







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
nadwrażliwością na substancje czynne lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1 (patrz punkty 4.4 i 4.8);
każdym rodzajem ostrej kwasicy metabolicznej (takiej jak kwasica mleczanowa, cukrzycowa kwasica ketonowa);
cukrzycowym stanem przedśpiączkowym;
ciężką niewydolnością nerek (GFR < 30 ml/min) (patrz punkt 4.4);
ostrymi stanami mogącymi zmieniać czynność nerek, jak na przykład:
odwodnienie,
ciężkie zakażenie,
wstrząs,
donaczyniowe podanie środków kontrastowych zawierających jod (patrz punkt 4.4);
ostrą lub przewlekłą chorobą, która może spowodować niedotlenienie tkanek, taką jak:
niewydolność serca lub układu oddechowego,
niedawno przebyty zawał mięśnia sercowego,
wstrząs;
zaburzeniami czynności wątroby;
ostrym zatruciem alkoholowym, alkoholizmem;
karmiących piersią.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
zmniejszając wytwarzanie glukozy w wątrobie poprzez hamowanie glukoneogenezy i glikogenolizy;
umiarkowanie zwiększając wrażliwość na insulinę w tkance mięśniowej, co poprawia obwodowy wychwyt glukozy i jej zużycie;
opóźniając wchłanianie glukozy w jelitach.
Metformina pobudza wewnątrzkomórkową syntezę glikogenu, działając na syntazę glikogenową. Metformina zwiększa zdolności transportowe określonych rodzajów transporterów glukozy w błonie komórkowej (GLUT-1 i GLUT-4).
Skuteczność kliniczna i bezpieczeństwo stosowania
U ludzi, niezależnie od wpływu na glikemię, metformina korzystnie wpływa na metabolizm lipidów. W kontrolowanych średnio- i długoterminowych badaniach klinicznych wykazano, że metformina w dawkach terapeutycznych zmniejsza stężenie cholesterolu całkowitego, cholesterolu frakcji
LDL oraz trójglicerydów.
W prospektywnym badaniu z randomizacją (UKPDS) stwierdzono długotrwałe korzyści intensywnej kontroli glikemii w cukrzycy typu 2. Analiza wyników uzyskanych u pacjentów z nadwagą leczonych metforminą, po nieskutecznym leczeniu samą dietą, wykazała:
istotne zmniejszenie bezwzględnego ryzyka powikłań związanych z cukrzycą w grupie leczonej metforminą (29,8 zdarzeń/1000 pacjentolat) w porównaniu z grupą leczoną samą dietą
(43,3 zdarzeń /1000 pacjentolat), p=0,0023, oraz w porównaniu z łącznymi wynikami dla grup pacjentów leczonych pochodnymi sulfonylomocznika i insuliną w monoterapii (40,1 zdarzeń
/1000 pacjentolat), p=0,0034;
istotne zmniejszenie bezwzględnego ryzyka śmiertelności związanej z cukrzycą: metformina 7,5 zdarzeń /1000 pacjentolat, sama dieta 12,7 zdarzeń /1000 pacjentolat, p=0,017;
istotne zmniejszenie bezwzględnego ryzyka śmiertelności ogólnej: metformina 13,5 zdarzeń
/1000 pacjentolat w porównaniu z samą dietą: 20,6 zdarzeń /1000 pacjentolat, (p=0,011) oraz w porównaniu z łącznymi wynikami dla grup pacjentów leczonych pochodnymi sulfonylomocznika i insuliną w monoterapii 18,9 zdarzeń /1000 pacjentolat (p=0,021);
istotne zmniejszenie bezwzględnego ryzyka zawału mięśnia sercowego: metformina 11 zdarzeń
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Metformax Combi, 50 mg + 850 mg, tabletki powlekane Metformax Combi, 50 mg + 1000 mg, tabletki powlekane
Metformax Combi, 50 mg + 850 mg, tabletki powlekane
Każda tabletka zawiera sytagliptyny chlorowodorek jednowodny, co odpowiada 50 mg sytagliptyny i 850 mg metforminy chlorowodorku.
Substancja pomocnicza o znanym działaniu:
Każda tabletka zawiera 13,02 mg laktozy jednowodnej.
Metformax Combi, 50 mg + 1000 mg, tabletki powlekane
Każda tabletka zawiera sytagliptyny chlorowodorek jednowodny, co odpowiada 50 mg sytagliptyny i 1000 mg metforminy chlorowodorku.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana
Metformax Combi, 50 mg + 850 mg to różowe, owalne, obustronnie wypukłe, tabletki powlekane o wymiarach około 20,5 mm x 9,5 mm, z wytłoczeniem “S476” po jednej stronie.
Metformax Combi, 50 mg + 1000 mg to brązowe, owalne, obustronnie wypukłe, tabletki powlekane o wymiarach około 21,5 mm x 10,0 mm, z wytłoczeniem “S477” po jednej stronie.
U dorosłych pacjentów z cukrzycą typu 2:
Metformax Combi wskazany jest do stosowania, oprócz diety i ćwiczeń fizycznych, w celu poprawy kontroli glikemii u pacjentów z glikemią niedostatecznie wyrównaną podczas stosowania maksymalnej tolerowanej dawki metforminy w monoterapii, lub u pacjentów już leczonych sytagliptyną
w skojarzeniu z metforminą.
Metformax Combi wskazany jest do stosowania w skojarzeniu z pochodną sulfonylomocznika (tj. w leczeniu potrójnie skojarzonym), oprócz diety i ćwiczeń fizycznych, u pacjentów z glikemią niedostatecznie wyrównaną podczas stosowania maksymalnej tolerowanej dawki metforminy i pochodnej sulfonylomocznika.
Metformax Combi jest wskazany w leczeniu potrójnie skojarzonym z agonistą receptora aktywowanego przez proliferatory peroksysomów typu gamma (PPAR) (np. tiazolidynedionem) oprócz diety i ćwiczeń
fizycznych, u pacjentów z glikemią niedostatecznie wyrównaną podczas stosowania maksymalnej tolerowanej dawki metforminy i agonisty receptora PPAR.
Metformax Combi jest także wskazany do stosowania, jako lek uzupełniający podanie insuliny (tj. w leczeniu potrójnie skojarzonym), pomocniczo wraz z dietą i ćwiczeniami fizycznymi w celu poprawy kontroli glikemii u pacjentów, u których stosowanie stałej dawki insuliny i metforminy nie zapewnia odpowiedniej kontroli glikemii.
Dawkowanie
Dawkę przeciwcukrzycowego produktu leczniczego Metformax Combi, należy każdorazowo dostosować na podstawie aktualnego schematu leczenia oraz skuteczności i tolerancji, przy czym nie należy przekraczać maksymalnej zalecanej dawki dobowej wynoszącej 100 mg sytagliptyny.
Dorośli z prawidłową czynnością nerek (GFR ≥ 90 ml/min)
Pacjenci z niedostatecznie wyrównaną glikemią, stosujący maksymalne tolerowane dawki metforminy w monoterapii
U pacjentów z glikemią niedostatecznie wyrównaną podczas stosowania metforminy w monoterapii, zwykle stosowana dawka początkowa powinna zapewniać dostarczanie 50 mg sytagliptyny dwa razy na dobę (całkowita dawka dobowa 100 mg) oraz przyjmowanej już dawki metforminy.
Pacjenci już przyjmujący jednocześnie sytagliptynę i metforminę
W przypadku pacjentów przyjmujących jednocześnie sytagliptynę i metforminę, Metformax Combi powinien być włączany do leczenia w dawce odpowiadającej przyjmowanym już dawkom sytagliptyny i metforminy.
Pacjenci z niedostatecznie wyrównaną glikemią podczas skojarzonego leczenia maksymalną tolerowaną dawką metforminy i pochodną sulfonylomocznika
Dawka powinna zapewniać dostarczanie 50 mg sytagliptyny dwa razy na dobę (całkowita dawka dobowa 100 mg) oraz dawki metforminy podobnej do dawki przyjmowanej poprzednio. W przypadku stosowania produktu leczniczego Metformax Combi w skojarzeniu z pochodną sulfonylomocznika, może być konieczne obniżenie dawki pochodnej sulfonylomocznika w celu zmniejszenia ryzyka hipoglikemii (patrz punkt 4.4).
Pacjenci z niedostatecznie wyrównaną glikemią podczas skojarzonego leczenia maksymalną tolerowaną dawką metforminy i agonisty receptora PPAR
Dawka powinna zapewniać dostarczanie 50 mg sytagliptyny dwa razy na dobę (całkowita dawka dobowa 100 mg) oraz dawki metforminy podobnej do dawki przyjmowanej poprzednio.
Pacjenci z niedostatecznie wyrównaną glikemią podczas skojarzonego leczenia insuliną i maksymalną tolerowaną dawką metforminy
Dawka powinna zapewniać dostarczanie 50 mg sytagliptyny dwa razy na dobę (całkowita dawka dobowa 100 mg) oraz dawki metforminy podobnej do dawki przyjmowanej poprzednio. W przypadku stosowania produktu leczniczego Metformax Combi w skojarzeniu z insuliną, może być konieczne obniżenie dawki insuliny w celu zmniejszenia ryzyka hipoglikemii (patrz punkt 4.4).
Dla różnych dawek metforminy, Metformax Combi jest dostępny w tabletkach zawierających 50 mg sytagliptyny oraz 850 mg metforminy chlorowodorku lub 1000 mg metforminy chlorowodorku.
Wszyscy pacjenci powinni kontynuować zalecaną dietę o odpowiednim rozkładzie spożycia węglowodanów w ciągu dnia.
Szczególne populacje
Zaburzenia czynności nerek
Nie ma konieczności dostosowania dawki u pacjentów z łagodnymi zaburzeniami czynności nerek (współczynnik przesączania kłębuszkowego [ang. GFR] ≥ 60 ml/min). Wartość GFR należy oznaczyć przed rozpoczęciem leczenia produktem leczniczym zawierającym metforminę, a następnie co najmniej raz na rok. U pacjentów ze zwiększonym ryzykiem dalszego pogorszenia czynności nerek oraz u pacjentów w podeszłym wieku czynność nerek należy oceniać częściej, np. co 3–6 miesięcy.
Maksymalna dawka dobowa metforminy powinna być optymalnie podzielona na 2 lub 3 dawki na dobę. Przed rozważeniem rozpoczęcia leczenia metforminą u pacjentów z wartością GFR < 60 ml/min, należy przeanalizować czynniki mogące zwiększyć ryzyko kwasicy mleczanowej (patrz punkt 4.4).
Jeśli produkt leczniczy Metformax Combi o odpowiedniej mocy jest niedostępny należy zastosować jego poszczególne składniki osobno zamiast produktu złożonego o ustalonej dawce.
GFR ml/min | Metformina | Sytagliptyna |
60-89 | Maksymalna dawka dobowa wynosi 3000 mg. Można rozważyć zmniejszenie dawki w reakcji na pogarszającą się czynność nerek. | Maksymalna dawka dobowa wynosi 100 mg. |
45-59 | Maksymalna dawka dobowa wynosi 2000 mg. Dawka początkowa nie jest większa niż połowa dawki maksymalnej. | Maksymalna dawka dobowa wynosi 100 mg. |
30-44 | Maksymalna dawka dobowa wynosi 1000 mg. Dawka początkowa nie jest większa niż połowa dawki maksymalnej. | Maksymalna dawka dobowa wynosi 50 mg. |
< 30 | Metformina jest przeciwwskazana. | Maksymalna dawka dobowa wynosi 25 mg. |
Zaburzenia czynności wątroby
Nie wolno stosować produktu leczniczego Metformax Combi u pacjentów z zaburzeniami czynności wątroby (patrz punkt 5.2).
Pacjenci w podeszłym wieku
Ponieważ metformina i sytagliptyna są wydalane przez nerki, należy zachować ostrożność podczas stosowania produktu leczniczego Metformax Combi u starszych pacjentów. Pomocniczo w zapobieganiu związanej z przyjmowaniem metforminy kwasicy mleczanowej, zwłaszcza u osób w podeszłym wieku, konieczne jest monitorowanie czynności nerek (patrz punkty 4.3 i 4.4).
Dzieci i młodzież
Produktu leczniczego Metformax Combi nie należy stosować u dzieci i młodzieży w wieku od 10 do 17 lat ze względu na niewystarczającą skuteczność. Aktualne dane przedstawiono w punktach 4.8, 5.1 i 5.2. Nie przeprowadzono badań dotyczących stosowania produktu leczniczego Metformax Combi u dzieci w wieku poniżej 10 lat.
Sposób podawania
Metformax Combi należy przyjmować dwa razy na dobę podczas posiłku w celu ograniczenia działań niepożądanych ze strony przewodu pokarmowego, związanych z przyjmowaniem metforminy.
Metformax Combi przeciwwskazany jest u pacjentów z:
Ogólne
Nie należy stosować produktu leczniczego Metformax Combi u pacjentów z cukrzycą typu 1 i nie wolno go stosować w leczeniu kwasicy ketonowej w przebiegu cukrzycy.
Ostre zapalenie trzustki
Stosowanie inhibitorów DPP-4 wiąże się z ryzykiem wystąpienia ostrego zapalenia trzustki. Należy poinformować pacjentów o charakterystycznym objawie ostrego zapalenia trzustki: uporczywym, silnym bólu brzucha. Po odstawieniu sytagliptyny (z leczeniem wspomagającym lub bez) zaobserwowano ustąpienie zapalenia trzustki, ale odnotowano bardzo rzadkie przypadki martwiczego lub krwotocznego zapalenia trzustki i (lub) zgonu. W przypadku podejrzenia zapalenia trzustki należy odstawić produkt leczniczy Metformax Combi oraz inne potencjalnie budzące wątpliwości produkty lecznicze. W przypadku potwierdzenia ostrego zapalenia trzustki nie należy ponownie rozpoczynać leczenia produktem leczniczym Metformax Combi. Należy zachować ostrożność u pacjentów z zapaleniem trzustki w wywiadzie.
Kwasica mleczanowa
Kwasica mleczanowa, rzadkie ale ciężkie powikłanie metaboliczne, występuje najczęściej w ostrym pogorszeniu czynności nerek, chorobach układu krążenia lub chorobach układu oddechowego, lub posocznicy. W przypadkach nagłego pogorszenia czynności nerek dochodzi do kumulacji metforminy, co zwiększa ryzyko kwasicy mleczanowej.
W przypadku odwodnienia (ciężkie wymioty, biegunka, gorączka lub zmniejszona podaż płynów) należy tymczasowo wstrzymać stosowanie metforminy i zalecane jest zwrócenie się do lekarza.
U pacjentów leczonych metforminą należy ostrożnie rozpoczynać leczenie produktami leczniczymi, które mogą ciężko zaburzyć czynność nerek (takimi jak leki przeciwnadciśnieniowe, moczopędne lub NLPZ). Inne czynniki ryzyka kwasicy mleczanowej to nadmierne spożycie alkoholu, niewydolność wątroby, źle kontrolowana cukrzyca, ketoza, długotrwałe głodzenie i wszelkie stany związane
z niedotlenieniem, jak również jednoczesne stosowanie produktów leczniczych mogących wywołać kwasicę mleczanową (patrz punkty 4.3 i 4.5).
Pacjentów i (lub) ich opiekunów należy poinformować o ryzyku wystąpienia kwasicy mleczanowej. Kwasicę mleczanową charakteryzuje występowanie duszności kwasiczej, bólu brzucha, skurczów mięśni, astenii i hipotermii, po której następuje śpiączka. W razie wystąpienia podejrzanych objawów pacjent powinien odstawić metforminę i szukać natychmiastowej pomocy medycznej. Odchylenia od wartości prawidłowych w wynikach badań laboratoryjnych obejmują zmniejszenie wartości pH krwi (< 7,35), zwiększenie stężenia mleczanów w osoczu (> 5 mmol/l) oraz zwiększenie luki anionowej
i stosunku mleczanów do pirogronianów.
Czynność nerek
Wartość GFR powinna być oznaczona przed rozpoczęciem leczenia, a następnie w regularnych odstępach czasu (patrz punkt 4.2). Produkt leczniczy Metformax Combi jest przeciwwskazany u pacjentów z GFR < 30 ml/min i należy go tymczasowo odstawić w razie występowania stanów wpływających na czynność nerek (patrz punkt 4.3).
Hipoglikemia
U pacjentów przyjmujących Metformax Combi w skojarzeniu z pochodną sulfonylomocznika lub insuliną może wystąpić ryzyko hipoglikemii. Konieczne może być zatem zmniejszenie dawki pochodnej sulfonylomocznika lub insuliny.
Reakcje nadwrażliwości
Po wprowadzeniu produktu leczniczego do obrotu zgłaszano występowanie ciężkich reakcji nadwrażliwości u pacjentów leczonych sytagliptyną. Reakcje te obejmują anafilaksję, obrzęk naczynioruchowy oraz złuszczające choroby skóry, w tym zespół Stevensa-Johnsona. Początek tych reakcji występował w ciągu pierwszych 3 miesięcy po rozpoczęciu leczenia sytagliptyną,
a w przypadku kilku zgłoszeń po pierwszej dawce. W przypadku podejrzenia reakcji nadwrażliwości należy przerwać stosowanie produktu leczniczego Metformax Combi, ocenić czy są możliwe inne przyczyny zdarzenia oraz zastosować alternatywną metodę leczenia cukrzycy (patrz punkt 4.8).
Pemfigoid pęcherzowy
W okresie po wprowadzeniu produktu leczniczego do obrotu zgłaszano występowanie pemfigoidu pęcherzowego u pacjentów przyjmujących inhibitory DPP-4, w tym sytagliptynę. W przypadku podejrzenia pemfigoidu pęcherzowego należy przerwać stosowanie produktu leczniczego Metformax Combi.
Zabieg chirurgiczny
Podawanie produktu leczniczego Metformax Combi musi być przerwane bezpośrednio przed zabiegiem chirurgicznym w znieczuleniu ogólnym, podpajęczynówkowym lub zewnątrzoponowym. Leczenie można wznowić nie wcześniej niż po 48 godzinach po zabiegu chirurgicznym lub wznowieniu odżywiania doustnego oraz dopiero po ponownej ocenie czynności nerek i stwierdzeniu, że jest stabilna.
Podawanie środków kontrastowych zawierających jod
Donaczyniowe podanie środków kontrastowych zawierających jod może doprowadzić do nefropatii wywołanej środkiem kontrastowym, powodując kumulację metforminy i zwiększenie ryzyka kwasicy mleczanowej. Należy przerwać stosowanie produktu leczniczego Metformax Combi przed badaniem lub podczas badania obrazowego i nie stosować go przez co najmniej 48 godzin po badaniu, po czym można wznowić podawanie produktu leczniczego Metformax Combi pod warunkiem ponownej oceny czynności nerek i stwierdzeniu, że jest ona stabilna (patrz punkty 4.3 i 4.5).
Zmiana stanu klinicznego pacjentów z uprzednio kontrolowaną cukrzycą typu 2
Pacjenci z cukrzycą typu 2 uprzednio dobrze wyrównaną w wyniku stosowania produktu leczniczego Metformax Combi, u których wystąpią nieprawidłowe wyniki badań laboratoryjnych lub choroby kliniczne (zwłaszcza niesprecyzowane i słabo określone) powinni zostać niezwłocznie poddani badaniu na obecność oznak kwasicy ketonowej lub kwasicy mleczanowej. Badanie powinno obejmować stężenie elektrolitów i ketonów w surowicy, stężenie glukozy we krwi oraz, w przypadku wskazań, odczyn pH krwi i stężenia mleczanów, pirogronianów i metforminy. Jeśli wystąpi którykolwiek z wymienionych dwóch rodzajów kwasicy, należy natychmiast odstawić leczenie i zastosować odpowiednie środki zaradcze.
Metformax Combi zawiera sód:
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu na tabletkę, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
Metformax Combi, 50 mg + 850 mg zawiera laktozę:
Tabletki powlekane 50 mg + 850 mg zawierają laktozę. Produkt leczniczy nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Równoczesne podawanie dawek wielokrotnych sytagliptyny (50 mg dwa razy na dobę) i metforminy (1000 mg dwa razy na dobę) pacjentom z cukrzycą typu 2 nie zmieniało w istotny sposób farmakokinetyki sytagliptyny ani metforminy.
Nie przeprowadzono farmakokinetycznych badań dotyczących interakcji produktu leczniczego Metformax Combi z lekami, jednak badania takie przeprowadzono dla poszczególnych substancji czynnych, sytagliptyny i metforminy.
Niezalecane jednoczesne stosowanie
Alkohol
Zatrucie alkoholem związane jest ze zwiększonym ryzykiem kwasicy mleczanowej, szczególnie w przypadkach głodzenia, niedożywienia lub zaburzeń czynności wątroby.
Środki kontrastowe zawierające jod
Stosowanie produktu leczniczego Metformax Combi musi być przerwane przed badaniem lub podczas badania obrazowego; nie wolno wznawiać jego stosowania przez co najmniej 48 godzin po badaniu, po czym można wznowić podawanie produktu leczniczego Metformax Combi pod warunkiem ponownej oceny czynności nerek i stwierdzeniu, że jest ona stabilna (patrz punkty 4.3 i 4.4).
Skojarzenia leków wymagające środków ostrożności podczas stosowania
Pewne produkty lecznicze mogą wywierać niekorzystne działanie na czynność nerek, co może zwiększać ryzyko kwasicy mleczanowej, np. niesteroidowe leki przeciwzapalne (NLPZ), w tym selektywne inhibitory cyklooksygenazy (COX) 2, inhibitory ACE, antagoniści receptora angiotensyny II i leki moczopędne, w szczególności pętlowe. W razie rozpoczynania stosowania lub stosowania takich produktów leczniczych w skojarzeniu z metforminą, konieczne jest dokładne monitorowanie czynności nerek.
Jednoczesne stosowanie leków, które wpływają na wspólny układ transportu w kanalikach nerkowych biorących udział w wydalaniu metforminy przez nerki (np. inhibitory transportera kationów organicznych 2 [ang. OCT2] / transportera usuwania wielu leków i toksyn [ang. MATE], takich jak ranolazyna, wandetanib, dolutegrawir oraz cymetydyna) może zwiększać ogólnoustrojową ekspozycję na metforminę oraz ryzyko wystąpienia kwasicy mleczanowej. Należy rozważyć korzyści
i zagrożenia związane z jednoczesnym stosowaniem leków. W przypadku jednoczesnego stosowania tych produktów leczniczych należy rozważyć ścisłe monitorowanie kontroli glikemii, dostosowanie dawki w ramach zalecanego dawkowania oraz zmiany w leczeniu cukrzycy.
Glikokortykosteroidy (podawane ogólnie lub miejscowo), agoniści receptorów beta-2- adrenergicznych i diuretyki wykazują aktywność hiperglikemiczną. Należy poinformować o tym pacjentów i częściej kontrolować stężenie glukozy we krwi, zwłaszcza na początku leczenia wyżej wymienionymi produktami leczniczymi. Jeśli okaże się to konieczne, należy dostosować dawkę leku przeciwcukrzycowego w trakcie leczenia innym produktem leczniczym i podczas jego odstawiania.
Inhibitory ACE mogą obniżać stężenie glukozy we krwi. Jeśli okaże się to konieczne, należy dostosować dawkę leku przeciwcukrzycowego w trakcie leczenia innym produktem leczniczym i podczas jego odstawiania.
Wpływ innych produktów leczniczych na sytagliptynę
Dane z badań w warunkach in vitro oraz dane kliniczne opisane poniżej wskazują, że ryzyko wystąpienia znaczących klinicznie interakcji w następstwie jednoczesnego podania innych produktów leczniczych jest niewielkie.
Badania w warunkach in vitro wykazały, że głównym enzymem odpowiedzialnym za ograniczenie metabolizmu sytagliptyny jest CYP3A4, ze współudziałem CYP2C8. U pacjentów z prawidłową czynnością nerek, metabolizm, także przy udziale CYP3A4, ma tylko niewielki wpływ na klirens sytagliptyny. Metabolizm może mieć bardziej istotny wpływ na eliminację sytagliptyny w przypadku ciężkiego zaburzenia czynności nerek lub schyłkowej niewydolności nerek (ang.
ESRD). Z tej przyczyny silnie działające inhibitory CYP3A4 (np. ketokonazol, itrakonazol, rytonawir, klarytromycyna) mogą zmieniać farmakokinetykę sytagliptyny u pacjentów z ciężkim zaburzeniem czynności nerek lub schyłkową niewydolnością nerek. Wpływ silnie działających inhibitorów CYP3A4 w przypadku zaburzenia czynności nerek nie był oceniany w badaniach klinicznych.
Badania transportu leku w warunkach in vitro wykazały, że sytagliptyna jest substratem dla glikoproteiny p oraz transportera anionów organicznych 3 (ang. OAT3). Transport sytagliptyny,
w którym pośredniczy OAT3, hamowany był w warunkach in vitro przez probenecyd, chociaż ryzyko wystąpienia znaczących klinicznie interakcji uznawane jest za niewielkie. Jednoczesne stosowanie inhibitorów OAT3 nie było oceniane w warunkach in vivo.
Cyklosporyna: Przeprowadzono badanie w celu określenia wpływu cyklosporyny, silnego inhibitora glikoproteiny p, na farmakokinetykę sytagliptyny. Jednoczesne podanie sytagliptyny w pojedynczej dawce doustnej wynoszącej 100 mg z cyklosporyną w pojedynczej dawce doustnej wynoszącej
600 mg zwiększało wartość AUC oraz Cmax sytagliptyny odpowiednio o około 29% i 68%. Takich zmian farmakokinetyki sytagliptyny nie uznano za istotne klinicznie. Klirens nerkowy sytagliptyny nie uległ znaczącej zmianie. Z tego względu nie należy spodziewać się znaczących interakcji z innymi inhibitorami glikoproteiny p.
Wpływ sytagliptyny na inne produkty lecznicze
Digoksyna: Sytagliptyna miała niewielki wpływ na stężenie digoksyny w osoczu krwi. W wyniku stosowania przez 10 dni digoksyny w dawce 0,25 mg jednocześnie z sytagliptyną w dawce 100 mg na dobę osoczowe AUC dla digoksyny zwiększyło się przeciętnie o 11%, a osoczowe wartości Cmax
o 18%. Nie zaleca się dostosowywania dawki digoksyny. Jednak w przypadku jednoczesnego stosowania sytagliptyny i digoksyny należy monitorować pacjentów, u których istnieje ryzyko zatrucia digoksyną.
Dane z badań in vitro wskazują, że sytagliptyna nie hamuje ani nie indukuje izoenzymów CYP450. W badaniach klinicznych sytagliptyna nie powodowała znaczących zmian farmakokinetyki metforminy, gliburydu, symwastatyny, rozyglitazonu, warfaryny czy doustnych
środków antykoncepcyjnych, wskazując na niewielką możliwość wchodzenia w interakcje z substratami CYP3A4, CYP2C8, CYP2C9 i transporterem kationów organicznych (ang. OCT) w warunkach in vivo. Sytagliptyna w warunkach in vivo może być słabym inhibitorem glikoproteiny p.
Ciąża
Brak jest wystarczających danych dotyczących stosowania sytagliptyny u kobiet w okresie ciąży. Badania na zwierzętach wykazały szkodliwy wpływ sytagliptyny na reprodukcję w przypadku stosowania dużych dawek (patrz punkt 5.3).
Ograniczona ilość danych sugeruje, że stosowanie metforminy u kobiet w okresie ciąży nie zwiększa ryzyka wystąpienia wad wrodzonych. Badania na zwierzętach dotyczące stosowania metforminy nie wykazały szkodliwego wpływu na przebieg ciąży, rozwój zarodka lub płodu, przebieg porodu lub rozwój pourodzeniowy (patrz również punkt 5.3).
Produktu leczniczego Metformax Combi nie należy stosować w okresie ciąży. Jeśli pacjentka planuje zajść w ciążę lub w przypadku gdy zajdzie w ciążę, należy odstawić leczenie i jak najszybciej przejść na leczenie insuliną.
Karmienie piersią
Nie prowadzono badań na zwierzętach w okresie laktacji otrzymujących substancje czynne zawarte w tym produkcie leczniczym. W badaniach prowadzonych dla poszczególnych substancji czynnych stwierdzono, że zarówno sytagliptyna jak i metformina przenikają do mleka samic szczura w okresie laktacji. Metformina przenika do mleka ludzkiego w niewielkich ilościach. Nie wiadomo, czy sytagliptyna przenika do mleka ludzkiego. W związku z powyższym nie wolno stosować produktu leczniczego Metformax Combi u kobiet karmiących piersią (patrz punkt 4.3).
Płodność
W badaniach na zwierzętach nie obserwowano wpływu na płodność u samców i samic zwierząt. Brak danych dotyczących wpływu sytagliptyny na płodność u ludzi.
Metformax Combi nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Niemniej jednak, podczas prowadzenia pojazdów i obsługiwania maszyn, należy wziąć pod uwagę, że w przypadku stosowania sytagliptyny zgłaszano występowanie zawrotów głowy i senności.
Ponadto należy ostrzec pacjentów o ryzyku wystąpienia hipoglikemii podczas stosowania produktu leczniczego Metformax Combi w skojarzeniu z pochodnymi sulfonylomocznika lub z insuliną.
Podsumowanie profilu bezpieczeństwa
Nie prowadzono badań klinicznych produktu leczniczego zawierającego sytagliptynę i metforminę w tabletkach, jednak wykazano biorównoważność referencyjnego produktu leczniczego zawierającego sytagliptynę i metforminę z podawaniem sytagliptyny jednocześnie z metforminą (patrz punkt 5.2). Zgłaszano poważne działania niepożądane, w tym zapalenie trzustki i reakcje nadwrażliwości. Zgłaszano występowanie hipoglikemii w przypadku leczenia skojarzonego z pochodną sulfonylomocznika (13,8%) i insuliną (10,9%).
Sytagliptyna i metformina
Wykaz działań niepożądanych w ujęciu tabelarycznym
Działania niepożądane podano poniżej zgodnie z MedDRA, według klasyfikacji układów i narządów oraz całkowitej częstości występowania (Tabela 1). Częstości występowania określono jako występujące: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); niezbyt często (≥ 1/1000 do
< 1/100); rzadko (≥ 1/10 000 do < 1/1000); bardzo rzadko (< 1/10 000) i częstość nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 1: Częstość występowania działań niepożądanych dla sytagliptyny i metforminy stosowanych w monoterapii w kontrolowanych placebo badaniach klinicznych i po wprowadzeniu produktu leczniczego do obrotu
Działanie niepożądane | Częstość występowania działania niepożądanego |
Zaburzenia krwi i układu chłonnego | |
trombocytopenia | rzadko |
Zaburzenia układu immunologicznego | |
reakcje nadwrażliwości, w tym odpowiedzi anafilaktyczne*,† | częstość nieznana |
Zaburzenia metabolizmu i odżywiania | |
hipoglikemia† | często |
Zaburzenia układu nerwowego | |
senność | niezbyt często |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
śródmiąższowa choroba płuc* | częstość nieznana |
Zaburzenia żołądka i jelit | |
biegunka | niezbyt często |
nudności | często |
wzdęcia | często |
zaparcia | niezbyt często |
ból w górnej części brzucha | niezbyt często |
wymioty | często |
ostre zapalenie trzustki*,†,‡ | częstość nieznana |
martwicze i krwotoczne zapalenie trzustki ze skutkiem śmiertelnym lub bez*,† | częstość nieznana |
Zaburzenia skóry i tkanki podskórnej | |
świąd* | niezbyt często |
obrzęk naczynioruchowy*,† | częstość nieznana |
wysypka*,† | częstość nieznana |
pokrzywka*,† | częstość nieznana |
zapalenie naczyń skóry*,† | częstość nieznana |
złuszczające choroby skóry, w tym zespół Stevensa-Johnsona*,† | częstość nieznana |
pemfigoid pęcherzowy* | częstość nieznana |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
bóle stawów* | częstość nieznana |
bóle mięśni* | częstość nieznana |
ból kończyn | częstość nieznana |
ból pleców* | częstość nieznana |
artropatia* | częstość nieznana |
Zaburzenia nerek i dróg moczowych | |
zaburzenia czynności nerek* | częstość nieznana |
ostra niewydolność nerek* | częstość nieznana |
*Działania niepożądane zgłaszane po wprowadzeniu produktu leczniczego do obrotu.
†Patrz punkt 4.4.
‡ Patrz Badanie bezpieczeństwa sercowo-naczyniowego TECOS poniżej.
Opis wybranych działań niepożądanych
Obserwowano większą częstość występowania niektórych działań niepożądanych przy stosowaniu sytagliptyny i metforminy w skojarzeniu z innymi przeciwcukrzycowymi produktami leczniczymi w porównaniu z badaniami sytagliptyny i metforminy stosowanych w monoterapii. Obejmowały one hipoglikemię (działanie niepożądane występowało bardzo często w leczeniu skojarzonym
z pochodnymi sulfonylomocznika lub insuliną), zaparcia (często przy stosowaniu z pochodnymi sulfonylomocznika), obrzęki obwodowe (często przy stosowaniu z pioglitazonem) i ból głowy oraz suchość w ustach (niezbyt często przy stosowaniu z insuliną).
Sytagliptyna
W badaniach dotyczących porównania sytagliptyny stosowanej w monoterapii w dawce 100 mg raz na dobę do placebo, do odnotowanych działań niepożądanych leku należały: ból głowy, hipoglikemia, zaparcia oraz zawroty głowy.
U tych pacjentów działania niepożądane zgłaszane niezależnie od związku przyczynowego
z produktem leczniczym występowały z częstością co najmniej 5%, w tym zakażenia górnych dróg oddechowych oraz zapalenie błony śluzowej nosogardła. Ponadto, występowanie zapalenia kości
i stawów oraz bólu kończyn zgłaszano niezbyt często (z częstością > 0,5% większą u pacjentów leczonych sytagliptyną niż w grupie kontrolnej).
Metformina
Objawy ze strony układu pokarmowego zgłaszano bardzo często w badaniach klinicznych metforminy i po wprowadzeniu metforminy do obrotu. Objawy ze strony układu pokarmowego, takie jak nudności, wymioty, biegunka, ból brzucha i utrata apetytu, występowały najczęściej na początku leczenia i w większości przypadków ustępowały samoistnie. Dodatkowe działania niepożądane związane ze stosowaniem metforminy to: metaliczny posmak w ustach (często), kwasica mleczanowa, zaburzenia czynności wątroby, zapalenie wątroby, pokrzywka, rumień i świąd (bardzo rzadko). Długotrwałe leczenie metforminą wiązało się ze zmniejszeniem wchłaniania witaminy B12, co bardzo rzadko może prowadzić do istotnego klinicznie niedoboru witaminy B12 (np. niedokrwistość megaloblastyczna).
Kategorie częstości występowania oparte są na informacjach zawartych w dostępnej w UE Charakterystyce Produktu Leczniczego dla metforminy.
Dzieci i młodzież
W badaniach klinicznych prowadzonych z zastosowaniem referencyjnego produktu leczniczego zawierającego sytagliptynę i metforminę u dzieci i młodzieży z cukrzycą typu 2 w wieku od 10 do 17 lat profil działań niepożądanych był zasadniczo porównywalny z profilem obserwowanym u dorosłych. U dzieci i młodzieży przyjmujących lub nieprzyjmujących insulinę podstawową, stosowanie sytagliptyny wiązało się ze zwiększonym ryzykiem hipoglikemii.
Badanie bezpieczeństwa sercowo-naczyniowego TECOS
Do badania TECOS (ang. TECOS, Trial Evaluating Cardiovascular Outcomes with Sitagliptin), oceniającego wpływ sytagliptyny na układ sercowo-naczyniowy włączono 7332 pacjentów leczonych sytagliptyną w dawce 100 mg na dobę (lub 50 mg na dobę, jeśli wartość początkowa eGFR wynosiła
≥ 30 i < 50 ml/min/1,73 m2) oraz 7339 pacjentów z populacji wyodrębnionej zgodnie z zaplanowanym leczeniem otrzymujących placebo. Obie metody leczenia stosowano jednocześnie z zazwyczaj stosowanym leczeniem zmierzającym do regionalnych docelowych wartości dla HbA1c i czynników ryzyka sercowo-naczyniowego (ang. CV, Cardiovascular). Całkowita częstość występowania ciężkich działań niepożądanych u pacjentów otrzymujących sytagliptynę była podobna do tej obserwowanej
u pacjentów otrzymujących placebo.
W populacji wyodrębnionej zgodnie z zaplanowanym leczeniem wśród pacjentów, którzy w chwili rozpoczęcia badania stosowali insulinę i (lub) sulfonylomocznik, częstość występowania ciężkiej hipoglikemii w grupie pacjentów leczonych sytagliptyną wynosiła 2,7% i 2,5% w grupie pacjentów otrzymujących placebo. Wśród pacjentów, którzy w chwili rozpoczęcia badania nie stosowali insuliny i (lub) sulfonylomocznika, częstość występowania ciężkiej hipoglikemii w grupie pacjentów leczonych sytagliptyną wynosiła 1,0% i 0,7% w grupie pacjentów otrzymujących placebo. Częstość występowania obiektywnie potwierdzonych przypadków zapalenia trzustki w grupie pacjentów leczonych sytagliptyną wynosiła 0,3% i 0,2% w grupie pacjentów otrzymujących placebo.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C, 02-222 Warszawa,
Tel.: + 48 22 49 21 301, Faks: + 48 22 49 21 309, strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
W badaniach klinicznych z udziałem zdrowych osób i z grupą kontrolną sytagliptyna była podawana w pojedynczych dawkach wynoszących do 800 mg. W jednym badaniu z zastosowaniem sytagliptyny w dawce wynoszącej 800 mg obserwowano minimalne wydłużenie odstępu QTc, nieuznane za istotne klinicznie. Nie ma doświadczeń dotyczących stosowania w badaniach klinicznych dawek większych niż 800 mg. W badaniach I fazy z zastosowaniem dawek wielokrotnych nie stwierdzono zależnych od dawki klinicznych działań niepożądanych podczas stosowania sytagliptyny w dawkach do 600 mg na dobę przez okres do 10 dni i 400 mg na dobę przez okres do 28 dni.
Znaczne przedawkowanie metforminy (lub współistniejące ryzyko kwasicy mleczanowej) może prowadzić do wystąpienia kwasicy mleczanowej, która jest stanem zagrożenia życia i musi być leczona w szpitalu. Najskuteczniejszą metodą usunięcia mleczanów i metforminy jest hemodializa.
W badaniach klinicznych podczas hemodializy trwającej 3 do 4 godzin usunięto około 13,5% podanej dawki. W określonych stanach klinicznych można rozważyć zastosowanie przedłużonej hemodializy. Nie wiadomo, czy sytagliptynę można usunąć za pomocą dializy otrzewnowej.
W przypadku przedawkowania uzasadnione jest zastosowanie zwykłych środków wspomagających, np. usunięcie niewchłoniętego produktu leczniczego z przewodu pokarmowego, obserwacja kliniczna (w tym wykonanie elektrokardiogramu) oraz, jeśli zajdzie potrzeba, leczenie objawowe w warunkach szpitalnych.
Grupa farmakoterapeutyczna: leki stosowane w cukrzycy. Złożone doustne leki hipoglikemizujące, kod ATC: A10BD07.
Metformax Combi zawiera dwa leki przeciwcukrzycowe o komplementarnych mechanizmach działania, w celu zapewnienia lepszej kontroli glikemii u pacjentów z cukrzycą typu 2: sytagliptyny chlorowodorek, będący inhibitorem dipeptydylopeptydazy 4 (DPP-4) oraz metforminy chlorowodorek, należący do leków z grupy biguanidów.
Sytagliptyna
Mechanizm działania
Sytagliptyny chlorowodorek po podaniu doustnym jest aktywnym, silnym i wysoce selektywnym inhibitorem enzymu dipeptydylopeptydazy 4 (DPP-4), stosowanym w leczeniu cukrzycy typu 2. Inhibitory DPP-4 są substancjami o działaniu wzmacniającym działanie inkretyn. Hamując enzym DPP-4, sytagliptyna zwiększa stężenia dwóch znanych czynnych inkretyn – glukagonopodobnego peptydu-1 (GLP-1) i zależnego od glukozy polipeptydu insulinotropowego (GIP). Inkretyny stanowią część systemu endogennego uczestniczącego w fizjologicznej kontroli homeostazy glukozy. Kiedy stężenie glukozy we krwi jest prawidłowe lub podwyższone, GLP-1 oraz GIP zwiększają syntezę insuliny i uwalnianie jej z komórek beta trzustki. GLP-1 obniża również wydzielanie glukagonu przez komórki alfa trzustki, prowadząc do zmniejszenia wytwarzania glukozy w wątrobie. Kiedy stężenie glukozy we krwi jest niskie, nie następuje wzmocnienie uwalniania insuliny ani hamowanie wydzielania glukagonu.
Sytagliptyna jest silnym i wysoce selektywnym inhibitorem enzymu DPP-4 i w stężeniach stosowanych terapeutycznie nie hamuje blisko spokrewnionych z nim enzymów DPP-8 lub DPP-9. Sytagliptyna ma inną strukturę chemiczną i działanie farmakologiczne niż analogi GLP-1, insulina, pochodna sulfonylomocznika lub meglitynidy, biguanidy, agoniści aktywowanych przez proliferatory peroksysomów receptorów gamma (PPAR), inhibitory alfaglukozydazy i analogi amyliny.
W trwającym dwa dni badaniu z udziałem zdrowych osób sama sytagliptyna zwiększała stężenia aktywnej postaci GLP-1, natomiast sama metformina zwiększała w podobnym stopniu stężenia aktywnej postaci i całkowitego GLP-1. Jednoczesne podawanie sytagliptyny i metforminy wywiera addytywny wpływ na stężenie aktywnej postaci GLP-1. Sytagliptyna powodowała zwiększenie stężenia aktywnej postaci GIP, natomiast metformina nie.
Skuteczność kliniczna i bezpieczeństwo stosowania
Ogólnie, sytagliptyna poprawiała kontrolę glikemii, gdy była stosowana w monoterapii lub w leczeniu skojarzonym u dorosłych pacjentów z cukrzycą typu 2.
W badaniach klinicznych monoterapia sytagliptyną polepszyła kontrolę glikemii, istotnie obniżając stężenia hemoglobiny A1c (HbA1c) oraz glukozy na czczo i po posiłku. Obniżenie stężenia glukozy w osoczu na czczo odnotowywano po upływie 3 tygodni, tj. w pierwszym punkcie czasowym,
w którym dokonywano pomiaru stężenia glukozy na czczo. Obserwowana częstość przypadków hipoglikemii u pacjentów leczonych sytagliptyną była podobna, jak u pacjentów przyjmujących placebo. Podczas leczenia sytagliptyną nie miał miejsca przyrost masy ciała w porównaniu
z poziomem wyjściowym. Zaobserwowano poprawę wartości markerów zastępczych czynności komórek beta, w tym wskaźników modelu oceny homeostazy HOMA-β, stosunku stężeń proinsuliny do insuliny oraz reaktywności komórek beta w teście tolerancji glukozy z częstym pobieraniem próbek.
Badania dotyczące stosowania sytagliptyny jednocześnie z metforminą
Trwające 24 tygodnie badanie kliniczne z grupą kontrolną placebo miało na celu ocenę skuteczności i bezpieczeństwa dodania sytagliptyny w dawce 100 mg, raz na dobę, do już trwającego leczenia metforminą. Dodanie sytagliptyny wiązało się z istotną poprawą parametrów
glikemii w porównaniu do grupy przyjmującej placebo. Zmiana masy ciała w porównaniu z poziomem wyjściowym u pacjentów leczonych sytagliptyną była podobna, jak u pacjentów otrzymujących placebo. W omawianym badaniu częstość przypadków hipoglikemii zgłaszanych u pacjentów leczonych sytagliptyną była podobna, jak u pacjentów otrzymujących placebo.
W trwającym 24 tygodnie badaniu czynnikowym z grupą kontrolną placebo dotyczącym leczenia początkowego, stosowanie sytagliptyny w dawce 50 mg, dwa razy na dobę w skojarzeniu z metforminą (500 mg lub 1000 mg, dwa razy na dobę) wiązało się z istotną poprawą parametrów glikemii
w porównaniu z monoterapią każdym z tych leków. Zmniejszenie masy ciała u pacjentów leczonych sytagliptyną w skojarzeniu z metforminą było podobne do obserwowanego u pacjentów leczonych jedynie metforminą lub przyjmujących placebo; nie stwierdzono zmiany masy ciała w porównaniu
z poziomem wyjściowym u pacjentów leczonych jedynie sytagliptyną. Częstość występowania hipoglikemii była zbliżona we wszystkich grupach leczonych.
Badanie dotyczące stosowania sytagliptyny jednocześnie z metforminą i pochodną sulfonylomocznika Trwające 24 tygodnie badanie z grupą kontrolną placebo miało na celu ocenę skuteczności
i bezpieczeństwa stosowania sytagliptyny (100 mg, raz na dobę) dodanej do schematu leczenia glimepirydem (w monoterapii lub w skojarzeniu z metforminą). Dodanie sytagliptyny do glimepirydu i metforminy wiązało się z istotną poprawą parametrów glikemii. U pacjentów leczonych sytagliptyną stwierdzono umiarkowany przyrost masy ciała (+1,1 kg) w porównaniu z pacjentami przyjmującymi placebo.
Badanie dotyczące stosowania sytagliptyny jednocześnie z metforminą i agonistą receptora PPAR Trwające 26 tygodni badanie z grupą kontrolną placebo miało na celu ocenę skuteczności
i bezpieczeństwa stosowania sytagliptyny (100 mg raz na dobę) dodanej do terapii skojarzonej pioglitazonem i metforminą. Dodanie sytagliptyny do pioglitazonu i metforminy wiązało się z istotną poprawą parametrów glikemii. Zmiana masy ciała w stosunku do poziomu wyjściowego była zbliżona u pacjentów leczonych sytagliptyną i przyjmujących placebo. Częstość występowania hipoglikemii była zbliżona u pacjentów leczonych sytagliptyną i przyjmujących placebo.
Badanie dotyczące stosowania sytagliptyny w skojarzeniu z metforminą i insuliną Trwające 24 tygodnie badanie z grupą kontrolną placebo miało na celu ocenę skuteczności
i bezpieczeństwa dodania sytagliptyny (100 mg raz na dobę) do leczenia insuliną (w stabilnej dawce przez przynajmniej 10 tygodni) z metforminą lub bez (przynajmniej 1500 mg). U pacjentów przyjmujących gotową mieszankę insulinową średnia dawka dobowa wynosiła 70,9 jednostek insuliny/dobę. U pacjentów przyjmujących mieszankę insulinową nieprzygotowaną (o pośrednim lub przedłużonym czasie działania) średnia dawka dobowa wynosiła 44,3 jednostki insuliny/dobę.
W Tabeli 2 przedstawiono dane 73% pacjentów, którzy przyjmowali metforminę. Dodanie sytagliptyny do insuliny spowodowało znaczącą poprawę parametrów glikemii. W żadnej z grup nie zaobserwowano znaczącej zmiany masy ciała w porównaniu z poziomem wyjściowym.
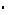
Tabela 2: Wyniki HbA1c uzyskane w kontrolowanych placebo badaniach dotyczących leczenia sytagliptyną w skojarzeniu z metforminą*
Badanie | Średnia początkowa wartość HbA1c (%) | Średnia zmiana HbA1c (%) w stosunku do wartości początkowych | Korygowana dla placebo średnia zmiana HbA1c (%) (95% CI) |
Sytagliptyna 100 mg jeden raz na dobę dodana do trwającego leczenia metforminą (N=453) | 8,0 | -0,7† | -0,7†,‡ (-0,8; -0,5) |
Sytagliptyna 100 mg jeden raz na dobę dodana do trwającego leczenia glimepiryd + metformina (N=115) | 8,3 | -0,6† | -0,9†,‡ (-1,1; -0,7) |
Sytagliptyna 100 mg jeden raz na dobę dodana do trwającego leczenia pioglitazon + metformina¶ (N=152) | 8,8 | -1,2† | -0,7†,‡ (-1,0; -0,5) |
Sytagliptyna 100 mg jeden raz na dobę dodana do trwającego leczenia insulina + metformina (N=223) | 8,7 | -0,7§ | -0,5§,‡ (-0,7; -0,4) |
Leczenie początkowe (dwa razy na dobę) : Sytagliptyna 50 mg + metformina 500 mg (N=183) | 8,8 | -1,4† | -1,6†,‡ (-1,8; -1,3) |
Leczenie początkowe (dwa razy na dobę) : Sytagliptyna 50 mg + metformina 1000 mg (N=178) | 8,8 | -1,9† | -2,1†,‡ (-2,3; -1,8) |
* Wszyscy pacjenci leczonej populacji (analiza zgodna z zaplanowanym leczeniem).
† Średnia najmniejszych kwadratów skorygowana dla stanu uprzedniego leczenia przeciwcukrzycowego i wartości początkowej.
‡ p<0,001 w porównaniu do placebo lub placebo + leczenie skojarzone.
" HbA1c (%) w 24. Tygodniu.
¶ HbA1c (%) w 26. Tygodniu.
§ Średnia najmniejszych kwadratów, skorygowana względem stosowania insuliny w trakcie Wizyty 1. (gotowa mieszanka insulinowa wobec insuliny niemieszanej [o pośrednim lub przedłużonym czasie działania]) oraz względem wartości początkowej.
W trwającym 52 tygodnie badaniu porównującym skuteczność działania i bezpieczeństwo stosowania jako leku dodatkowego sytagliptyny w dawce 100 mg raz na dobę lub glipizydu (pochodna sulfonylomocznika) u pacjentów, u których kontrola glikemii w przypadku stosowania metforminy w monoterapii była niewystarczająca, sytagliptyna wykazywała podobne działanie do glipizydu w zakresie zmniejszania wartości HbA1c (średnia zmiana w porównaniu z poziomem wyjściowym -0,7% w tygodniu 52., przy wyjściowym poziomie HbA1c wynoszącym w obu grupach około 7,5%). Średnia dawka glipizydu stosowana w grupie porównawczej wynosiła 10 mg na dobę, z czego około 40% pacjentów wymagających dawki glipizydu wynoszącej ≤ 5 mg/dobę przez cały czas trwania badania. Niemniej jednak, więcej pacjentów przerwało leczenie z powodu braku skuteczności w grupie otrzymującej sytagliptynę niż w grupie przyjmującej glipizyd. U pacjentów leczonych sytagliptyną stwierdzono istotne średnie zmniejszenie masy ciała (-1,5 kg) względem wartości wyjściowych w porównaniu z istotnym zwiększeniem masy ciała u pacjentów otrzymujących glipizyd (+1,1 kg). W badaniu tym stosunek proinsuliny do insuliny, który jest markerem skuteczności syntezy i uwalniania insuliny, był korzystniejszy w przypadku podawania sytagliptyny i mniej korzystny w przypadku leczenia glipizydem. Częstość występowania hipoglikemii w grupie otrzymującej sytagliptynę (4,9%) była znacząco mniejsza niż w grupie stosującej glipizyd (32,0%).
Trwające 24 tygodnie badanie z grupą kontrolną placebo, w którym uczestniczyło 660 pacjentów, zaprojektowano w celu oceny efektu oszczędzania insuliny oraz bezpieczeństwa stosowania sytagliptyny (w dawce 100 mg raz na dobę) jako uzupełnienia terapii insuliną glargine wraz z metforminą (w dawce co najmniej 1500 mg) lub bez metforminy podczas intensyfikacji insulinoterapii. U pacjentów przyjmujących metforminę wartość wyjściowa HbA1c wynosiła 8,70%, a dawka wyjściowa insuliny wynosiła 37 j.m./dobę. Pacjentów poinstruowano, aby zwiększali dawkę insuliny glargine na podstawie wyniku oznaczenia stężenia glukozy na czczo z użyciem próbki krwi z opuszki palca. Wśród pacjentów przyjmujących metforminę, w 24. tygodniu, dobowa dawka insuliny wzrosła o 19 j.m./dobę u pacjentów leczonych sytagliptyną, a u pacjentów otrzymujących placebo – o 24 j.m./dobę.
Wartość HbA1c u pacjentów przyjmujących sytagliptynę, metforminę i insulinę uległa zmniejszeniu o -1,35% w porównaniu do -0,90% w przypadku pacjentów
przyjmujących placebo, metforminę i insulinę (różnica -0,45% [95% CI: -0,62; -0,29]). Częstość występowania hipoglikemii wynosiła 24,9% u pacjentów leczonych sytagliptyną, metforminą i insuliną oraz 37,8% u pacjentów przyjmujących placebo, metforminę i insulinę. Różnica wynikała głównie
z większego odsetka pacjentów w grupie placebo, u których hipoglikemia wystąpiła 3 razy lub częściej (9,1% w stosunku do 19,8%). Nie stwierdzono różnicy w częstości występowania ciężkiej hipoglikemii.
Metformina
Mechanizm działania
Metformina jest biguanidem o właściwościach przeciwcukrzycowych, który zmniejsza stężenia glukozy w osoczu, na czczo i po posiłku. Metformina nie pobudza wydzielania insuliny i dzięki temu nie powoduje hipoglikemii.
Metformina może działać w trzech mechanizmach:
/1000 pacjentolat, sama dieta 18 zdarzeń /1000 pacjentolat, (p=0,01).
Badanie TECOS było randomizowanym badaniem z udziałem 14 671 pacjentów w populacji wyodrębnionej zgodnie z zaplanowanym leczeniem z wartością HbA1c wynoszącą ≥ 6,5 do 8,0% i rozpoznaną chorobą CV, którzy otrzymywali sytagliptynę (7332) w dawce100 mg na dobę (lub
50 mg na dobę, jeśli wartość początkowa eGFR wynosiła ≥ 30 i < 50 ml/min/1,73 m2) lub placebo (7339) jednocześnie z zazwyczaj stosowanym leczeniem zmierzającym do regionalnych docelowych wartości dla HbA1c i czynników ryzyka CV. Pacjentów, u których wartość eGFR wynosiła
< 30 ml/min/1,73 m2 nie włączono do badania. Populacja badania liczyła 2004 pacjentów w wieku
≥ 75 lat oraz 3324 pacjentów z zaburzeniami czynności nerek (eGFR < 60 ml/min/1,73 m2).
W trakcie badania całkowita średnia szacunkowa (ang. SD) różnica wartości HbA1c w grupach leczonych sytagliptyną i otrzymujących placebo wynosiła 0,29% (0,01), 95%CI (-0,32; -0,27);
p < 0,001.
Pierwszorzędowym sercowo-naczyniowym punktem końcowym była składowa pierwszego wystąpienia zgonu z przyczyn sercowo-naczyniowych, zawału mięśnia sercowego niezakończonego zgonem, udaru mózgu niezakończonego zgonem lub niestabilnej dławicy piersiowej
wymagającej hospitalizacji. Do drugorzędowych sercowo-naczyniowych punktów końcowych włączono pierwsze wystąpienie zgonu z przyczyn sercowo-naczyniowych, zawału mięśnia sercowego niezakończonego zgonem lub udaru mózgu niezakończonego zgonem; pierwsze wystąpienie poszczególnych składowych pierwszorzędowego złożonego punktu końcowego; zgon z jakiejkolwiek przyczyny oraz zastoinową niewydolność serca wymagającą hospitalizacji.
Po okresie obserwacji, którego mediana wynosiła 3 lata, sytagliptyna podawana jednocześnie
z zazwyczaj stosowanym leczeniem nie powodowała zwiększenia ryzyka poważnych sercowo- naczyniowych działań niepożądanych lub ryzyka niewydolności serca wymagającej hospitalizacji w porównaniu z zazwyczaj stosowanym leczeniem bez sytagliptyny u pacjentów z cukrzycą typu 2 (Tabela 3).
Tabela 3: Wskaźniki występowania złożonych zdarzeń sercowo-naczyniowych i głównych zdarzeń drugorzędowych
Sytagliptyna 100 mg | Placebo | Współczynnik ryzyka (95% CI) | wartość-p† | |||
N (%) | Wskaźni k częstości występo wania na 100 pacjentol at* | N (%) | Wskaźni k częstości występow ania na 100 pacjentol at* | |||
Analiza w populacji wyodrębnionej zgodnie z zaplanowanym leczeniem | ||||||
Liczba pacjentów | 7332 | 7339 | 0,98 (0,89–1,08) | < 0,001 | ||
Pierwszorzędowy złożony punkt końcowy (Zgon z przyczyn sercowo- naczyniowych, zawał mięśnia sercowego niezakończony zgonem, udar mózgu niezakończony zgonem lub niestabilna dławica piersiowa wymagająca hospitalizacji) | 839 (11,4) | 4,1 | 851 (11,6) | 4,2 | ||
Drugorzędowy złożony punkt końcowy (Zgon z przyczyn sercowo- naczyniowych, zawał mięśnia sercowego niezakończony zgonem lub udar mózgu niezakończony zgonem) | 745 (10,2) | 3,6 | 746 (10,2) | 3,6 | 0,99 (0,89–1,10) | < 0,001 |
Zdarzenie drugorzędowe | ||||||
Zgon z przyczyn sercowo- naczyniowych | 380 (5,2) | 1,7 | 366 (5,0) | 1,7 | 1,03 (0,89–1,19) | 0,711 |
Zawał mięśnia sercowego (zakończony zgonem i niezakończony zgonem) | 300 (4,1) | 1,4 | 316 (4,3) | 1,5 | 0,95 (0,81–1,11) | 0,487 |
Udar mózgu (zakończony zgonem i niezakończony zgonem) | 178 (2,4) | 0,8 | 183 (2,5) | 0,9 | 0,97 (0,79–1,19) | 0,760 |
Niestabilna dławica piersiowa wymagająca hospitalizacji | 116 (1,6) | 0,5 | 129 (1,8) | 0,6 | 0,90 (0,70–1,16) | 0,419 |
Zgon niezależnie od przyczyny | 547 (7,5) | 2,5 | 537 (7,3) | 2,5 | 1,01 (0,90–1,14) | 0,875 |
Niewydolność serca wymagająca hospitalizacji‡ | 228 (3,1) | 1,1 | 229 (3,1) | 1,1 | 1,00 (0,83–1,20) | 0,983 |
* Współczynnik częstości występowania na 100 pacjentolat oblicza się za pomocą następującego wzoru: 100 × (całkowita liczba pacjentów, u których w analizowanym okresie ekspozycji wystąpiło ≥ 1 zdarzenie na całkowitą liczbę pacjentolat obserwacji).
† Wg stratyfikowanego modelu Coxa na podstawie regionu. Dla złożonych punktów końcowych wartości-p odpowiadają testowi równoważności, który ma wykazać, że współczynnik ryzyka wynosi mniej niż 1,3. Dla wszystkich pozostałych punktów końcowych wartości-p odpowiadają testowi różnic we współczynnikach ryzyka.
‡ Analizę hospitalizacji ze względu na niewydolność serca dostosowano do niewydolność serca w wywiadzie w chwili rozpoczęcia badania.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego sytagliptynę i metforminę we wszystkich podgrupach populacji dzieci i młodzieży w leczeniu cukrzycy typu 2 (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Bezpieczeństwo stosowania i skuteczność sytagliptyny w leczeniu uzupełniającym u dzieci
i młodzieży w wieku od 10 do 17 lat z cukrzycą typu 2 i niedostateczną kontrolą glikemii pomimo leczenia metforminą podawaną w skojarzeniu z insuliną lub bez insuliny oceniano w dwóch badaniach trwających ponad 54 tygodnie. Dodanie sytagliptyny (podawanej w schemacie sytagliptyna + metformina lub sytagliptyna + metformina o przedłużonym uwalnianiu - ang. extended release XR) porównywano z dodaniem placebo do metforminy lub metforminy XR.
Chociaż w łącznej analizie danych z tych dwóch badań po 20 tygodniach wykazano skuteczniejsze obniżenie wartości HbA1c w przypadku stosowania schematów sytagliptyna + metformina / sytagliptyna + metformina XR niż w przypadku stosowania samej metforminy, wyniki poszczególnych badań były niespójne. Ponadto, po upływie 54 tygodni nie stwierdzono większej skuteczności schematów sytagliptyna + metformina / sytagliptyna + metformina XR w porównaniu z samą metforminą. Dlatego produktu leczniczego Metformax Combi nie należy stosować u dzieci i młodzieży w wieku od 10 do 17 lat ze względu na niewystarczającą skuteczność (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Połączenie sytagliptyny i metforminy w tabletkach
W badaniu biorównoważności prowadzonym z udziałem osób zdrowych wykazano, że stosowanie tabletek referencyjnego produktu leczniczego zawierającego sytagliptynę i metforminę jest biologicznie równoważne równoczesnemu podawaniu sytagliptyny i metforminy chlorowodorku w postaci oddzielnych tabletek.
Poniżej znajduje się opis właściwości farmakokinetycznych poszczególnych substancji czynnych wchodzących w skład produktu leczniczego Metformax Combi.
Sytagliptyna
Wchłanianie
Po podaniu doustnie dawki wynoszącej 100 mg osobom zdrowym sytagliptyna była szybko wchłaniana, a jej stężenie w osoczu krwi osiągało maksymalne wartości (mediana Tmax) w ciągu 1 do 4 godzin po podaniu, średnie osoczowe AUC dla sytagliptyny wynosiło 8,52 M•h, a Cmax wynosiło 950 nM. Bezwzględna biodostępność sytagliptyny wynosi około 87%. Ponieważ przyjmowanie sytagliptyny podczas posiłku bogatego w tłuszcze nie wpływało na farmakokinetykę, sytagliptyna może być podawana z jedzeniem lub niezależnie od posiłków.
Osoczowe AUC dla sytagliptyny zwiększało się w sposób proporcjonalny do dawki leku. W przypadku Cmax i C24h nie określono proporcjonalności względem dawki leku (wzrost Cmax był większy niż zależny od dawki, a wzrost C24h mniejszy niż zależny od dawki).
Dystrybucja
Po podaniu pojedynczej dawki dożylnej wynoszącej 100 mg sytagliptyny osobom zdrowym średnia objętość dystrybucji w stanie równowagi wynosi około 198 litrów. Frakcja sytagliptyny związana w sposób odwracalny z białkami osocza jest mała (38%).
Metabolizm
Sytagliptyna jest w przeważającej mierze eliminowana z moczem w postaci niezmienionej, a metabolizm leku ma drugorzędne znaczenie. Około 79% sytagliptyny wydalane jest z moczem w postaci niezmienionej.
Po podaniu doustnym [14C]sytagliptyny około 16% dawki radioaktywnej wydalane było w postaci metabolitów sytagliptyny. Wykryto sześć metabolitów w stężeniach śladowych i można spodziewać się, że nie odpowiadają one za działanie sytagliptyny hamujące aktywność DPP-4 w osoczu krwi.
Wyniki badań in vitro wskazują na to, że głównym enzymem odpowiedzialnym za ograniczony metabolizm sytagliptyny jest CYP3A4 przy współudziale CYP2C8.
Dane z badań in vitro wykazały, że sytagliptyna nie jest inhibitorem izoenzymów CYP: CYP3A4, 2C8, 2C9, 2D6, 1A2, 2C19 czy 2B6 i nie indukuje CYP3A4 i CYP1A2.
Eliminacja
Po podaniu doustnym [14C]sytagliptyny osobom zdrowym około 100% podanej dawki radioaktywnej eliminowane było z kałem (13%) lub moczem (87%) w okresie jednego tygodnia od podania.
Rzeczywisty końcowy okres półtrwania t½ po podaniu doustnym 100 mg sytagliptyny wynosił około 12,4 godziny. Sytagliptyna jedynie w minimalnym stopniu ulega kumulacji po podaniu w dawkach wielokrotnych. Klirens nerkowy wynosił około 350 ml/min.
Eliminacja sytagliptyny następuje głównie w wyniku wydalania przez nerki z udziałem aktywnego wydzielania kanalikowego. Sytagliptyna jest substratem dla ludzkiego transportera anionów organicznych-3 (hOAT-3), który może uczestniczyć w eliminacji sytagliptyny przez nerki. Znaczenie kliniczne hOAT-3 w transporcie sytagliptyny nie zostało ustalone. Sytagliptyna jest także substratem dla glikoproteiny p, która także może pośredniczyć w eliminacji sytagliptyny przez nerki. Jednak cyklosporyna, inhibitor glikoproteiny p, nie zmniejsza klirensu nerkowego sytagliptyny. Sytagliptyna nie jest substratem dla transporterów OCT2, OAT1 czy PEPT1/2. W warunkach in vitro sytagliptyna nie hamuje transportu, w którym pośredniczy OAT3 (IC50=160 M) lub glikoproteina p (do 250
M), w istotnych terapeutycznie stężeniach w osoczu. W badaniu klinicznym sytagliptyna miała niewielki wpływ na stężenie digoksyny w osoczu krwi, co wskazuje na to, że może być słabym inhibitorem glikoproteiny p.
Charakterystyka leku u pacjentów
U osób zdrowych i u pacjentów z cukrzycą typu 2 farmakokinetyka sytagliptyny była na ogół podobna.
Zaburzenia czynności nerek
Przeprowadzono badanie otwarte z użyciem dawki jednorazowej w celu oceny farmakokinetyki zmniejszonej dawki sytagliptyny (50 mg) u pacjentów z przewlekłymi zaburzeniami czynności nerek o różnym nasileniu w porównaniu z grupą kontrolną złożoną ze zdrowych osób. Do badania włączono pacjentów z łagodnymi, umiarkowanymi oraz ciężkimi zaburzeniami czynności nerek, jak również pacjentów poddawanych hemodializie ze schyłkową niewydolnością nerek (ESRD).
Ponadto wpływ zaburzenia czynności nerek na właściwości farmakokinetyczne sytagliptyny u pacjentów z cukrzycą typu 2 oraz łagodnymi, umiarkowanymi lub ciężkimi zaburzeniami czynności nerek (w tym ESRD) oceniano na podstawie analiz farmakokinetyki populacyjnej.
W porównaniu do zdrowych osób stanowiących grupę kontrolną, AUC sytagliptyny w osoczu było zwiększone odpowiednio około 1,2-krotnie i 1,6-krotnie u pacjentów z łagodnymi zaburzeniami czynności nerek (GFR ≥ 60 do < 90 ml/min) oraz u pacjentów z umiarkowanymi zaburzeniami czynności nerek (GFR ≥ 45 do < 60 ml/min). Ponieważ zwiększenie to nie jest klinicznie znaczące, dostosowanie dawkowania w tej grupie pacjentów nie jest konieczne.
U pacjentów z umiarkowanymi zaburzeniami czynności nerek (GFR ≥ 30 do < 45 ml/min) oraz u pacjentów z ciężkimi zaburzeniami czynności nerek (GFR < 30 ml/min), w tym u pacjentów poddawanych hemodializie ze schyłkową niewydolnością nerek, obserwowano odpowiednio około 2-krotne i około 4-krotne zwiększenie AUC sytagliptyny w osoczu.
Sytagliptyna usuwana była w stopniu umiarkowanym za pomocą hemodializy (13,5% w ciągu 3 do 4 godzin hemodializy, począwszy od 4. godziny po podaniu dawki).
Zaburzenia czynności wątroby
U pacjentów z zaburzeniami czynności wątroby w stopniu łagodnym do umiarkowanego ( 9 punktów wg skali Child-Pugha) nie jest wymagane dostosowywanie dawki sytagliptyny. Nie ma doświadczenia klinicznego dotyczącego stosowania u pacjentów z ciężkimi zaburzeniami czynności wątroby (> 9 punktów wg skali Child-Pugha). Jednak ze względu na to, że sytagliptyna jest eliminowana głównie przez nerki, nie należy się spodziewać, aby ciężkie zaburzenia czynności wątroby miały wpływ na farmakokinetykę sytagliptyny.
Pacjenci w podeszłym wieku
Nie jest wymagane dostosowywanie dawki ze względu na wiek. Na podstawie wyników analizy farmakokinetyki w populacji przeprowadzonej z wykorzystaniem danych uzyskanych w fazie I i w fazie II badań stwierdzono, że wiek pacjenta nie miał istotnego klinicznie wpływu na farmakokinetykę sytagliptyny. U osób w podeszłym wieku (65 do 80 lat) stężenie
sytagliptyny w osoczu krwi było o około 19% wyższe niż u osób młodszych.
Dzieci i młodzież
Farmakokinetykę sytagliptyny (w dawce pojedynczej wynoszącej 50 mg, 100 mg lub 200 mg) zbadano u dzieci i młodzieży (w wieku od 10 do 17 lat) z cukrzycą typu 2. W przypadku dawki wynoszącej
100 mg, w tej populacji pacjentów wartość AUC sytagliptyny w osoczu skorygowana względem dawki była o około 18% niższa niż u dorosłych pacjentów z cukrzycą typu 2. Nie przeprowadzono badań dotyczących stosowania sytagliptyny u dzieci w wieku <10 lat.
Inne charakterystyczne grupy pacjentów
Nie jest konieczne dostosowywanie dawki ze względu na płeć, rasę lub wskaźnik masy ciała (BMI). Na podstawie łącznej analizy danych dotyczących farmakokinetyki uzyskanych w fazie I oraz analizy danych dotyczących farmakokinetyki w populacji w fazie I i w fazie II badań stwierdzono, że cechy te nie mają istotnego klinicznie wpływu na farmakokinetykę sytagliptyny.
Metformina
Wchłanianie
Po przyjęciu doustnej dawki metforminy, Tmax zostaje osiągnięty w czasie 2,5 h. Bezwzględna biodostępność tabletki zawierającej 500 mg metforminy wynosi u zdrowych osób około 50-60%. Po przyjęciu dawki doustnej niewchłonięta część dawki odzyskana z kału stanowiła 20-30% dawki podanej.
Wchłanianie metforminy po podaniu doustnym jest wysycalne i niepełne. Przypuszcza się, że farmakokinetyka wchłaniania metforminy jest nieliniowa. W przypadku stosowania zwykle zalecanych dawek i schematów podawania metforminy, stężenia w stanie stacjonarnym w osoczu są osiągane po upływie 24 do 48 godzin po podaniu dawki i na ogół nie przekraczają 1 µg/ml.
W kontrolowanych badaniach klinicznych maksymalne stężenie metforminy w osoczu (Cmax) nie przekraczało 5 µg/ml, nawet po zastosowaniu maksymalnych dawek.
Przyjmowanie pokarmów zmniejsza stopień wchłaniania metforminy i powoduje jego niewielkie opóźnienie. Po podaniu dawki 850 mg obserwowano szczytowe stężenia w osoczu niższe o 40%, 25% zmniejszenie wartości AUC oraz wydłużenie czasu do osiągnięcia stężenia szczytowego o 35 minut. Znaczenie kliniczne obserwowanego spadku wartości pozostaje nieznane.
Dystrybucja
Metformina w nieznacznym stopniu wiąże się z białkami osocza. Metformina przenika do erytrocytów. Maksymalne stężenie metforminy we krwi jest mniejsze niż w osoczu i występuje mniej więcej w tym samym czasie. Krwinki czerwone są prawdopodobnie drugim kompartmentem dystrybucji metforminy. Średnia objętość dystrybucji Vd wynosiła 63 – 276 L.
Metabolizm
Metformina jest wydalana z moczem w postaci niezmienionej. Nie stwierdzono występowania metabolitów metforminy u ludzi.
Eliminacja
Klirens nerkowy metforminy wynosi > 400 ml/min, co wskazuje na jej wydalanie w drodze przesączania kłębkowego i wydzielania kanalikowego. Po podaniu doustnym pozorny okres półtrwania w końcowej fazie eliminacji wynosi około 6,5 h. W przypadku upośledzenia czynności nerek następuje zmniejszenie klirensu nerkowego proporcjonalnie do klirensu kreatyniny, przez co okres półtrwania eliminacji ulega wydłużeniu, co z kolei prowadzi do zwiększenia stężeń metforminy w osoczu.
Nie przeprowadzono badań na zwierzętach z wykorzystaniem tabletek sytagliptyny + metforminy.
W trwających 16 tygodni badaniach, w których psy leczone były albo wyłącznie metforminą, albo metforminą w skojarzeniu z sytagliptyną, nie obserwowano dodatkowego działania toksycznego
w terapii skojarzonej. W omawianych badaniach obserwowano NOEL przy narażeniu na sytagliptynę przekraczającym około 6 razy poziom narażenia człowieka oraz przy narażeniu na metforminę przekraczającym około 2,5 razy poziom narażenia człowieka.
Poniższe dane przedstawiają obserwacje pochodzące z badań wykonywanych oddzielnie dla sytagliptyny lub metforminy.
Sytagliptyna
U gryzoni obserwowano toksyczne działanie sytagliptyny na wątrobę i nerki przy narażeniu organizmu przekraczającym 58 razy poziom narażenia człowieka, natomiast w przypadku narażenia 19 razy większym niż narażenie człowieka, nie obserwowano żadnego wpływu. Przy narażeniu przekraczającym 67 razy narażenie w warunkach klinicznych, obserwowano nieprawidłowości
siekaczy u szczurów; w trwającym 14 tygodni badaniu u szczurów nie stwierdzono żadnego wpływu na zęby przy narażeniu 58-krotnym. Znaczenie tych obserwacji dla ludzi nie jest znane. U psów przy narażeniu około 23 razy przekraczającym narażenie w warunkach klinicznych obserwowano przemijające, związane z leczeniem objawy fizykalne. Niektóre z nich, takie jak oddychanie
z otwartym pyskiem, ślinienie się, białe, pieniste wymioty, ataksja, drżenie, ograniczenie aktywności i (lub) zgarbiona postawa wskazywały na toksyczne uszkodzenie nerwów. Ponadto, w badaniach histologicznych obserwowano także zwyrodnienie mięśni szkieletowych w stopniu nieznacznym lub niewielkim w przypadku dawek powodujących narażenie ustrojowe na poziomie przekraczającym około 23 razy narażenie człowieka. Nie stwierdzono żadnego wpływu na te parametry podczas narażenia 6-krotnie przekraczającego narażenie w warunkach klinicznych.
W badaniach przedklinicznych nie wykazano genotoksyczności sytagliptyny. Sytagliptyna nie miała działania rakotwórczego u myszy. U szczurów stwierdzono zwiększenie częstości występowania gruczolaków i raków wątroby podczas narażenia ustrojowego przekraczającego 58 razy narażenie człowieka. Ponieważ wykazano korelację działania hepatotoksycznego z wywoływaniem nowotworów wątroby u szczurów, zwiększona częstość występowania guzów wątroby była prawdopodobnie zjawiskiem wtórnym do przewlekłego działania hepatotoksycznego przy stosowaniu dużych dawek. Ze względu na szeroki margines bezpieczeństwa (poziom, na którym lek nie wywiera wpływu stanowi w tym przypadku dziewiętnastokrotność narażenia w warunkach klinicznych) obserwowane zmiany nowotworowe nie są uznawane za znaczące w odniesieniu do ludzi.
Nie obserwowano związanego z leczeniem niepożądanego wpływu na płodność u samic i samców szczurów w przypadku podawania sytagliptyny przed i podczas parzenia się.
W badaniu dotyczącym rozwoju przed-/pourodzeniowego szczurów, sytagliptyna nie wywierała żadnych działań niepożądanych.
W badaniach dotyczących toksycznego wpływu na reprodukcję wykazano niewielkie, związane z leczeniem, zwiększenie częstości występowania zniekształceń żeber u płodów szczurów (brak, niedorozwój i falistość żeber) przy narażeniu organizmu większym niż 29-krotne narażenie człowieka. U królików obserwowano toksyczny wpływ na matkę, przy narażeniu większym niż 29-krotne narażenie człowieka. Ze względu na szeroki margines bezpieczeństwa, powyższe obserwacje nie wskazują na istotne zagrożenie rozrodczości u ludzi. Sytagliptyna przenika w znacznej ilości do mleka karmiących samic szczurów (wskaźnik mleko/osocze wynosi 4:1).
Metformina
Dane przedkliniczne dla metforminy, wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, badań toksyczności po podaniu wielokrotnym, genotoksyczności, potencjalnego działania rakotwórczego oraz toksycznego wpływu na rozród, nie ujawniają szczególnego zagrożenia dla człowieka.
Rdzeń tabletki
celuloza mikrokrystaliczna (typ 102) powidon (K29/32)
sodu laurylosiarczan sodu stearylofumaran
Otoczka tabletki 50 mg + 850 mg tytanu dwutlenek (E 171)
żelaza tlenek czerwony (E 172) laktoza jednowodna hypromeloza 2910
triacetyna
Otoczka tabletki 50 mg + 1000 mg tytanu dwutlenek (E 171)
żelaza tlenek czerwony (E 172) alkohol poliwinylowy makrogol 3350
talk
żelaza tlenek czarny (E 172)
Nie dotyczy.
2 lata.
Nie przechowywać w temperaturze powyżej 30°C.
Blistry PVC/PVDC/aluminium w tekturowym pudełku
Opakowania po 14, 28, 30, 56, 60, 98, 112, 196 tabletek powlekanych. Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Teva Pharmaceuticals Polska Sp. z o.o., ul. Emilii Plater 53,
00-113 Warszawa
Metformax Combi, 50 mg + 850 mg: Metformax Combi, 50 mg + 1000 mg:
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu:








