







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
gdy nie doszło do normalizacji wydzielania po leczeniu chirurgicznym i (lub)
radioterapii;
u pacjentów, dla których leczenie chirurgiczne nie jest odpowiednie;
u pacjentów napromienianych, aż do chwili, gdy radioterapia osiągnie skuteczność.
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
uwalnianie GH stymulowane argininą, wysiłkiem fizycznym i hipoglikemią wywołaną
przez podanie insuliny;
poposiłkowe uwalnianie insuliny, glukagonu, gastryny i innych peptydów wytwarzanych przez wewnątrzwydzielniczy układ GEP oraz uwalnianie insuliny i glukagonu stymulowane przez argininę;
uwalnianie hormonu tyreotropowego (TSH) stymulowane przez hormon uwalniający tyreotropinę (TRH).
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania produktu
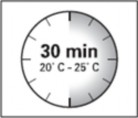
Zestaw do wstrzyknięć musi osiągnąć temperaturę pokojową. Należy wyjąć zestaw do wstrzyknięć z lodówki i przed odtworzeniem leku pozostawić go na minimum 30 minut w temperaturze pokojowej, jednak nie dłużej niż na 24 godziny.
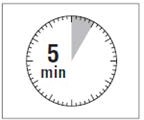
Po dodaniu rozpuszczalnika należy odstawić fiolkę na 5 minut, aby zapewnić pełne
nasycenie proszku roztworem.
Po nasyceniu należy wstrząsać fiolką z umiarkowaną siłą w płaszczyźnie poziomej przez minimum 30 sekund, do utworzenia jednolitej zawiesiny. Zawiesina produktu leczniczego Okteva musi zostać przygotowana bezpośrednio przed podaniem.
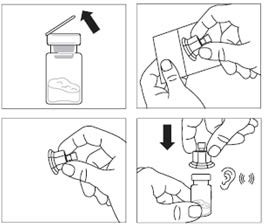
Produkt leczniczy Okteva może być podawany wyłącznie przez wyszkolony personel medyczny.
Krok 1
UWAGA: Bardzo istotne jest, by rozpocząć proces odtwarzania dopiero, gdy zestaw do wstrzyknięć osiągnie temperaturę pokojową. Należy pozostawić zestaw w temperaturze pokojowej przez minimum 30 minut przed odtworzeniem, jednak nie na dłużej niż przez 24 godziny.
Uwaga: Zestaw do wstrzyknięć można w razie
potrzeby ponownie włożyć do lodówki.
Krok 2
Wyjąć zestaw do wstrzyknięć z produktem Okteva z lodówki, w której był przechowywany.
Zdjąć z fiolki plastikowe wieczko i przetrzeć gumowy korek fiolki wacikiem nasączonym alkoholem.
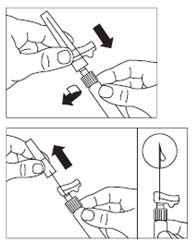
Oderwać folię pokrywającą opakowanie łącznika fiolki i wyjąć łącznik fiolki z opakowania, trzymając pomiędzy białym łącznikiem Luera a kołnierzem. NIE DOTYKAĆ końcówki łącznika w żadnym punkcie.
Postawić fiolkę na płaskiej powierzchni. Umieścić łącznik fiolki na fiolce i docisnąć do końca tak, by
wsunął się na miejsce z głośnym
„kliknięciem”.
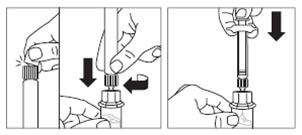
Przetrzeć końcówkę łącznika fiolki wacikiem nasączonym alkoholem.
Zdjąć osłonkę końcówki z ampułko-strzykawki
Powoli wcisnąć tłok do samego końca, aby cały roztwór rozpuszczalnika znalazł się w fiolce.
Na tym etapie należy przygotować pacjenta do wykonania wstrzyknięcia.
Po czasie wystarczającym do nasycenia proszku, należy sprawdzić, czy tłok jest dociśnięty w dół do samego końca strzykawki.
Odwrócić strzykawkę z fiolką dnem do góry i powoli odciągając tłok pobrać całą zawartość fiolki do strzykawki.
Odkręcić strzykawkę od łącznika fiolki.
Przygotować miejsce wstrzyknięcia przecierając je wacikiem nasączonym alkoholem
Przykręcić bezpieczną igłę do wstrzyknięć do strzykawki.
Jeżeli natychmiastowe podanie nie jest możliwe, ponownie delikatnie wstrząsać strzykawką, aby uzyskać mleczną jednorodną zawiesinę tuż przed podaniem.
Zdjąć nakładkę ochronną z igły prostym
Delikatnie opukać palcami strzykawkę powodując przesunięcie widocznych pęcherzyków powietrza ku górze, a następnie usunąć je ze strzykawki.
Przejść natychmiast do punktu Krok 8. Każde opóźnienie może spowodować powstanie osadu.
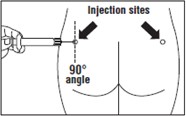
Produkt leczniczy Okteva musi być podawany wyłącznie w głębokim wstrzyknięciu do mięśnia pośladka, NIGDY dożylnie.
Wprowadzić całą igłę w lewy lub prawy pośladek pod kątem 90o do powierzchni skóry.
Powoli odciągnąć tłok ampułko-strzykawki, aby upewnić się, że igła nie znajduje się w naczyniu krwionośnym (zmienić położenie igły, jeśli znajduje się ona w naczyniu krwionośnym).
Powoli, ze stałym naciskiem przesuwać tłok, aż do opróżnienia strzykawki. Wyjąć
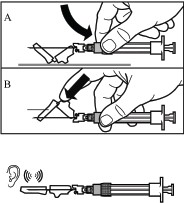
Uruchomić nakładkę ochronną igły, stosując jedną z 2 następujących metod:
docisnąć składaną część osłonki do
twardej powierzchni (rysunek A)
lub przycisnąć składaną część osłonki
palcem (rysunek B).
Prawidłowe uruchomienie zostanie potwierdzone głośnym kliknięciem.
Uwaga: należy zapisać miejsce wstrzyknięcia na karcie pacjenta, zmieniając je co miesiąc.
Strzykawkę natychmiast wyrzucić (do odpowiedniego pojemnika na ostre odpady).
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Okteva, 10 mg, proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o
przedłużonym uwalnianiu
Okteva, 20 mg, proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o
przedłużonym uwalnianiu
Okteva, 30 mg, proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o
przedłużonym uwalnianiu
Każda fiolka zawiera 10 mg oktreotydu (w postaci oktreotydu octanu). Każda fiolka zawiera 20 mg oktreotydu (w postaci oktreotydu octanu). Każda fiolka zawiera 30 mg oktreotydu (w postaci oktreotydu octanu).
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Proszek i rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o przedłużonym
uwalnianiu.
Proszek: biały lub prawie biały proszek bez widocznych cząstek obcych. Rozpuszczalnik do sporządzania zawiesiny do wstrzykiwań o przedłużonym uwalnianiu: przejrzysty, bezbarwny roztwór praktycznie bez widocznych cząstek stałych.
Leczenie pacjentów z akromegalią, u których leczenie chirurgiczne jest niewskazane lub nieskuteczne, albo pacjentów będących w okresie przejściowym, przed wystąpieniem całkowitego efektu działania radioterapii (patrz punkt 4.2).
Leczenie objawów u pacjentów z hormonalnie czynnymi guzami żołądka, jelit i trzustki, np. rakowiakami z cechami zespołu rakowiaka (patrz punkt 5.1).
Leczenie pacjentów z zaawansowanymi guzami neuroendokrynnymi wywodzącymi się ze środkowej części prajelita lub o nieznanym ognisku pierwotnym, w przypadku których wykluczono ognisko pierwotne nieznajdujące się w środkowej części prajelita.
Leczenie gruczolaków przysadki wydzielających TSH:
Dawkowanie
Akromegalia
Zaleca się rozpoczęcie leczenia od podawania produktu Okteva w dawce 20 mg co 4 tygodnie przez 3 miesiące. Pacjenci przyjmujący podskórnie (sc.) oktreotyd mogą rozpocząć leczenie produktem Okteva następnego dnia po ostatnim podaniu podskórnym oktreotydu. Następnie należy dostosować dawkę produktu na podstawie stężenia w surowicy hormonu wzrostu (ang. GH - growth hormone) i insulinopodobnego czynnika wzrostu 1/somatomedyny C
(ang, IGF-1- insulin-like growth factor) oraz objawów klinicznych.
U pacjentów, u których po 3 miesiącach objawy kliniczne i parametry biochemiczne (GH; IGF-1) nie są całkowicie kontrolowane (stężenia GH nadal są większe niż 2,5 mikrograma/l), dawkę można zwiększyć do 30 mg co 4 tygodnie. Jeśli po 3 miesiącach parametry GH, IGF-1 i (lub) inne objawy nadal nie są właściwie kontrolowane podczas podawania dawki 30 mg, dawkę można zwiększyć do 40 mg co 4 tygodnie.
U pacjentów, u których stężenie GH utrzymuje się stale poniżej 1 mikrograma/l, a stężenie IGF-1 w surowicy uległo normalizacji oraz u których większość odwracalnych objawów przedmiotowych i podmiotowych akromegalii cofnęła się po 3 miesiącach leczenia dawką 20 mg, można zastosować produkt leczniczy Okteva w dawce 10 mg co 4 tygodnie. Jednakże, szczególnie w tej grupie pacjentów wskazane jest ścisłe monitorowanie skuteczności leczenia przez oznaczanie stężeń GH i IGF-1 w surowicy oraz ocenę klinicznych objawów przedmiotowych i podmiotowych podczas leczenia tą małą dawką produktu Okteva.
U pacjentów otrzymujących ustaloną dawkę produktu leczniczego Okteva należy określać stężenie GH i IGF-1 co 6 miesięcy.
Hormonalnie czynne guzy żołądka, jelit i trzustki
Leczenie pacjentów z objawami związanymi z hormonalnie czynnymi guzami neuroendokrynnymi żołądka, jelit i trzustki
Zaleca się rozpoczynanie leczenia od podawania dawki 20 mg produktu leczniczego Okteva co 4 tygodnie. Pacjenci przyjmujący podskórnie oktreotyd powinni kontynuować to leczenie w dawce uprzednio skutecznej przez 2 tygodnie po pierwszym wstrzyknięciu produktu Okteva.
U pacjentów, u których po 3 miesiącach leczenia wystąpiło zadowalające złagodzenie objawów i wskaźników biologicznych, dawkę produktu leczniczego Okteva można zmniejszyć do 10 mg podawanych co 4 tygodnie.
U pacjentów, u których po 3 miesiącach leczenia wystąpiło tylko częściowe złagodzenie objawów można zwiększyć dawkę produktu leczniczego Okteva do 30 mg podawanych co 4 tygodnie.
W dniach, gdy pomimo leczenia produktem Okteva objawy związane z guzami żołądka, jelit i trzustki są nasilone, zaleca się podać dodatkowo podskórnie oktreotyd w dawce stosowanej przed wprowadzeniem produktu leczniczego Okteva. Może się to zdarzyć szczególnie
podczas pierwszych 2 miesięcy leczenia, zanim zostanie osiągnięte terapeutyczne stężenie oktreotydu.
Leczenie pacjentów z zaawansowanymi guzami neuroendokrynnymi wywodzącymi się ze środkowej części prajelita lub o nieznanym ognisku pierwotnym, w przypadku których wykluczono ognisko pierwotne nieznajdujące się w środkowej części prajelita
Zalecana dawka produktu leczniczego Okteva wynosi 30 mg, podawane co 4 tygodnie (patrz punkt 5.1). Leczenie produktem Okteva w celu zahamowania rozwoju guza należy kontynuować w sytuacji braku progresji guza.
Leczenie gruczolaków wydzielających TSH
Leczenie produktem Okteva należy rozpoczynać od dawki 20 mg podawanej co 4 tygodnie i kontynuować przez 3 miesiące przed ewentualnym dostosowaniem dawki. Następnie dawkę można dostosować w zależności od wartości TSH i odpowiedzi hormonu tarczycy.
Stosowanie u pacjentów z zaburzeniami czynności nerek
Zaburzenia czynności nerek nie wpływały na wielkość pola pod krzywą (AUC) oktreotydu podanego podskórnie, dlatego nie ma konieczności dostosowywania dawki produktu leczniczego Okteva.
Stosowanie u pacjentów z zaburzeniami czynności wątroby
Badania oktreotydu podawanego podskórnie i dożylnie wykazały, że możliwości eliminacji mogą być zmniejszone u pacjentów z marskością wątroby, lecz nie są zmniejszone u pacjentów ze stłuszczeniem wątroby. W pewnych przypadkach pacjenci z zaburzeniami czynności wątroby mogą wymagać dostosowania dawki.
Stosowanie u pacjentów w podeszłym wieku
W badaniu z oktreotydem podawanym podskórnie nie było konieczne dostosowanie dawki u pacjentów w wieku ≥ 65 lat, dlatego nie ma konieczności dostosowania dawki produktu leczniczego Okteva w tej grupie pacjentów.
Stosowanie u dzieci
Doświadczenie ze stosowaniem produktu leczniczego Okteva u dzieci jest ograniczone.
Sposób podawania
Produkt leczniczy Okteva może być podawany wyłącznie w głębokim wstrzyknięciu domięśniowym. Kolejne wstrzyknięcia domięśniowe należy wykonywać naprzemiennie w lewy lub prawy mięsień pośladkowy (patrz punkt 6.6).
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Ogólne
Guzy przysadki mózgowej wydzielające hormon wzrostu mogą się czasami rozrastać, powodując poważne powikłania (np. ubytki w polu widzenia), dlatego ważne jest, aby
każdego pacjenta dokładnie obserwować. Jeżeli pojawią się cechy rozrostu guza, należy rozważyć zastosowanie innych metod leczenia.
Terapeutyczne korzyści z leczenia polegające na zmniejszeniu stężenia hormonu wzrostu (GH) oraz normalizacji insulinopodobnego czynnika wzrostu 1 (IGF-1) u pacjentek z akromegalią mogą potencjalnie przywracać płodność. Pacjentki w wieku rozrodczym należy poinformować o potrzebie stosowania odpowiedniej antykoncepcji podczas leczenia oktreotydem, o ile jest to konieczne (patrz punkt 4.6).
U pacjentów poddanych długotrwałemu leczeniu oktreotydem należy kontrolować czynność tarczycy.
U pacjentów leczonych oktreotydem należy kontrolować czynność wątroby.
Zdarzenia dotyczące układu sercowo-naczyniowego
Obserwowano częste przypadki bradykardii. Konieczne może być dostosowanie dawki takich produktów leczniczych jak antagoniści receptorów beta-adrenergicznych, antagoniści kanałów wapniowych czy leki stosowane do utrzymania równowagi wodno-elektrolitowej (patrz punkt 4.5).
Objawy związane z pęcherzykiem żółciowym
Leczenie oktreotydem często powoduje kamicę żółciową, co może wiązać się z zapaleniem
pęcherzyka żółciowego i rozszerzeniem przewodu żółciowego (patrz punkt 4.8).
Ponadto po wprowadzeniu produktu leczniczego do obrotu zgłaszano przypadki zapalenia dróg żółciowych jako powikłania kamicy żółciowej u pacjentów przyjmujących oktreotyd we wstrzyknięciach o przedłużonym uwalnianiu.
Zaleca się badanie ultrasonograficzne pęcherzyka żółciowego przed leczeniem i co mniej
więcej 6 miesięcy podczas leczenia wstrzyknięciami oktreotydu o przedłużonym uwalnianiu .
Metabolizm glukozy
Z powodu hamującego działania na hormon wzrostu, glukagon i insulinę, produkt leczniczy Okteva może wpływać na regulację stężenia glukozy. Może wystąpić upośledzona poposiłkowa tolerancja glukozy. Jak zgłaszano u pacjentów leczonych oktreotydem podawanym podskórnie, w niektórych przypadkach może rozwinąć się stan trwałej hiperglikemii w wyniku długotrwałego podawania. Zgłaszano również przypadki hipoglikemii.
U pacjentów ze współistniejącą cukrzycą typu I, produkt leczniczy Okteva może wpływać na regulację stężenia glukozy i zapotrzebowanie na insulinę może się zmniejszyć. U pacjentów bez cukrzycy oraz pacjentów z cukrzycą typu II z częściowo zachowanymi rezerwami insuliny, oktreotyd podawany podskórnie może zwiększać poposiłkowe stężenie glukozy we krwi. Dlatego też zaleca się monitorowanie tolerancji glukozy i leczenia przeciwcukrzycowego.
U pacjentów z guzem insulinowym oktreotyd może nasilać hipoglikemię i przedłużać czas jej trwania z powodu silniejszego niż insulina hamowania wydzielania hormonu wzrostu i glukagonu, a także z powodu krócej trwającego działania hamującego na insulinę. Pacjentów tych należy starannie kontrolować.
Czynność trzustki
U niektórych pacjentów leczonych oktreotydem w związku z neuroendokrynnymi guzami przewodu pokarmowego i trzustki obserwowano zewnątrzwydzielniczą niewydolność trzustki (PEI, ang. pancreatic exocrine insufficiency). Objawy PEI mogą obejmować biegunkę tłuszczową, luźne stolce, wzdęcia brzucha i zmniejszenie masy ciała. U pacjentów, u których wystąpiły objawy choroby, należy rozważyć badania przesiewowe i odpowiednie leczenie PEI zgodnie z wytycznymi klinicznymi.
Odżywianie
Oktreotyd może u niektórych pacjentów zmieniać wchłanianie spożywanych tłuszczów.
U niektórych pacjentów przyjmujących oktreotyd obserwowano zmniejszenie stężenia witaminy B12 i nieprawidłowe wyniki testu Schillinga. Zaleca się monitorowanie stężenia witaminy B12 w czasie leczenia produktem Okteva u pacjentów, u których występował niedobór witaminy B12 w wywiadzie.
Zawartość sodu
Produkt leczniczy Okteva zawiera mniej niż 1 mmol (23 mg) sodu na dawkę, tzn. zasadniczo jest „wolny od sodu”.
Podczas jednoczesnego stosowania produktu leczniczego Okteva może zajść konieczność dostosowania dawek takich produktów leczniczych jak środki beta-adrenolityczne, antagoniści wapnia lub leki stosowane do utrzymania równowagi wodno-elektrolitowej (patrz punkt 4.4).
Podczas jednoczesnego stosowania produktu leczniczego Okteva może zajść konieczność dostosowania dawki insuliny i leków przeciwcukrzycowych (patrz punkt 4.4).
Stwierdzono, że oktreotyd zmniejsza jelitowe wchłanianie cyklosporyny i opóźnia wchłanianie cymetydyny.
Jednoczesne podanie oktreotydu i bromokryptyny zwiększa biodostępność bromokryptyny.
Ograniczone opublikowane dane wskazują, że analogi somatostatyny mogą spowalniać metabolizm związków, które są metabolizowane przez enzymy cytochromu P450. Może to być spowodowane zmniejszonym wydzielaniem hormonu wzrostu. Ponieważ nie można wykluczyć, że oktreotyd wywiera takie działanie, inne leki metabolizowane głównie przez CYP3A4, które mają wąski przedział terapeutyczny należy stosować z zachowaniem ostrożności (np. chinidyna, terfenadyna).
Jednoczesne stosowanie z radioaktywnymi analogami somatostatyny
Somatostatyna i jej analogi, takie jak oktreotyd, konkurencyjnie wiążą się z receptorami somatostatyny i mogą wpływać na skuteczność radioaktywnych analogów somatostatyny. Należy unikać podawania produktu leczniczego Okteva, przez co najmniej 4 tygodnie przed podaniem lutetu (177 Lu) oksodotreotydu, radiofarmaceutyku wiążącego się z receptorami somatostatyny. W razie potrzeby, pacjenci mogą być leczeni krótko działającymi analogami somatostatyny do 24 godzin przed podaniem lutetu oksodotreotydu (177Lu).
Po podaniu lutetu oksodotreotydu (177Lu) leczenie produktem leczniczym Okteva można wznowić w ciągu 4 do 24 godzin i należy je ponownie przerwać 4 tygodnie przed kolejnym podaniem lutetu (177Lu) oksodotreotydu.
Ciąża
Istnieją tylko ograniczone dane (mniej niż 300 przypadków ciąży) dotyczące stosowania oktreotydu u kobiet ciężarnych, przy czym w około jednej trzeciej przypadków wyniki ciąży nie są znane. Większość zgłoszeń otrzymano po wprowadzeniu oktreotydu do obrotu, a ponad 50% pacjentek w ciąży narażonych na działanie leku było pacjentkami z akromegalią.
Większość kobiet była narażona na oktreotyd w pierwszym trymestrze ciąży, w zakresie dawek 100 do 1200 mikrogramów oktreotydu podawanego podskórnie lub 10 do 40 mg na miesiąc oktreotydu we wstrzyknięciach o przedłużonym uwalnianiu. Wady wrodzone zgłoszono w około 4% ciąż o znanym wyniku. Nie podejrzewano, by w tych przypadkach istniał związek przyczynowy z oktreotydem.
Badania na zwierzętach nie wykazały bezpośredniego lub pośredniego toksycznego wpływu
na reprodukcję (patrz punkt 5.3).
W ramach środków ostrożności należy unikać stosowania produktu leczniczego Okteva podczas ciąży (patrz punkt 4.4).
Karmienie piersią
Nie wiadomo, czy oktreotyd przenika do mleka ludzkiego. Badania na zwierzętach wykazały przenikanie oktreotydu do mleka karmiących zwierząt. Podczas leczenia produktem leczniczym Okteva nie należy karmić piersią.
Płodność
Nie wiadomo, czy oktreotyd ma wpływ na płodność ludzi. U męskiego potomstwa samic leczonych w okresie ciąży i laktacji stwierdzono późne zstąpienie jąder. Oktreotyd nie zaburzał jednak płodności u szczurów obu płci po podaniu dawek do 1 mg/kg mc. na dobę (patrz punkt 5.3).
Okteva nie ma wpływu lub wywiera znikomy wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Należy doradzić pacjentom zachowanie ostrożności podczas prowadzenia pojazdów i obsługiwania maszyn, jeśli podczas leczenia produktem leczniczym Okteva wystąpią u nich zawroty głowy, osłabienie/uczucie zmęczenia lub ból głowy.
Podsumowanie profilu bezpieczeństwa
Do najczęstszych działań niepożądanych zgłaszanych podczas leczenia oktreotydem należą zaburzenia żołądka i jelit, zaburzenia układu nerwowego, zaburzenia wątroby i dróg żółciowych oraz zaburzenia metabolizmu i odżywiania.
Do działań niepożądanych zgłaszanych najczęściej podczas badań klinicznych z zastosowaniem oktreotydu należały: biegunka, bóle brzucha, nudności, wzdęcia, bóle głowy,
kamica żółciowa, hiperglikemia i zaparcia. Innymi, często zgłaszanymi działaniami niepożądanymi były zawroty głowy, miejscowy ból, błotko żółciowe, zaburzenia czynności tarczycy (np. zmniejszenie aktywności hormonu pobudzającego tarczycę [TSH], zmniejszenie całkowitej T4 oraz zmniejszenie wolnej T4), luźne stolce, zaburzenia
tolerancji glukozy, wymioty, osłabienie i hipoglikemia.
Tabelaryczny wykaz działań niepożądanych
Działania niepożądane wymienione w Tabeli 1 zostały zebrane w czasie badań klinicznych z zastosowaniem oktreotydu:
Działania niepożądane (Tabela 1) uszeregowano zgodnie z częstością występowania, jako pierwsze – najczęściej występujące, stosując następującą konwencję: bardzo często (≥1/10); często (≥1/100, <1/10); niezbyt często (≥1/1 000, <1/100); rzadko (≥1/10 000, <1/1 000) bardzo rzadko (<1/10 000), w tym pojedyncze doniesienia. W obrębie każdej grupy o określonej częstości występowania objawy niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 1 Działania niepożądane zgłaszane w badaniach klinicznych
Zaburzenia żołądka i jelit | |
Bardzo często: | Biegunka, bóle brzucha, nudności, zaparcia, gazy. |
Często: | Niestrawność, wymioty, wzdęcia, stolce tłuszczowe, luźne stolce, odbarwienie kału. |
Zaburzenia układu nerwowego | |
Bardzo często: | Bóle głowy |
Często: | Zawroty głowy |
Zaburzenia endokrynologiczne | |
Często: | Niedoczynność tarczycy, zaburzenia czynności tarczycy (np. zmniejszenie aktywności TSH, zmniejszenie całkowitej T4 oraz zmniejszenie stężenia wolnej T4) |
Zaburzenia wątroby i dróg żółciowych | |
Bardzo często: | Kamica żółciowa |
Często: | Zapalenie pęcherzyka żółciowego, błotko żółciowe, hiperbilirubinemia |
Zaburzenia metabolizmu i odżywiania | |
Bardzo często: | Hiperglikemia |
Często: | Hipoglikemia, zaburzenia tolerancji glukozy, brak łaknienia |
Niezbyt często: | Odwodnienie |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często: | Reakcje w miejscu wstrzyknięcia |
Często: | Osłabienie |
Badania diagnostyczne | |
Często: | Zwiększenie aktywności aminotransferaz |
Zaburzenia skóry i tkanki podskórnej | |
Często: | Świąd, wysypka, łysienie |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Często: | Duszność |
Zaburzenia serca | |
Często: | Bradykardia |
Niezbyt często: | Tachykardia |
Dane po wprowadzeniu produktu leczniczego do obrotu
O spontanicznie zgłaszanych działaniach niepożądanych, przedstawionych w Tabeli 2 informowano na zasadzie dobrowolności, a rzetelne ustalenie częstości ich występowania oraz związku przyczynowego z narażeniem na działanie leku nie zawsze jest możliwe.
Tabela 2 Działania niepożądane pochodzące ze zgłoszeń spontanicznych
Zaburzenia krwi i układu chłonnego |
Małopłytkowość |
Zaburzenia układu immunologicznego |
Anafilaksja, reakcje alergiczne/nadwrażliwość. |
Zaburzenia skóry i tkanki podskórnej |
Pokrzywka |
Zaburzenia wątroby i dróg żółciowych |
Ostre zapalenie trzustki, ostre zapalenie wątroby bez zastoju żółci, cholestatyczne zapalenie wątroby, zastój żółci, żółtaczka, żółtaczka cholestatyczna |
Zaburzenia serca |
Arytmia |
Badania diagnostyczne |
Zwiększenie aktywności fosfatazy zasadowej, zwiększenie aktywności gamma glutamylotransferazy. |
Opis wybranych działań niepożądanych
Objawy związane z pęcherzykiem żółciowym
Wykazano, że analogi somatostatyny hamują kurczliwość pęcherzyka żółciowego i zmniejszają wydzielanie żółci, co może prowadzić do nieprawidłowości pęcherzyka żółciowego lub powstania błotka żółciowego. U 15 do 30% pacjentów, którym długotrwale podawano podskórnie oktreotyd opisano powstawanie kamieni żółciowych. W populacji ogólnej częstość występowania kamieni żółciowych (w grupie wiekowej 40 do 60 lat) wynosi około 5 do 20%. Długotrwałe podawanie wstrzyknięć oktreotydu o przedłużonym uwalnianiu pacjentom z akromegalią lub hormonalnie czynnymi guzami żołądka, jelit i trzustki wskazuje, że wstrzyknięcia oktreotydu o przedłużonym uwalnianiu nie zwiększają częstości tworzenia się kamieni żółciowych, w porównaniu z leczeniem podskórnym.
Powstające kamienie żółciowe zwykle nie wywoływały objawów klinicznych; kamicę żółciową objawową należy leczyć metodą rozpuszczania kamieni żółciowych z wykorzystaniem kwasów żółciowych lub chirurgicznie.
Zaburzenia żołądka i jelit
W rzadkich przypadkach niepożądane objawy ze strony żołądka i jelit mogą przypominać ostrą niedrożność przewodu pokarmowego z postępującym rozdęciem brzucha, silnym bólem w nadbrzuszu, tkliwością brzucha i obroną mięśniową.
Wiadomo, że częstość występowania zdarzeń niepożądanych ze strony żołądka i jelit zmniejsza się z upływem czasu, w miarę kontynuowania leczenia.
Nadwrażliwość i reakcje anafilaktyczne
Po wprowadzeniu produktu leczniczego do obrotu zgłaszano występowanie reakcji nadwrażliwości i reakcji alergicznych. Najczęściej dotyczyły one reakcji skórnych, rzadziej ust i dróg oddechowych. Zgłaszano pojedyncze przypadku wstrząsu anafilaktycznego.
Reakcje w miejscu wstrzyknięcia
U pacjentów otrzymujących wstrzykiwania oktreotydu o przedłużonym uwalnianiu często zgłaszano reakcje w miejscu wstrzyknięcia, w tym ból, zaczerwienienie, krwotok, świąd, obrzęk lub stwardnienie; jednak w większości przypadków zdarzenia te nie wymagały interwencji klinicznej.
Zaburzenia metabolizmu i odżywiania
Chociaż może zwiększyć się wydzielanie tłuszczu w kale, brak jest danych wskazujących, że długotrwałe leczenie oktreotydem może prowadzić do niedoborów odżywiania z powodu złego wchłaniania pokarmu.
Enzymy trzustkowe
Bardzo rzadko obserwowano ostre zapalenie trzustki, które występowało w pierwszych godzinach lub dniach podawania podskórnego oktreotydu i ustępowało po zaprzestaniu podawania leku. Ponadto, u pacjentów leczonych długotrwale oktreotydem podawanym podskórnie obserwowano zapalenie trzustki wywołane kamicą żółciową.
Zaburzenia serca
Częstym niepożądanym działaniem analogów somatostatyny jest bradykardia. Zarówno u pacjentów z akromegalią, jak i z rakowiakami obserwowano zmiany w EKG, takie jak wydłużenie odstępu QT, zmiany osi serca, wczesną repolaryzację, niski woltaż, zmiany załamka R, przejście załamka R w S, wczesną progresję załamków R oraz niespecyficzne zmiany odcinka ST-T. Jednakże nie udowodniono związku tych zmian ze stosowaniem octanu oktreotydu, gdyż u wielu z tych pacjentów występują choroby serca, które mogą być przyczyną takich zmian (patrz punkt 4.4).
Małopłytkowość
Po wprowadzeniu produktu do obrotu zgłaszano małopłytkowość, zwłaszcza podczas leczenia wstrzykiwaniami oktreotydu (i.v.) u pacjentów z marskością wątroby oraz podczas leczenia wstrzykiwaniami oktreotydu o przedłużonym uwalnianiu. Jest to odwracalne po przerwaniu leczenia.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C, 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Zgłaszano ograniczoną liczbę przypadków niezamierzonego przedawkowania wstrzykiwań oktreotydu o przedłużonym uwalnianiu. Dawki wahały się od 100 mg do 163 mg na miesiąc oktreotydu o przedłużonym uwalnianiu we wstrzyknięciu. Jedynym zgłaszanym zdarzeniem niepożądanym były uderzenia gorąca.
Zgłaszano przypadki pacjentów z chorobą nowotworową otrzymujących dawki oktreotydu o
przedłużonym uwalnianiu we wstrzyknięciu do 60 mg/miesiąc oraz do 90 mg/2 tygodnie. Dawki te były na ogół dobrze tolerowane; zgłaszano jednak następujące zdarzenia niepożądane: częste oddawanie moczu, uczucie zmęczenia, depresję, niepokój i brak koncentracji.
W przypadku przedawkowania stosuje się leczenie objawowe.
Grupa farmakoterapeutyczna: somatostatyna i jej analogi, kod ATC: H01CB02
Mechanizm działania
Oktreotyd jest syntetycznym oktapeptydem, pochodną naturalnie występującej somatostatyny, o podobnych właściwościach farmakologicznych, ale znacząco dłuższym okresie działania.
Hamuje on patologicznie zwiększone wydzielanie hormonu wzrostu (GH) oraz peptydów i serotoniny, uwalnianych przez wewnątrzwydzielniczy układ żołądkowo-jelitowo-trzustkowy (GEP).
U zwierząt oktreotyd silniej niż somatostatyna hamuje uwalnianie hormonu wzrostu, glukagonu i insuliny, wykazując większą selektywność w hamowaniu hormonu wzrostu i glukagonu.
U zdrowych ochotników wykazano, że oktreotyd, podobnie jak somatostatyna, hamuje:
W przeciwieństwie do somatostatyny, oktreotyd hamuje przede wszystkim uwalnianie GH i w mniejszym stopniu insuliny, a jego podanie nie powoduje efektu z odbicia wynikającego z hipersekrecji hormonów (tj. GH u pacjentów z akromegalią).
U pacjentów z akromegalią produkt leczniczy Okteva, postać farmaceutyczna oktreotydu przeznaczona do podawania w odstępach 4 tygodni, zapewnia stałe stężenie terapeutyczne oktreotydu w surowicy, dzięki czemu równomiernie zmniejsza stężenie hormonu wzrostu i normalizuje stężenie IGF 1 w surowicy u większości pacjentów. U większości pacjentów oktreotyd w postaci wstrzykiwań o przedłużonym uwalnianiu znacznie zmniejsza nasilenie objawów chorobowych, takich jak: bóle głowy, nadmierne pocenie się, parestezje, zmęczenie,
bóle kości i stawów i zespół cieśni nadgarstka. W przypadku uprzednio nieleczonej akromegalii u pacjentów z gruczolakiem przysadki wydzielającym hormon wzrostu podawanie oktreotydu w postaci wstrzykiwań o przedłużonym uwalnianiu
prowadziło do zmniejszenia objętości guza o >20% u znacznego odsetka pacjentów (50%).
Zgłaszano, że u poszczególnych pacjentów z gruczolakiem przysadki wydzielającym GH wstrzyknięcia oktreotydu o przedłużonym uwalnianiu powodowały obkurczenie guza (przed zabiegiem chirurgicznym). Nie należy jednak opóźniać leczenia chirurgicznego.
U pacjentów z hormonalnie czynnymi guzami żołądka, jelit i trzustki leczenie wstrzyknięciami oktreotydem o przedłużonym uwalnianiu prowadziło do trwałego opanowania objawów związanych z chorobą podstawową.
Działanie oktreotydu stosowanego w leczeniu różnych typów guzów żołądka, jelit i trzustki jest następujące:
Rakowiaki
Stosowanie oktreotydu może doprowadzić do zmniejszenia objawów choroby, szczególnie takich, jak nagłe zaczerwienienia skóry twarzy i biegunka. W wielu przypadkach towarzyszy temu zmniejszenie stężenia serotoniny w osoczu i zmniejszenie wydalania kwasu 5- hydroksyindolooctowego z moczem.
VIPoma
Pod względem biochemicznym guzy te charakteryzują się nadmiernym wytwarzaniem wazoaktywnego peptydu jelitowego (ang. VIP - vasoactive intestinal peptide). W większości przypadków stosowanie oktreotydu łagodzi przebieg ostrej biegunki wydzielniczej, będącej typowym objawem tej choroby, co w rezultacie daje poprawę jakości życia. Jednocześnie obserwuje się zmniejszenie zaburzeń elektrolitowych, np. hipokaliemii, co umożliwia zaprzestanie podawania płynów i elektrolitów, zarówno dojelitowo, jak i pozajelitowo. U niektórych pacjentów tomografia komputerowa wskazuje na spowolnienie lub zatrzymanie wzrostu guza, a nawet zmniejszenie jego rozmiarów, co dotyczy zwłaszcza przerzutów do wątroby.
Poprawie klinicznej z reguły towarzyszy zmniejszenie w osoczu stężenia wazoaktywnego peptydu jelitowego, które może obniżyć się do wartości prawidłowych.
Glukagonoma
U większości pacjentów podawanie oktreotydu powoduje wyraźną poprawę zmian skórnych, występujących w postaci charakterystycznego dla tej choroby nekrolitycznego rumienia wędrującego. Oktreotyd w niewielkim stopniu wpływa na pojawiającą się często w tych przypadkach łagodną cukrzycę i zazwyczaj jego stosowanie nie zmniejsza zapotrzebowania na insulinę lub doustne leki przeciwcukrzycowe. Oktreotyd łagodzi przebieg biegunki, co sprzyja zwiększeniu masy ciała. Podanie oktreotydu często prowadzi do natychmiastowego zmniejszenia stężenia glukagonu w osoczu, jednak działanie to na ogół nie utrzymuje się
w czasie długotrwałego leczenia, mimo stałej poprawy klinicznej.
Gastrinoma/zespół Zollingera-Ellisona
Podawanie leków z grupy inhibitorów pompy protonowej lub leków blokujących receptor H2 pozwala na ogół opanować nadmierne wydzielanie kwasu żołądkowego. Jednak biegunka, będąca częstym objawem może nie być wystarczająco łagodzona przez inhibitory pompy protonowej lub leki blokujące receptor H2. Produkt Okteva może być pomocny w dalszym zmniejszaniu nadmiernego wydzielania kwasu żołądkowego i łagodzeniu objawów choroby,
w tym biegunki, ponieważ u niektórych pacjentów zmniejsza podwyższoną aktywność
gastryny.
Insulinoma
Stosowanie oktreotydu prowadzi do zmniejszenia stężenia insuliny immunoreaktywnej. U pacjentów z guzami, zakwalifikowanych do leczenia operacyjnego, oktreotyd może być pomocny w przywracaniu i utrzymywaniu prawidłowego stężenia glukozy we krwi przed operacją. U pacjentów z łagodnymi lub złośliwymi nieoperacyjnymi guzami można skuteczniej utrzymywać właściwe stężenie glukozy we krwi, nawet bez jednoczesnego trwałego zmniejszenia stężenia insuliny we krwi.
Zaawansowane guzy neuroendokrynne wywodzące się ze środkowej części prajelita lub o nieznanym ognisku pierwotnym, w przypadku których wykluczono ognisko pierwotne nieznajdujące się w środkowej części prajelita
Randomizowane, podwójnie ślepe badanie III fazy kontrolowane placebo (PROMID) wykazało, że wstrzyknięcia oktreotydu o przedłużonym uwalnianiu hamują rozwój guza u pacjentów z zaawansowanymi guzami neuroendokrynnymi środkowego odcinka prajelita. 85 pacjentów zrandomizowano do grupy otrzymującej oktreotyd w postaci wstrzyknięć o przedłużonym uwalnianiu w dawce 30 mg podawanej co 4 tygodnie (n=42) lub placebo (n=43) przez 18 miesięcy, bądź do stwierdzenia progresji guza lub zgonu pacjenta.
Głównymi kryteriami włączenia były: brak wcześniejszego leczenia; histologicznie potwierdzone, miejscowo nieoperacyjne lub z przerzutami, dobrze zróżnicowane, hormonalnie czynne lub nieczynne guzy/raki neuroendokrynne; z pierwotnym ogniskiem guza zlokalizowanym w środkowym odcinku prajelita lub guzy nieznanego pochodzenia, o których uważa się, że pochodzą ze środkowego odcinka jelita, jeśli wykluczono obecność ogniska pierwotnego w trzustce, klatce piersiowej lub w innej lokalizacji.
Pierwszorzędowym punktem końcowym był czas do progresji guza (time to progression,
TTP) lub zgon związany z guzem.
W analizie populacji intent-to-treat (ITT) (wszyscy zrandomizowani pacjenci) odnotowano 26 i 41 progresji lub zgonów związanych z guzem odpowiednio w grupie otrzymującej wstrzyknięcia oktreotydu o przedłużonym uwalnianiu i placebo (HR = 0,32; 95% CI, 0,19 do 0,55; wartość p =0,000015).
W konserwatywnej analizie populacji ITT (cITT), z której wykluczono 3 pacjentów w momencie randomizacji, odnotowano 26 i 40 progresji lub zgonów związanych z guzem odpowiednio w grupie otrzymującej oktreotyd w postaci wstrzyknięć o przedłużonym uwalnianiu i placebo (HR=0,34; 95% CI, 0,20 do 0,59; wartość p =0,000072; Ryc. 1).
Mediana czasu do progresji guza wyniosła 14,3 miesięcy (95% CI, 11,0 do 28,8 miesięcy) w grupie otrzymującej wstrzyknięcia oktreotydu o przedłużonym uwalnianiu oraz 6,0 miesięcy (95% CI, 3,7 do 9,4 miesięcy) w grupie placebo.
W analizie populacji spełniającej kryteria protokołu badania (PP), z której pod koniec terapii wykluczono dodatkowych pacjentów, progresję lub zgon związany z guzem odnotowano w 19 i 38 przypadkach odpowiednio w grupie otrzymującej oktreotyd w postaci wstrzyknięć o przedłużonym uwalnianiu i w grupie placebo (HR = 0,24; 95% CI, 0,13 do 0,45; wartość p
=0,0000036).
Rycina 1 Estymatory Kaplana-Meiera dla czasu do progresji guza, porównujące Okteva z placebo (populacja cITT)
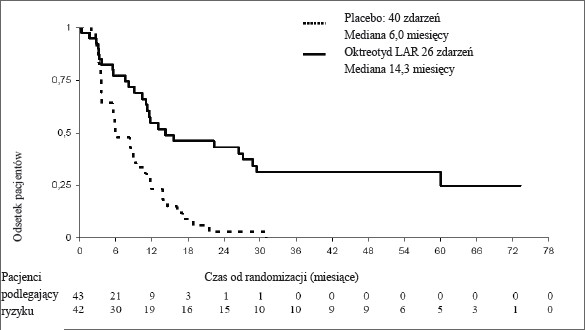
*Logarytmiczny test rang ze stratyfikacją uwzgledniającą aktywność hormonalną: p=0,000072, HR= 0,34 [95% CI, 0,20-0,59]
Tabela 3 Wyniki dotyczące czasu do progresji guza (TTP) w zależności od analizowanej
populacji
Zdarzenia TTP | Mediana TTP w miesiącach [95% C.I.] | HR [95% C.I.] wartość p* | |||
Oktreotyd o przedłużonym uwalnianiu we wstrzyknięciu | Placebo | Oktreotyd o przedłużonym uwalnianiu we wstrzyknięciu | Placebo | ||
ITT | 26 | 41 | NR | NR | 0,32 |
[95% CI, | |||||
0,19 do | |||||
0,55] | |||||
p=0,000015 | |||||
cITT | 26 | 40 | 14,3 | 6,0 | 0,34 |
[95% CI, 11,0 | [95% CI, 3,7 | [95% CI, | |||
do 28,8] | do | 0,20 do | |||
9,4] | 0,59] | ||||
p=0,000072 | |||||
PP | 19 | 38 | NR | NR | 0,24 |
[95% CI, 0,13 do 0,45] p=0,0000036 | |||||
NR=niezgłaszane; HR=współczynnik ryzyka; TTP=czas do progresji guza; ITT=analiza populacji intention to treat; cITT=konserwatywna analiza ITT; PP=analiza populacji spełniającej kryteria protokołu *Logarytmiczny test rang ze stratyfikacją uwzgledniającą aktywność hormonalną | |||||
Obserwowano podobny wynik leczenia u pacjentów z hormonalnie czynnymi (HR = 0,23; 95% CI, 0,09 do 0,57) i nieczynnymi guzami (HR = 0,25; 95% CI, 0,10 do 0,59).
Po 6 miesiącach leczenia stabilizację choroby obserwowano u 66% pacjentów z grupy otrzymującej wstrzyknięcia oktreotydu o przedłużonym uwalnianiu i 37% pacjentów z grupy placebo.
Z uwagi na istotne korzyści kliniczne wstrzyknięć oktreotydu o przedłużonym uwalnianiu obserwowane w analizie przeprowadzonej przed planowaną analizą okresową, rekrutacja została przerwana.
Bezpieczeństwo stosowania wstrzyknięć oktreotydu o przedłużonym uwalnianiu w tym
badaniu było zgodne z ustalonym profilem bezpieczeństwa produktu.
Leczenie gruczolaków przysadki wydzielających TSH
Wykazano, że podawanie wstrzyknięć oktreotydu o przedłużonym uwalnianiu, w dawce jedno wstrzyknięcie domięśniowe co 4 tygodnie, zmniejsza podwyższone aktywności hormonów tarczycy, normalizuje TSH oraz powoduje poprawę klinicznych przedmiotowych i podmiotowych objawów nadczynności tarczycy u pacjentów z gruczolakami wydzielającymi TSH. Wpływ leczenia oktreotydem w postaci wstrzyknięć o przedłużonym uwalnianiu osiągnął znamienność statystyczną po 28 dniach, w porównaniu ze stanem początkowym, a korzyści z leczenia utrzymywały się przez okres do 6 miesięcy.
Wchłanianie
Po jednorazowym domięśniowym podaniu wstrzyknięcia oktreotydu o przedłużonym uwalnianiu, stężenie oktreotydu w surowicy osiąga przemijającą początkową maksymalną wartość w ciągu 1 godziny. W ciągu 24 godzin następuje stopniowe zmniejszenie stężenia oktreotydu do małych, nieoznaczalnych wartości. Po tym początkowym maksimum pierwszego dnia, stężenie oktreotydu pozostaje na poziomie subterapeutycznym u większości pacjentów przez następne 7 dni. Następnie stężenie oktreotydu ponownie zwiększa się, osiągając stan równowagi około 14. dnia i pozostając na tym względnie stałym poziomie przez następne 3 do 4 tygodni. Stężenie maksymalne, występujące 1. dnia, jest mniejsze niż stężenie w stanie równowagi, a ilość leku uwalnianego w ciągu pierwszego dnia nie przekracza 0,5% całej uwalnianej dawki. Po około 42 dniach stężenie oktreotydu zmniejsza się powoli, jednocześnie z końcową fazą rozpadu matrycy polimerowej podanej postaci leku.
U pacjentów z akromegalią średnie stężenie oktreotydu w stanie równowagi po podaniu
pojedynczego wstrzyknięcia w dawce 10 mg, 20 mg lub 30 mg oktreotydu o przedłużonym
uwalnianiu wynosi odpowiednio 358 ng/l, 926 ng/l lub 1 710 ng/l. Stężenia oktreotydu w surowicy w stanie równowagi, uzyskiwane po 3 iniekcjach podawanych co 4 tygodnie
są około 1,6 do 1,8 razy większe i wynoszą 1 557 ng/l i 2 384 ng/l po wielokrotnym podaniu wstrzyknięć oktreotydu o przedłużonym uwalnianiu w dawkach odpowiednio 20 mg i 30 mg.
U pacjentów z rakowiakiem średnie wartości (oraz mediana) stężeń oktreotydu w surowicy w stanie równowagi, po wielokrotnym wstrzyknięciu oktreotydu o przedłużonym uwalnianiu w dawkach 10 mg, 20 mg i 30 mg co 4 tygodnie również zwiększały się wprost proporcjonalnie do dawki i wynosiły odpowiednio 1 231 (894) ng/l, 2 620 (2 270) ng/l i 3 928 (3 010) ng/l.
W czasie stosowania 28 comiesięcznych wstrzykiwań oktreotydu w postaci wstrzyknięć o przedłużonym uwalnianiu nie zaobserwowane zjawiska nagromadzenia się oktreotydu, z wyjątkiem spodziewanego, spowodowanego powolnym uwalnianiem się leku.
Dystrybucja i metabolizm
Profil farmakokinetyczny oktreotydu po wstrzyknięciu oktreotydu o przedłużonym uwalnianiu odpowiada profilowi uwalniania z matrycy polimerowej i jego biodegradacji. Po uwolnieniu do krwi krążącej dystrybucja oktreotydu następuje zgodnie ze znanymi właściwościami farmakokinetycznymi, opisanymi dla podania podskórnego. Objętość dystrybucji oktreotydu w stanie stacjonarnym wynosi 0,27 l/kg, a całkowity klirens - 160 ml/min. Z białkami osocza wiąże się do 65% oktreotydu i lek w zasadzie nie wiąże
się z komórkami krwi.
Dane farmakologiczne z ograniczonej liczby próbek krwi pobranych od dzieci i młodzieży w wieku 7–17 lat, chorych na otyłość podwzgórzową, którym podawano oktreotyd w postaci wstrzyknięć o przedłużonym uwalnianiu w dawce 40 mg raz na miesiąc wykazały, że najmniejsze stężenie oktreotydu w osoczu wynosi średnio 1395 ng/l po pierwszym wstrzyknięciu oraz 2973 ng/l w stanie stacjonarnym. Obserwuje się dużą zmienność międzyosobniczą.
Najmniejsze stężenia oktreotydu w stanie stacjonarnym nie korelowały z wiekiem i wartością BMI, ale umiarkowanie korelowały z masą ciała (52,3–133 kg) i różniły się znamiennie w zależności od płci pacjentów, tj. były o około 17% większe u dziewcząt.
Badania toksyczności ostrej i po podaniu wielokrotnych dawek, badania genotoksyczności, rakotwórczości i toksycznego wpływu na reprodukcję prowadzone na zwierzętach nie wykazały szczególnego zagrożenia dla bezpieczeństwa ludzi.
Badania reprodukcji prowadzone na zwierzętach nie wykazały teratogennego działania oktreotydu, ani jego wpływu na zarodek/płód lub innego wpływu na reprodukcję zwierząt po podaniu dawek wynoszących maksymalnie 1 mg/kg mc./dobę. Nieznaczne, przemijające opóźnienia fizjologicznego wzrostu, zaobserwowane u potomstwa szczurów były wynikiem zahamowania wydzielania hormonu wzrostu, które zostało spowodowane nadmierną aktywnością farmakodynamiczną leku (patrz punkt 4.6).
Nie przeprowadzono specyficznych badań na młodych szczurach. W badaniach rozwoju przed- i pourodzeniowego obserwowano spowolniony wzrost i dojrzewanie u potomstwa F1
samic, którym oktreotyd podawano przez całą ciążę i laktację. U męskiego potomstwa F1 obserwowano opóźnione zstąpienie jąder, jednak płodność tych osobników pozostała prawidłowa. Z tego względu, wymienione wyżej objawy były przejściowe i uważane za skutek zahamowania wydzielania hormonu wzrostu.
Proszek (fiolka):
Kopolimer D,L-laktydu i glikolidu (55:45) Mannitol (E 421)
Rozpuszczalnik (ampułko-strzykawka):
Karmeloza sodowa Mannitol (E 421)
Poloksamer 188 Woda do wstrzykiwań
Nie mieszać produktu leczniczego z innymi produktami leczniczymi, ponieważ nie wykonywano badań dotyczących zgodności.
3 lata
Nie przechowywać produktu leczniczego po odtworzeniu, (zużyć natychmiast).
Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Przechowywać w lodówce (2°C – 8°C). Nie zamrażać.
W dniu wykonania wstrzyknięcia produkt Okteva można przechowywać w temperaturze poniżej 25oC.
Warunki przechowywania po odtworzeniu, patrz punkt 6.3.
Okteva 10 mg:
Zestaw: opakowanie jednostkowe zawiera jedną szklaną fiolkę zamkniętą korkiem z gumy chlorobutylowej z aluminiowym uszczelnieniem z ciemnoniebiskim wieczkiem typu flip-off, zawierającą proszek do sporządzania zawiesiny do wstrzykiwań, i jedną ampułko-strzykawkę z bezbarwnego szkła z tłokiem z gumy bromobutylowej pełniącym również rolę korka zawierającą 2 ml rozpuszczalnika, łącznie zapakowane na plastikowej tacce wraz z łącznikiem fiolki oraz jedną bezpieczną igłą do wstrzykiwań.
Dostępne wielkości opakowań: 1 opakowanie jednostkowe i 3 opakowania jednostkowe.
Okteva 20 mg:
Zestaw: Opakowanie jednostkowe zawiera jedną szklaną fiolkę zamkniętą korkiem z gumy chlorobutylowej, i aluminiowym uszczelnieniem z pomarańczowym wieczkiem typu flip-off, zawierającą proszek do sporządzania zawiesiny do wstrzykiwań, i jedną ampułko-strzykawkę z bezbarwnego szkła z tłokiem z gumy bromobutylowej pełniącym również rolę korka zawierającą 2 ml rozpuszczalnika, łącznie zapakowane na plastikowej tacce wraz z łącznikiem fiolki oraz jedną bezpieczną igłą do wstrzykiwań.
Dostępne wielkości opakowań: 1 opakowanie jednostkowe i 3 opakowania jednostkowe. Okteva 30 mg:
Zestaw: Opakowanie jednostkowe zawiera jedną szklaną fiolkę zamkniętą korkiem z gumy chlorobutylowej, i aluminiowym uszczelnieniem zciemnoczerwonym wieczkiem typu flip- off, zawierającą proszek do sporządzania zawiesiny do wstrzykiwań, i jedną ampułko- strzykawkę z bezbarwnego szkła z tłokiem z gumy bromobutylowej pełniącym również rolę korka j zawierającą 2 ml rozpuszczalnika, łącznie zapakowane na plastikowej tacce wraz z łącznikiem fiolki oraz jedną bezpieczną igłą do wstrzykiwań.
Dostępne wielkości opakowań: 1 opakowanie jednostkowe i 3 opakowania jednostkowe. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
leczniczego do stosowania
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Instrukcja przygotowania i domięśniowego podania produktu leczniczego Okteva
WYŁĄCZNIE DO GŁĘBOKIEGO WSTRZYKNIĘCIA W MIĘŚNIE POŚLADKA
Zestaw:
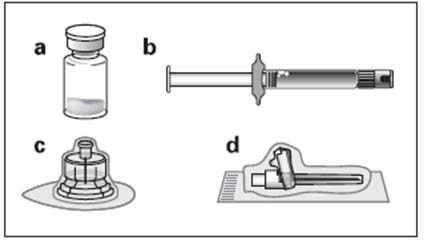
a Jedna fiolka zawierająca produkt Okteva w postaci proszku
b Jedna ampułko-strzykawka zawierająca rozpuszczalnik do sporządzenia zawiesiny
c Jeden łącznik fiolki do odtworzenia produktu leczniczego
d Jedna bezpieczna igła do wstrzyknięć
Należy postępować zgodnie z podaną poniżej instrukcją, aby właściwie odtworzyć produkt leczniczy Okteva przed wykonaniem głębokiego wstrzyknięcia do mięśnia pośladka.
Dla prawidłowego odtworzenia produktu leczniczego Okteva krytyczne znaczenie mają 3 wymagania. Niespełnienie tych wymagań może być przyczyną niewłaściwego podania leku.
Krok 3 wypełnionej rozpuszczalnikiem i przykręcić strzykawkę do łącznika fiolki. |
Krok 4 UWAGA: Bardzo istotne jest, by odstawić fiolkę na 5 minut, aby zapewnić całkowite nasycenie proszku rozpuszczalnikiem. Uwaga: Tłok może zostać odepchnięty z powrotem do góry, co jest normalne z powodu niewielkiego nadciśnienia w fiolce. |
Krok 5 UWAGA: Trzymając tłok dociśnięty do samego końca, umiarkowanie wstrząsać fiolką w płaszczyźnie poziomej przez minimum 30 sekund, tak aby cały proszek utworzył zawiesinę (jednorodna mleczna zawiesina). Jeśli część proszku nie połączyła się z roztworem, powtórzyć umiarkowane wstrząsanie przez kolejne 30 sekund. |
Krok 6 |
Krok 7 ruchem do góry. |
Krok 8 |
igłę z miejsca wkłucia i uruchomić nakładkę ochronną igły (zgodnie ze wskazówkami podanymi w punkcie Krok 9).
Krok 9
Teva B.V.
Swensweg 5
2031 GA Haarlem Holandia
Pozwolenie nr 25591, 25592, 25593
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 2019.10.15
2022.03.04







