







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Łagodne zaburzenia czynności wątroby (A wg skali Child-Pugh) – zalecana dawka to 7,5 mg na dobę.
Umiarkowane zaburzenia czynności wątroby (B wg skali Child-Pugh) – zalecana dawka to 5 mg na dobę.
Ciężkie zaburzenia czynności wątroby (C wg skali Child-Pugh) – stosowanie leku Everolimus Genthon jest zalecane tylko wtedy, kiedy pożądane korzyści z leczenia przewyższają ryzyko. W takiej sytuacji nie należy przekraczać dawki 2,5 mg na dobę.
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Everolimus Genthon, 2,5 mg, tabletki Everolimus Genthon, 5 mg, tabletki Everolimus Genthon, 10 mg, tabletki
Everolimus Genthon 2,5 mg tabletki
Jedna tabletka zawiera 2,5 mg ewerolimusu.
Substancja pomocnicza o znanym działaniu:
Jedna tabletka zawiera 74,3 mg laktozy.
Everolimus Genthon 5 mg tabletki
Jedna tabletka zawiera 5 mg ewerolimusu.
Substancja pomocnicza o znanym działaniu:
Jedna tabletka zawiera 148,5 mg laktozy.
Everolimus Genthon 10 mg tabletki
Jedna tabletka zawiera 10 mg ewerolimusu.
Substancja pomocnicza o znanym działaniu:
Jedna tabletka zawiera 297,0 mg laktozy.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka.
2,5 mg tabletka: Białe lub białawe, owalne, obustronnie wypukłe tabletki (około 10 mm x 5 mm) z wytłoczonym napisem E9VS na jednej stronie i 2.5 na drugiej stronie.
5 mg tabletka: Białe lub białawe, owalne, obustronnie wypukłe tabletki (około 13 mm x 6 mm) z wytłoczonym napisem E9VS 5 na jednej stronie.
10 mg tabletka: Białe lub białawe, owalne, obustronnie wypukłe tabletki (około 16 mm x 8 mm) z wytłoczonym napisem E9VS 10 na jednej stronie.
Zaawansowany rak piersi z ekspresją receptorów hormonalnych
Produkt leczniczy Everolimus Genthon jest wskazany w leczeniu zaawansowanego raka piersi z ekspresją receptorów hormonalnych, bez nadekspresji HER2/neu, w skojarzeniu z eksemestanem u kobiet po menopauzie bez objawowego zajęcia narządów wewnętrznych, po wystąpieniu wznowy lub progresji po leczeniu niesteroidowym inhibitorem aromatazy.
Nowotwory neuroendokrynne trzustki
Everolimus Genthon jest wskazany w leczeniu nieoperacyjnych lub z przerzutami wysoko lub średnio zróżnicowanych nowotworów neuroendokrynnych trzustki u dorosłych pacjentów z chorobą o przebiegu postępującym.
Nowotwory neuroendokrynne układu pokarmowego lub płuc
Everolimus Genthon jest wskazany w leczeniu nieoperacyjnych lub z przerzutami, wysoko zróżnicowanych (stopień G1 lub G2), hormonalnie nieczynnych nowotworów neuroendokrynnych układu pokarmowego lub płuc u dorosłych pacjentów z chorobą o przebiegu postępującym (patrz punkt 4.4 i 5.1).
Rak nerkowokomórkowy
Everolimus Synhton jest wskazany w leczeniu pacjentów z zaawansowanym rakiem nerkowokomórkowym, u których postęp choroby nastąpił w trakcie lub po przebytej terapii anty- VEGF (czynnik wzrostu śródbłonka naczyniowego).
Leczenie produktem leczniczym Everolimus Genthon powinno być rozpoczęte i nadzorowane przez lekarza doświadczonego w stosowaniu leków przeciwnowotworowych.
Dawkowanie
W celu umożliwienia odpowiedniego dawkowania, produkt leczniczy Everolimus Genthon jest dostępny w postaci tabletek w dawkach 2,5 mg, 5 mg i 10 mg.
Zalecana dawka ewerolimusu wynosi 10 mg raz na dobę. Leczenie należy kontynuować tak długo, jak długo obserwuje się korzyści kliniczne lub do wystąpienia objawów niemożliwej do zaakceptowania toksyczności.
W przypadku pominięcia dawki, nie należy przyjmować dodatkowej dawki, ale przyjąć kolejną dawkę o zwykłej porze.
Dostosowanie dawkowania ze względu na działania niepożądane
W przypadku wystąpienia ciężkich i (lub) niemożliwych do zaakceptowania działań niepożądanych, może zajść konieczność zmniejszenia dawkowania i (lub) tymczasowego przerwania leczenia produktem leczniczym Everolimus Genthon. Dostosowanie dawki zazwyczaj nie jest wymagane w przypadku działań niepożądanych 1. stopnia. Jeżeli konieczne jest zmniejszenie dawki, zalecana dawka to 5 mg na dobę i nie może być mniejsza niż 5 mg na dobę.
W Tabeli 1 zestawiono zalecenia dotyczące dostosowania dawki w przypadku różnych działań niepożądanych (patrz także punkt 4.4).
Tabela 1 Zalecenia dotyczące dostosowania dawki produktu leczniczego Everolimus Genthon
Działanie niepożądane | Nasilenie1 | Zalecenia dotyczące dostosowania dawki produktu leczniczego Everolimus Genthon |
Nieinfekcyjne zapalenie płuc | 2. stopień | Rozważyć przerwanie leczenia, aż do czasu złagodzenia objawów do ≤1 stopnia. Rozważyć wznowienie leczenia w dawce 5 mg. Jeśli toksyczność 3. stopnia wystąpi ponownie, należy rozważyć zakończenie leczenia. |
3. stopień | Przerwać leczenie do czasu złagodzenia objawów do ≤1 stopnia. Rozważyć wznowienie leczenia w dawce 5 mg. Jeśli toksyczność 3. stopnia wystąpi ponownie, należy rozważyć zakończenie | |
4. stopień | Zakończyć leczenie. | |
Zapalenie jamy ustnej | 2. stopień | Okresowo przerwać podawanie leku aż do czasu złagodzenia objawów do ≤1 stopnia. Wznowić leczenie w tej samej dawce. W przypadku nawrotu zapalenia jamy ustnej 2. stopnia przerwać podawanie produktu leczniczego, aż do czasu złagodzenia objawów do ≤1 stopnia. Wznowić leczenie w dawce 5 mg. |
3. stopień | Okresowo przerwać podawanie produktu leczniczego, aż do złagodzenia objawów do ≤1 stopnia. | |
4. stopień | Zakończyć leczenie. | |
Inne niehematologiczne objawy toksyczności (z wyjątkiem zaburzeń metabolicznych) | 2. stopień | Jeśli objawy toksyczności są tolerowane, nie ma konieczności dostosowania dawki. Jeśli objawy toksyczności nie są tolerowane, należy okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤1 stopnia. Wznowić leczenie w tej samej dawce. Jeśli objawy toksyczności 2. stopnia wystąpią ponownie, należy przerwać leczenie, aż do czasu złagodzenia objawów do ≤1 stopnia. Wznowić leczenie w dawce 5 mg. |
3. stopień | Okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤1 stopnia. Rozważyć wznowienie leczenia w dawce 5 mg. Jeśli toksyczność 3. stopnia wystąpi ponownie, należy rozważyć zakończenie leczenia. | |
stopień | Zakończyć leczenie. | |
Zaburzenia metaboliczne (np. hiperglikemia dyslipidemia) | 2. stopień | Brak konieczności dostosowania dawki. |
3. stopień | Okresowo przerwać leczenie. Wznowić leczenie w dawce 5 mg. | |
4. stopień | Zakończyć leczenie. | |
Trombocytopenia | 2. stopień (<75, ≥50x109/l) | Okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤1 stopnia (≥75x109/l). Wznowić leczenie w tej samej dawce. |
3. i 4. stopień (<50x109/l) | Okresowo przerwać leczenie, aż do złagodzenia objawów do ≤1 stopnia (≥75x109/l). Wznowić leczenie w dawce 5 mg. | |
Neutropenia | 2. stopień (≥1x109/l) | Brak konieczności dostosowania dawki. |
3. stopień (<1, ≥0,5x109/l) | Okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤2 stopnia (≥1x109/l). Wznowić leczenie w tej samej dawce. | |
4. stopień (<0,5x109/l) | Okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤2 stopnia (≥1x109/l). Wznowić leczenie w dawce 5 mg. | |
Gorączka neutropeniczna | 3. stopień | Okresowo przerwać leczenie, aż do czasu złagodzenia objawów do ≤2 stopnia (≥1,25x109/l) i ustąpienia gorączki. Wznowić leczenie w dawce 5 mg. |
4. stopień | Zakończyć leczenie. | |
1 Stopniowanie nasilenia objawów według National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) wersja 3.0 | ||
Szczególne grupy pacjentów
Osoby w podeszłym wieku (≥65 lat)
Dostosowanie dawkowania nie jest konieczne (patrz punkt 5.2).
Pacjenci z zaburzeniami czynności nerek
Dostosowanie dawkowania nie jest konieczne (patrz punkt 5.2).
Pacjenci z zaburzeniami czynności wątroby
Jeśli w trakcie leczenia stan wątroby pacjenta (wg skali Child-Pugh) ulegnie zmianie, dawkowanie należy dostosować (patrz także punkty 4.4 i 5.2).
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego Everolimus Genthon u dzieci w wieku 0 do 18 lat. Dane nie są dostępne.
Sposób podawania
Everolimus Genthon powinien być podawany doustnie, raz na dobę o tej samej porze, z posiłkiem lub bez (patrz punkt 5.2). Everolimus Genthon w postaci tabletek należy połykać w całości popijając szklanką wody. Tabletek nie należy żuć ani rozgryzać.
Nadwrażliwość na substancję czynną, na inne pochodne rapamycyny lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Nieinfekcyjne zapalenie płuc
Nieinfekcyjne zapalenie płuc jest efektem klasowym pochodnych rapamycyny, w tym ewerolimusu. Przypadki wystąpienia nieinfekcyjnego zapalenia płuc (m.in. śródmiąższowego zapalenia płuc) były często zgłaszane u pacjentów przyjmujących produkt leczniczy Everolimus Genthon (patrz punkt 4.8). Niektóre przypadki były niekiedy ciężkie, a w rzadkich przypadkach, choroba prowadziła do śmierci. Rozpoznanie nieinfekcyjnego zapalenia płuc należy rozważyć u pacjentów z niespecyficznymi objawami przedmiotowymi i podmiotowymi ze strony układu oddechowego, takimi jak niedotlenienie, wysięk do opłucnej, kaszel lub duszność oraz u pacjentów, u których wykluczono, za pomocą odpowiednich metod diagnostycznych, zakaźne, nowotworowe lub niemedyczne przyczyny występujących objawów. W diagnostyce różnicowej nieinfekcyjnego zapalenia płuc należy wykluczyć zakażenia oportunistyczne, takie jak pneumocystozowe zapalenie płuc wywołane przez Pneumocystis jiroveci (carinii) (PJP, PCP) (patrz punkt „Zakażenia” poniżej). Pacjentów należy poinformować o konieczności natychmiastowego zgłaszania nowych lub pogorszeniu istniejących objawów ze strony układu oddechowego.
Jeżeli u pacjentów wystąpią zmiany w obrazie radiologicznym sugerujące nieinfekcyjne zapalenie płuc, a jednocześnie występują nieliczne objawy kliniczne lub objawy nie występują, podawanie produktu leczniczego Everolimus Genthon można kontynuować bez konieczności zmiany dawkowania. W przypadku wystąpienia objawów o umiarkowanym nasileniu (2. stopnia) lub ciężkim nasileniu (3. stopnia) wskazane może być podawanie kortykosteroidów, aż do momentu ustąpienia objawów klinicznych.
U pacjentów, u których konieczne jest podanie kortykosteroidów w leczeniu nieinfekcyjnego zapalenia płuc należy rozważyć profilaktykę pneumocystozowego zapalenia płuc (PJP, PCP).
Zakażenia
Ewerolimus ma właściwości immunosupresyjne i może zwiększać podatność pacjentów na zakażenia bakteryjne, grzybicze, wirusowe lub pierwotniakowe, w tym zakażenia patogenami oportunistycznymi (patrz punkt 4.8). U pacjentów przyjmujących ewerolimus występowały zakażenia miejscowe i ogólnoustrojowe, w tym zapalenie płuc, inne zakażenia bakteryjne, inwazyjne zakażenia grzybicze, takie jak aspergiloza, kandydoza lub pneumocystozowe zapalenie płuc wywołane przez Pneumocystis jirovecii (carinii) (PJP. PCP) i zakażenia wirusowe, w tym reaktywacja wirusowego zapalenia wątroby typu B. Niektóre z wyżej wymienionych zakażeń charakteryzował ciężki przebieg (np. wystąpienie posocznicy, niewydolności oddechowej lub niewydolności wątroby), a część z nich prowadziła do śmierci.
Lekarze i pacjenci powinni być świadomi zwiększonego ryzyka wystąpienia zakażenia w trakcie leczenia produktem leczniczym Everolimus Genthon. Przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon należy całkowicie wyleczyć wszystkie istniejące zakażenia. Podczas podawania produktu leczniczego Everolimus Genthon, należy uważnie obserwować, czy nie pojawiają się podmiotowe i przedmiotowe objawy zakażenia: jeśli rozpoznano zakażenie, należy natychmiast rozpocząć właściwe leczenie i rozważyć przerwanie lub zaprzestanie podawania produktu leczniczego Everolimus Genthon.
Jeżeli rozwinie się inwazyjne zakażenie grzybicze, należy natychmiast przerwać na stałe podawanie produktu leczniczego Everolimus Genthon, a pacjentowi podać odpowiednie leki przeciwgrzybicze.
U pacjentów otrzymujących ewerolimus zgłaszano przypadki pneumocystozowego zapalenia płuc wywołanego przez Pneumocystis jirovecii (carinii) (PJP, PCP), z których część zakończyła się zgonem pacjenta. Występowanie PJP/PCP może być związane z jednoczesnym stosowaniem kortykosteroidów lub innych leków immunosupresyjnych. Należy rozważyć profilaktykę PJP, PCP u pacjentów wymagających jednoczesnego stosowania kortykosteroidów lub innych leków immunosupresyjnych.
Reakcje nadwrażliwości
U pacjentów stosujących ewerolimus obserwowano reakcje nadwrażliwości, obejmujące m. in. anafilaksję, duszność, zaczerwienienie twarzy, ból w klatce piersiowej lub obrzęk naczynioruchowy (np. obrzęk dróg oddechowych lub języka z zaburzeniami oddychania lub bez tych zaburzeń) (patrz punkt 4.3).
Jednoczesne stosowanie inhibitorów konwertazy angiotensyny (ACE)
U pacjentów leczonych jednocześnie inhibitorem ACE (np. ramiprylem) może wystąpić zwiększone ryzyko obrzęku naczynioruchowego (np. obrzęk dróg oddechowych lub języka z zaburzeniami układu oddechowego lub bez zaburzeń) (patrz punkt 4.5).
Zapalenie jamy ustnej
Zapalenie jamy ustnej, w tym owrzodzenia jamy ustnej i zapalenie błony śluzowej jamy ustnej jest najczęściej zgłaszanym działaniem niepożądanym obserwowanym u pacjentów leczonych ewerolimusem (patrz punkt 4.8). Zapalenie jamy ustnej występuje głównie w ciągu pierwszych 8 tygodni leczenia. Wyniki badania z jedną grupą terapeutyczną z udziałem pacjentek po menopauzie z rakiem piersi leczonych ewerolimusem w skojarzeniu z eksemestanem sugerowały, że stosowanie bezalkoholowego doustnego roztworu kortykosteroidu podawanego jako płyn do płukania jamy ustnej w pierwszych 8 tygodniach leczenia może zmniejszyć częstość występowania i nasilenie zapalenia jamy ustnej (patrz punkt 5.1). Postępowanie w zapaleniu jamy ustnej może zatem obejmować profilaktyczne i (lub) terapeutyczne stosowanie terapii miejscowych, takich jak bezalkoholowy doustny roztwór kortykosteroidu w postaci płynu do płukania jamy ustnej. Należy jednak unikać stosowania produktów zawierających alkohol, nadtlenek wodoru, jod i wyciągi z tymianku, ponieważ mogą one zaostrzać objawy. Zaleca się monitorowanie w celu wykrycia zakażenia grzybiczego i jego
leczenia, zwłaszcza u pacjentów otrzymujących leki na bazie steroidów. Nie należy stosować leków przeciwgrzybiczych, jeżeli nie zdiagnozowano zakażenia grzybiczego (patrz punkt 4.5).
Przypadki niewydolności nerek
U pacjentów leczonych ewerolimusem obserwowano przypadki niewydolności nerek (w tym ostrą niewydolność nerek), niektóre ze skutkiem śmiertelnym (patrz punkt 4.8). Należy kontrolować czynność nerek szczególnie u pacjentów, u których występują inne czynniki ryzyka, które dodatkowo mogą zaburzać czynność nerek.
Badania laboratoryjne
Czynność nerek
Zgłaszano zwiększone stężenie kreatyniny w surowicy, najczęściej niewielkie, i białkomocz (patrz punkt 4.8). Zaleca się, aby przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon oraz okresowo w trakcie leczenia kontrolować czynność nerek, w tym stężenie azotu mocznikowego we krwi (BUN), stężenie białka w moczu i stężenie kreatyniny w surowicy.
Stężenie glukozy we krwi
Obserwowano przypadki hiperglikemii (patrz punkt 4.8). Zaleca się, aby przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon oraz okresowo w trakcie leczenia kontrolować stężenie glukozy w surowicy na czczo. Częstsze kontrolowanie zaleca się, kiedy produkt leczniczy Everolimus Genthon jest podawany jednocześnie z innymi produktami leczniczymi, które mogą spowodować hiperglikemię. Jeśli jest to możliwe, należy dążyć do uzyskania optymalnej kontroli glikemii przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon.
Stężenie lipidów we krwi
U pacjentów przyjmujących produkt leczniczy ewerolimus obserwowano przypadki dyslipidemii (w tym hipercholesterolemii i hipertriglicerydemii). Zaleca się, aby przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon oraz okresowo w trakcie leczenia, kontrolować stężenie cholesterolu i triglicerydów we krwi, a także zastosować odpowiednie leczenie.
Parametry hematologiczne
Zgłaszano zmniejszonie stężenia hemoglobiny i liczby limfocytów, neutrofilii i płytek krwi (patrz punkt 4.8). Zaleca się, aby przed rozpoczęciem podawania produktu leczniczego Everolimus Genthon oraz okresowo w trakcie leczenia kontrolować wyniki morfologii krwi.
Hormonalnie czynne rakowiaki
W randomizowanym, podwójnie zaślepionym, wieloośrodkowym badaniu z udziałem pacjentów z hormonalnie czynnymi rakowiakami, ewerolimus w skojarzeniu z długo działającym oktreotydem porównywano z placebo w skojarzeniu z długo działającym oktreotydem. W badaniu nie uzyskano pierwszorzędowego punktu końcowego skuteczności (PFS, ang. progression free survival), a pośrednia analiza (OS, ang. overall survival) wykazała liczbową przewagę na korzyść grupy otrzymującej placebo w skojarzeniu z długo działającym oktreotydem. Z tego względu, bezpieczeństwo stosowania i skuteczność ewerolimusu u pacjentów z hormonalnie czynnymi rakowiakami nie zostały ustalone.
Czynniki prognostyczne w nowotworach neuroendokrynnych układu pokarmowego lub płuc
U pacjentów z hormonalnie nieczynnymi nowotworami neuroendokrynnymi układu pokarmowego lub płuc i dobrymi wyjściowymi czynnikami prognostycznymi, np. guzem pierwotnym umiejscowionym w jelicie krętym i prawidłowymi wartościami chromograniny A lub bez zajęcia kości, należy dokonać indywidualnej oceny korzyści względem ryzyka przed rozpoczęciem leczenia produktem leczniczym Everolimus Genthon. Odnotowano ograniczone dowody korzyści w zakresie PFS w podgrupie chorych na nowotwory neuroendokrynne wywodzące się z jelita krętego (patrz punkt 5.1).
Interakcje
Należy unikać jednoczesnego podawania inhibitorów i induktorów CYP3A4 i (lub) wielolekowej pompy glikoproteiny P (PgP) z produktem leczniczym Everolimus Genthon. Jeśli nie można uniknąć jednoczesnego podawania umiarkowanych inhibitorów lub induktorów CYP3A4 i (lub) PgP, należy
rozważyć dostosowanie dawki produktu leczniczego Everolimus Genthon w oparciu o przewidywane AUC (patrz punkt 4.5).
Jednoczesne podawanie z silnymi inhibitorami CYP3A4 skutkuje dramatycznym wzrostem stężenia ewerolimusu w osoczu (patrz punkt 4.5). Obecnie nie ma wystarczających danych pozwalających na dostosowanie dawkowania w takiej sytuacji. Dlatego jednoczesne podawanie produktu leczniczego Everolimus Genthon i silnych inhibitorów nie jest zalecane.
Należy zachować ostrożność stosując produkt leczniczy Everolimus Genthon w skojarzeniu z doustnymi substratami CYP3A4 o wąskim indeksie terapeutycznym z uwagi na możliwość wystąpienia interakcji. Jeśli produkt leczniczy Everolimus Genthon jest podawany z doustnymi substratami CYP3A4 o wąskim indeksie terapeutycznym (np. pimozydem, terfenadyną, astemizolem, cisaprydem, chinidyną lub pochodnymi alkaloidów sporyszu), pacjenta należy monitorować w kierunku działań niepożądanych opisanych w drukach informacyjnych doustnych substratów CYP3A4 (patrz punkt 4.5).
Zaburzenia czynności wątroby
Ekspozycja na ewerolimus była zwiększona u pacjentów z łagodnymi (A wg skali Child-Pugh), umiarkowanymi (B wg skali Child-Pugh) i ciężkimi (C wg skali Child-Pugh) zaburzeniami czynności wątroby (patrz punkt 5.2).
Stosowanie produktu Everolimus Genthon jest zalecane wyłącznie u pacjentów z ciężkim zaburzeniem czynności wątroby (C wg skali Child-Pugh), jeśli potencjalna korzyść z leczenia przewyższa ryzyko (patrz punkty 4.2 i 5.2).
Aktualnie brak dostępnych danych dotyczących bezpieczeństwa lub skuteczności klinicznej potwierdzających zalecenia dotyczące dostosowania dawkowania w przypadku działań niepożądanych u pacjentów z zaburzeniami czynności wątroby.
Szczepienia
Należy unikać podawania żywych szczepionek w trakcie leczenia produktem leczniczym Everolimus Genthon (patrz punkt 4.5).
Komplikacje związane z gojeniem się ran
Utrudnione gojenie się ran to efekt klasowy pochodnych rapamycyny, w tym ewerolimusu. Należy zachować ostrożność podczas przyjmowania produktu leczniczego Everolimus Genthon w okresie okołooperacyjnym.
Ostrzeżenia związane z substancją pomocniczą
Lek nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Ewerolimus jest substratem CYP3A4. Jest również substratem i umiarkowanym inhibitorem PgP. Tym samym, substancje, które działają na CYP3A4 i (lub) PgP wpływają na wchłanianie i wydalanie ewerolimusu. W warunkach in vitro ewerolimus jest kompetencyjnym inhibitorem CYP3A4 oraz mieszanym inhibitorem CYP2D6.
W Tabeli 2 poniżej przedstawiono zarówno znane, jak i teoretycznie możliwe interakcje z wybranymi inhibitorami i induktorami CYP3A4 i PgP.
Inhibitory CYP3A4 i PgP zwiększające stężenie ewerolimusu
Substancje będące inhibitorami CYP3A4 lub PgP mogą zwiększać stężenie ewerolimusu we krwi poprzez spowolnienie jego metabolizmu lub przenikanie ewerolimusu z komórek jelita.
Induktory CYP3A4 i PgP zmniejszające stężenie ewerolimusu
Substancje będące induktorami CYP3A4 i PgP mogą zmniejszać stężenie ewerolimusu we krwi poprzez przyspieszenie jego metabolizmu lub nasilenie przenikania ewerolimusu z komórek jelita.
Tabela 2 Wpływ innych substancji czynnych na ewerolimus
Substancja czynna i rodzaj interakcji | Interakcja – Zmiana AUC/Cmax Ewerolimusu Stosunek średnich geometrycznych (obserwowany zakres) | Zalecenia dotyczące leczenia skojarzonego |
Silne inhibitory CYP3A4/PgP | ||
Ketokonazol | AUC ↑15,3-krotnie | Jednoczesne podawanie produktu |
(zakres 11,2-22,5) | leczniczego Everolimus Genthon i silnych | |
Cmax ↑4,1-krotnie | inhibitorów nie jest zalecane. | |
(zakres 2,6-7,0) | ||
Itrakonazol, posakonazol, | Nie badano. Należy spodziewać się | |
worikonazol | dużego wzrostu stężenia ewerolimusu. | |
Telitromycyna, | ||
klarytromycyna | ||
Nefazodon | ||
Rytonawir, atazanawir, | ||
sakwinawir, darunawir, | ||
indinawir, nelfinawir | ||
Umiarkowane inhibitory CYP3A4/PgP | ||
Erytromycyna | AUC ↑4,4-krotnie (zakres 2,0-12,6) Cmax ↑2,0-krotnie (zakres 0,9-3,5) | Należy zachować ostrożność, kiedy nie można uniknąć jednoczesnego podawania umiarkowanych inhibitorów CYP3A4 lub PgP. U pacjentów wymagających jednoczesnego podania umiarkowanego inhibitora CYP3A4 lub PgP, należy rozważyć zmniejszenie dawki do 5 mg na dobę lub 2,5 mg na dobę. Nie ma danych klinicznych dotyczących takiego dostosowania dawkowania. W związku z różnicami pomiędzy pacjentami, zalecane dostosowanie dawkowania może nie być optymalne dla wszystkich pacjentów, dlatego też zaleca się ścisłą obserwację działań niepożądanych. W przypadku odstawienia umiarkowanego inhibitora CYP3A4 należy wziąć pod uwagę okres wypłukiwania leku z organizmu, trwający co najmniej 2 do 3 dni (przeciętny czas eliminacji większości najczęściej stosowanych umiarkowanych inhibitorów CYP3A4) przed przywróceniem dawki produktu leczniczego Everolimus Genthon do wielkości stosowanej przed rozpoczęciem leczenia skojarzonego. |
Imatinib | AUC ↑ 3.7-krotnie | |
Cmax t 2.2-krotnie | ||
Werapamil | AUC ↑3,5-krotnie (zakres 2,2-6,3) Cmax ↑2,3-krotnie (zakres 1,3-3.8) | |
Cyklosporyna, doustnie | AUC ↑2,7-krotnie (zakres 1,5-4,7) Cmax ↑1,8-krotnie (zakres 1,3-2,6) | |
Flukonazol | Nie badano. Przewidywany wzrost AUC. | |
Diltiazem | ||
Dronedaron | Nie badano. Przewidywany wzrost AUC. | |
Amprenawir, fosamprenawir | Nie badano. Przewidywany wzrost AUC. . | |
Sok grejpfrutowy lub inne pokarmy wpływające na CYP3A4/PgP | Nie badano. Przewidywany wzrost AUC (znaczne zróżnicowanie wpływu). | Należy unikać jednoczesnego leczenia. |
Silne i umiarkowane induktory CYP3A4/PgP | ||
Ryfampicyna | AUC ↓63% | Należy unikać jednoczesnego podawania |
(zakres 0-80%) | produktu leczniczego Everolimus | |
Cmax ↓58% | Genthon z silnymi induktorami CYP3A4. | |
(zakres 10-70%) | Jeśli u pacjenta konieczne jest podanie | |
produktu leczniczego ewerolimus z silnym induktorem CYP3A4 należy rozważyć zwiększenie dawki produktu leczniczego ewerolimus z 10 mg/dobę nawet do 20 mg na dobę, etapami co 5 mg | ||
Deksametazon | Nie badano. Przewidywane zmniejszenie AUC. | |
Karbamazepina, fenobarbital, fenytoina | Nie badano. Przewidywane zmniejszenie AUC. | |
lub mniej, podawanej w 4 i 8 dniu następującej po podaniu induktora. Przewiduje się, że taka dawka produktu | ||
Efawirenz, newirapina | Nie badano. Przewidywane zmniejszenie AUC. | |
leczniczego Everolimus Genthon | ||
spowoduje modyfikację AUC do zakresu | ||
wielkości obserwowanych bez podawania | ||
induktorów CYP3A4. Brak jest jednak | ||
danych klinicznych dotyczących takiej | ||
modyfikacji dawkowania. W razie | ||
przerwania leczenia induktorem CYP3A4 | ||
należy wziąć pod uwagę okres | ||
wypłukiwania leku z organizmu, trwający | ||
co najmniej 3 do 5 dni (uzasadniony czas | ||
odwrócenia indukcji enzymów) przed | ||
przywróceniem dawki produktu | ||
leczniczego Everolimus Genthon do | ||
wielkości sprzed skojarzonego leczenia. | ||
Dziurawiec (Hypericum Perforatum) | Nie badano. Przewidywane duże zmniejszenie stężenia ewerolimusu. | Podczas leczenia ewerolimusem nie należy stosować preparatów zawierających dziurawiec. |
Leki, których stężenie w osoczu może ulec zmianie pod wpływem ewerolimusu
Na podstawie badań in vitro stężenia ogólnoustrojowe uzyskane po podaniu doustnych dawek w wysokości 10 mg na dobę nie powinny spowodować zahamowania aktywności PgP, CYP3A4 i CYP2D6. Nie można jednak wykluczyć zahamowania CYP3A4 i PgP w jelicie. Badanie interakcji u osób zdrowych wykazało, że jednoczesne podanie doustnej dawki midazolamu, przykładowego wrażliwegeo substratu CYP3A, z ewerolimusem spowodowało 25% wzrost wartości Cmax midazolamu i 30% wzrost AUC(0-inf) midazolamu. Efekt ten jest prawdopodobnie wynikiem zahamowania CYP3A4 w jelicie przez ewerolimus. Dlatego ewerolimus może wpływać na dostępność biologiczną jednocześnie stosowanych doustnych substratów CYP3A4. Nie należy się jednak spodziewać wystąpienia klinicznie istotnego wpływu na ekspozycję na substraty CYP3A4 podawane ogólnoustrojowo (patrz punkt 4.4).
Jednoczesne podawanie ewerolimusu i długo działającego oktreotydu zwiększyło Cmin oktreotydu, przy stosunku średniej geometrycznej (ewerolimus/placebo) wynoszącej 1,47. Nie udało się ustalić, czy ma to istotny klinicznie wpływ na skuteczność odpowiedzi na leczenie ewerolimusem pacjentów z zaawansowanymi nowotworami neuroendokrynnymi.
Jednoczesne podawanie ewerolimusu i eksemestanu spowodowało zwiększenie Cmin i C2h eksemestanu odpowiednio o 45% i 64%. Jednak odpowiednie stężenia estradiolu w stanie stacjonarnym (4 tygodnie) nie różniły się w obu grupach leczenia. U pacjentek z zaawansowanym rakiem piersi z ekspresją receptorów hormonalnych, otrzymujacych to leczenie skojarzone nie obserwowano częściej zdarzeń niepożądanych związanych z podawaniem eksemestanu. Jest mało prawdopodobne, by zwiększenie stężeń eksemestanu miało wpływ na skuteczność lub bezpieczeństwo stosowania leku.
Jednoczesne stosowanie inhibitorów konwertazy angiotensyny (ACE)
U pacjentów leczonych jednocześnie inhibitorem ACE (np. ramiprylem) może wystąpić zwiększone ryzyko obrzęku naczynioruchowego (patrz punkt 4.4).
Szczepienia
W trakcie leczenia produktem leczniczym Everolimus Genthon odpowiedź immunologiczna na szczepienie może być zmieniona i dlatego szczepionka może być mniej skuteczna. Należy unikać stosowania żywych szczepionek w czasie podawania produktu leczniczego Everolimus Genthon (patrz punkt 4.4). Przykłady żywych szczepionek to: donosowa szczepionka przeciw grypie, szczepionki przeciw odrze, nagminnemu zapaleniu przyusznic, różyczce, doustna szczepionka przeciw polio, BCG (Bacillus Calmette-Guérin, szczepionka przeciw gruźlicy), przeciw żółtej febrze, ospie wietrznej i szczepionki przeciw durowi brzusznemu TY21.
Kobiety w wieku rozrodczym/Antykoncepcja u mężczyzn i kobiet
Kobiety w wieku rozrodczym w trakcie przyjmowania ewerolimusu i w okresie do 8 tygodni po zakończeniu leczenia muszą stosować wysoce skuteczną antykoncepcję (np. doustną, iniekcyjną, lub implantowaną niezawierającą estrogenów hormonalną metodę kontroli narodzin, antykoncepcję opartą na progesteronie, histerektomię, podwiązanie jajowodów, całkowitą abstynencję, metody barierowe, wkładkę wewnątrzmaciczną i (lub) sterylizację kobiet /mężczyzn). W przypadku mężczyzn nie ma przeciwwskazań do poczęcia dziecka w trakcie leczenia ewerolimusem.
Ciąża
Nie ma wystarczających danych na temat stosowania ewerolimusu u kobiet w ciąży. Badania na zwierzętach wykazały toksyczny wpływ na reprodukcję, w tym działanie toksyczne na zarodek i płód (patrz punkt 5.3). Potencjalne zagrożenie dla człowieka nie jest znane.
Nie zaleca się podawania ewerolimusu kobietom w ciąży i kobietom w okresie rozrodczym, które nie stosują antykoncepcji.
Karmienie piersią
Brak danych na temat przenikania ewerolimusu do mleka kobiet karmiących. U samic szczurów ewerolimus i (lub) jego metabolity z łatwością przenikają do mleka (patrz punkt 5.3). Z tego względu kobiety przyjmujące ewerolimus nie powinny karmić piersią podczas leczenia i przez 2 tygodnie po przyjęciu ostatniej dawki.
Płodność
Nie wiadomo, czy ewerolimus powoduje niepłodność u mężczyzn i kobiet, jednak u pacjentek obserwowano brak miesiączki (wtórny brak miesiączki i inne zaburzenia menstruacyjne) i związane z tym zaburzenia równowagi hormonu luteinizującego (LH)/hormonu folikulotropowego (FSH). Na podstawie obserwacji nieklinicznych stwierdza się możliwość upośledzenia płodności mężczyzn i kobiet w trakcie podawania ewerolimusu (patrz punkt 5.3).
Everolimus Genthon może wywierać niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Pacjentów należy poinformować, że powinni zachować ostrożność podczas prowadzenia pojazdów i obsługi maszyn, jeśli w trakcie leczenia produktem Everolimus Genthon będą odczuwać zmęczenie.
Podsumowanie profilu bezpieczeństwa
Profil bezpieczeństwa jest oparty o dane zgromadzone od 2 672 pacjentów biorących udział w dziesięciu badaniach klinicznych z zastosowaniem ewerolimusu, w skład których wchodziło pięć randomizowanych, z podwójnie zaślepioną próbą, kontrolowanych placebo badań III fazy oraz pięć otwartych badań I fazy i II fazy w ramach zatwierdzonych wskazań.
Najczęstszymi działaniami niepożądanymi (występującymi ≥1/10) zgromadzonymi z danych dotyczących bezpieczeństwa stosowania są (w malejącej kolejności): zapalenie jamy ustnej, wysypka, zmęczenie, biegunka, zakażenia, mdłości, zmniejszenie apetytu, niedokrwistość, zaburzenie smaku, zapalenie płuc, obrzęk obwodowy, hiperglikemia, osłabienie, świąd, zmniejszenie wagii ciała, hipercholesterolemia, krwawienie z nosa, kaszel i ból głowy.
Najczęstsze działania niepożądane stopnia 3-4 (częstość występowania ≥1/100 do <1/10) to zapalenie jamy ustnej, niedokrwistość, hiperglikemia, zakażenia, zmęczenie, biegunka, zapalenie płuc, osłabienie, małopłytkowość, neutropenia, duszność, białkomocz, limfopenia, krwotoki, hipofosfatemia, wysypka, nadciśnienie, zapalenie płuc, wzrost aktywności aminotransferazy alaninowej (ALT), wzrost aktywności aminotransferazy asparaginianowej (AST) i cukrzyca. Nasilenie działań niepożądanych stopniowano wg CTCAE, wersja 3.0 i 4.03.
Tabelaryczny spis działań niepożądanych
W Tabeli 3 przedstawiono częstość występowania działań niepożądanych na podstawie zbiorczych danych w łącznej populacji ocenianej pod względem bezpieczeństwa. Wszystkie działania niepożądane, zgłaszane w badaniu głównym, oparto na częstości występowania, zaczynając od najczęściej występującego. Działania niepożądane zostały przedstawione zgodnie z klasyfikacją układów i narządów MedDRA oraz klasyfikacją częstości. Kategorie częstości określone są zgodnie z następującą konwencją: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1 000 do
<1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000). W obrębie każdej grupy o określonej częstości występowania objawy niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 3 Działanie niepożądane zgłoszone w badaniach klinicznych
Zakażenia i zarażenia pasożytnicze | |
Bardzo często | Zakażenia a, * |
Zaburzenia krwi i układu chłonnego | |
Bardzo często | Niedokrwistość |
Często | Małopłytkowość, neutropenia, leukopenia, limfopenia |
Niezbyt często | Pancytopenia |
Rzadko | Wybiórcza aplazja czerwonokrwinkowa |
Zaburzenia układu immunologicznego | |
Niezbyt często | Nadwrażliwość |
Zaburzenia metabolizmu i odżywiania | |
Bardzo często | Zmniejszenie apetytu, hiperglikemia, hypercholesterolemia |
Często | Hipertriglicerydemia, hipofosfatemia, cukrzyca, hiperlipidemia, hipokaliemia, odwodnienie, hipokalcemia |
Zaburzenia psychiczne | |
Często | Bezsenność |
Zaburzenia układu nerwowego | |
Bardzo często | Zaburzenia smaku, ból głowy |
Niezbyt często | Brak smaku |
Zaburzenia oka | |
Często | Obrzęk powiek |
Niezbyt często | Zapalenie spojówek |
Zaburzenia serca | |
Niezbyt często | Zastoinowa niewydolność serca |
Zaburzenia naczyniowe | |
Często | Krwotok b, nadciśnienie |
Niezbyt często | Uderzenia gorąca, zakrzepica żył głębokich |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Bardzo często | Zapalenie płuc c, krwawienie z nosa, kaszel |
Często | Duszność |
Niezbyt często | Krwioplucie, zatorowość płucna |
Rzadko | Zespół ostrej niewydolności oddechowej |
Zaburzenia żołądka i jelit | |
Bardzo często | Zapalenie jamy ustnej d, biegunka, mdłości |
Często | Wymioty, suchość jamy ustnej, ból brzucha, zapalenie błon śluzowych, ból jamy usnej, niestrawność, utrudnienia połykania |
Zaburzenia wątroby i dróg żółciowych | |
Często | Zwiększona aktywność aminotransferazy asparaginianowej, zwiększona aktywność aminotransferazy alaninowej |
Zaburzenia skóry i tkanki podskórnej | |
Bardzo często | Wysypka, świąd |
Często | Suchość skóry, zmiany w obrębie paznokci, łysienie o łagodnym nasileniu, trądzik, rumień, łamliwość paznokci, zespół ręka-stopa, złuszczanie skóry, zmiany skórne |
Rzadko | Obrzęk naczynioruchowy |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często | Ból stawów |
Zaburzenia nerek i dróg moczowych | |
Często | Białkomocz*, zwiększenie stężenia kreatyniny we krwi, niewydolność nerek* |
Niezbyt często | Zwiększona częstość oddawania moczu w ciągu dnia, ostra niewydolność nerek* |
Zaburzenia układu rozrodczego i piersi | |
Często | Nieregularne miesiączkowanie e |
Niezbyt często | Brak miesiączki e |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często | Zmęczenie, osłabienie, obrzęk obwodowy |
Często | Gorączka |
Niezbyt często | Ból w klatce piersiowej nie pochodzenia sercowego, utrudnione gojenie się ran |
Badania diagnostyczne | |
Bardzo często | Zmniejszenie masy ciała |
* Patrz także podpunkt „Opis wybranych działań niepożądanych” a W tym wszystkie działania uwzględnione w klasyfikacji “zakażenia i zarażenia pasożytnicze”, w tym (często) zapalenie płuc, zakażenie układu moczowego; (niezbyt często) zapalenie oskrzeli, półpasiec, posocznica, ropnie i pojedyncze przypadki zakażeń oportunistycznych [np. aspergiloza, kandydoza, pneumocystozowe zapalenie płuc wywołane przez Pneumocystis jirovecii (carinii) (PJP, PCP) i zapalenie wątroby typu B (patrz także punkt 4.4)] oraz (rzadko) wirusowe zapalenie mięśnia sercowego b W tym inne krwawienia z różnych miejsc niewymienione osobno c W tym (często) zapalenie płuc, śródmiąższowe zapalenie płuc, nacieki w płucach i (rzadko) krwotok do pęcherzyków płucnych, reakcje toksyczne ze strony płuc, zapalenie pęcherzyków płucnych d W tym (bardzo często) zapalenie jamy ustnej, (często) aftowe zapalenie jamy ustnej, owrzodzenie jamy ustnej i języka i (niezbyt często) ból języka, zapalenie języka e Częstość w oparciu o liczbę kobiet w wieku od 10 do 55 lat w danych zbiorczych | |
Opis wybranych działań niepożądanych
W badaniach klinicznych oraz na podstawie zgłoszeń spontanicznych po wprowadzeniu produktu leczniczego do obrotu, podawanie ewerolimusu wiązano z ciężkimi przypadkami reaktywacji wirusowego zapalenia wątroby typu B, włącznie z przypadkami prowadzącymi do zgonu.
Reaktywacja zakażenia jest spodziewanym działaniem w okresie występowania immunosupresji.
W badaniach klinicznych oraz na podstawie zgłoszeń spontanicznych po wprowadzeniu produktu leczniczego do obrotu, stwierdzono związek ewerolimusu z przypadkami niewydolności nerek (włącznie z przypadkami prowadzącymi do śmierci) i białkomoczem. Zaleca się kontrolowanie czynności nerek (patrz punkt 4.4).
W badaniach klinicznych i na podstawie zgłoszeń spontanicznych zebranych po wprowadzeniu produktu leczniczego do obrotu, podawanie ewerolimusu wiązano z przypadkami braku miesiączki (wtórnego braku miesiączki i innych zaburzeń menstruacyjnych).
W badaniach klinicznych i spontanicznych doniesieniach po wprowadzeniu produktu leczniczego do obrotu stwierdzono związek ewerolimusu z przypadkami pneumocystozowego zapalenia płuc wywołanego przez Pneumocystis jirovecii (carinii) (PJP, PCP), z których część zakończyła się zgonem pacjenta (patrz punkt 4.4).
W badaniach klinicznych i spontanicznych doniesieniach po wprowadzeniu produktu leczniczego do obrotu zgłaszano występowanie obrzęku naczynioruchowego, wraz z jednoczesnym stosowaniem inhibitorów ACE i bez (patrz punkt 4.4).
Osoby w podeszłym wieku
W łączonej populacji, ocenianej pod względem bezpieczeństwa, 37% pacjentów leczonych ewerolimusem było w wieku ≥65 lat. Liczba pacjentów, u których wystąpiły działania niepożądane prowadzące do zakończenia leczenia była wyższa w grupie pacjentów w wieku ≥65 lat (20% vs. 13%). Najczęstszymi działaniami niepożądanymi prowadzącymi do zakończenia leczenia były: zapalenie płuc (w tym choroba śródmiąższowa płuc), zapalenie jamy ustnej, uczucie zmęczenia i duszność.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem krajowego systemu zgłaszania wymienionego w Departament Monitorowania Niepożądanych, Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C , PL-02 222 Warszawa, Tel.: + 48 22 49 21 301, Faks: + 48 22 49 21 309 e-mail: ndl@urpl.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Dane na temat przedawkowania u ludzi są bardzo ograniczone. Po podaniu pojedynczej dawki do 70 mg obserwowano akceptowalną tolerancję. W każdym przypadku przedawkowania należy stosować ogólne środki wspomagające.
Grupa farmakoterapeutyczna: Leki przeciwnowotworowe, inne leki przeciwnowotworowe, inhibitory kinaz białkowych, kod ATC: L01XE10
Mechanizm działania
Ewerolimus jest selektywnym inhibitorem mTOR (mammalian target of rapamycin). mTOR jest kluczową kinazą serynowo-treoninową, której aktywność jest nasilona w wielu ludzkich nowotworach złośliwych. Ewerolimus wiąże się z międzykomórkowym białkiem FKBP-12, tworząc kompleks, który hamuje działanie kompleksu 1 kinazy mTOR (mTORC1). Zahamowanie szlaku przekazywania sygnałów mTORC1 zaburza translację i syntezę białek, hamując działanie rybosomalnej kinazy S6
(S6K1) oraz 4EBP1 (białka wiążącego eukariotyczny czynnik elongacyjny 4E), które regulują aktywność białek uczestniczących w cyklu komórkowym, angiogenezie i glikolizie. Uważa się, że substrat S6K1 fosforyluje domenę aktywacyjną 1 receptora estrogenowego, odpowiedzialną za aktywację receptora niezależną od ligandu. Ewerolimus zmniejsza stężenie czynnika wzrostu śródbłonka naczyniowego (VEGF), nasilającego procesy angiogenezy guza. Ewerolimus jest silnym inhibitorem wzrostu i namnażania komórek guza, komórek śródbłonka, fibroblastów i komórek mięśni gładkich naczyń krwionośnych. Wykazano również, że ewerolimus hamuje glikolizę guzów litych in vitro i in vivo.
Skuteczność kliniczna i bezpieczeństwo stosowania
Zaawansowany rak piersi z z ekspresją receptorów hormonalnych
BOLERO-2 (badanie CRAD001Y2301) było randomizowanym, z podwójnie ślepą próbą, wieloośrodkowym badaniem III fazy z zastosowaniem ewerolimusu w skojarzeniu z eksemestanem w porównaniu do placebo w skojarzeniu z eksemestanem, w którym uczestniczyły kobiety po menopauzie z zaawansowanym rakiem piersi z ekspresją receptorów estrogenowych, bez nadekspresjii receptora HER2/neu, u których wystąpiła wznowa lub progresja choroby po wcześniejszym leczeniu letrozolem lub anastrozolem. Randomizacja była stratyfikowana względem udokumentowanej wrażliwości na wcześniej stosowaną terapię hormonalną oraz obecności przerzutów w narządach wewnętrznych. Wrażliwość na wcześniej stosowaną terapię hormonalną definiowano jako (1) udokumentowaną korzyść kliniczną (odpowiedź całkowita [CR], odpowiedź częściowa [PR], stabilizacja choroby ≥24 tygodnie) w trakcie stosowania co najmniej jednej terapii hormonalnej w chorobie zaawansowanej lub (2) co najmniej 24 miesiące hormonalnej terapii uzupełniającej przed wystąpieniem wznowy.
Pierwszorzędowym punktem końcowym badania było przeżycie bez progresji choroby (progression – free survival, PFS) oceniane wg kryteriów RECIST (Kryteria Odpowiedzi na Leczenie w Guzach Litych) na podstawie oceny badacza (badanie radiologiczne oceniane lokalnie). Dodatkowe analizy PFS prowadzono w oparciu o niezależną centralną ocenę radiologiczną.
Do drugorzędowych punktów końcowych należało przeżycie całkowite (OS), wskaźnik obiektywnej odpowiedzi na leczenie, wskaźnik korzyści klinicznej, bezpieczeństwo, zmiana jakości życia (QoL) oraz czas do pogorszenia stanu sprawnościowego wg ECOG (Eastern Cooperative Oncology Group).
Łącznie 724 pacjentki zostały zrandomizowane w stosunku 2:1 do grupy leczenia skojarzonego przyjmującej ewerolimus (10 mg na dobę) + eksemestan (25 mg na dobę) (n=485) lub do grupy przyjmującej placebo + eksemestan (25 mg na dobę) (n=239). W momencie przeprowadzania ostatecznej analizy OS, mediana czasu trwania leczenia ewerolimusem wyniosła 24,0 tygodnie (zakres 1,0–199,1 tygodni). Mediana czasu trwania leczenia eksemestanem była dłuższa w grupie przyjmującej ewerolimus + eksemestan i wyniosła 29,5 tygodni (1,0-199,1) w porównaniu do 14,1 tygodni (1,0-156,0) w grupie przyjmującej placebo + eksemestan.
W odniesieniu do pierwszorzędowych punktów końcowych wyniki dotyczące skuteczności otrzymano po przeprowadzeniu ostatecznej analizy PFS (patrz Tabela 4 i Rycina 1). W chwili progresji pacjentki z grupy otrzymującej placebo + eksemestan nie przechodziły do grupy przyjmującej ewerolimus.
Tabela 4 Wyniki dotyczące skuteczności w badaniu BOLERO-2
Analiza | Ewerolimus a n=485 | Placeboa n=239 | Współczynni k ryzyka | Wartość p |
Mediana przeżycia bez progresji choroby (miesiące) (95% CI) | ||||
Lokalna ocena radiologiczna | 7,8 | 3,2 | 0,45 | <0,0001 |
(6,9 do 8,5) | (2,8 do 4,1) | (0,38 do 0,54) | ||
Niezależna ocena | 11,0 | 4,1 | 0,38 | <0,0001 |
radiologiczna | (9,7 do 15,0) | (2,9 do 5,6) | (0,31 do 0,48) | |
Mediana całkowitego przeżycia (miesiące) (95% CI) | ||||
Mediana całkowitego | 31,0 | 26,6 | 0,89 | 0,1426 |
przeżycia | (28,0 – 34,6) | (22,6 – 33,1) | (0,73 – 1,10) | |
Najlepsza odpowiedź całkowita (%) (95% CI) | ||||
Wskaźnik odpowiedzi obiektywnejb | 12,6% (9,8 do 15,9) | 1,7% (0,5 do 4,2) | n/ad | <0,0001e |
Wskaźnik korzyści klinicznejc | 51,3% (46,8 do 55,9) | 26,4% (20,9 do 32,4) | n/ad | <0,0001e |
a W skojarzeniu z eksemestanem b Wskaźnik odpowiedzi obiektywnej = odsetek pacjentek z odpowiedzią całkowitą lub częściową c Wskaźnik korzyści klinicznej = odsetek pacjentek z odpowiedzią całkowitą lub częściową, lub chorobą stabilną ≥24 tygodnie d Nie dotyczy e Wartość p jest otrzymywana na podstawie dokładnego testu Cochrana-Mantela-Haenszela z użyciem stratyfikowanej wersji testu permutacji Cochrana-Armitage’a. | ||||
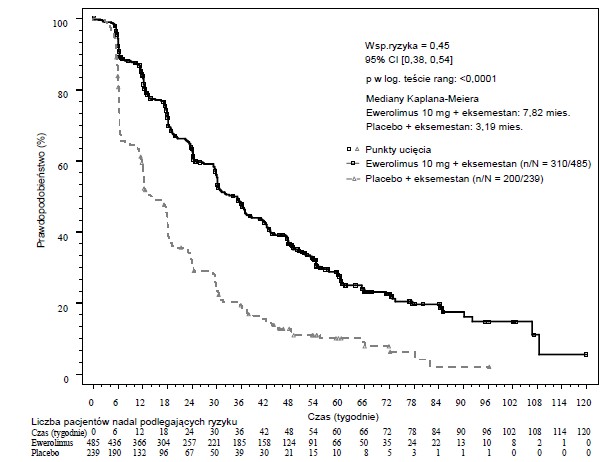
Ryc. 1 Krzywe Kaplana-Meiera dotyczące przeżycia bez progresji choroby w badaniu BOLERO-2 (ocena wyniku badania radiologicznego dokonana przez badacza)
<Wchłanianie>
<Dystrybucja>
<Metabolizm>
<Eliminacja>
<Liniowość lub nieliniowość>
<Zależności farmakokinetyczno-farmakodynamiczne>
<Dane niekliniczne wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, badań toksyczności po podaniu wielokrotnym, genotoksyczności, potencjalnego działania rakotwórczego oraz toksycznego wpływu na rozród i rozwój potomstwa nie ujawniają żadnego szczególnego zagrożenia dla człowieka.>
<W badaniach nieklinicznych działanie toksyczne obserwowano jedynie w przypadku narażenia przekraczającego maksymalną ekspozycję u człowieka, co wskazuje na niewielkie znaczenie tych obserwacji w praktyce klinicznej.>
<Działania niepożądane, których nie obserwowano w badaniach klinicznych, a które występowały u zwierząt po narażeniu podobnym do występującego w warunkach klinicznych i które mogą mieć znaczenie w praktyce klinicznej, były następujące:>
<Ocena ryzyka dla środowiska>
Szacowany wpływ leczenia na PFS został potwierdzony wynikami planowanej analizy podgrup w odniesieniu do PFS wg oceny badacza. We wszystkich analizowanych podgrupach (wiek, wrażliwość na wcześniej stosowaną terapię hormonalną, liczba zajętych narządów, obecność zmian zlokalizowanych wyłącznie w kościach oraz przerzuów do narządów wewnętrznych oraz występowanie we wszystkich najważniejszych podgrupach demograficznych i prognostycznych) pozytywny wpływ leczenia obserwowano w grupie terapii skojarzonej ewerolimusem z eksemestanem z szacowanym współczynnikiem ryzyka wynoszącym od 0,25 do 0,60 w porównaniu z grupą otrzymującą placebo z eksemestanem.
Nie obserwowano różnic w czasie do pogorszenia (≥5%) wyników funkcjonalnych domen QLQ- C30 w obu ramionach.
Zaawansowane nowotwory neuroendokrynne trzustki (pNET)
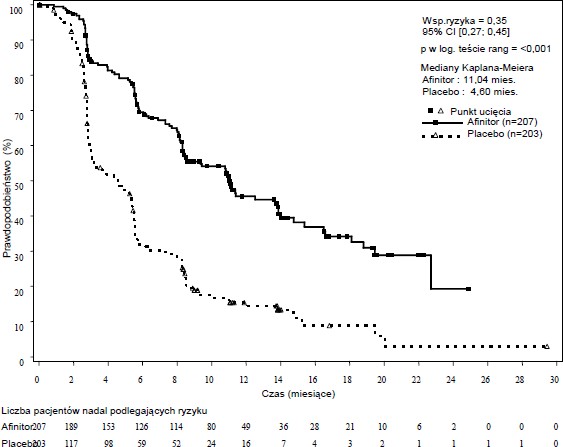
RADIANT-3 (badanie CRAD001C2324), wieloośrodkowe, randomizowane badanie III fazy, prowadzone metodą podwójnie ślepej próby z zastosowaniem ewerolimusu plus najlepsze leczenie wspomagające (BSC, ang. best supportive care) w porównaniu do zastosowania placebo plus BSC u pacjentów z zaawansowanymi nowotworami neuroendokrynnymi trzustki, wykazało istotną statystycznie korzyść kliniczną ewerolimusu nad placebo, przejawiającą się 2,4 –krotnym wydłużeniem mediany przeżycia wolnego od progresji choroby (PFS, ang. progression free-survival) (11,04 miesiąca wobec 4,6 miesiąca), (HR 0,35; 95% CI; 0,27, 0,45; p<0,0001) (patrz Tabela 5 i
Rycina 2).
W badaniu RADIANT-3 uczestniczyli pacjenci z wysoko lub średnio zróżnicowanymi nowotworami neuroendokrynnymi trzustki, u których wystąpiła progresja choroby w ciągu 12 miesięcy. Dozwolone było leczenie analogami somatostatyny jako leczenie w ramach BSC.
Pierwszorzędowym punktem końcowym badania był PFS, oceniony za pomocą kryteriów RECIST (Kryteria Odpowiedzi na Leczenie w przypadku Guzów Litych). Po udokumentowaniu radiologicznym progresji choroby, badacz mógł ujawnić, jaki produkt leczniczy przyjmują pacjenci. Pacjenci zrandomizowani do grupy przyjmującej placebo mogli następnie przyjmować ewerolimus w otwartej fazie badania.
Drugorzędowe punkty końcowe obejmowały: bezpieczeństwo stosowania, wskaźnik obiektywnej odpowiedzi na leczenie, czas trwania odpowiedzi na leczenie oraz przeżycie całkowite (OS).
Łącznie 410 pacjentów zostało randomizowanych w stosunku 1:1 do grupy przyjmującej Ewerolimus (n=207) lub placebo (n=203). Dane dotyczące charakterystyki demograficznej pacjentów były dobrze zrównoważone (mediana wieku 58 lat, 55% mężczyzn, 78,5% pacjentów rasy białej). Pięćdziesiąt osiem procent pacjentów w obu ramionach badania otrzymało wcześniejsze leczenie systemowe.
Mediana czasu trwania zaślepionego leczenia w badaniu wyniosła 37,8 tygodni (zakres 1,1-129,9 tygodni) dla pacjentów otrzymujących ewerolimus oraz 16,1 tygodni (zakres 0,4-147,0 tygodni) dla pacjentów otrzymujących placebo.
Po wystąpieniu progresji choroby lub po rozkodowaniu badania, 172 z 203 pacjentów (84,7%) pierwotnie zrandomizowanych do grupy placebo przeszło do grupy otrzymującej otwarte leczenie ewerolimusem. Mediana czasu trwania leczenia otwartego wyniosła 47,7 tygodni u wszystkich pacjentów; 67,1 tygodni u 53 pacjentów zrandomizowanych do grupy otrzymującej ewerolimus, którzy przeszli na otwarte leczenie ewerolimusem oraz 44,1 tygodni u 172 pacjentów zrandomizowanych do grupy placebo, którzy przeszli do grupy otwartego leczenia ewerolimusem.
Tabela 5 Badanie RADIANT-3 - Wyniki w zakresie skuteczności
Populacja | Ewerolimus n=207 | Placebo n=203 | Współczynnik ryzyka (95% CI) | Wartośćp |
Mediana przeżycia wolnego od progresji (miesiące) (95% CI) | ||||
Ocena | 11,04 | 4,60 | 0,35 | <0,0001 |
radiologiczna | (8,41, 13,86) | (3,06, 5,39) | (0,27, 0,45) | |
przeprowadzona | ||||
przez badacza | ||||
Niezależna | 13,67 | 5,68 | 0,38 | <0,0001 |
ocena | (11,17, 18,79) | (5,39, 8,31) | (0,28, 0,51) | |
radiologiczna | ||||
Mediana całkowitego przeżycia (miesiące) (95% CI) | ||||
Mediana | 44,02 | 37,68 | 0,94 | 0,300 |
całkowitego | (35,61, 51,75) | (29,14, 45,77) | (0,73, 1,20) | |
przeżycia | ||||
Rycina 2 Badanie RADIANT-3 - Krzywe Kaplana-Meiera przeżycia wolnego do progresji (ocena radiologiczna przeprowadzona przez badacza)
Zaawansowane nowotwory neuroendokrynne układu pokarmowego lub płuc
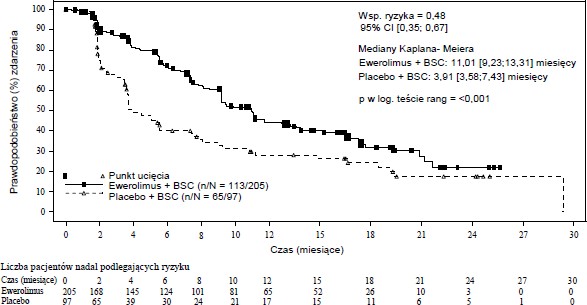
RADIANT-4 (badanie CRAD001T2302), wieloośrodkowe, randomizowane badanie III fazy, prowadzone metodą podwójnie ślepej próby z zastosowaniem ewerolimusu plus najlepsze leczenie wspomagające (BSC) w porównaniu do zastosowania placebo plus BSC zostało przeprowadzone z udziałem chorych na zaawansowane, wysoko zróżnicowane (stopnia 1 lub stopnia 2), hormonalnie nieczynne nowotwory neuroendokrynne układu pokarmowego lub płuc, bez wcześniejszego, ani obecnego występowania aktywnych objawów związanych z zespołem rakowiaka.
Pierwszorzędowym punktem końcowym badania było przeżycie wolne od progresji (PFS), oceniane za pomocą Kryteriów Odpowiedzi na Leczenie w przypadku Guzów Litych (RECIST), na podstawie niezależnej oceny radiologicznej. Analiza wspomagająca PFS opierała się na ocenie dokonywanej lokalnie przez badacza. Drugorzędowe punkty końcowe obejmowały: przeżycie całkowite (OS), całkowity wskaźnik odpowiedzi, stopień kontroli choroby, bezpieczeństwo stosowania, zmianę w jakości życia (FACT-G) oraz czas do pogorszenia stanu sprawności wg Światowej Organizacji Zdrowia (WHO PS).
Łącznie 302 pacjentów zostało zrandomizowanych w stosunku 2:1 do grupy otrzymującej ewerolimus (10 mg na dobę) (n=205) lub placebo (n=97). Dane demograficzne i dane dotyczące choroby były na ogół zrównoważone (mediana wieku wynosiła 63 lata [zakres 22 do 86], 76% stanowiły osoby rasy białej, stosowanie analogu somatostatyny [SSA] w wywiadzie). Mediana czasu trwania zaślepionego leczenia w badaniu wyniosła 40,4 tygodni dla pacjentów otrzymujących ewerolimus oraz 19,6 tygodni dla pacjentów otrzymujących placebo. Pacjenci z grupy placebo nie przeszli do grupy otrzymującej ewerolimus w chwili progresji.
Wyniki dotyczące skuteczności w odniesieniu do pierwszorzędowego punktu końcowego otrzymano po przeprowadzeniu ostatecznej analizy PFS (patrz Tabela 6 i Rycina 3).
Tabela 6 RADIANT-4 – Wyniki w zakresie przeżycia wolnego od progresji
Populacja | Ewerolimus n=205 | Placebo n=97 | Współczynnik ryzyka (95% CI) | Wartość pa |
Mediana przeżycia wolnego od progresji (miesiące) (95% CI) | ||||
Niezależna ocena | 11,01 | 3,91 | 0,48 | <0,0001 |
radiologiczna | (9,2, 13,3) | (3,6, 7,4) | (0,35, 0,67) | |
Ocena | 13,96 | 5,45 | 0,39 | <0,0001 |
radiologiczna | (11,2, 17,7) | (3,7, 7,4) | (0,28, 0,54) | |
przeprowadzona | ||||
przez badacza | ||||
a Jednostronna wartość p ze stratyfikowanego logarytmicznego testu rang | ||||
Rycina 3 RADIANT-4 – Krzywe Kaplana-Meiera przeżycia wolnego do progresji (niezależna ocena radiologiczna)
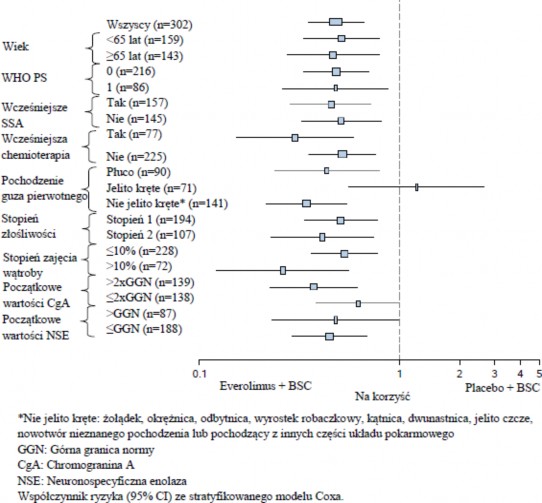
W analizach wspomagających obserwowano pozytywny wpływ leczenia we wszystkich podgrupach z wyjątkiem podgrupy pacjentów z guzem pierwotnym pochodzącym z jelita krętego (jelito kręte: HR=1,22 [95% CI: 0,56 do 2,65]; inne miejsca poza jelitem krętym: HR=0,34 [95% CI: 0,22 do0,54];
Płuco: HR=0,43 [95% CI: 0,24 do 0,79]) (patrz Rycina 4).
Rycina 4 RADIANT-4 – Wyniki dotyczące przeżycia bez progresji choroby z uwzględnieniem predefiniowanych podgrup pacjentów (niezależna ocena radiologiczna)
Zaplanowana wcześniej analiza etapowa OS po 101 zgonach (ze 191 wymaganych do przeprowadzenia analizy końcowej) i 33 miesiącach obserwacji dała wyniki na korzyść grupy stosującej ewerolimus; nie odnotowano jednak statystycznie znamiennej różnicy w OS (HR=0,73 [95% CI: 0,48 do 1,11; p=0,071]).
Nie obserwowano różnic pomiędzy dwiema grupami w zakresie czasu do definitywnego pogorszenia stanu sprawności wg WHO (≥1 punkt) i czasu do definitywnego pogorszenia jakości życia (całkowitego wyniku FACT-G ≥7 punktów).
Zaawansowany rak nerkowokomórkowy
RECORD-1 (badanie CRAD001C2240), wieloośrodkowe, międzynarodowe, randomizowane badanie III fazy, z podwójnie ślepą próbą, w którym porównywano podawanie ewerolimusu w dawce 10 mg na dobę i placebo, w połączeniu z najlepszym leczeniem wspomagającym pacjentów raka nerkowokomórkowego z przerzutami, u których nastąpił postęp choroby podczas lub po leczeniu inhibitorami kinazy tyrozynowej receptora naczyniowo-śródbłonkowego czynnika wzrostu (VEGFR- TKI, vascular endothelial growth factor receptor tyrosine kinase inhibitor), (sunitynibem, sorafenibem lub zarówno sunitynibem, jak i sorafenibem). Dozwolone było również wcześniejsze leczenie bewacyzumabem i interferonem alfa. Pacjentów stratyfikowano według skali prognostycznej Memorial Sloan-Kettering Cancer Center – MSKCC (rokowanie dobre – pośrednie – niekorzystne) i według rodzaju przebytego leczenia przeciwnowotworowego (1 lub 2 leki z grupy VEGFR-TKI).
Pierwszorzędowym punktem końcowym było przeżycie bez progresji choroby, oceniane na podstawie kryteriów RECIST (Response Evaluation Criteria in Solid Tumours, kryteria oceny odpowiedzi na
leczenie guzów litych) i które oceniano w ogólnym, niezależnym przeglądzie ze ślepą próbą. Drugorzędowe punkty końcowe badania to bezpieczeństwo stosowania, wskaźnik obiektywnych odpowiedzi guza na leczenie, przeżywalność całkowita, objawy związane z chorobą i jakość życia. Po potwierdzonej radiologicznie progresji choroby badacz mógł ujawnić przynależność pacjenta do grupy: pacjentom, którzy zostali randomizowani do grupy przyjmującej placebo, mogło być zaoferowane leczenie ewerolimusem w dawce 10 mg/dobę. Niezależna Komisja ds. Monitorowania Danych (ang. Independent Data Monitoring Committee) zaleciła zakończenie badań w czasie drugiej analizy w trakcie trwania badania, gdyż osiągnięto pierwszorzędowy punkt końcowy.
Ogółem, 416 pacjentów zrandomizowano w stosunku 2:1 do grupy przyjmującej ewerolimus (n=277) lub placebo (n=139). Pacjenci byli odpowiednio dobrani pod względem demograficznym (zbiorcza mediana wieku: 61 lat; zakres 27-85, 78% mężczyzn, 88% rasy białej, liczba wcześniejszych terapii VEGFR-TKI: [1-74%, 2-26%]. Mediana czasu trwania leczenia zaślepionego wyniosła 141 dni (zakres 19-451 dni) dla pacjentów otrzymujących ewerolimus oraz 60 dni (zakres 21-295 dni) dla pacjentów otrzymujących placebo.
Ewerolimus wykazał przewagę w porównaniu do placebo, jeśli chodzi o pierwszorzędowy punkt końcowy, czyli przeżywalność bez progresji choroby, ze statystycznie istotnym 67% zmniejszeniem ryzyka progresji choroby lub śmierci (patrz Tabela 7 i Rycina 5).
Tabela 7 RECORD-1 – Wyniki przeżywalności bez progresji choroby
Populacja | n | Ewerolimus n=277 | Placebo n=139 | Współczynni k ryzyka (95%CI) | Wartość p |
Średni czas przeżycia bez progresji (miesiące) (95% CI) | |||||
Analiza pierwszorzędowa | |||||
Cała (ogólna niezależna analiza zaślepionej grupy pacjentów) | 416 | 4,9 (4,0-5,5) | 1,9 (1,8-1,9) | 0,33 (0,25- 0,43) | <0,0001a |
Analizy wspomagające/czułości | |||||
Cała (przeglądy częściowe dokonane przez badaczy) | 416 | 5,5 (4,6-5,8) | 1,9 (1,8-2,2) | 0,32 (0,25- 0,41) | <0,0001a |
Skala prognostyczna MSKCC ( zaślepione niezależne przeglądy dokonane centralnie) | |||||
Małe ryzyko | 120 | 5,8 (4,0-7,4) | 1,9 (1,9-2,8) | 0,31 (0,19- 0,50) | <0,0001 |
Umiarkowane ryzyko | 235 | 4,5 (3,8-5,5) | 1,8 (1,8-1,9) | 0,32 (0,22- 0,44) | <0,0001 |
Duże ryzyko | 61 | 3,6 (1,9-4,6) | 1,8 (1,8-3,6) | 0,44 (0,22- 0,85) | 0,007 |
a Logarytmiczny test rang z warstwowym doborem prób | |||||
Rycina 5 RECORD-1 – Krzywe Kaplana-Meiera przeżywalności bez postępu choroby (niezależna ocena centralna)
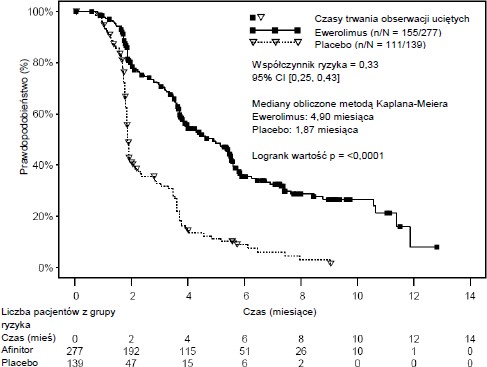
Wskaźniki przeżywalności bez progresji choroby w okresie sześciu miesięcy wyniosły 36% w grupie przyjmującej produkt leczniczy Ewerolimus w porównaniu z 9% w grupie przyjmującej placebo.
Potwierdzoną odpowiedź obiektywną zaobserwowano u 5 pacjentów (2%) przyjmujących Ewerolimus, podczas gdy nie zaobserwowano jej w ogóle w grupie przyjmującej placebo. Tym samym, przeżywalność bez progresji choroby dotyczy głównie populacji z ustabilizowaną chorobą (odpowiadającej 67% grupy przyjmującej Ewerolimus).
Nie zaobserwowano statystycznie istotnej różnicy w przeżywalności ogólnej (współczynnik ryzyka 0,87; przedział ufności: 0,65–1,17; p=0,177). Po stwierdzeniu progresji choroby i podaniu ewerolimusu pacjentom przydzielonym pierwotnie do grupy placebo, wykazano brak różnic w ogólnej przeżywalności związanych z rodzajem leczenia.
Inne badania
Zapalenie jamy ustnej jest najczęściej zgłaszanym działaniem niepożądanym u pacjentów leczonych ewerolimusem (patrz punkt 4.4 i 4.8). W badaniu z jedną grupą terapeutyczną przeprowadzonym po wprowadzeniu leku do obrotu z udziałem kobiet po menopauzie z zaawansowanym rakiem piersi (n=92) stosowano miejscowe leczenie bezalkoholowym doustnym roztworem deksametazonu o stężeniu 0,5 mg/5 ml w postaci płynu do płukania jamy ustnej (4 razy na dobę przez pierwsze 8 tygodni), od chwili rozpoczynania leczenia ewerolimusem (w dawce 10 mg na dobę) w skojarzeniu z eksemestanem (w dawce 25 mg na dobę), aby zmniejszyć częstość występowania i nasilenie zapalenia jamy ustnej. Częstość występowania zapalenia jamy ustnej stopnia ≥2. po 8 tygodniach wyniosła 2,4% (n=2/85 pacjentek, u których możliwe było dokonanie oceny), co było wartością mniejszą niż odsetek zgłaszany w przeszłości. Częstość występowania zapalenia jamy ustnej stopnia 1. wyniosła 18,8% (n=16/85) i nie zgłaszano żadnych przypadków zapalenia jamy ustnej o 3. lub 4. stopniu nasilenia.
Ogólny profil bezpieczeństwa stosowania w tym badaniu był spójny z profilem bezpieczeństwa ustalonym dla ewerolimusu we wskazaniach onkologicznych i w stwardnieniu guzowatym (TSC, ang. tuberous sclerosis complex), z wyjątkiem nieznacznie zwiększonej częstości występowania kandydozy jamy ustnej, którą zgłaszano u 2,2% (n=2/92) pacjentek.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego ewerolimus we wszystkich podgrupach populacji dzieci i młodzieży z nowotworami neuroendokrynnymi trzustki, nowotworami neuroendokrynnymi klatki piersiowej i z rakiem nerkowokomórkowym (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Wchłanianie
Mediana czasu do osiągnięcia szczytowych wartości stężeń ewerolimusu Cmax po podaniu doustnym w dawce 5 mg lub 10 mg na czczo lub z lekkim posiłkiem beztłuszczowym wynosiła 1 godzinę. Wartość Cmax jest proporcjonalna do dawki w zakresie dawek od 5 mg do 10 mg. Ewerolimus jest substratem i umiarkowanym inhibitorem PgP.
Wpływ pokarmu
U zdrowych pacjentów, pokarmy bogate w tłuszcze zmniejszały ogólnoustrojową ekspozycję na ewerolimus w dawce 10 mg (mierzoną wartością pola pod krzywą stężeń, AUC) o 22%, a maksymalne stężenie w osoczu Cmax o 54%. Pokarmy ubogie w tłuszcze zmniejszały AUC o 32%, a Cmax o 42%. Mimo to, jedzenie nie miało wyraźnego wpływu na profil zależności stężenia od czasu po fazie wchłaniania.
Dystrybucja
Stosunek stężenia ewerolimusu we krwi do stężenia w osoczu, zależny od stężenia w zakresie dawek od 5 do 5 000 ng/ml, wyniósł od 17% do 73%. Około 20% stężenia ewerolimusu we krwi pełnej zawiera się w osoczu pacjentów z rakiem leczonych ewerolimusem w dawce 10 mg na dobę. Wiązanie z białkami osocza wynosi około 74% zarówno u zdrowych osób, jak i pacjentów z umiarkowanym zaburzeniem czynności wątroby. U pacjentów z zaawansowanymi guzami litymi, objętość dystrybucji Vd wyniosła 191 l dla kompartmentu centralnego i 517 l dla kompartmentu obwodowego.
Metabolizm
Ewerolimus jest substratem CYP3A4 i PgP. Po podaniu doustnym, ewerolimus jest głównym związkiem obecnym we krwi. W ludzkiej krwi zidentyfikowano sześć głównych metabolitów substancji ewerolimus, m.in. trzy monohydroksylowe pochodne ewerolimusu, dwa produkty hydrolityczne z otwartymi łańcuchami i postać ewerolimusu sprzężonego z fosfatydylocholiną. Wymienione metabolity wykryto również u zwierząt w badaniach toksyczności. Wykazywały one aktywność około 100 razy mniejszą niż ewerolimus. W związku z tym, uważa się, że ewerolimus jest głównie odpowiedzialny za całą farmakologiczną aktywność ewerolimusu.
Eliminacja
Średni klirens po podaniu doustnym (CL/F) ewerolimusu w dawce 10 mg/dobę u pacjentów z zaawansowanym nowotworem litym wyniósł 24,5 l/h. Średni okres półtrwania ewerolimusu wynosi około 30 godzin.
Nie przeprowadzono specjalnych badań dotyczących wydalania ewerolimusu u pacjentów z rakiem, jednak dostępne są dane z badań przeprowadzonych z udziałem pacjentów po przeszczepach. Po podaniu jednej dawki ewerolimusu znakowanego izotopowo w połączeniu z cylosporyną 80% radioaktywnej substancji znaleziono w kale, a 5% zostało wydalone z moczem. W moczu i kale nie wykryto leku w postaci niezmienionej.
Właściwości farmakokinetyczne w stanie stacjonarnym
Po podaniu ewerolimusu pacjentom z zaawansowanymi guzami litymi AUC0-γ w stanie stacjonarnym było proporcjonalne do dawki w zakresie dawek od 5 mg do 10 mg na dobę. Stan stacjonarny osiągano w ciągu 2 tygodni. Wartości Cmax są proporcjonalne do dawki w zakresie od 5 mg do 10 mg. Tmax występuje od 1 do 2 godzin po podaniu. Stwierdzono znaczącą korelację pomiędzy AUC0-γ oraz minimalnym stężeniem w stanie stacjonarnym, przed podaniem kolejnej dawki leku.
Szczególne grupy pacjentów
Zaburzenia czynności wątroby
Bezpieczeństwo stosowania, tolerancja i farmakokinetyka ewerolimusu były oceniane w dwóch badaniach z podaniem pojedynczej doustnej dawki produktu leczniczego Ewerolimus w postaci tabletek 8 i 34 pacjentom z zaburzeniami czynności wątroby i porównane z działaniem leku u osób z prawidłową czynnością wątroby.
W pierwszym badaniu średnia AUC ewerolimusu podanego 8 pacjentom z umiarkowanymi zaburzeniami czynności wątroby (B w skali Child-Pugh) wzrosła dwukrotnie w porównaniu z 8 pacjentami z prawidłową czynnością wątroby.
W drugim badaniu z udziałem 34 pacjentów z innym zaburzeniem czynności wątroby w porównaniu z osobami zdrowymi odnotowano 1,6-krotny; 3,3-krotny oraz 3,6-krotny wzrost ekspozycji na lek (tzn. AUC0-inf) odpowiednio u pacjentów z łagodnymi (A wg skali Child-Pugh), umiarkowanymi (B wg skali Child-Pugh) i ciężkimi (C wg skali Child-Pugh) zaburzeniami czynności wątroby.
Symulacje dotyczące farmakokinetyki po podaniu wielokrotnych dawek leku uzasadniają zalecenia dotyczące dawkowania u pacjentów z zaburzeniami czynności wątroby na podstawie klasyfikacji w skali Child-Pugh.
Na podstawie wyników osiągniętych z dwóch badań, u pacjentów z zaburzeniami czynności wątroby zaleca się dostosowanie dawki leku (patrz punkty 4.2 i 4.4).
Zaburzenia czynności nerek
W populacyjnej analizie farmakokinetycznej przeprowadzonej u 170 pacjentów z zaawansowanym rakiem litym nie wykryto znaczącego wpływu klirensu kreatyniny (25-178 ml/min) na klirens ewerolimusu. Zaburzenia czynności nerek po przeszczepie (klirens kreatyniny w zakresie 11-107 ml/min) nie wpływały na właściwości farmakokinetyczne ewerolimusu u pacjentów po przebytym przeszczepie.
Pacjenci w podeszłym wieku
W badaniu właściwości farmakokinetycznych u pacjentów z rakiem nie wykryto znaczącego wpływu wieku (27-85 lat) na klirens ewerolimusu podanego doustnie.
Niekliniczny profil bezpieczeństwa ewerolimusu oceniano u myszy, szczurów, miniaturowych świń, małp i królików. Głównymi narządami docelowymi były układy rozrodcze samców i samic (zwyrodnieniowe zmiany w jądrach, zmniejszona zawartość plemników w najądrzach i zanik macicy) u kilku gatunków; płuca (zwiększona liczba makrofagów w pęcherzykach płucnych) szczurów i myszy; trzustka (degranulacja i wakuolizacja komórek zewnątrzwydzielniczych odpowiednio u małp i świnek miniaturowych oraz zwyrodnienie komórek Langerhansa u małp) oraz oczy (zmętnienia przednich szwów soczewki) - tylko u szczurów. U szczurów (nasilenie powstawania lipofuscyny wraz z wiekiem w nabłonku cewek nerkowych zwiększa się w wodonerczu) i u myszy (zaostrzenie stanu wcześniej istniejących zmian) zaobserwowano nieznaczne zmiany w obrębie nerek. Nie obserwowano toksyczności w obrębie nerek u małp i miniaturowych świń.
Ewerolimus wydaje się zaostrzać stan chorób współistniejących (przewlekłe zapalenie mięśnia sercowego u szczurów, zakażenie osocza i serca wirusem Coxsackie u małp, zakażenie żołądka i jelit miniaturowych świń kokcydiami, zmiany skórne u myszy i małp). Opisane wyżej przypadki obserwowano na ogół przy ekspozycji ogólnoustrojowej w zakresie dawek terapeutycznych lub powyżej tego zakresu, poza przypadkami u szczurów, które wystąpiły poniżej dawek terapeutycznych ze względu na wysoką dystrybucje wewnątrz tkanek.
W badaniu płodności samców szczurów, morfologia jąder była zmieniona przy dawce 0,5 mg/kg mc. i większej; przy 5 mg/kg mc. zmniejszona była ruchliwość plemników, liczba plemników, stężenie
testosteronu w osoczu. Dawki te ograniczały płodność samców. Udowodniono odwracalność tego stanu.
Badania rozrodczości u zwierząt nie wykazały wpływu na płodność samic. Jednak doustne dawki ewerolimusu podane samicom szurów wynoszące ≥0,1 mg/kg (około 4% AUC0-24h u pacjentów otrzymujących dawkę dobową 10 mg) skutkowały zwiększeniem ilości poronień.
Ewerolimus przenikał przez łożysko i wykazywał działanie toksyczne na płód. U szczurów ewerolimus wykazywał toksyczność dla embrionu i płodu przy ekspozycji ogólnoustrojowej poniżej zakresu terapeutycznego, co odzwierciedlała śmiertelność i zmniejszona masa płodów. Ryzyko wystąpienia zmian szkieletowych i wad wrodzonych (np. rozszczepu mostka) wzrosło przy dawce 0,3 i 0,9 mg/kg mc. U królików działanie toksyczne na embriony objawiało się zwiększeniem późnych resorpcji.
W badaniach genotoksyczności spełniających kryteria zakończenia badań w związku z genotoksycznością nie zaobserwowano działania klastogennego i mutagennego. Podawanie ewerolimusu myszom i szczurom przez okres do 2 lat nie spowodowało działania rakotwórczego nawet przy najwyższych dawkach, będących odpowiednio 3,9 i 0,2 razy większe od szacowanej ekspozycji u ludzi.
Butylohydroksytoluen (E321) Hypromeloza (E464) ( Laktoza
Laktoza jednowodna Krospowidon (E1202) Magnezu stearynian (E470b)
Nie dotyczy.
2 lata
Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Brak specjalnych zaleceń dotyczących temperatury przechowywania produktu leczniczego.
Blister OPA/Aluminium/PVC/Aluminium
Everolimus Genthon 2,5 mg jest dostępny w opakowania zawierające 30 lub 90 tabletek.
Everolimus Genthon 5 mg i Everolimus Genthon 10 mg są dostępne w opakowaniach zawierających 10, 30 lub 90 tabletek.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Genthon BV Microweg 22
6545 CM Nijmegen Holandia
2,5 mg:
5 mg:
10 mg:
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:








