







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY I TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
w leczeniu chorych na akromegalię, gdy stężenie krążącego hormonu wzrostu (GH) i (lub) insulinopodobnego czynnika wzrostu (IGF-1) pozostają nieprawidłowe po operacji i (lub) radioterapii oraz u pacjentów, u których z innego powodu jest konieczne zastosowanie leczenia farmakologicznego,
w leczeniu guzów neuroendokrynnych żołądkowo-jelitowo-trzustkowych (GEP-NET) G1 i części guzów G2 (indeks Ki67 do maksymalnie 10%) środkowej części prajelita, trzustki lub nieznanego pochodzenia, po wykluczeniu ognisk pierwotnych w końcowej części prajelita, u dorosłych pacjentów z nieoperacyjnymi guzami miejscowo zaawansowanymi lub z przerzutami (patrz punkt 5.1),
w leczeniu objawów związanych z guzami neuroendokrynnymi (zwłaszcza rakowiakami).
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
lata
Po otwarciu aluminiowej torebki ochronnej produkt leczniczy należy podać niezwłocznie.
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
Burlington Road
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY I TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Myrelez, 60 mg, roztwór do wstrzykiwań w ampułko-strzykawce Myrelez, 90 mg, roztwór do wstrzykiwań w ampułko-strzykawce Myrelez, 120 mg, roztwór do wstrzykiwań w ampułko-strzykawce
Lanreotyd 60 mg, 90 mg, 120 mg (w postaci lanreotydu octanu).
Każda fabrycznie napełniona ampułko-strzykawka zawiera przesycony roztwór lanreotydu octanu, o stężeniu odpowiadającym 0,246 mg lanreotydu w postaci zasady na 1 mg roztworu, co zapewnia rzeczywistą dawkę lanreotydu w jednej iniekcji wynoszącą odpowiednio 60 mg, 90 mg lub 120 mg.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Roztwór do wstrzykiwań w ampułko-strzykawce.
Roztwór biały do jasnożółtego, o półstałej konsystencji, praktycznie wolny od cząstek obcych.
Produkt leczniczy Myrelez jest wskazany:
Dawkowanie
Akromegalia
Zalecana dawka początkowa wynosi od 60 do 120 mg podawana co 28 dni.
Dawkę można zmieniać w zależności od odpowiedzi pacjenta (ocenianej na podstawie objawów i (lub) efektu biochemicznego) lub na podstawie możliwego doświadczenia pacjenta z analogami somatostatyny.
Na przykład u pacjentów leczonych uprzednio lanreotydem w dawce 30 mg co 14 dni, początkowa dawka produktu leczniczego Myrelez powinna wynosić 60 mg co 28 dni, u pacjentów leczonych uprzednio lanreotydem w dawce 30 mg co 10 dni, początkowa dawka produktu leczniczego Myrelez powinna wynosić 90 mg co 28 dni.
Następnie należy modyfikować dawkę leku w zależności od odpowiedzi pacjenta (mierzonej złagodzeniem objawów i (lub) obniżeniem stężeń GH i (lub) IGF-1).
W przypadku pacjentów, których objawy kliniczne i parametry biochemiczne nie są odpowiednio kontrolowane, dawkę produktu leczniczego Myrelez można zwiększyć do maksymalnie 120 mg co 28 dni.
W przypadku uzyskania odpowiedzi całkowitej (manifestującej się obniżeniem stężeń GH poniżej 1 ng/ml, normalizacją stężeń IGF-1 i (lub) ustąpieniem objawów) dawkę leku można zmniejszyć.
U pacjentów, u których analog somatostatyny zapewnia dobrą kontrolę objawów choroby, produkt leczniczy Myrelez można wstrzykiwać w dawce 120 mg co 42-56 dni (od 6 do 8 tygodni).
Długoterminowe monitorowanie objawów oraz stężeń GH i IGF-1 należy prowadzić zgodnie ze wskazaniami klinicznymi.
Leczenie guzów neuroendokrynnych żołądkowo-jelitowo-trzustkowych (GEP-NET) G1 i części guzów G2 (indeks Ki67 do maksymalnie 10%) środkowej części prajelita, trzustki lub nieznanego pochodzenia, po wykluczeniu ognisk pierwotnych w końcowej części prajelita,
u dorosłych pacjentów z nieoperacyjnymi guzami miejscowo zaawansowanymi lub z przerzutami.
Zalecana dawka to jedno wstrzyknięcie produktu leczniczego Myrelez 120 mg podawane co 28 dni. Leczenie produktem Myrelez 120 mg należy prowadzić tak długo, jak jest to konieczne w celu kontroli guza.
Leczeniu objawów związanych z guzami neuroendokrynnymi
Zalecana dawka początkowa wynosi od 60 do 120 mg podawana co 28 dni.
Następnie należy modyfikować dawkę leku w zależności od odpowiedzi pacjenta mierzonej złagodzeniem objawów.
Zaburzenie czynności nerek i (lub) wątroby
U pacjentów z zaburzeniem czynności nerek lub wątroby nie jest konieczne dostosowanie dawki ze względu na szeroki zakres dawek terapeutycznych lanreotydu (patrz punkt 5.2).
Pacjenci w podeszłym wieku
U pacjentów w podeszłym wieku nie jest konieczne dostosowanie dawki ze względu na szeroki zakres dawek terapeutycznych lanreotydu (patrz punkt 5.2).
Dzieci i młodzież
Nie zaleca się stosowania produktu leczniczego Myrelez u dzieci i młodzieży z powodu braku danych dotyczących bezpieczeństwa i skuteczności.
Sposób podawania
Produkt leczniczy Myrelez należy wstrzykiwać głęboko podskórnie w górny zewnętrzny kwadrant pośladka lub w górną zewnętrzną część uda.
W przypadku pacjentów, którzy otrzymują stabilną dawkę produktu leczniczego Myrelez oraz po odpowiednim przeszkoleniu, produkt leczniczy może być podany samodzielnie przez pacjenta lub przez przeszkoloną osobę. W przypadku samodzielnego podania leku iniekcje należy podawać
w górną, zewnętrzną powierzchnię uda.
Decyzję dotyczącą tego, czy pacjent może wykonywać iniekcje samodzielnie, czy powinna je podawać przeszkolona osoba, powinien podjąć lekarz.
Niezależnie od miejsca wykonania iniekcji nie należy przy tym tworzyć fałdu skóry, a igłę należy wprowadzać z sposób zdecydowany, na całą jej długość, prostopadle do powierzchni skóry.
Iniekcje należy podawać naprzemiennie po lewej i prawej stronie ciała.
Nadwrażliwość na lanreotyd, somatostatynę, pochodne peptydy lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Lanreotyd może hamować motorykę pęcherzyka żółciowego i przyczyniać się do powstawania kamieni żółciowych. Dlatego też pacjenci mogą wymagać okresowego monitorowania. Istnieją doniesienia po wprowadzeniu produktu leczniczego do obrotu o tworzeniu się kamieni żółciowych u pacjentów przyjmujących lanreotyd, prowadzącym do powikłań, w tym zapalenia pęcherzyka żółciowego, zapalenia dróg żółciowych oraz zapalenia trzustki, wymagającym cholecystektomii. W razie podejrzenia wystąpienia powikłań w postaci kamicy żółciowej należy przerwać podawanie lanreotydu i zastosować odpowiednie leczenie.
Badania farmakologiczne przeprowadzone na zwierzętach i u ludzi wykazały, że lanreotyd, podobnie jak somatostatyna i jej analogi, może hamować wydzielanie insuliny i glukagonu. W związku z tym u pacjentów leczonych lanreotydem może wystąpić hipoglikemia lub hiperglikemia Rozpoczynając leczenie lanreotydem lub przy zmianie dawki należy kontrolować stężenie glukozy we krwi,
a u pacjentów chorych na cukrzycę należy odpowiednio zmodyfikować leczenie przeciwcukrzycowe.
W trakcie leczenia lanreotydem pacjentów z akromegalią obserwowano niewielkie zahamowanie czynności tarczycy, chociaż wystąpienie klinicznej niedoczynności tarczycy jest rzadkie (<1%). O ile jest to wskazanie kliniczne, należy przeprowadzić testy oceniające czynność tarczycy.
Podczas leczenia lanreotydem u pacjentów bez wcześniejszych zaburzeń kardiologicznych może dojść do zwolnienia akcji serca, które jednak nie musi prowadzić do przekroczenia progu bradykardii.
U pacjentów z chorobą serca przed włączeniem lanreotydu może wystąpić bradykardia zatokowa. U pacjentów z bradykardią należy zachować ostrożność przy rozpoczynaniu leczenia lanreotydem (patrz punkt 4.5).
Działanie lanreotydu na układ pokarmowy może skutkować zmniejszeniem wchłaniania jelitowego innych leków stosowanych jednocześnie, w tym cyklosporyny. Jednoczesne podawanie cyklosporyny i lanreotydu może zmniejszać względną biodostępność cyklosporyny i dlatego może wymagać dostosowania dawki cyklosporyny dla zachowania jej stężeń terapeutycznych.
Interakcje z lekami silnie wiążącymi się z białkami osocza są mało prawdopodobne ze względu na umiarkowane wiązanie się lanreotydu z białkami surowicy.
Ograniczone dane z literatury wskazują, że jednoczesne podawanie analogów somatostatyny i bromokryptyny może zwiększać dostępność bromokryptyny.
Jednoczesne podawanie leków indukujących bradykardię (np. beta-adrenolityków) może wywierać addytywny efekt na czynność serca, nieco zwolnioną wskutek podawania lanreotydu. Konieczne może być dostosowanie dawki tych leków przy ich jednoczesnym stosowaniu.
Ograniczone dane z literatury wskazują, że analogi somatostatyny mogą zmniejszać klirens metaboliczny związków metabolizowanych przez enzymy cytochromu P450, co może wynikać z supresji hormonu wzrostu. Ponieważ nie można wykluczyć, że lanreotyd może wywierać takie
działanie, inne leki o wąskim indeksie terapeutycznym, metabolizowane głównie przez CYP3A4 (np. chinidyna, terfenadyna) należy stosować z zachowaniem ostrożności.
Ciąża
Badania na zwierzętach nie wykazały działania teratogennego w okresie organogenezy, związanego ze stosowaniem lanreotydu.
Dane pochodzące z obserwacji ograniczonej liczby ciężarnych z ekspozycją na lanreotyd nie wykazały niekorzystnego działania lanreotydu na przebieg ciąży oraz zdrowie płodu i (lub) noworodka.
Dotychczas nie uzyskano innych, odpowiednich danych epidemiologicznych.
Ponieważ badania na zwierzętach nie zawsze pozwalają przewidzieć odpowiedź na produkt leczniczy u ludzi, lanreotyd można podawać kobietom w ciąży tylko w przypadku wyraźnych wskazań.
Karmienie piersią
Nie wiadomo, czy lanreotyd przenika do mleka kobiet karmiących piersią.
Ponieważ wiele produktów leczniczych przenika do mleka ludzkiego, należy zachować ostrożność przy podawaniu lanreotydu kobietom karmiącym piersią.
Płodność
U samic szczura przy podawaniu dawek przekraczających dawki uzyskiwane u ludzi przy dawkach terapeutycznych obserwowano obniżoną płodność, spowodowaną zahamowania wydzielania GH.
Produkt leczniczy Myrelez wywiera niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania urządzeń mechanicznych. Nie przeprowadzono badań oceniających wpływ produktu leczniczego na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Jednak donoszono o zawrotach głowy podczas stosowania produktu leczniczego Myrelez (patrz punkt 4.8). Jeżeli takie działanie niepożądane wystąpi u pacjenta, nie powinien on prowadzić pojazdów ani obsługiwać maszyn.
Działania niepożądane, zgłaszane w badaniach klinicznych u pacjentów z akromegalią oraz guzami neuroendokrynnymi (GEP-NET), leczonych lanreotydem, wymieniono w tabeli z podziałem na odpowiednie układy i narządy, zgodnie z następującą klasyfikacją:
bardzo często (≥ 1/10); często (od ≥ 1/100 do <1/10), niezbyt często (od ≥ 1/1 000 do <1/100).
Działania niepożądane występujące najczęściej podczas leczenia lanreotydem to: zaburzenia ze strony przewodu pokarmowego (najczęściej zgłaszano przemijającą biegunkę i ból brzucha, zwykle
o nasileniu łagodnym do umiarkowanego), kamica żółciowa (często bezobjawowa) oraz reakcje w miejscu podania (ból, guzek lub stwardnienie).
Profil działań niepożądanych jest podobny jak przy stosowaniu tego leku we wszystkich wskazaniach.
Klasyfikacja układów i narządów | Bardzo często (≥1/10) | Często (od ≥1/100 do <1/10) | Niezbyt często (od ≥1/1 000 do <1/100) | Doświadczenie dotyczące bezpieczeństwa po wprowadzeniu leku do obrotu (częstość nieznana) |
Zakażenia i zakażenia pasożytnicze | Ropień w miejscu wstrzyknięcia | |||
Zaburzenia metabolizmu i odżywania | Hipoglikemia, zmniejszenie apetytu**, hiperglikemia, cukrzyca | |||
Zaburzenia psychiczne | Bezsenność* | |||
Zaburzenia układu nerwowego | Zawroty głowy, ból głowy, ospałość** | |||
Zaburzenia serca | Bradykardia zatokowa | |||
Zaburzenia naczyniowe | Uderzenia gorąca* | |||
Zaburzenia żołądka i jelit | Biegunka, wolne stolce*, ból brzucha | Nudności, wymioty, zaparcia, wzdęcia, uczucie pełności, uczucie dyskomfortu w brzuchu, dyspepsja, biegunka tłuszczowa** | Nieprawidłowe zabarwienie stolca* | Zapalenie trzustki |
Zaburzenia wątroby i dróg żółciowych | Kamica żółciowa | Poszerzenie przewodów żółciowych* | Zapalenie pęcherzyka żółciowego i dróg żółciowych | |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Ból mięśniowo-szkieletowy**, ból mięśniowy** | |||
Zaburzenia skóry i tkanki podskórnej | Łysienie, hipotrychoza* | |||
Zaburzenia ogólne i stany w miejscu podania | Astenia, zmęczenie, reakcje w miejscu podania (ból, zgrubienie, stwardnienie, guzek, świąd) | |||
Badania diagnostyczne | Wzrost aktywności AlAT*, nieprawidłowa aktywność AspAT*, nieprawidłowa aktywność AlAT*, wzrost stężenia bilirubiny we krwi*, wzrost stężenia glukozy we krwi*, wzrost stężenia hemoglobiny glikozylowanej*, zmniejszenie masy ciała, zmniejszenie aktywności enzymów trzustkowych** | Wzrost aktywności AspAT*, wzrost aktywności fosfatazy zasadowej we krwi*, nieprawidłowe stężenie bilirubiny we krwi*, spadek stężenia sodu we krwi* | ||
Zaburzenia układu immunologicznego | Reakcje alergiczne (w tym obrzęk naczynioruchowy, anafilaksja, nadwrażliwość) |
* na podstawie badań przeprowadzonych z udziałem pacjentów z akromegalią
** na podstawie badań przeprowadzonych z udziałem pacjentów z guzami GEP-NET
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: +48 22 49 21 301
Faks: +48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W przypadku przedawkowania zaleca się leczenie objawowe.
Grupa farmakoterapeutyczna: hormony podwzgórza; somatostatyna i analogi, kod ATC: H01C B03 Lanreotyd jest oktapeptydem, analogiem naturalnie występującej w organizmie somatostatyny.
Podobnie jak somatostatyna, lanreotyd jest inhibitorem różnych funkcji endokrynnych, neuroendokrynnych, egzokrynnych i parakrynnych Wykazuje wysokie powinowactwo do ludzkich receptorów somatostatyny (SSTR) 2 i 5, oraz obniżone powinowactwo do ludzkich receptorów SSTR 1, 3 i 4. Uważa się, że działanie produktu leczniczego na receptory SSTR 2 i 5 jest głównym mechanizmem odpowiedzialnym za zahamowanie wydzielania GH. Lanreotyd wykazuje wyższą aktywność niż naturalna somatostatyna oraz cechuje się dłuższym czasem działania.
Lanreotyd, podobnie jak somatostatyna, wykazuje ogólne działanie hamujące czynność zewnątrzwydzielniczą. Hamuje podstawowe wydzielanie motyliny, GIP (żołądkowego peptydu hamującego) oraz polipeptydu trzustkowego, natomiast nie ma istotnego wpływu na wydzielanie sekretyny i gastryny na czczo. Lanreotyd zmniejsza także stężenie chromograniny A w osoczu
i 5-HIAA (kwas 5-hydroksyindolooctowy) w moczu u pacjentów z guzami GEP-NET i podwyższonym stężeniem tych markerów nowotworowych. Lanreotyd wyraźnie hamuje poposiłkowy wzrost przepływu w tętnicy krezkowej górnej i żyle wrotnej. Lanreotyd znacznie obniża stymulowane prostaglandyną E1 wydzielanie wody, sodu, potasu i chlorków do światła jelita czczego. Lanreotyd obniża stężenie prolaktyny u przewlekle leczonych pacjentów chorych na akromegalię.
W badaniu otwartym podawano lanreotyd w dawce 120 mg co 28 dni przez okres 48 tygodni grupie 90 nieleczonych wcześniej pacjentów z akromegalią ze zdiagnozowanym makrogruczolakiem przysadki Z badania wykluczeni zostali pacjenci, u których planowano przeprowadzenie operacji przysadki lub radioterapii w okresie trwania badania.
Zmniejszenie objętości guza o ≥20% obserwowano u 63% pacjentów (95% CI: 52% - 73%).
W 48. tygodniu średnie procentowe zmniejszenie objętości guza wyniosło 26,8%, poziomy GH były poniżej 2,5 µg/l u 77,8% pacjentów, a poziomy IGF-1 znormalizowane u 50%. Znormalizowane poziomy IGF-1 w połączeniu z poziomami GH poniżej 2,5 µg/l obserwowano u 43,5% pacjentów. Większość pacjentów zgłaszała wyraźne ustąpienie objawów akromegalii, takich jak zmęczenie, nadmierne pocenie się, bóle stawów i obrzęk tkanek miękkich. Zarówno wczesne, jak i trwałe zmniejszenie objętości guza oraz poziomów GH i IGF-1 obserwowano od 12. tygodnia.
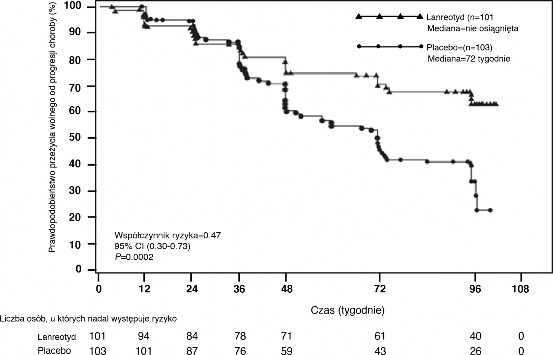
96-tygodniowe, o wyznaczonym czasie trwania, randomizowane, podwójnie zaślepione, wieloośrodkowe, kontrolowane placebo badanie kliniczne fazy III z zastosowaniem lanreotydu przeprowadzono z udziałem pacjentów z guzami neuroendokrynnymi żołądkowo-jelitowo- trzustkowymi w celu oceny antyproliferacyjnego działania lanreotydu.
Pacjentów zrandomizowano w stosunku 1:1 do grupy przyjmującej lanreotyd 120 mg co 28 dni (n=101) lub do grupy placebo (n=103). Randomizację stratyfikowano w zależności od uprzedniego leczenia w chwili przystąpienia do badania oraz obecności/braku progresji w chwili rozpoczęcia badania wg kryteriów RECIST 1.0 (Response Evaluation Criteria in Solid Tumours) podczas fazy przesiewowej trwającej od 3 do 6 miesięcy.
U pacjentów występowały przerzuty oraz (lub) miejscowo zaawansowane, nieoperacyjne guzy o histologicznie potwierdzonym wysokim lub umiarkowanie wysokim stopniu zróżnicowania,
umiejscowione przede wszystkim w trzustce (44,6% pacjentów), środkowej części prajelita (35,8% pacjentów), końcowej części prajelita (6,9% pacjentów) lub w innej/nieznanej lokalizacji pierwotnej (12,7% pacjentów).
U 69% pacjentów z guzami GEP-NET stwierdzono 1. stopień zaawansowania choroby (G1), zdefiniowany jako indeks proliferacyjny Ki67 ≤ 2% (50,5% całkowitej populacji pacjentów) lub indeks mitotyczny <2 mitozy/10 HPF (18,5% całkowitej populacji pacjentów), a u 30% pacjentów stopień zaawansowania guzów GEP-NET był niższy i mieścił się w dolnym zakresie 2. stopnia (G2) (zdefiniowany jako indeks Ki67 > 2% - ≤ 10%). W przypadku 1% pacjentów informacja dotycząca stopnia zaawansowania guza była niedostępna. Do badania nie włączono pacjentów z guzami GEP-
NET G2 z wyższym indeksem proliferacji komórkowej (Ki 67 >10% - ≤ 20%) oraz pacjentów z guzami neuroendokrynnymi GEP G3 (indeks Ki 67 > 20%).
Ogółem, u 52,5% pacjentów stopień zajęcia wątroby wynosił ≤10%, u 14,5% > 10 i ≤25%, a u 33% pacjentów wynosił on >25%.
Pierwszorzędowym punktem końcowym był czas wolny od progresji (PFS) zdefiniowany jako czas do wystąpienia progresji wg kryteriów RECIST 1.0 lub zgonu w okresie 96 tygodni od pierwszego podania produktu leczniczego. Analiza PFS opierała się na niezależnej, radiologicznej ocenie progresji przeprowadzonej centralnie.
Tabela 1: Dane skuteczności z badania fazy III
Mediana czasu wolnego od progresji (tygodnie) | Współczynnik ryzyka (95% CI) | Zmniejszenie ryzyka progresji lub zgonu | Wartość p | |
lanreotyd (n=101) | Placebo (n=103) | |||
> 96 tygodni | 72,00 tygodnie (95% CI: 48,57, 96,00) | 0,470 (0,304, 0,729) | 53% | 0,0002 |
Rycina 1: Estymatory Kaplana-Meiera dla czasu do progresji guza
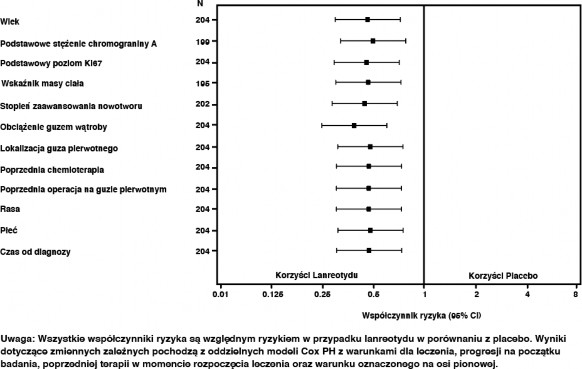
Korzystne działanie lanreotydu w zmniejszeniu ryzyka progresji lub zgonu było spójne niezależnie od umiejscowienia guza pierwotnego, stopnia zajęcia wątroby, uprzedniej chemioterapii, wyjściowej wartości Ki67, stopnia zaawansowania guza lub innych parametrów przedstawionych na Ryc. 2.
Klinicznie istotne korzyści z leczenia lanreotydem obserwowano u pacjentów z guzami trzustki, środkowej części prajelita i innym/nieznanym ogniskiem pierwotnym, jak i w całej populacji badania. Ograniczona liczba pacjentów z guzami końcowej części prajelita (14/204) przyczyniała się do trudności w interpretacji wyników w tej podgrupie. Dostępne dane wskazują na brak korzyści ze stosowania lanreotydu u tych pacjentów
Rycina 2: Wyniki analizy PFS metodą z zastosowaniem modelu proporcjonalnego hazardu Coxa
Zmiana terapii z placebo na lanreotyd w otwartym przedłużeniu badania miała miejsce u 45,6% (47/103) pacjentów
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego lanreotyd we wszystkich podgrupach populacji dzieci i młodzieży
w leczeniu akromegalii i gigantyzmu przysadkowego (stosowanie u dzieci i młodzieży, patrz punkt 4.2). Europejska Agencja Leków wymieniła guzy neuroendokrynne żołądkowo-jelitowo-trzustkowe (z wyłączeniem nerwiaka płodowego, nerwiaka płodowego zwojowego i guza chromochłonnego nadnerczy) w wykazie klas objętych zwolnieniem.
Po dożylnym podaniu lanreotydu zdrowym ochotnikom obserwuje się ograniczoną dystrybucję pozanaczyniową, przy czym objętość dystrybucji w stanie równowagi wynosiła 16,1 l. Całkowity klirens wynosił 23,7 l/h, końcowy okres półtrwania – 1,14 godziny, a średni czas pozostawania lanreotydu w organizmie – 0,68 godziny.
Badania oceniające wydalanie wskazują, że mniej niż 5% lanreotydu wydalane jest z moczem i mniej niż 0,5% w kale, co wskazuje na niewielkie wydalanie z żółcią.
Po głębokim podskórnym podaniu lanreotydu 60, 90 i 120 mg zdrowym ochotnikom stężenia lanreotydu wzrosły, osiągając przeciętne maksymalne stężenia w surowicy wynoszące odpowiednio 4,25, 8,39 i 6,79 ng/ml. Takie wartości Cmax osiągnięto w pierwszym dniu po podaniu leku po
8, 12 i 7 godzinach (wartości średnie). Od szczytowych stężeń lanreotydu w surowicy jego stężenia zmniejszają się powoli według kinetyki pierwszego rzędu, z połowiczym okresem końcowej eliminacji, wynoszącym odpowiednio 23,3, 27,4 i 30,1 dni. 4 tygodnie po podaniu produktu średnie stężenia lanreotydu w surowicy wynosiły odpowiednio 0,9, 1,11 i 1,69 ng/ml. Bezwzględna biodostępność wynosiła odpowiednio 73,4, 69,0 i 78,4%.
Po głębokim podskórnym podaniu lanreotydu 60, 90 i 120 mg zdrowym ochotnikom stężenia lanreotydu wzrosły, osiągając przeciętne maksymalne stężenia w surowicy wynoszące odpowiednio 1,6, 3,5 i 3,1 ng/ml. Takie wartości Cmax osiągnięto w pierwszym dniu po podaniu leku po
6, 6 i 24 godzinach. Od szczytowych stężeń lanreotydu w surowicy jego stężenia zmniejszają się powoli według kinetyki pierwszego rzędu. 4 tygodnie po podaniu średnie stężenia lanreotydu
w surowicy wynosiły odpowiednio 0,7, 1,0 i 1,4 ng/ml.
Stabilne stężenie lanreotydu zostaje osiągnięte przeciętnie po podaniu 4 iniekcji co 4 tygodnie. Po podaniu powtarzanych dawek co 4 tygodnie przeciętne wartości Cmax w stanie stabilnym wynosiły 3,8, 5,7 i 7,7 ng/ml dla odpowiednio 60, 90 i 120 mg, przeciętne uzyskane wartości Cmin wynosiły 1,8, 2,5 i 3,8 ng/ml. Wskaźnik fluktuacji “peak trough” był umiarkowany i mieścił się w zakresie od 81 do 108%.
Obserwowano liniową kinetykę uwalniania lanreotydu po głębokim podskórnym podaniu produktu lanreotydu 60, 90, 120 mg u pacjentów z akromegalią.
Najniższe stężenie lanreotydu w surowicy uzyskane po trzech głębokich podskórnych podaniach lanreotydu w dawce 60 mg, 90 mg, 120 mg, podawanych co 28 dni, jest podobne do najniższego stężenia lanreotydu uzyskanego u chorych na akromegalię wcześniej leczonych domięśniowym podaniem lanreotydu 30 mg w postaci mikrocząsteczek o przedłużonym uwalnianiu odpowiednio co 14, 10, 7 dni.
W populacyjnej analizie farmakokinetycznej 290 pacjentów z guzami GEP-NET przyjmujących lanreotyd w dawce 120 mg obserwowano początkowe szybkie uwalnianie ze średnimi wartościami Cmax wynoszącymi 7,49 ± 7,58 ng/ml już w pierwszym dniu po wykonaniu jednej iniekcji. Stężenia w fazie stacjonarnej były osiągane po wykonaniu 5 iniekcji lanreotydu 120 mg co 28 dni
i utrzymywały się aż do końcowej oceny (maksymalnie 96 tygodni po pierwszej iniekcji). W stanie stacjonarnym średnie wartości Cmax wynosiły 13,9 ± 7,44 ng/ml, a średnie minimalne stężenia
w surowicy wynosiły 6,56 ± 1,99 ng/ml. Średni pozorny okres półtrwania wynosił 49,8 ± 28,0 dni.
Niewydolność nerek i (lub) wątroby
U osób z ciężką niewydolnością nerek stwierdza się około 2-krotny spadek całkowitego klirensu lanreotydu w surowicy, a w konsekwencji wzrost okresu półtrwania i AUC. U osób z umiarkowaną lub ciężką niewydolnością wątroby obserwuje się redukcję klirensu (30%). U osób z niewydolnością wątroby o wszystkich stopniach zaawansowania wzrasta objętość dystrybucji i średni czas obecności leku w organizmie.
Nie zaobserwowano wpływu na klirens lanreotydu w populacyjnej analizie farmakokinetycznej pacjentów z guzami GEP-NET, w tym 165 osób z łagodnymi i umiarkowanymi zaburzeniami czynności nerek (odpowiednio 106 i 59) leczonych lanreotydem. Nie badano pacjentów z guzami GEP-NET z ciężką niewydolnością nerek.
Nie badano pacjentów z guzami GEP-NET z niewydolnością wątroby (wg klasyfikacji Child-Pugh).
U pacjentów z niewydolnością nerek lub wątroby nie jest konieczna zmiana dawki początkowej, gdyż oczekiwane w tych populacjach stężenia lanreotydu w surowicy mieszczą się w zakresie bezpiecznie tolerowanym przez osoby zdrowe.
Pacjenci w podeszłym wieku
U osób w podeszłym wieku stwierdza się wydłużenie okresu półtrwania i średniego czasu obecności leku w organizmie, w porównaniu do młodych, zdrowych osób. U pacjentów w podeszłym wieku nie jest konieczna zmiana dawki początkowej, gdyż oczekiwane w tych populacjach stężenia lanreotydu w surowicy mieszczą się w zakresie bezpiecznie tolerowanym przez osoby zdrowe.
Nie zaobserwowano wpływu wieku na klirens i objętość dystrybucji lanreotydu w populacyjnej analizie farmakokinetycznej pacjentów z guzami GEP-NET, w tym 122 osób w wieku 65–85 lat.
W badaniach nieklinicznych działanie toksyczne obserwowano jedynie w przypadku narażenia przekraczającego maksymalną ekspozycję u człowieka, co wskazuje na niewielkie znaczenie tych obserwacji w praktyce klinicznej.
W biologicznych badaniach karcinogenności przeprowadzanych na szczurach i myszach przy dawkach przekraczających dawki uzyskiwanych przy dawkach leczniczych u ludzi nie obserwowano zmian nowotworowych. Zaobserwowano zwiększoną częstość występowania guzów podskórnych
w miejscach iniekcji, prawdopodobnie wskutek zwiększonej częstości dawkowania u zwierząt (codziennie), w porównaniu do dawkowania co miesiąc u ludzi; stąd obserwacja ta może nie być istotna klinicznie.
W standardowych zestawach testów in vitro i in vivo nie stwierdzono potencjału genotoksycznego lanreotydu.
Woda do wstrzykiwań
Kwas octowy lodowaty (do ustalenia pH)
Nie dotyczy.
Przechowywać w lodówce w temperaturze 2 – 8 °C oryginalnym opakowaniu w celu ochrony przed światłem.
Produkt można przechowywać w lodówce do późniejszego użycia pod warunkiem, że był przechowywany w zamkniętej torebce w maksymalnej temperaturze 40 °C nie dłużej niż 24 godziny (liczba takich wahań temperatury nie może przekraczać trzech).
Produkt leczniczy Myrelez jest dostarczany w ampułko-strzykawce (polipropylen z końcówką tłoka z termoplastycznej gumy elastomerowej z nasadką polipropylenową) umieszczoną na plastikowej tacce i zamkniętej w torebce aluminiowej wraz z zapakowanym oddzielnie automatycznym urządzeniem zabezpieczającym igłę do jednorazowego użytku. Ampułko-strzykawka i urządzenie są zapakowane w pudełko tekturowe.
Pudełko zawierające jedną strzykawkę o pojemności 0,5 ml z jedną bezpieczną igłą (1,2 mm x 20 mm) zapakowane razem.
Opakowanie zbiorcze z 3 pudełkami, z których każde zawiera jedną strzykawkę o pojemności 0,5 ml z jedną bezpieczną igłą (1,2 mm x 20 mm) zapakowane razem.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Roztwór do wstrzykiwań w ampułko-strzykawce jest gotowy do użycia.
Do natychmiastowego jednorazowego podania po pierwszym otwarciu opakowania. Nie używać, jeśli torebka zostało otwarte lub uszkodzone.
Ważne, aby iniekcje produktu leczniczego wykonywać dokładnie według instrukcji podanych w ulotce informacyjnej.
Zużyte urządzenie do wstrzykiwań należy wyrzucić do przeznaczonego na nie pojemnika na ostre odpady medyczne.
Wszelkie niewykorzystane resztki produktu lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Amdipharm Limited
Dublin 4, Irlandia
Amdipharm Limited wchodzi w skład grupy ADVANZ PHARMA.
Myrelez 60 mg – pozwolenie nr Myrelez, 90 mg – pozwolenie nr Myrelez, 120 mg – pozwolenie nr
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:







