieć znaczenie kliniczne.
Badania in vitro wykazały, że imatynib jest kompetycyjnym inhibitorem standardowych substratów CYP2C9, CYP2D6 i CYP3A4/5. Wartości Ki w mikrosomach ludzkiej wątroby wynosiły, odpowiednio, 27; 7,5 i 7,9 μmol/l. Maksymalne stężenie imatynibu w osoczu pacjentów wynosi
2-4 μmol/l, dlatego możliwe jest zahamowanie metabolizmu leków podawanych jednocześnie z imatynibem i metabolizowanych przez CYP2D6 i (lub) CYP3A4/5. Imatynib nie wpływał na metabolizm 5-fluorouracylu, ale hamował metabolizm paklitakselu w wyniku hamowania kompetycyjnego CYP2C8 (Ki = 34,7 μmol/l). Ta wartość Ki jest dużo większa niż oczekiwane
stężenie imatynibu w osoczu pacjentów i dlatego nie jest spodziewana interakcja po jednoczesnym podaniu 5-fluorouracylu lub paklitakselu z imatynibem.
Wydalanie
Po doustnym podaniu imatynibu znakowanego 14C stwierdzono, że w ciągu 7 dni około 81% dawki wykrywane jest w kale (68%) i w moczu (13%). Niezmieniony imatynib stanowił 25% podanej dawki (5% w moczu, 20% w kale), zaś pozostałą część stanowią metabolity.
Właściwości farmakokinetyczne w osoczu
Po doustnym podaniu leku zdrowym ochotnikom okres półtrwania (t½) wynosił około 18 godzin, co wskazywałoby zasadność dawkowania raz na dobę. Stwierdzono, że po podaniu doustnym imatynibu zwiększenie średnich wartości AUC wraz z dawką było liniowe i proporcjonalne do dawki w zakresie od 25 mg do 1000 mg. Nie odnotowano zmian farmakokinetyki imatynibu po wielokrotnym podawaniu, a kumulacja leku w organizmie była 1,5–2,5-krotnie większa w stanie stacjonarnym, gdy imatynib podawano raz na dobę.
Właściwości farmakokinetyczne u pacjentów z GIST
U pacjentów z GIST ekspozycja w stanie równowagi po podaniu tych samych dawek (400 mg na dobę) była 1,5 raza większa niż obserwowana u pacjentów z CML. Na podstawie wstępnej oceny właściwości farmakokinetycznych w populacji pacjentów z GIST znaleziono trzy wskaźniki
(albuminy, liczba krwinek białych i bilirubina), które miały statystycznie istotny wpływ na farmakokinetykę imatynibu. Zmniejszone stężenie albumin spowodowało obniżenie klirensu (CL/f); zwiększona liczba krwinek białych prowadzi do obniżenia CL/f. Jednakże zależności te nie są wystarczająco wyrażone, aby stanowiły podstawę do zmiany dawkowania. W tej populacji pacjentów, występowanie przerzutów nowotworowych w wątrobie może potencjalnie prowadzić do niewydolności wątroby i zmniejszyć metabolizm.
Farmakokinetyka populacyjna
W oparciu o analizę farmakokinetyki w populacji pacjentów z CML stwierdzono, że wiek pacjentów miał niewielki wpływ na objętość dystrybucji (12% zwiększenie u pacjentów w wieku >65 lat).
Zmiany tej nie uważa się za istotną klinicznie. Wpływ masy ciała na klirens imatynibu jest następujący: u pacjentów o masie ciała 50 kg średni klirens powinien wynosić 8,5 l/h, podczas gdy u pacjentów o masie ciała 100 kg klirens zwiększy się do 11,8 l/h. Uważa się, że zmiany te nie wymagają dostosowania dawkowania w zależności od masy ciała pacjenta. Płeć pacjentów nie ma wpływu na kinetykę imatynibu.
Farmakokinetyka u dzieci
Tak jak u dorosłych pacjentów, imatynib był szybko wchłaniany po doustnym podaniu dzieciom
i młodzieży w ramach badań I i II fazy. Ekspozycja po podaniu dawek od 260 do 340 mg/m2 pc. była taka sama, jak po zastosowaniu u dorosłych dawek, odpowiednio, 400 mg i 600 mg. Porównanie AUC(0-24) w 8. i 1. dniu podawania dawki 340 mg/m2 pc. wykazało 1,7-krotną kumulację po wielokrotnym podaniu raz na dobę.
W oparciu o zbiorczą analizę farmakokinetyki w populacji dzieci i młodzieży z zaburzeniami hematologicznymi (CML, Ph+ ALL lub innymi zaburzeniami hematologicznymi leczonymi imatynibem) stwierdzono, że klirens imatynibu zwiększa się wraz ze zwiększeniem powierzchni ciała (pc.). Po dokonaniu korekty względem pc., inne parametry demograficzne, takie jak wiek, masa ciała i wskaźnik masy ciała nie miały klinicznie istotnego wpływu na ekspozycję na imatynib. Analiza potwierdziła, że ekspozycja na imatynib u dzieci i młodzieży otrzymujących dawkę 260 mg/m2 pc. raz
na dobę (nie więcej niż 400 mg raz na dobę) lub 340 mg/m2 pc. raz na dobę (nie więcej niż 600 mg raz na dobę) była podobna do ekspozycji u dorosłych, którzy otrzymywali imatynib w dawce 400 mg lub 600 mg raz na dobę.
Zaburzenia czynności narządów
Imatynib i jego metabolity nie są w znaczącym stopniu wydalane przez nerki. Wydaje się, że
u pacjentów z lekkimi i umiarkowanymi zaburzeniami czynności nerek stężenie imatynibu w osoczu jest większe niż u pacjentów z prawidłową czynnością nerek. Ekspozycja ta jest zwiększona średnio 1,5 do 2 razy, co odpowiada 1,5-krotnemu zwiększeniu stężenia AGP w osoczu, białka, z którym silnie wiąże się imatynib. Ponieważ imatynib jest tylko w nieznacznym stopniu wydalany przez nerki, klirens wolnego leku u pacjentów z zaburzeniami czynności nerek jest prawdopodobnie zbliżony do tego, jaki notuje się u pacjentów z prawidłową czynnością nerek.
Wprawdzie wyniki analizy farmakokinetycznej wykazały istnienie znacznych różnic międzyosobniczych, średnia ekspozycja na imatynib nie zwiększyła się u pacjentów z zaburzeniami czynności wątroby różnego stopnia w porównaniu z pacjentami z prawidłową czynnością tego narządu (patrz punkty 4.2, 4.4 i 4.8).
Profil bezpieczeństwa imatynibu w badaniach nieklinicznych oceniano u szczurów, psów, małp
i królików.
Badania toksyczności po podaniu wielokrotnym wykazały lekkie do umiarkowanych zmiany hematologiczne u szczurów, psów i małp, którym towarzyszyły zmiany w szpiku u szczurów i psów.
U szczurów i psów narządem docelowym była wątroba. U obu gatunków obserwowano lekkie do umiarkowanego zwiększenie aktywności aminotransferaz i nieznaczne zmniejszenie stężenia
cholesterolu, triglicerydów, białka całkowitego i albumin. W wątrobie szczurów nie stwierdzono zmian histopatologicznych. U psów, którym podawano imatynib przez 2 tygodnie, obserwowano ciężkie uszkodzenie wątroby ze zwiększeniem aktywności enzymów wątrobowych, martwicą komórek wątrobowych, martwicą przewodów żółciowych i rozrostem w obrębie przewodów żółciowych.
U małp, którym podawano imatynib przez 2 tygodnie obserwowano uszkodzenie nerek z ogniskową mineralizacją, rozszerzeniem cewek nerkowych i zwyrodnieniem cewek nerkowych. U kilku małp stwierdzono zwiększenie stężenia azotu mocznikowego we krwi (BUN) i kreatyniny. U szczurów, po podaniu dawki ≥6 mg/kg przez 13 tygodni, obserwowano rozrost przejściowego nabłonka brodawek nerkowych i pęcherza moczowego, bez zmian wskaźników w surowicy i moczu. W czasie długotrwałego podawania imatynibu stwierdzono zwiększenie częstości zakażeń oportunistycznych.
W trwającym 39 tygodni badaniu na małpach nie ustalono wartości NOAEL (ang. no observed adverse effect level, czyli ekspozycji, przy której nie obserwowano działań niepożądanych) na poziomie najmniejszej dawki leku 15 mg/kg mc., stanowiącej około 1/3 maksymalnej dawki zalecanej u ludzi (800 mg) w przeliczeniu na powierzchnię ciała. Leczenie imatynibem powodowało pogorszenie zazwyczaj zahamowanego przewlekłego zakażenia malarią u tych zwierząt.
Imatynib nie miał działania genotoksycznego w badaniach in vitro z zastosowaniem komórek bakteryjnych (test Amesa), w badaniu in vitro z zastosowaniem komórek ssaków (chłoniaka mysiego) ani in vivo w mikrojądrowym teście u szczurów. Dodatnie efekty genotoksyczności uzyskano dla imatynibu w badaniu in vitro komórek ssaków (komórki jajnika chomików) wykrywającym działanie klastogenne (aberracje chromosomowe) przy aktywacji metabolicznej. Dwa z produktów pośrednich procesu wytwarzania, obecnych w produkcie końcowym, miało działanie mutagenne w teście Amesa. Jeden z nich miał również działanie mutagenne w teście z zastosowaniem komórek chłoniaka mysiego.
W badaniach wpływu na płodność wykazano zmniejszenie masy jąder i najądrzy oraz odsetka ruchliwych plemników u samców szczurów, którym przez 70 dni przed kojarzeniem podawano imatynib w dawce 60 mg/kg mc. (równej w przybliżeniu maksymalnej zalecanej dawce klinicznej wynoszącej 800 mg/dobę w przeliczeniu na powierzchnię ciała). Działania takiego nie obserwowano po podaniu dawek ≥20 mg/kg mc. Nieznaczne do umiarkowanego zmniejszenie spermatogenezy obserwowano również u psów po podaniu dawek doustnych ≥30 mg/kg mc. Podawanie imatynibu samicom szczura na 14 dni przed kojarzeniem do 6. dnia potencjalnej ciąży nie spowodowało wpływu na przebieg kojarzenia ani na liczbę ciężarnych samic. Po podaniu dawki 60 mg/kg mc. u samic szczurów stwierdzono istotne zwiększenie poimplantacyjnych utrat płodów i zmniejszenie liczby żywych płodów. Nie stwierdzono takiego działania po podaniu dawek ≥20 mg/kg.
W badaniu przed- i pourodzeniowego rozwoju u szczurów doustne podawanie imatynibu w dawce dobowej 45 mg/kg mc. powodowało wystąpienie czerwonej wydzieliny z pochwy w 14. lub 15. dniu ciąży. Po podaniu tej samej dawki liczba urodzonych martwych młodych oraz tych, które padły między 0. i 4. dniem po porodzie, była zwiększona. U młodych pokolenia F1 ta sama dawka spowodowała zmniejszenie średniej masy ciała od porodu do końca badania, a liczba młodych osiągających stadium odwiedzenia napletka była nieznacznie zmniejszona. Płodność w pokoleniu F1 nie była zmieniona, ale zwiększyła się liczba resorpcji i zmniejszyła liczba żywych płodów po podaniu dawki 45 mg/kg mc./dobę. Dawka NOEL (brak działań) zarówno dla matek, jak i ich potomstwa pokolenia F1 wynosiła 15 mg/kg mc./dobę (jedna czwarta maksymalnej dawki stosowanej u ludzi czyli 800 mg).
Imatynib podawany szczurom w okresie organogenezy w dawkach ≥100 mg/kg mc. wykazywał działanie teratogenne. Dawka ta jest zbliżona do maksymalnej dawki klinicznej (800 mg/dobę)
w przeliczeniu na powierzchnię ciała. Działanie teratogenne obejmowało częściowy lub całkowity brak kości czaszki, przepuklinę mózgową, nieobecność/redukcję kości czołowej i nieobecność kości ciemieniowej. Działania takiego nie obserwowano po zastosowaniu dawek ≥30 mg/kg.
W badaniu toksykologicznym dotyczącym rozwoju młodych szczurów (dzień 10-70 po urodzeniu) nie
wykazano żadnego nowego narządu docelowego w odniesieniu do znanych narządów docelowych
u dorosłych szczurów. W badaniu toksykologicznym na młodych osobnikach obserwowano wpływ na wzrost, opóźnienie otwarcia pochwy i separacji napletka, przy ekspozycji około 0,3 do 2 razy większej niż przeciętna pediatryczna ekspozycja po podaniu największej zalecanej dawki 340 mg/m2 pc.
Ponadto u młodych zwierząt (w fazie usamodzielniania się) obserwowano śmiertelność przy około
2-krotności przeciętnej pediatrycznej ekspozycji po podaniu największej zalecanej dawki 340 mg/m2.
W trwającym 2 lata badaniu rakotwórczości u szczurów podawanie imatynibu w dawkach 15, 30
i 60 mg/kg mc./dobę powodowało statystycznie istotne skrócenie czasu życia samców po dawkach 60 mg/kg mc./dobę i samic po dawkach ≥30 mg/kg mc./dobę. Badanie histopatologiczne martwych osobników wykazało jako główną przyczynę padnięcia lub powód uśmiercenia zwierząt laboratoryjnych kardiomiopatię (u szczurów obu płci), przewlekłą postępującą chorobę nerek
(u samic) oraz brodawczaka gruczołu napletkowego. Narządami docelowymi dla zmian nowotworowych były nerki, pęcherz moczowy, cewka moczowa, gruczoł napletkowy i łechtaczkowy, jelito cienkie, przytarczyce, nadnercza oraz dno żołądka.
Przypadki brodawczaka/raka gruczołów napletkowych/łechtaczkowych odnotowano po podaniu dawek od 30 mg/kg mc./dobę, odpowiadających około 0,5- lub 0,3-krotność dobowej ekspozycji na lek u ludzi (na podstawie AUC) po podaniu dawki 400 mg/dobę lub 800 mg/dobę i 0,4-krotność dobowej ekspozycji na lek u dzieci (na podstawie AUC) po podaniu dawki 340 mg/m2 pc./dobę.
Dawka NOEL wynosiła 15 mg/kg mc./dobę. Występowanie gruczolaka/raka nerek, brodawczaka pęcherza moczowego i cewki moczowej, gruczolakoraka jelita cienkiego, gruczolaków przytarczyc, łagodnych i złośliwych guzów części rdzennej nadnerczy oraz brodawczaków/raków dna żołądka odnotowano po dawce 60 mg/kg mc./dobę, co stanowiło odpowiednio około 1,7 lub 1 krotność dobowej ekspozycji na lek u ludzi (na podstawie AUC) po podaniu dawki 400 mg/dobę lub 800 mg/dobę oraz 1,2-krotność dobowej ekspozycji na lek u dzieci (na podstawie AUC) po podaniu dawki 340 mg/m2 pc./dobę. Dawka, po której nie ma objawów działań niepożądanych (NOEL) wynosiła
30 mg/kg mc./dobę.
Mechanizm oraz znaczenie dla ludzi wyników badań rakotwórczości na szczurach nie zostały jeszcze wyjaśnione.
Do zmian nienowotworowych nieobserwowanych we wcześniejszych badaniach nieklinicznych należały zmiany w układzie sercowo-naczyniowym, w trzustce, w narządach układu wewnątrzwydzielniczego i w zębach. Najważniejsze zmiany to przerost mięśnia sercowego i rozstrzeń jam serca, prowadzące u niektórych zwierząt do objawów niewydolności serca.
Substancja czynna, imatynib, wykazuje zagrożenie dla środowiska dla organizmów żyjących
w materiałach osadowych.
Celuloza mikrokrystaliczna Krospowidon (typ A) Hypromeloza
Magnezu stearynian
Krzemionka koloidalna bezwodna
Otoczka:
Żelaza tlenek czerwony (E 172) Żelaza tlenek żółty (E 172) Makrogol 4000
Talk Hypromeloza
Nie dotyczy.
3 lata
Nie przechowywać w temperaturze powyżej 30°C.
Imatinib Sandoz, 400 mg pakowany w blistry z folii PCV/PE/PVDC/Aluminium należy przechowywać w temperaturze poniżej 25°C.
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Tabletki powlekane pakowane są w blistry z folii PVC/Aluminium lub blistry z folii PVC/PE/PVDC/Aluminium i umieszczane w tekturowym pudełku.
Wielkość opakowań:
Imatinib Sandoz, 100 mg
Blistry z folii PVC/Aluminium
Blistry z folii PVC/PE/PVDC/Aluminium
20, 30, 50, 60, 80, 90 i 120 tabletek powlekanych
Imatinib Sandoz, 400 mg
Blistry z folii PVC/PE/PVDC/Aluminium
10, 30, 50, 60, 80 i 90 tabletek powlekanych
Nie wszystkie rodzaje i wielkości opakowań muszą znajdować się w obrocie.
stosowania
Wszelkie niewykorzystane resztki produktu lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
DO OBROTU
Sandoz GmbH
Biochemiestrasse 10
6250 Kundl, Austria
Imatinib Sandoz, 100 mg Pozwolenie nr 23317 Imatinib Sandoz, 400 mg Pozwolenie nr 23318
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 11.07.2016 r. Data ostatniego przedłużenia pozwolenia: 25.09.2020 r.
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
05.12.2022 r.








