







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
leczenia nowo rozpoznanego hormonowrażliwego raka gruczołu krokowego wysokiego ryzyka z przerzutami (ang. metastatic hormone sensitive prostate cancer, mHSPC) u dorosłych mężczyzn w skojarzeniu z terapią supresji androgenowej (ang. androgen deprivation therapy, ADT) (patrz punkt 5.1)
leczenia opornego na kastrację raka gruczołu krokowego z przerzutami (ang. metastatic castration resistant prostate cancer, mCRPC) u dorosłych mężczyzn, bez objawów lub z objawami o niewielkim nasileniu, po niepowodzeniu terapii supresji androgenowej, u których zastosowanie chemioterapii nie jest jeszcze wskazane klinicznie (patrz punkt 5.1)
leczenia mCRPC u dorosłych mężczyzn, u których choroba postępuje w trakcie lub po chemioterapii zawierającej docetaksel.
Dawkowanie i sposób podawania
Przeciwwskazania
Nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą wymienioną
w punkcie 6.1.
Kobiety, które są lub mogą prawdopodobnie być w ciąży (patrz punkt 4.6).
Ciężkie zaburzenia czynności wątroby [klasa C wg klasyfikacji Childa-Pugha (patrz punkty 4.2, 4.4 i 5.2)].
Stosowanie produktu leczniczego Abiraterone Pharmascience z prednizonem lub prednizolonem w skojarzeniu z Ra-223 jest przeciwwskazane.
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Zgłoszenia spontaniczne po wprowadzeniu abirateronu octanu do obrotu
Zwiększenie aktywności aminotransferazy alaninowej i (lub) zwiększenie aktywności aminotransferazy
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Wartość p pochodzi z logarytmicznego testu rang stratyfikowanego według punktacji ECOG PS (0/1 lub 2) i
zmian trzewnych (nieobecność lub obecność).
Współczynnik ryzyka pochodzi ze stratyfikowanego proporcjonalnego modelu ryzyka. Współczynnik ryzyka
wartość p pochodzi z logarytmicznego testu rang stratyfikowanego wg punktacji skali sprawności ECOG
(0 lub 1)
** Współczynnik ryzyka < 1 na korzyść abirateronu octanu.
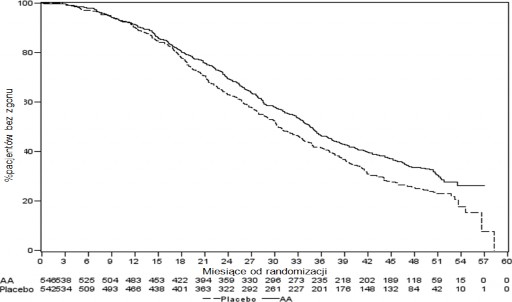
Wykres 3: Krzywe Kaplana-Meiera przeżycia bez progresji radiograficznej u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii
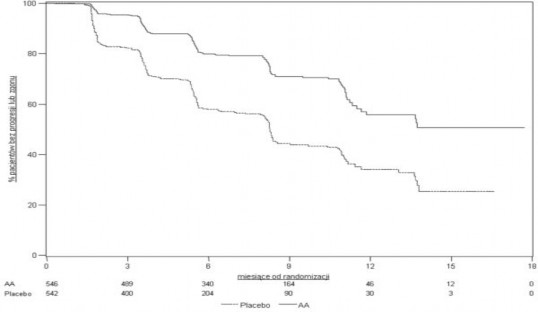
AA = abirateronu octan
Nadal kontynuowano zbieranie danych od osób badanych do daty drugiej analizy pośredniej całkowitego przeżycia (ang. Overall survival, OS). Radiograficzny przegląd rPFS, przeprowadzony przez badaczy jako kontynuacja analizy czułości przedstawia Tabela 5 i Wykres 4.
607 badanych miało progresję radiograficzną lub zmarło: 271 (50%) w grupie abirateronu octanu i 336 (62%) w grupie placebo. Leczenie abirateronu octanem zmniejszyło ryzyko progresji radiograficznej lub zgonu o 47% w porównaniu z placebo (HR=0,530; 95% CI: [0,451; 0,623]; p < 0,0001).
Mediana rPFS wyniosła 16,5 miesięcy w grupie abirateronu octanu i 8,3 miesięcy w grupie placebo.
Tabela 5: Badanie 302: Przeżycie bez progresji radiograficznej u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii (Podczas drugiej analizy pośredniej OS-Rewizja Badacza)
Abirateronu octan (N=546)
Placebo (N=542)
Przeżycie bez radiograficznej progresji (rPFS)
Progresja lub zgon
271 (50%)
336 (62%)
Mediana rPFS w miesiącach (95% CI)
16,5 (13,80; 16,79)
8,3 (8,05; 9,43)
wartość p*
< 0,0001
Współczynnik ryzyka ** (95%CI)
0,530 (0,451; 0,623)
wartość p pochodzi z logarytmicznego testu rang stratyfikowanego wg punktacji skali sprawności ECOG
(0 lub 1)
** Współczynnik ryzyka < 1 na korzyść abirateronu octanu.
Wykres 4: Krzywe Kaplana-Meiera przeżycia bez progresji radiograficznej u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii (Podczas drugiej analizy pośredniej OS-Rewizja Badacza)
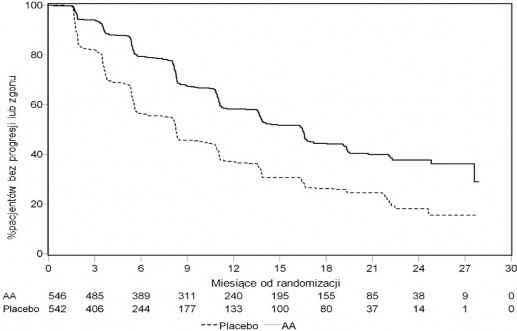
AA = abirateronu octan
Planową analizę pośrednią (ang. interim analysis, IA) OS przeprowadzono po stwierdzeniu 333
zgonów. Badanie zostało odkodowane na podstawie ważności stwierdzonych korzyści klinicznych,
a pacjentom z grupy placebo zaproponowano leczenie abirateronu octanem. Całkowity czas przeżycia był dłuższy dla abirateronu octanu niż placebo, z 25% redukcją ryzyka zgonu (HR = 0,752; 95% CI: [0,606; 0,934], p = 0,0097), lecz nie osiągnięto OS, a wyniki pośrednie nie osiągnęły zamierzonej wartości granicznej dla znamienności statystycznej (patrz Tabela 4). Przeżycie będzie nadal obserwowane po tej analizie pośredniej.
Planową analizę końcową OS przeprowadzono po stwierdzeniu 741 zgonów (mediana obserwacji – 49 miesięcy). Zmarło 65% (354 z 546) pacjentów leczonych abirateronu octanem, w porównaniu z 71% (387 z 542) pacjentów otrzymujących placebo. Wykazano statystycznie znamienną różnicę w OS na korzyść grupy leczonej abirateronu octanu z 19,4% zmniejszeniem ryzyka zgonu (HR = 0,806; 95% CI: [0,697; 0,931], p = 0,0033) oraz poprawą średniego OS o 4,4 miesiące (abirateronu octan 34,7 miesięcy, placebo 30,3 miesięcy), (patrz Tabela 6 i Wykres 5). Tę poprawę wykazano także u 44% pacjentów z grupy placebo, którzy następnie otrzymywali abirateronu octan.
Tabela 6: Badanie 302: całkowite przeżycie u pacjentów otrzymujących abirateronu octan
lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH
lub po wcześniejszej orchidektomii
Abirateronu octan (N=546)
Placebo (N=542)
Analiza pośrednia przeżycia
Zgony (%)
147 (27%)
186 (34%)
Mediana przeżycia w miesiącach (95% CI)
Nie oceniono (NE; NE)
27,2 (25,95; NE)
wartość p*
0,0097
Współczynnik ryzyka ** (95% CI)b
0,752 (0,606; 0,934)
Analiza końcowa przeżycia
Zgony (%)
354 (65%)
387 (71%)
Mediana przeżycia w miesiącach (95% CI)
34,7 (32,7; 36,8)
30,3 (28,7; 33,3)
wartość p*
0,0033
Współczynnik ryzyka ** (95% CI)b
0,806 (0,697; 0,931)
NE = nie oceniono.
wartość p pochodzi z logarytmicznego testu rang stratyfikowanego wg punktacji skali sprawności ECOG
wartość p pochodzi z logarytmicznego testu rang stratyfikowanego wg punktacji skali wydolności ECOG (0-1 vs. 2), punktacji bólu (nieobecny vs. obecny), liczby wcześniejszych schematów chemioterapii (1 vs. 2), i rodzaju progresji choroby (tylko PSA vs. radiograficzna).
Współczynnik ryzyka pochodzi ze stratyfikowanego proporcjonalnego modelu ryzyka. Współczynnik ryzyka
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
Abiraterone Pharmascience, 500 mg, tabletki powlekane
Każda tabletka powlekana zawiera 500 mg abirateronu octanu, co jest równoważne 446 mg abirateronu.
Substancje pomocnicze o znanym działaniu
Każda tabletka powlekana zawiera 245 mg laktozy jednowodnej.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka powlekana
Fioletowe, o owalnym kształcie, (20 mm długości x 10 mm szerokości) obustronnie wypukłe tabletki powlekane ze ściętymi krawędziami z wytłoczonym oznakowaniem „A” po jednej stronie i „500”
po drugiej stronie.
Produkt leczniczy Abiraterone Pharmascience jest wskazany w skojarzeniu z prednizonem lub prednizolonem do:
Ten produkt leczniczy powinien być zalecany przez lekarza z odpowiednią specjalizacją.
Dawkowanie
Zalecana dawka wynosi 1000 mg (dwie tabletki 500 mg) podawana jako pojedyncza dawka raz na dobę. Produktu leczniczego nie wolno przyjmować razem z jedzeniem (patrz poniżej „Sposób podawania”).
Przyjmowanie produktu leczniczego z jedzeniem zwiększa całkowite narażenie organizmu na
abirateron (patrz punkty 4.5 i 5.2).
Dawkowanie prednizonu lub prednizolonu
W leczeniu mHSPC produkt leczniczy Abiraterone Pharmascience stosuje się w skojarzeniu z 5 mg
prednizonu lub prednizolonu na dobę.
W leczeniu mCRPC produkt leczniczy Abiraterone Pharmascience stosuje się w skojarzeniu z 10 mg
prednizonu lub prednizolonu na dobę.
U pacjentów niekastrowanych chirurgicznie należy w trakcie leczenia kontynuować farmakologiczną kastrację analogami gonadoliberyny (ang. luteinising hormone releasing hormone, LHRH).
Zalecana obserwacja
Należy oceniać aktywność aminotransferaz w surowicy przed rozpoczęciem leczenia, co dwa tygodnie przez pierwsze trzy miesiące leczenia, a następnie co miesiąc. Ciśnienie tętnicze krwi, stężenie potasu w surowicy i zastój płynów należy oceniać co miesiąc. Jednakże, pacjentów z istotnym ryzykiem zastoinowej niewydolności serca należy badać co 2 tygodnie przez pierwsze 3 miesiące terapii, a następnie co miesiąc (patrz punkt 4.4).
U pacjentów z występującą wcześniej hipokaliemią lub z hipokaliemią, która rozwinęła się w trakcie leczenia produktem leczniczym Abiraterone Pharmascience, należy utrzymywać stężenie potasu na poziomie ≥ 4,0 mM.
U pacjentów, u których wystąpią objawy toksyczności stopnia ≥ 3. w tym nadciśnienie, hipokaliemia, obrzęk i inne działania toksyczne niezwiązane z mineralokortykosteroidami, należy wstrzymać leczenie i wdrożyć odpowiednie postępowanie. Nie należy wznawiać leczenia produktem Abiraterone Pharmascience, aż nasilenie objawów toksyczności zmniejszy się do stopnia 1. lub wartości wyjściowych.
W przypadku pominięcia dawki dobowej, zarówno produktu leczniczego Abiraterone Pharmascience, jak i prednizonu lub prednizolonu, należy wznowić leczenie zwykle stosowaną dawką dobową następnego dnia.
Hepatotoksyczność
U pacjentów, u których wystąpi działanie hepatotoksyczne podczas leczenia (zwiększy się aktywność aminotransferazy alaninowej [AlAT] lub zwiększy się aktywność aminotransferazy asparaginianowej [AspAT] ponad pięciokrotnie powyżej górnej granicy normy [GGN]), należy natychmiast wstrzymać leczenie (patrz punkt 4.4). Można wznowić leczenie po powrocie parametrów badań czynnościowych wątroby do wartości wyjściowych, stosując zmniejszoną dawkę 500 mg (jedna tabletka) raz na dobę. U pacjentów, u których wznowiono leczenie, należy badać aktywność aminotransferaz w surowicy przynajmniej co dwa tygodnie przez trzy miesiące, a następnie co miesiąc. W razie nawrotu hepatotoksyczności podczas stosowania zmniejszonej dawki 500 mg na dobę, należy przerwać leczenie.
Jeśli u pacjentów wystąpi ciężka hepatotoksyczność (aktywność AlAT lub AspAT zwiększona ponad 20 razy powyżej GGN) kiedykolwiek podczas terapii, należy przerwać leczenie i nie rozpoczynać go ponownie.
Zaburzenia czynności wątroby
Nie jest konieczne dostosowanie dawki u pacjentów z występującymi wcześniej łagodnymi zaburzeniami czynności wątroby, klasy A wg klasyfikacji Childa-Pugha. U pacjentów z umiarkowanymi zaburzeniami czynności wątroby (klasa B wg klasyfikacji Childa-Pugha) wykazano około 4-krotne zwiększenie całkowitego wpływu abirateronu na organizm po jednorazowej dawce doustnej 1000 mg abirateronu octanu (patrz punkt 5.2). Brak danych dotyczących bezpieczeństwa klinicznego i skuteczności wielokrotnych dawek abirateronu octanu podawanych pacjentom z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby (klasa B lub C wg klasyfikacji Childa- Pugha). Nie można określić zaleceń dotyczących dostosowania dawki. Zastosowanie produktu leczniczego Abiraterone Pharmascience należy rozważnie ocenić u pacjentów z umiarkowanymi zaburzeniami czynności wątroby, u których korzyści powinny wyraźnie przeważać nad możliwym ryzykiem (patrz punkty 4.2 i 5.2). Nie należy stosować produktu leczniczego Abiraterone Pharmascience u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkty 4.3, 4.4 i 5.2).
Zaburzenia czynności nerek
Nie jest konieczne dostosowanie dawki u pacjentów z zaburzeniami czynności nerek (patrz punkt 5.2). Brak danych klinicznych u pacjentów z rakiem gruczołu krokowego i ciężkimi zaburzeniami czynności nerek. Należy zachować ostrożność u tych pacjentów (patrz punkt 4.4).
Dzieci i młodzież
Produkt leczniczy Abiraterone Pharmascience nie ma odpowiedniego zastosowania w populacji dzieci
i młodzieży.
Sposób podawania
Produkt leczniczy Abiraterone Pharmascience podaje się doustnie.
Tabletki należy przyjmować jako pojedynczą dawkę raz na dobę na pusty żołądek. Produkt leczniczy Abiraterone Pharmascience należy przyjmować co najmniej dwie godziny po jedzeniu i nie wolno spożywać posiłków przez co najmniej jedną godzinę po przyjęciu tego produktu. Tabletki należy połykać w całości, popijając wodą.
Nadciśnienie, hipokaliemia, zastój płynów i niewydolność serca wynikające z nadmiaru
mineralokortykosteroidów
Produkt leczniczy Abiraterone Pharmascience może powodować nadciśnienie, hipokaliemię i zastój płynów (patrz punkt 4.8) jako następstwa zwiększenia stężeń mineralokortykosteroidów, wynikającego z hamowania CYP17 (patrz punkt 5.1).
Jednoczesne podawanie kortykosteroidu hamuje wydzielanie hormonu adrenokortykotropowego (ACTH), co skutkuje zmniejszeniem częstości i nasilenia tych działań niepożądanych. Należy zachować ostrożność podczas leczenia pacjentów, u których stan schorzeń współistniejących może ulec pogorszeniu w wyniku zwiększenia ciśnienia tętniczego, hipokaliemii (np. u stosujących glikozydy nasercowe) lub zastoju płynów (np. u pacjentów z niewydolnością serca, ciężką lub niestabilną dławicą piersiową, niedawno przebytym zawałem serca lub arytmią komorową oraz u pacjentów z ciężkimi zaburzeniami czynności nerek).
Produkt leczniczy Abiraterone Pharmascience należy stosować z ostrożnością u pacjentów z chorobami sercowo-naczyniowymi w wywiadzie. Badania fazy 3 abirateronu octanu nie dotyczyły pacjentów z niepoddającym się leczeniu nadciśnieniem tętniczym, istotną klinicznie chorobą serca, potwierdzoną zawałem mięśnia sercowego lub tętniczymi zdarzeniami zakrzepowymi w okresie ostatnich 6 miesięcy, z ciężką lub niestabilną dławicą piersiową lub niewydolnością serca klasy III lub IV wg NYHA (ang. New York Heart Association) (badanie 301) lub niewydolnością serca klasy II do IV (badania 0311 i 302) lub pacjentów z frakcją wyrzutową serca < 50%. Z badań 3011 i 302 wykluczono pacjentów z migotaniem przedsionków lub innymi arytmiami komorowymi, które wymagają leczenia. Nie określono bezpieczeństwa u pacjentów z frakcją wyrzutową lewej komory (ang. Left Ventricular Ejection Fraction, LVEF) < 50% lub z niewydolnością serca klasy III lub IV wg NYHA (w badaniu 301) lub niewydolnością serca klasy II do IV (w badaniach 3011 i 302) (patrz punkty 4.8 i 5.1).
Przed rozpoczęciem leczenia pacjentów z istotnym ryzykiem zastoinowej niewydolności serca (np. niewydolność serca w wywiadzie, nieopanowane nadciśnienie lub zdarzenia sercowe, takie jak choroba niedokrwienna serca) należy rozważyć wykonanie badań oceniających czynność serca
(np. echokardiografię). Przed rozpoczęciem terapii produktem leczniczym Abiraterone Pharmascience należy leczyć niewydolność serca i zoptymalizować czynność serca. Należy wyrównać i kontrolować nadciśnienie, hipokaliemię i zastój płynów. Podczas leczenia należy co 2 tygodnie przez 3 miesiące, a następnie co miesiąc monitorować ciśnienie krwi, stężenie potasu w osoczu, zastój płynów (zwiększenie masy ciała, obrzęki obwodowe) i inne objawy przedmiotowe i podmiotowe zastoinowej niewydolności serca i korygować nieprawidłowości. U pacjentów z hipokaliemią, podczas leczenia abirateronem stwierdzano wydłużenie odstępu QT. Należy oceniać czynność serca zgodnie ze wskazaniami klinicznymi, ustalić właściwe postępowanie i rozważyć zaprzestanie tego leczenia, gdy nastąpi znaczne pogorszenie czynności serca (patrz punkt 4.2).
Hepatotoksyczność i zaburzenia czynności wątroby
W kontrolowanych badaniach klinicznych stwierdzono znaczne zwiększenie aktywności enzymów wątrobowych, prowadzące do przerwania leczenia lub zmiany dawki (patrz punkt 4.8). Należy oceniać aktywność aminotransferaz w surowicy przed rozpoczęciem leczenia, co dwa tygodnie przez pierwsze trzy miesiące leczenia, a następnie co miesiąc. Jeśli kliniczne objawy podmiotowe i przedmiotowe wskazują na hepatotoksyczność, należy natychmiast dokonać pomiaru aktywności aminotransferaz w surowicy. Jeśli kiedykolwiek aktywność AlAT lub AspAT zwiększy się ponad 5-krotnie powyżej GGN, należy natychmiast przerwać leczenie i szczegółowo monitorować czynność wątroby. Wznowić leczenie w zmniejszonej dawce można tylko po powrocie wyników badań czynnościowych wątroby do wartości wyjściowych (patrz punkt 4.2).
Jeśli u pacjentów kiedykolwiek podczas terapii wystąpi ciężka hepatotoksyczność (aktywność AlAT lub AspAT zwiększona ponad 20 razy powyżej GGN), należy przerwać leczenie i nie rozpoczynać terapii ponownie.
Pacjentów z czynnym lub objawowym wirusowym zapaleniem wątroby nie włączono do badań klinicznych; dlatego nie ma danych potwierdzających celowość zastosowania produktu leczniczego Abiraterone Pharmascience w tej populacji.
Brak danych dotyczących bezpieczeństwa klinicznego i skuteczności wielokrotnych dawek abirateronu octanu stosowanego u pacjentów z umiarkowanymi lub ciężkimi zaburzeniami czynności wątroby (Klasa B lub C wg klasyfikacji Childa-Pugha). Zastosowanie produktu leczniczego Abiraterone Pharmascience należy rozważnie ocenić u pacjentów z umiarkowanymi zaburzeniami czynności wątroby, u których korzyści powinny wyraźnie przeważać nad możliwym ryzykiem (patrz punkty 4.2 i 5.2). Nie należy stosować produktu leczniczego Abiraterone Pharmascience u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkty 4.2, 4.3 i 5.2).
Po wprowadzeniu abirateronu octanu do obrotu zgłaszano rzadkie przypadki ostrej niewydolności wątroby i nadostrego zapalenia wątroby, niektóre zakończone zgonem (patrz punkt 4.8).
Odstawianie kortykosteroidów i zabezpieczenie sytuacji stresogennych
Zaleca się zachowanie ostrożności i obserwację w kierunku występowania objawów niewydolności nadnerczy, gdy pacjentom odstawia się prednizon lub prednizolon. Jeśli stosowanie produktu leczniczego Abiraterone Pharmascience jest kontynuowane po odstawieniu kortykosteroidów, pacjentów należy obserwować w kierunku występowania objawów nadmiaru mineralokortykosteroidów (patrz informacja powyżej).
U pacjentów przyjmujących prednizon lub prednizolon narażonych na wyjątkowy stres, może być wskazane zwiększenie dawki kortykosteroidów przed, w trakcie i po sytuacji stresogennej.
Gęstość kości
U mężczyzn z zaawansowanym rakiem gruczołu krokowego z przerzutami może wystąpić zmniejszenie gęstości kości. Stosowanie produktu leczniczego Abiraterone Pharmascience w skojarzeniu z glikokortykosteroidami może nasilić to działanie.
Wcześniejsze stosowanie ketokonazolu
U pacjentów, którzy stosowali wcześniej ketokonazol w leczeniu raka gruczołu krokowego można spodziewać się słabszej odpowiedzi na leczenie.
Hiperglikemia
Stosowanie glikokortykosteroidów może nasilać hiperglikemię, dlatego u pacjentów z cukrzycą należy często badać stężenie glukozy we krwi.
Hipoglikemia
Zgłaszano przypadki hipoglikemii po jednoczesnym podaniu abirateronu octanu z prednizonem/ prednizolonem pacjentom z istniejącą wcześniej cukrzycą, otrzymującym pioglitazon lub repaglinid (patrz punkt 4.5); dlatego u pacjentów z cukrzycą należy monitorować stężenie cukru we krwi.
Stosowanie podczas chemioterapii
Nie określono bezpieczeństwa ani skuteczności abirateronu octanu stosowanego jednocześnie
z cytotoksyczną chemioterapią (patrz punkt 5.1).
Nietolerancja substancji pomocniczych
Ten produkt leczniczy zawiera laktozę. Produkt leczniczy nie powinien być stosowany u pacjentów
z rzadko występującą dziedziczną nietolerancją galaktozy, brakiem laktazy (typu Lapp) lub zespołem złego wchłaniania glukozy-galaktozy.
Ten produkt leczniczy zawiera mniej niż 1 mmol (23 mg) sodu w dwóch tabletkach, to znaczy produkt leczniczy uznaje się za „wolny od sodu”.
Ryzyko związane ze stosowaniem
U mężczyzn z rakiem gruczołu krokowego z przerzutami, w tym u pacjentów przyjmujących produkt leczniczy Abiraterone Pharmascience, może wystąpić niedokrwistość i zaburzenia czynności seksualnych.
Wpływ na mięśnie szkieletowe
U pacjentów leczonych abirateronu octanem zgłaszano przypadki miopatii i rabdomiolizy. Większość zdarzeń wystąpiła w ciągu pierwszych 6 miesięcy leczenia, a po odstawieniu abirateronu octanu rabdomioliza ustąpiła. Należy zachować ostrożność u pacjentów leczonych jednocześnie produktami leczniczymi związanymi z występowaniem miopatii i (lub) rabdomiolizy.
Interakcje z innymi produktami leczniczymi
Ze względu na ryzyko zmniejszenia ekspozycji na abirateron należy unikać jednoczesnego stosowania silnych induktorów CYP3A4, chyba że nie istnieje alternatywne leczenie (patrz punkt 4.5).
Skojarzenie abirateronu i prednizonu/prednizolonu z Ra-223
W badaniach klinicznych stwierdzono, że leczenie abirateronem i prednizonem/prednizolonem,
w skojarzeniu z Ra-223 jest przeciwwskazane (patrz punkt 4.3) ze względu na zwiększone ryzyko złamań i tendencję do zwiększonej śmiertelności u pacjentów bezobjawowych lub z niewielkimi objawami z rakiem gruczołu krokowego. Zaleca się, aby kolejna terapia Ra-223 nie była rozpoczynana przez co najmniej 5 dni po ostatnim podaniu produktu leczniczego Abiraterone Pharmascience w skojarzeniu z prednizonem/prednizolonem.
Wpływ jedzenia na abirateronu octan
Podawanie z jedzeniem znacząco zwiększa wchłanianie abirateronu octanu. Nie ustalono skuteczności i bezpieczeństwa jego stosowania razem z jedzeniem, dlatego nie wolno produktu leczniczego Abiraterone Pharmascience przyjmować razem z jedzeniem (patrz punkty 4.2 i 5.2).
Interakcje z innymi produktami leczniczymi
Możliwość wpływu innych produktów leczniczych na ekspozycję na abirateron
W badaniu klinicznym interakcji farmakokinetycznych, przeprowadzonym u zdrowych osób otrzymujących wcześniej ryfampicynę – silny induktor CYP3A4 w dawce 600 mg na dobę przez
6 dni, podanie pojedynczej dawki 1000 mg abirateronu octanu, skutkowało zmniejszeniem średniego
AUC∞ abirateronu w osoczu o 55%.
Podczas terapii należy unikać stosowania silnych induktorów CYP3A4 [np.: fenytoiny, karbamazepiny, ryfampicyny, ryfabutyny, ryfapentyny, fenobarbitalu, ziela dziurawca zwyczajnego (Hypericum perforatum)], chyba że nie istnieje alternatywne leczenie.
W innym badaniu klinicznym interakcji farmakokinetycznych przeprowadzonym u zdrowych osób, jednoczesne podawanie ketokonazolu, silnego inhibitora CYP3A4, nie miało istotnego klinicznego wpływu na farmakokinetykę abirateronu.
Możliwość wpływu na ekspozycję na inne produkty lecznicze
Abirateron jest inhibitorem metabolizmu wątrobowych enzymów CYP2D6 i CYP2C8.
W badaniu określającym wpływ abirateronu octanu (w skojarzeniu z prednizonem) na pojedynczą dawkę dekstrometorfanu będącego substratem CYP2D6, całkowite narażenie na dekstrometorfan (AUC) zwiększyło się około 2,9 razy. AUC24 dekstrorfanu, czynnego metabolitu dekstrometorfanu, zwiększyło się o około 33%.
Zaleca się zachowanie ostrożności podczas jednoczesnego podawania z produktami leczniczymi aktywowanymi lub metabolizowanymi przez CYP2D6, szczególnie z produktami leczniczymi z wąskim indeksem terapeutycznym. Należy rozważyć zmniejszenie dawki produktów leczniczych z wąskim indeksem terapeutycznym metabolizowanych przez CYP2D6. Przykłady produktów leczniczych metabolizowanych przez CYP2D6: metoprolol, propranolol, dezypramina, wenlafaksyna, haloperydol, rysperydon, propafenon, flekainid, kodeina, oksykodon i tramadol (ostatnie trzy substancje czynne wymagają CYP2D6 do wytworzenia metabolitów czynnych przeciwbólowo).
W badaniu interakcji lekowych dotyczących cytochromu CYP2C8, przeprowadzonym u zdrowych osób, AUC pioglitazonu zwiększyło się o 46%, a AUC czynnych metabolitów pioglitazonu M-III i M-IV zmniejszyły się o 10%, gdy pioglitazon podawano z pojedynczą dawką 1000 mg abirateronu octanu. Należy obserwować pacjentów czy nie występują u nich objawy toksyczności, związane z jednocześnie stosowanymi substratami CYP2C8 z wąskim indeksem terapeutycznym. Przykładami leków metabolizowanych przez CYP2C8 są pioglitazon i repaglinid (patrz punkt 4.4).
W warunkach in vitro, główne metabolity to abirateronu siarczan i N-tlenku abirateronu siarczan, hamowały wychwyt wątrobowy nośnikiem OATP1B1, co może skutkować zwiększeniem stężeń leków eliminowanych przez OATP1B1. Brak dostępnych danych klinicznych potwierdzających interakcje z tym nośnikiem.
Stosowanie z produktami leczniczymi, które mogą wydłużać odstęp QT
Ponieważ terapia supresji androgenowej może powodować wydłużenie odstępu QT, należy zachować ostrożność podczas stosowania produktu leczniczego Abiraterone Pharmascience z produktami, które mogą wydłużać odstęp QT, lub z produktami leczniczymi, które mogą wywoływać częstoskurcz komorowy typu torsades de pointes, takimi jak: leki przeciwarytmiczne klasy IA (np. chinidyna, dyzopiramid) lub klasy III (np. amiodaron, sotalol, dofetylid, ibutylid), metadon, moksyfloksacyna, leki przeciwpsychotyczne, itp.
Stosowanie ze spironolaktonem
Spironolakton wiąże się z receptorem androgenowym i może zwiększać stężenie swoistego antygenu gruczołu krokowego (ang. prostate specific antygen, PSA). Nie zaleca się jego stosowania z produktem leczniczym Abiraterone Pharmascience (patrz punkt 5.1).
Kobiety w wieku rozrodczym
Brak danych dotyczących stosowania abirateronu octanu u kobiet w ciąży. Produktu leczniczego Abiraterone Pharmascience nie stosuje się u kobiet w wieku rozrodczym.
Antykoncepcja mężczyzn i kobiet
Nie wiadomo czy abirateron lub jego metabolity są obecne w nasieniu. Konieczne jest stosowanie prezerwatywy podczas aktywności seksualnej pacjenta z kobietą będącą w ciąży. W okresie aktywności seksualnej pacjenta z kobietą w okresie rozrodczym, konieczne jest stosowanie prezerwatywy jednocześnie z inną skuteczną metodą antykoncepcyjną.
Badania na zwierzętach wykazały szkodliwy wpływ na reprodukcję (patrz punkt 5.3).
Ciąża
Produktu leczniczego Abiraterone Pharmascience nie stosuje się u kobiet i jest on przeciwwskazany u
kobiet w ciąży lub, które mogą zajść w ciążę (patrz punkty 4.3 i 5.3).
Karmienie piersią
Produktu leczniczego Abiraterone Pharmascience nie stosuje się u kobiet.
Płodność
Abirateron wpływał na płodność u samców i samic szczurów, lecz te działania były całkowicie przemijające (patrz punkt 5.3).
Produkt leczniczy Abiraterone Pharmascience nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Charakterystyka profilu bezpieczeństwa
W analizie działań niepożądanych ze złożonych badań fazy 3 abirateronu octanu, działania niepożądane, które stwierdzono u ≥10% pacjentów to: obrzęk obwodowy, hipokaliemia, nadciśnienie tętnicze, zakażenia dróg moczowych i zwiększenie aktywności aminotransferazy alaninowej i (lub) zwiększenie aktywności aminotransferazy asparaginianowej. Inne ważne działania niepożądane to: choroby serca, hepatotoksyczność, złamania i alergiczne zapalenie pęcherzyków płucnych.
Produkt leczniczy Abiraterone Pharmascience może wywoływać nadciśnienie tętnicze, hipokaliemię i zastój płynów w następstwie swojego mechanizmu działania. W badaniach fazy 3 oczekiwane działania niepożądane mineralokortykosteroidowe stwierdzano częściej u pacjentów leczonych abirateronu octanem niż u pacjentów otrzymujących placebo odpowiednio: hipokaliemia 18% vs. 8%, nadciśnienie tętnicze 22% vs. 16% i zastój płynów (obrzęk obwodowy) 23% vs. 17%. U pacjentów leczonych abirateronu octanem w porównaniu z pacjentami otrzymującymi placebo: stwierdzano hipokaliemię stopnia 3. i 4. wg CTCAE (wersja 4.0) (ang. Common Terminology Criteria for Adverse Events) u odpowiednio 6% vs. 1%, nadciśnienie tętnicze stopnia 3. i 4. wg CTCAE (wersja 4.0) u odpowiednio 7% i 5% oraz zastój płynów (obrzęk obwodowy) stopnia 3. i 4. u odpowiednio 1% vs.
1%. Reakcje mineralokortykosteroidowe zwykle można było skutecznie leczyć. Jednoczesne zastosowanie kortykosteroidów zmniejsza częstość i nasilenie tych działań niepożądanych (patrz punkt 4.4).
Tabelaryczne zestawienie działań niepożądanych
W badaniach klinicznych, pacjentom z zaawansowanym rakiem gruczołu krokowego z przerzutami, którzy stosowali analogi LHRH lub byli wcześniej leczeni za pomocą orchidektomii, podawano abirateronu octan w dawce 1000 mg na dobę, w skojarzeniu z małą dawką prednizonu lub prednizolonu (5 lub 10 mg na dobę, w zależności od wskazania).
Działania niepożądane zgłaszane podczas badań klinicznych oraz po wprowadzeniu abirateronu octanu do obrotu przedstawiono poniżej wg kategorii częstości występowania. Kategorie częstości zdefiniowano następująco: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), niezbyt często
(≥ 1/1 000 do < 1/100), rzadko (≥1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000) i częstość
nieznana (nie można określić częstości na podstawie dostępnych danych).
W obrębie każdej kategorii o określonej częstości występowania, działania niepożądane są
wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 1: Działania niepożądane stwierdzone podczas badań klinicznych oraz po wprowadzeniuabirateronuoctanu do obrotu
Klasyfikacja układów i narządów | Działanie niepożądane i częstość |
Zakażenia i zarażenia pasożytnicze | bardzo często: zakażenie dróg moczowych często: posocznica |
Zaburzenia układu immunologicznego | częstość nieznana: reakcje anafilaktyczne |
Zaburzenia endokrynologiczne | niezbyt często: niewydolność nadnerczy |
Zaburzenia metabolizmu i odżywiania | bardzo często: hipokaliemia często: hipertriglicerydemia |
Zaburzenia serca | często: niewydolność serca*, dławica piersiowa, migotanie przedsionków, częstoskurcz niezbyt często: inne zaburzenia rytmu serca częstość nieznana: zawał mięśnia sercowego, wydłużenie odstępu QT (patrz punkty 4.4 i 4.5) |
Zaburzenia naczyniowe | bardzo często: nadciśnienie tętnicze krwi |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | rzadko: alergiczne zapalenie pęcherzyków płucnycha |
Zaburzenia żołądka i jelit | bardzo często: biegunka często: niestrawność |
Zaburzenia wątroby i dróg żółciowych | bardzo często: zwiększenie aktywności aminotransferazy alaninowej i (lub) zwiększenie aktywności aminotransferazy asparaginianowejb rzadko: nadostre zapalenie wątroby, ostra niewydolność wątroby |
Zaburzenia skóry i tkanki podskórnej | często: wysypka |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | niezbyt często: miopatia, rabdomioliza |
Zaburzenia nerek i dróg moczowych | często: krwiomocz |
Zaburzenia ogólne i stany w miejscu podania | bardzo często: obrzęk obwodowy |
Urazy, zatrucia i powikłania po zabiegach | często: złamania** |
* Niewydolność serca obejmuje także: zastoinową niewydolność serca, dysfunkcję lewej komory i
zmniejszenie frakcji wyrzutowej
** Złamania obejmują osteoporozę i wszystkie złamania poza złamaniami patologicznymi
asparaginianowej obejmuje zwiększenie aktywności AlAT, AspAT i nieprawidłową czynność wątroby.
Następujące działania niepożądane stopnia 3. wg CTCAE (wersja 4.0) wystąpiły u pacjentów leczonych abirateronu octanem: hipokaliemia u 5%; zakażenia dróg moczowych u 2%; zwiększenie aktywności AlAT i (lub) AspAT u 4%; nadciśnienie tętnicze u 6%; złamania u 2%; oraz następujące
u 1% pacjentów: obrzęk obwodowy, niewydolność serca i migotanie przedsionków. Hipertriglicerydemia stopnia 3. wg CTCAE (wersja 4.0) i dławica piersiowa wystąpiły u < 1% pacjentów. Zakażenia dróg moczowych stopnia 4. wg CTCAE (wersja 4.0), zwiększenie aktywności AlAT i (lub) AspAT, hipokaliemia, niewydolność serca, migotanie przedsionków i złamania wystąpiły u < 1% pacjentów.
Większą częstość nadciśnienia tętnicze i hipokaliemii obserwowano w populacji wrażliwej na hormony (badanie 3011). Nadciśnienie tętnicze stwierdzono u 36,7% pacjentów w populacji wrażliwej na hormony (badanie 3011) w porównaniu do 11,8% i 20,2%, odpowiednio, w badaniach 301 i 302.
Hipokaliemię zaobserwowano u 20,4% pacjentów w populacji wrażliwej na hormony (badanie 3011), w porównaniu do 19,2% i 14,9%, odpowiednio, w badaniach 301 i 302.
Częstość i nasilenie działań niepożądanych były większe w podgrupie pacjentów z wyjściowym statusem wydolności ECOG2, a także u pacjentów w podeszłym wieku (≥75 lat).
Opis wybranych działań niepożądanych
Sercowo-naczyniowe działania niepożądane
Trzy badania fazy 3 przeprowadzono z wyłączeniem pacjentów z niepoddającym się leczeniu nadciśnieniem tętniczym, istotną klinicznie chorobą serca, potwierdzoną zawałem mięśnia sercowego lub tętniczymi zdarzeniami zakrzepowymi w okresie ostatnich 6 miesięcy, z ciężką lub niestabilną dławicą piersiową lub niewydolnością serca klasy III lub IV (badanie 301) lub niewydolnością serca klasy II do IV (badania 3011 i 302) wg NYHA lub frakcją wyrzutową serca wynoszącą < 50%.
Wszyscy włączeni pacjenci (zarówno w grupie czynnie leczonej oraz w grupie placebo) otrzymywali jednocześnie supresję androgenową, głównie z zastosowaniem analogów LHRH, których stosowanie wiązało się z wystąpieniem cukrzycy, zawału mięśnia sercowego, incydentów mózgowo- naczyniowych i nagłego zgonu z przyczyn kardiologicznych. Częstość sercowo-naczyniowych działań niepożądanych w badaniach 3 fazy u pacjentów stosujących abirateronu octan, w porównaniu
z pacjentami przyjmującymi placebo była następująca: migotanie przedsionków 2,6% vs. 2,0%, tachykardia 1,9% vs. 1,0%, dławica piersiowa 1,7% vs. 0,8%, niewydolność serca 0,7% vs. 0,2%
i arytmia 0,7% vs. 0,5%.
Hepatotoksyczność
U pacjentów stosujących abirateronu octan stwierdzano hepatotoksyczność ze zwiększoną aktywnością AlAT, AspAT i stężenia całkowitego bilirubiny. W badaniach klinicznych fazy 3, stwierdzano hepatotoksyczność stopnia 3 i 4 (np. AlAT lub AspAT zwiększone o > 5 x powyżej górnej granicy normy [GGN] lub bilirubina zwiększona o > 1,5 x GGN) u około 6% pacjentów, którzy otrzymywali abirateronu octan, zwykle podczas pierwszych 3 miesięcy od rozpoczęcia terapii.
W badaniu 3011, stwierdzano hepatotoksyczność stopnia 3. lub 4. u 8,4% pacjentów leczonych abirateronu octanem. Dziesięciu pacjentów przerwało stosowanie abirateronu octanu z powodu hepatotoksyczności; dwóch miało hepatotoksyczność stopnia 2., sześciu hepatotoksyczność stopnia 3., a dwóch hepatotoksyczność stopnia 4. Nie było zgonu z powodu hepatotoksyczności w badaniu 3011. W badaniach fazy 3, pacjenci, u których wyjściowe wartości AlAT lub AspAT były podwyższone, częściej doświadczali zwiększenia wartości parametrów badań czynnościowych wątroby, niż u pacjenci rozpoczynający leczenie z prawidłowymi wartościami. Gdy stwierdzano zwiększenie AlAT lub AspAT o > 5 x GGN lub zwiększenie bilirubiny o > 3 x GGN, stosowanie abirateronu octanu było wstrzymywane lub przerywane. W dwóch przypadkach wystąpiło znaczne zwiększenie wyników testów czynnościowych wątroby (patrz punkt 4.4). U tych dwóch pacjentów z prawidłową wyjściową czynnością wątroby wystąpiło zwiększenie AlAT lub AspAT od 15 do 40 x GGN i zwiększenie bilirubiny od 2 do 6 x GGN. Po odstawieniu abirateronu octanu, u obu pacjentów testy czynnościowe wątroby powróciły do normy, a u jednego pacjenta wznowiono leczenie bez ponownego zwiększania się wyników testów. W badaniu 302 stwierdzono zwiększenie AlAT lub AspAT stopnia 3. lub 4. u 35 (6,5%) pacjentów leczonych abirateronu octanem.
Zwiększenie aktywności aminotransferaz ustąpiło u wszystkich z wyjątkiem 3 pacjentów (u 2
z nowymi przerzutami do wątroby, a u 1 ze zwiększeniem AspAT, po około 3 tygodniach od podania ostatniej dawki abirateronu octanu). W badaniach fazy 3, rezygnacje z leczenia z powodu zwiększenia AlAT i AspAT lub nieprawidłowej czynności wątroby stwierdzono u 1,1% pacjentów leczonych
abirateronu octanem i u 0,6% pacjentów otrzymujących placebo. Nie było przypadków zgonów
z powodu hepatotoksyczności.
Ryzyko hepatotoksyczności było zmniejszone w badaniach klinicznych wskutek wyłączenia pacjentów z wyjściowym zapaleniem wątroby lub znaczącymi odstępstwami od normy wartości parametrów badań czynnościowych wątroby. Z badania 3011 wykluczano pacjentów z wyjściowymi wartościami AlAT i AspaT ≥ 2,5 x GGN, bilirubiny > 1,5 x GGN oraz pacjentów z czynnym lub objawowym wirusowym zapaleniem wątroby lub przewlekłą chorobą wątroby; wodobrzusze lub zaburzenia krwotoczne jako następstwa zaburzeń czynności wątroby były wykluczane z badania.
Z badania 301 wykluczano pacjentów z wyjściowymi wartościami AlAT i AspAT ≥ 2,5 x GGN
w przypadku braku przerzutów do wątroby i > 5 x GGN w razie obecności przerzutów do wątroby. Z badania 302 wykluczano pacjentów z przerzutami do wątroby oraz z wyjściowymi wartościami AlAT i AspAT ≥ 2,5 x GGN. Wystąpienie nieprawidłowych wyników testów czynnościowych wątroby u pacjentów uczestniczących w badaniach klinicznych skutkowało zdecydowanym
postępowaniem z koniecznością przerwania leczenia i zezwoleniem na wznowienie terapii dopiero po powrocie wyników testów czynnościowych wątroby do wartości wyjściowych (patrz punkt 4.2).
Pacjentów ze zwiększeniem AlAT lub AspAT o > 20 x GGN nie leczono ponownie. Nieznane jest bezpieczeństwo ponownego rozpoczęcia terapii u tych pacjentów. Mechanizm hepatotoksyczności nie jest poznany.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, Al. Jerozolimskie 181C, 02-222 Warszawa
Tel.: + 48 22 49 21 301, Faks: + 48 22 49 21 309, strona internetowa: https://smz.ezdrowie.gov.pl. Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Dostępne są jedynie ograniczone dane dotyczące przedawkowania abirateronu octanu u ludzi.
Nie ma swoistego antidotum. W razie przedawkowania, leczenie należy przerwać i zastosować ogólne leczenie podtrzymujące, w tym obserwację czynności serca pod kątem niemiarowości, hipokaliemii
i objawów przedmiotowych i podmiotowych zastoju płynów. Należy również ocenić czynność wątroby.
Grupa farmakoterapeutyczna: Leki stosowane w terapii hormonalnej, inni antagoniści hormonów i ich
pochodne. Kod ATC: L02BX03
Mechanizm działania
Abirateronu octan jest przekształcany in vivo do abirateronu, inhibitora biosyntezy androgenów. Abirateron wybiórczo hamuje aktywność enzymu CYP17 (o aktywności 17α-hydroksylazy i C17,20-liazy). Enzym ten wykazuje swoje działanie i jest niezbędny do biosyntezy androgenów w jądrach, nadnerczach i tkankach nowotworowych gruczołu krokowego. CYP17 jest katalizatorem przemiany pregnenolonu i progesteronu do prekursorów testosteronu, DHEA i androstendionu, odpowiednio, w reakcji 17α-hydroksylacji i rozerwania wiązania C17, 20. Hamowanie CYP17 skutkuje także zwiększonym wytwarzaniem mineralokortykosteroidów w nadnerczach (patrz punkt 4.4).
Rak gruczołu krokowego, który jest wrażliwy na androgeny, reaguje na leczenie zmniejszające stężenia androgenów. Terapie supresji androgenowej, takie jak leczenie analogami LHRH lub orchidektomia, zmniejszają wytwarzanie androgenów w jądrach, lecz nie wpływają na wytwarzanie androgenów w nadnerczach lub przez nowotwór. Leczenie abirateronu octanem zmniejsza stężenie testosteronu w osoczu do wartości nieoznaczalnych (przy zastosowaniu testów komercyjnych),
gdy jest stosowany z analogami LHRH (lub orchidektomią).
Rezultat działania farmakodynamicznego
Abirateronu octan zmniejsza stężenie testosteronu i innych androgenów w surowicy do wartości niższych niż uzyskiwane po zastosowaniu samych analogów LHRH lub za pomocą orchidektomii. Wynika to z wybiórczego hamowania enzymu CYP17 niezbędnego do biosyntezy androgenów.
PSA służy jako biomarker u pacjentów z rakiem gruczołu krokowego. W badaniu klinicznym fazy 3 u pacjentów, którzy mieli niepowodzenie wcześniejszej chemioterapii z zastosowaniem taksanów,
38% pacjentów leczonych abirateronu octanem, vs. 10% otrzymujących placebo, uzyskało co najmniej
50% zmniejszenie wartości PSA w porównaniu do wartości wyjściowych.
Skuteczność kliniczna i bezpieczeństwo stosowania
Skuteczność oceniano w trzech randomizowanych, wieloośrodkowych badaniach klinicznych 3 fazy
z kontrolą placebo (badania 3011, 302 i 301) u pacjentów z mHSPC i mCRPC.
Do badania 3011 włączono pacjentów z nowym rozpoznaniem mHSPC (w okresie 3 miesięcy od randomizacji), którzy mieli czynniki prognostyczne wysokiego ryzyka. Czynniki wysokiego ryzyka określono jako posiadanie co najmniej 2 z 3 następujących czynników ryzyka: (1) suma Gleason’a ≥8;
(2) obecność 3 lub więcej zmian w RTG kości; (3) obecność mierzalnych przerzutów trzewnych
(z wyłączeniem węzłów chłonnych). W ramieniu z czynnym leczeniem abirateronu octanem w dawce 1000 mg na dobę, w skojarzeniu z małą dawką prednizonu 5 mg raz na dobę i supresją androgenową (agonista LHRH lub orchidektomia), co było standardem leczenia. Pacjenci w grupie kontrolnej otrzymywali supresję androgenową i placebo zamiast abirateronu octanu i prednizonu.
Do badania 302 włączono pacjentów, którzy wcześniej nie otrzymywali docetakselu; podczas, gdy do badania 301 włączono pacjentów, którzy wcześniej otrzymywali docetaksel. Pacjenci stosowali analogi LHRH lub mieli wykonaną wcześniej orchidektomię. W czynnie leczonej grupie badanych, abirateronu octan podawano w dawce 1000 mg na dobę w skojarzeniu z małą dawką prednizonu lub
prednizolonu 5 mg dwa razy na dobę. Grupa kontrolna otrzymywała placebo i małą dawkę prednizonu lub prednizolonu 5 mg dwa razy na dobę.
Zmiany stężenia PSA w osoczu, niezależnie od parametru, nie zawsze wskazują na korzystne rezultaty kliniczne. Dlatego we wszystkich badaniach zalecano, aby pacjenci kontynuowali leczenie do momentu spełnienia kryteriów wykluczenia, podanych poniżej dla każdego badania.
Stosowanie spironolaktonu było niedozwolone we wszystkich badaniach, gdyż spironolakton wiąże się z receptorem androgenowym i może zwiększać stężenie PSA.
Badanie 3011 (pacjenci z nowym rozpoznaniem mHSPC wysokiego ryzyka)
W badaniu 3011 (n=1199), mediana wieku pacjentów włączonych do badania wynosiła 67 lat. Liczba pacjentów leczonych abirateronu octanem wg grup rasowych była następująca: biała 832 (69,4%), azjatycka 246 (20,5%), czarna lub afroamerykańska 25 (2,1%), inna 80 (6,7%), nieznana/niezgłoszona
13 (1,1%) oraz Indianie amerykańscy lub rdzenni mieszkańcy Alaski 3 (0,3%).
Status wydolności ECOG wynosił 0 lub 1 u 97% pacjentów. Pacjenci ze stwierdzonymi przerzutami do mózgu, niekontrolowanym nadciśnieniem tętniczym, ciężką chorobą serca lub niewydolnością serca klasy II do IV NYHA zostali wykluczeni z badania. Pacjenci wcześniej leczeni na raka gruczołu krokowego farmakoterapią, radioterapią lub chirurgicznie, zostali wykluczeni z badania, z wyjątkiem tych poddanych terapii ADT do 3 miesięcy lub 1 cyklowi radioterapii paliatywnej lub terapii chirurgicznej, w celu leczenia objawów wynikających z przerzutów. Równorzędnymi, pierwszorzędowymi punktami końcowymi skuteczności były przeżycie całkowite (OS), przeżycie bez progresji radiograficznej (rPFS). Mediana wyjściowej skali bólu, oceniana za pomocą skróconego formularza bólu (BPI-SF), wyniosła 2,0 w obu grupach terapeutycznych i w grupie placebo. Oprócz powyższych punktów końcowych, oceniono także korzyści w zakresie czasu do wystąpienia zdarzenia
związanego z kośćcem (SRE), czasu do następnej terapii raka gruczołu krokowego, czasu do rozpoczęcia chemioterapii, czasu do progresji bólowej i czasu do progresji PSA. Leczenie kontynuowano do czasu progresji choroby, wycofania zgody na udział w badaniu, wystąpienia nietolerowanej toksyczności lub zgonu. Przeżycie bez progresji radiograficznej definiowano jako czas od randomizacji do wystąpienia progresji radiograficznej lub zgonu z jakiejkolwiek przyczyny.
Progresja radiograficzna obejmowała progresję w RTG kości (zgodnie ze zmodyfikowanym PCWG2) lub progresję zmian w tkankach miękkich w TK lun NMR (zgodnie z RECIST 1.1).
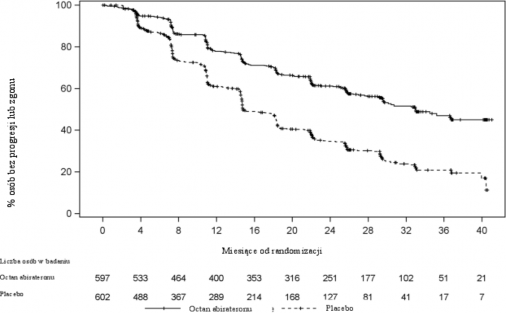
Stwierdzono istotną różnicę w rPFS pomiędzy grupami terapeutycznymi (patrz Tabela 2 i Wykres 1).
Tabela 2: Przeżycie bez progresji radiograficznej - analiza stratyfikacyjna: populacja z zamiarem leczenia (ITT) (badanie PCR 3011)
AA-P | Placebo | |
Osoby randomizowane | 597 | 602 |
Zdarzenie | 239 (40,0%) | 354 (58,8%) |
Ocenzurowano | 358 (60,0%) | 248 (41,2%) |
Czas do zdarzenia (miesiące) | ||
Mediana (95% CI) | 33,02 (29,57; NE) | 14,78 (14,69; 18,27) |
Zakres | (0,0+; 41,0+) | (0,0+; 40,6+) |
Wartość pa | < 0,0001 | |
Współczynnik ryzyka (95% CI)b | 0,466 (0,394; 0,550) | |
Uwaga: += obserwacja ocenzurowana, NE = brak możliwości oceny. Progresja radiograficzna i zgon były brane pod uwagę w definiowaniu zdarzenia rPFS. AA-P = osoby, które otrzymywały abirateronu octan i prednizon.
<1 na korzyść AA-P.
Wykres 1: Krzywa przeżycia Kaplana-Meiera bez progresji radiograficznej; populacja ITT (badanie PCR 3011)
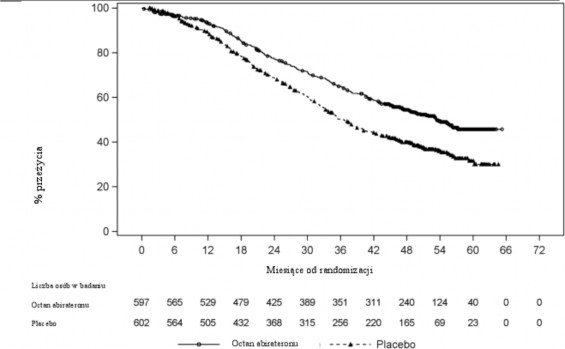
Stwierdzono statystycznie znamienną poprawę OS na korzyść AA-P plus ADT z 34% zmniejszeniem ryzyka zgonu w porównaniu do placebo plus ADT (HR=0,66; 95% CI: 0,56; 0,78; p<0,0001)
(patrz Tabela 3 i Wykres 2).
Tabela 3: Całkowite przeżycie pacjentów otrzymujących abirateronu octan lub placebo w badaniu PCR3011 (analiza z zamiarem leczenia)
Całkowite przeżycie | Abirateron z prednizonem (N=597) | Placebo (N=602) |
Zgony (%) | 275 (46%) | 343 (57%) |
Mediana przeżycia (miesiące) | 53,3 | 36,5 |
(95% CI) | (48,2; NE) | (33,5; 40,0) |
Współczynnik ryzyka (95% CI)1 | 0,66 (ʻ0,56ʼ, ʻ0,78ʼ) | |
NE = brak możliwości oceny
1 Współczynnik ryzyka pochodzi ze stratyfikowanego proporcjonalnego modelu ryzyka. Współczynnik ryzyka
<1 na korzyść abirateronu octanu z prednizonem.
Wykres 2: Krzywa Kaplana-Meiera całkowitego przeżycia; populacja ITT z analizy badania PCR3011
Analizy podgrup spójnie wykazują korzyści leczenia abirateronu octanem. Wyniki leczenia AA-P na rPFS i OS w ustalonych wcześniej podgrupach były korzystne i spójne z całkowitą populacją badania z wyjątkiem podgrupy z wynikiem 2 ECOG, gdzie nie stwierdzono korzystnego trendu, jednakże niewielka liczba badanych (n=40) ogranicza możliwości wyciągnięcia wiążących wniosków.
Oprócz stwierdzonej poprawy w przeżyciu całkowitym i rPFS, wykazano korzyści stosowania abirateronu octanu w porównaniu z placebo, we wszystkich prospektywnie zdefiniowanych drugorzędowych punktach końcowych.
Badanie 302 (pacjenci, którzy wcześniej nie otrzymywali chemioterapii)
Do badania włączono pacjentów bez objawów lub z objawami o łagodnym nasileniu, którzy wcześniej nie stosowali chemioterapii i u których zastosowanie chemioterapii nie było jeszcze wskazane klinicznie. Wynik 0-1 w skali BPI-SF (ang. Brief Pain Inventory-Short Form) dla najsilniejszego bólu w ciągu ostatnich 24 godzin uznawano za brak objawów, a wynik 2-3 uznawano za objawy
o łagodnym nasileniu.
W badaniu 302 (n=1088) mediana wieku włączonych pacjentów wyniosła 71 lat dla pacjentów leczonych abirateronu octanem z prednizonem lub prednizolonem oraz 70 lat dla pacjentów otrzymujących placebo z prednizonem lub prednizolonem. Liczby pacjentów leczonych abirateronu octanem wg ras były następujące: biała 520 (95,4%), czarna 15 (2,8%), azjatycka 4 (0,7%) i inne 6 (1,1%). Status wydolności ECOG (ang. Eastern Cooperative Oncology Group) wynosił 0 dla 76% pacjentów i 1 dla 24% pacjentów w obu grupach. 50% pacjentów miało tylko przerzuty do kości, dodatkowe 31% pacjentów miało przerzuty do kości i tkanek miękkich lub do węzłów chłonnych, a 19% pacjentów miało tylko przerzuty do tkanek miękkich lub do węzłów chłonnych. Wykluczano pacjentów z przerzutami trzewnymi. Współtowarzyszące pierwszorzędowe punkty końcowe to całkowite przeżycie i przeżycie bez radiograficznej progresji (rPFS). Ponadto, oprócz oceny współtowarzyszących pierwszorzędowych punktów końcowych, oceniano także korzyści z wykorzystaniem czasu do zastosowania opioidów w bólu nowotworowym, czasu do włączenia chemioterapii cytotoksycznej, czasu do pogorszenia punktacji wydolności ECOG o ≥1 punkt i czasu do progresji PSA, w oparciu o kryteria PCWG2 (ang. Prostate Cancer Working Group-2). Leczenie zakończono w momencie stwierdzenia ewidentnej progresji klinicznej. Leczenie mogło być także zakończone w momencie potwierdzonej progresji radiograficznej wg uznania badacza.
Przeżycie bez radiograficznej progresji (rPFS) oceniano z zastosowaniem sekwencyjnych badań obrazowych, definiowanych za pomocą kryteriów PCWG2 (dla uszkodzeń kości) i modyfikowanych kryteriów RECIST (Response Evaluation Criteria In Solid Tumors) (dla uszkodzeń tkanek miękkich). W analizie rPFS wykorzystywano rewidowaną centralnie radiograficzną ocenę progresji.
W zaplanowanej analizie rPFS było 401 zdarzeń, 150 (28%) pacjentów leczonych abirateronu octanem i 251 (46%) pacjentów otrzymujących placebo miało radiograficzne potwierdzenie progresji lub zmarło. Stwierdzono pomiędzy grupami istotne różnice w rPFS (patrz Tabela 4. i Wykres 3.).
Tabela 4: Badanie 302: Przeżycie bez progresji radiograficznej u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii
Abirateronu octan (N=546) | Placebo (N=542) | |
Przeżycie bez radiograficznej progresji (rPFS) | ||
Progresja lub zgon | 150 (28%) | 251 (46%) |
Mediana rPFS w miesiącach (95% CI) | Nie oceniono (11,66; NE) | 8,3 (8,12; 8,54) |
wartość p* | < 0,0001 | |
Współczynnik ryzyka**(95% CI) | 0,425 (0,347; 0,522) | |
NE = nie oceniono
(0 lub 1).
** Współczynnik ryzyka < 1 na korzyść abirateronu octanu.
Wykres 5: krzywe przeżycia Kaplana-Meiera u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii, analiza końcowa
AA = abirateronu octan
Oprócz stwierdzonej poprawy w całkowitym czasie przeżycia i rPFS, wykazano korzyści z terapii
abirateronem vs. placebo we wszystkich drugorzędowych punktach końcowych zgodnie z poniższym:
Czas do progresji PSA wg kryteriów PCWG2
Mediana czasu do progresji PSA wyniosła 11,1 miesięcy dla pacjentów otrzymujących abirateronu octan i 5,6 miesięcy dla pacjentów otrzymujących placebo (HR=0,488; 95% CI: [0,420, 0,568], p < 0,0001). Czas do progresji PSA był około dwukrotnie dłuższy podczas terapii abirateronu octanem (HR=0,488). Odsetek pacjentów z potwierdzoną odpowiedzią PSA był większy w grupie abirateronu octanu niż w grupie placebo (62% vs. 24%; p < 0,0001). U pacjentów z mierzalną chorobą tkanek miękkich, podczas leczenia abirateronu octanu zaobserwowano znaczące zwiększenie liczby całkowitych lub częściowych odpowiedzi guzów na leczenie.
Czas do zastosowania opioidów w bólu nowotworowym
Mediana czasu do zastosowania opioidów w bólu nowotworowym gruczołu krokowego w czasie analizy końcowej wyniosła 33,4 miesiące u pacjentów stosujących abirateronu octan, a u pacjentów otrzymujących placebo wyniosła 23,4 miesiące (HR=0,721; 95% CI: [0,614; 0,846]; p < 0,0001).
Czas do włączenia cytotoksycznej chemioterapii
Mediana czasu do włączenia cytotoksycznej chemioterapii wyniosła 25,2 miesięcy u pacjentów stosujących abirateronu octan, a u pacjentów otrzymujących placebo wyniosła 16,8 miesięcy (HR = 0,580; 95% CI: [0,487; 0,691], p < 0,0001).
Czas do pogorszenia punktacji wydolności ECOG o ≥ 1 punkt
Mediana czasu do pogorszenia punktacji wydolności ECOG o ≥ 1 punkt wyniosła 12,3 miesięcy u pacjentów stosujących abirateronu octan, a u pacjentów otrzymujących placebo wyniosła 10,9 miesięcy (HR = 0,821; 95% CI: [0,714; 0,943], p = 0,0053).
Następujące punkty końcowe wykazały statystycznie znamienną przewagę terapii abirateronu octanem:
Obiektywna odpowiedź
Obiektywną odpowiedź definiuje się jako proporcję badanych osób z mierzalną chorobą, uzyskujących całkowitą lub częściową odpowiedź zgodnie z kryteriami RECIST (wymagana wyjściowa wielkość węzła chłonnego ≥ 2 cm, by uznać ją za zmianę docelową). Odsetek badanych z mierzalną chorobą w punkcie wyjścia, którzy mieli obiektywną odpowiedź, wyniósł 36% w grupie abirateronu octanu i 16% w grupie placebo (p < 0,0001).
Ból
Leczenie abirateronu octanem znacząco zmniejszyło ryzyko progresji średniego nasilenia bólu o 18% w porównaniu z placebo (p = 0,0490). Mediana czasu do progresji wyniosła 26,7 miesięcy w grupie abirateronu octanu i 18,4 miesięcy w grupie placebo.
Czas do pogorszenia FACT-P (całkowitego wyniku)
Leczenie abirateronu octanem zmniejszyło ryzyko pogorszenia FACT-P (całkowitego wyniku) o 22% w porównaniu z placebo (p=0,0028). Mediana czasu do pogorszenia FACT-P (całkowitego wyniku) wyniosła 12,7 miesięcy w grupie abirateronu octanu i 8,3 miesięcy w grupie placebo.
Badanie 301 (pacjenci otrzymujący wcześniej chemioterapię)
Do badania 301 włączono pacjentów, którzy otrzymywali wcześniej docetaksel. Nie było wymagane, by pacjenci wykazywali progresję choroby w trakcie leczenia docetakselem, gdyż toksyczność tej chemioterapii może skutkować przerwaniem leczenia. U pacjentów kontynuowano podawanie badanych leków aż do progresji PSA (potwierdzone 25% zwiększenie wartości w stosunku do wartości wyjściowych/najniższych [nadir]) wspólnie z progresją radiograficzną, definiowaną wg protokołu i progresją w zakresie objawów klinicznych. Pacjentów z wcześniejszą terapią raka gruczołu krokowego ketokonazolem wyłączono z badania. Pierwszorzędowym punktem końcowym skuteczności był całkowity czas przeżycia.
Mediana wieku włączonych pacjentów wyniosła 69 lat (zakres 39 – 95). Liczba pacjentów leczonych abirateronu octanem wg grup rasowych była następująca: rasa biała 737 (93,2%), rasa czarna 28 (3,5%), rasa azjatycka 11 (1,4%) i inni 14 (1,8%). Jedenaście procent włączonych pacjentów miało punktację wydolności w skali ECOG wynoszącą 2; 70% miało wyniki radiograficzne wskazujące na postęp choroby z lub bez progresji PSA; 70% otrzymało wcześniej jeden cykl cytotoksycznej chemioterapii, a 30% otrzymało dwa cykle. U 11% pacjentów leczonych abirateronu octanem występowały przerzuty do wątroby.
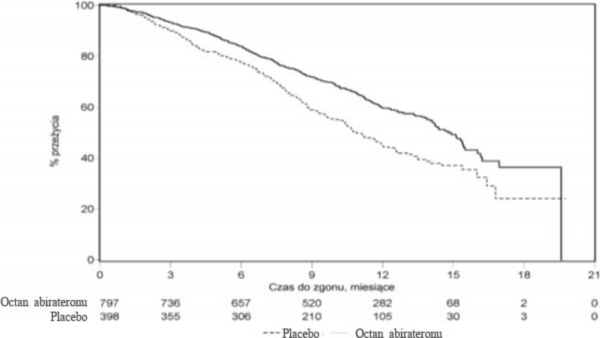
W planowej analizie przeprowadzonej po 552 zgonach stwierdzono, że zmarło 42% (333 z 797) pacjentów leczonych abirateronu octanem w porównaniu z 55% (219 z 398) pacjentów otrzymujących placebo. Wykazano istotną statystycznie poprawę w medianie całkowitego czasu przeżycia u pacjentów leczonych abirateronu octanem (patrz Tabela 7).
Tabela 7: Całkowity czas przeżycia pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii
Abirateronu octan (N=797) | Placebo (N=398) | |
Pierwotna analiza przeżywalności | ||
Zgony (%) | 333 (42%) | 219 (55%) |
Mediana przeżycia w miesiącach (95% CI) | 14,8 (14,1; 15,4) | 10,9 (10,2; 12,0) |
wartość pa | < 0,0001 | |
Współczynnik ryzyka (95% CI)b | 0,646 (0,543; 0,768) | |
Analiza końcowa przeżycia | ||
Zgony (%) | 501 (63%) | 274 (69%) |
Mediana przeżycia w miesiącach (95% CI) | 15,8 (14,8; 17,0) | 11,2 (10,4; 13,1) |
Współczynnik ryzyka (95% CI)b | 0,740 (0,638; 0,859) | |
< 1 na korzyść abirateronu octanu.
We wszystkich ocenianych punktach czasowych, po kilku początkowych miesiącach leczenia, większy odsetek pacjentów leczonych abirateronu octanem pozostawał przy życiu w porównaniu z pacjentami otrzymującymi placebo (patrz Wykres 6).
Wykres 6: Krzywe przeżycia Kaplana-Meiera u pacjentów otrzymujących abirateronu octan lub placebo w skojarzeniu z prednizonem lub prednizolonem oraz analogami LHRH lub po wcześniejszej orchidektomii
Analizy przeżycia w podgrupach zgodnie wykazały korzyści z terapii abirateronu octanem (patrz Wykres 7).
Wykres 7: Całkowity czas przeżycia w podgrupach: współczynnik ryzyka i 95% przedział
ufności
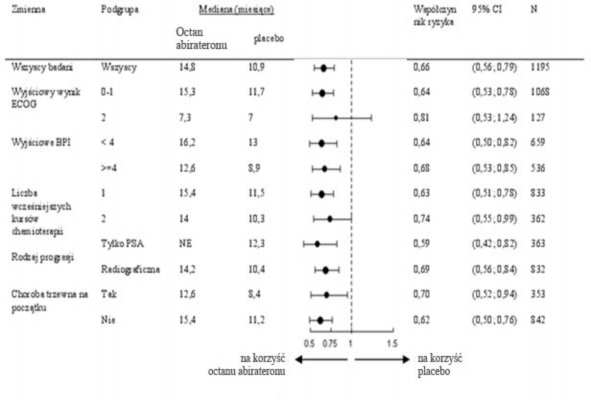
BPI = Skala bólu; CI = przedział ufności; ECOG = skala wydolności ECOG (ang. Eastern Cooperative Oncology Group); HR = współczynnik ryzyka; NE = brak możliwości oceny
Oprócz stwierdzonej poprawy całkowitego czasu przeżycia, wszystkie drugorzędowe punkty końcowe badania faworyzowały abirateronu octan i były znamienne statystycznie po dostosowaniu do testów wielokrotnych, jak następuje:
Pacjenci, którzy otrzymywali abirateronu octan wykazali znacznie większą całkowitą częstość odpowiedzi PSA (definiowaną jako ≥ 50% zmniejszenie z punktu wyjścia) w porównaniu do pacjentów otrzymujących placebo, 38% vs. 10%, p < 0,0001.
Mediana czasu do progresji PSA wynosiła 10,2 miesięcy dla pacjentów leczonych abirateronu octanem i 6,6 miesięcy dla pacjentów otrzymujących placebo (HR = 0,580; 95% CI: [0,462; 0,728],
p < 0,0001).
Mediana czasu przeżycia bez radiograficznej progresji wynosiła 5,6 miesięcy dla pacjentów leczonych abirateronu octanem i 3,6 miesięcy dla pacjentów otrzymujących placebo (HR = 0,673; 95% CI: [0,585; 0,776], p < 0,0001).
Ból
Odsetek pacjentów u których wystąpiło złagodzenie objawów bólowych był znamiennie statystycznie większy w grupie abirateronu octanu niż w grupie placebo (44% vs. 27%, p=0,0002). Pacjent odpowiadający na leczenie w zakresie złagodzenia objawów bólowych był definiowany jako pacjent, który uzyskiwał co najmniej 30% zmniejszenie wyniku BPI-SF nasilenia najgorszego bólu w porównaniu do wartości wyjściowych, w ciągu ostatnich 24 godzin, bez żadnego zwiększenia punktacji zastosowania leków przeciwbólowych, stwierdzane w dwóch następujących po sobie badaniach w odstępie czterotygodniowym. Łagodzenie objawów bólowych analizowano tylko u pacjentów (n=512) z wyjściową punktacją bólu ≥ 4 i co najmniej jednym wynikiem oceny bólu uzyskanym po badaniu wstępnym.
Mniejszy odsetek pacjentów leczonych abirateronu octanem miał progresję bólu w porównaniu do pacjentów otrzymujących placebo w 6 (22% vs. 28%), 12 (30% vs. 38%) i 18 miesiącu (35% vs. 46%). Progresję bólu definiowano jako zwiększenie z punktu wyjścia o ≥ 30% wyniku BPI-SF nasilenia najgorszego bólu w ciągu ostatnich 24 godzin, bez zmniejszenia punktacji zastosowania leków przeciwbólowych, stwierdzane w dwu następujących po sobie wizytach lub zwiększenie
o ≥ 30% punktacji zastosowania leków przeciwbólowych, stwierdzane w dwu następujących po sobie wizytach. Czas do progresji bólu w 25. percentylu wynosił 7,4 miesięcy w grupie abirateronu, w porównaniu do 4,7 miesięcy w grupie placebo.
Zdarzenia dotyczące kośćca
U mniejszego odsetka pacjentów w grupie abirateronu octanu występowały zdarzenia dotyczące kośćca w porównaniu do grupy placebo po 6 miesiącach (18% vs. 28%), 12 miesiącach (30% vs. 40%), i 18 miesiącach (35% vs. 40%) terapii. Czas do wystąpienia pierwszego zdarzenia dotyczącego kośćca w 25. percentylu w grupie abirateronu octanu był dwukrotnie dłuższy niż w grupie kontrolnej: 9,9 miesięcy w porównaniu do 4,9 miesięcy. Zdarzenia dotyczące kośćca to: złamania patologiczne, ucisk rdzenia kręgowego, paliatywną radioterapię kości lub zabiegi chirurgiczne kości.
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego we wszystkich podgrupach populacji dzieci i młodzieży w zaawansowanym raku gruczołu krokowego. Zastosowanie u dzieci i młodzieży patrz punkt 4.2.
Zbadano farmakokinetykę abirateronu i abirateronu octanu po podaniu abirateronu octanu u zdrowych
osób, pacjentów z zaawansowanym rakiem gruczołu krokowego z przerzutami oraz u osób bez raka z zaburzeniami czynności wątroby i nerek. Abirateronu octan jest szybko przekształcany in vivo do abirateronu, inhibitora biosyntezy androgenów (patrz punkt 5.1).
Wchłanianie
Po doustnym podaniu na czczo abirateronu octanu, czas do osiągnięcia maksymalnego stężenia
w osoczu wynosił około 2 godziny.
Podawanie abirateronu octanu z jedzeniem w porównaniu do podawania na czczo skutkowało nawet 10-krotnym zwiększeniem (AUC) i 17-krotnym (Cmax) zwiększeniem średniego całkowitego wpływu abirateronu na organizm, zależnego od zawartości tłuszczu w posiłku. Biorąc pod uwagę różnorodność zawartości i składu posiłków, przyjmowanie abirateronu octanu z posiłkami może potencjalnie skutkować dużą zmiennością ekspozycji. Dlatego produktu leczniczego Abiraterone Pharmascience nie wolno przyjmować razem z jedzeniem. Tabletki produktu leczniczego należy przyjmować jako pojedynczą dawkę raz na dobę na pusty żołądek. Produkt leczniczy Abiraterone Pharmascience należy stosować co najmniej dwie godziny po jedzeniu i nie wolno spożywać posiłków przez co najmniej jedną godzinę po przyjęciu tego produktu. Tabletki należy połykać w całości popijając wodą (patrz punkt 4.2).
Dystrybucja
Znakowany 14C-abirateron wiąże się z ludzkimi białkami osocza w 99,8%. Pozorna objętość dystrybucji wynosi około 5 630 l, co sugeruje, że abirateron podlega znacznej dystrybucji w tkankach obwodowych.
Metabolizm
Po doustnym podaniu znakowanego 14C- abirateronu octanu w kapsułkach, jest on hydrolizowany do abirateronu, który następnie podlega metabolizmowi m.in. sulfuryzacji, hydroksylacji i utlenianiu, głównie w wątrobie. Większość krążącej promieniotwórczości (około 92%) jest znajdowane w postaci metabolitów abirateronu. Z 15 wykrytych metabolitów, 2 podstawowe metabolity, abirateronu siarczan i N-tlenku abirateronu siarczan, stanowią około 43% całkowitej promieniotwórczości każdy.
Eliminacja
Na podstawie danych uzyskanych od zdrowych osób średni okres półtrwania abirateronu w osoczu wynosi około 15 godzin. Po doustnym podaniu dawki 1000 mg znakowanego 14C- abirateronu octanu około 88% dawki promieniotwórczej znajduje się w kale, a około 5% w moczu. Większość składników znalezionych w kale stanowi niezmieniony abirateronu octan i abirateron (odpowiednio około 55% i 22% podanej dawki).
Zaburzenia czynności wątroby
Zbadano farmakokinetykę abirateronu octanu u osób z istniejącymi wcześniej łagodnymi lub umiarkowanymi zaburzeniami czynności wątroby (odpowiednio klasa A i B wg klasyfikacji Childa- Pugha) oraz w grupie kontrolnej zdrowych osób. Całkowite narażenie organizmu na abirateron po pojedynczym doustnym podaniu dawki 1000 mg zwiększało się odpowiednio o 11% i 260% u osób z łagodnymi i umiarkowanymi zaburzeniami czynności wątroby. Średni okres półtrwania abirateronu wydłużył się do około 18 godzin u osób z łagodnymi zaburzeniami czynności wątroby i do około 19 godzin u osób z umiarkowanymi zaburzeniami czynności wątroby.
W innym badaniu oceniano farmakokinetykę abirateronu u 8 osób z wcześniej występującymi ciężkimi zaburzeniami wątroby (klasa C wg klasyfikacji Childa-Pugha) oraz w grupie kontrolnej u 8 zdrowych osób z prawidłową czynnością wątroby. AUC abirateronu zwiększyła się o około 600%, a wolna frakcja leku zwiększyła się o 80% u osób z ciężkimi zaburzeniami wątroby w porównaniu z osobami z prawidłową czynnością wątroby.
Nie jest konieczne dostosowywanie dawki u pacjentów z występującymi wcześniej łagodnymi zaburzeniami czynności wątroby. Zastosowanie abirateronu octanu należy rozważnie ocenić u pacjentów z umiarkowanymi zaburzeniami czynności wątroby, u których korzyści powinny wyraźnie przeważać nad ryzykiem (patrz punkty 4.2 i 4,4). Nie należy stosować abirateronu octanu u pacjentów z ciężkimi zaburzeniami czynności wątroby (patrz punkty 4.2, 4.3 i 4.4).
U pacjentów, u których wystąpi hepatotoksyczność podczas leczenia, może być konieczne
zawieszenie leczenia lub dostosowanie dawki (patrz punkty 4.2 i 4.4).
Zaburzenia czynności nerek
Porównano farmakokinetykę abirateronu octanu u pacjentów z krańcowym stadium choroby nerek, stabilnych na hemodializie z dopasowaną grupą kontrolną osób z prawidłową czynnością nerek.
Całkowite narażenie organizmu na abirateron po pojedynczym doustnym podaniu dawki 1000 mg nie zwiększyło się u dializowanych pacjentów z krańcowym stadium choroby nerek. Nie jest konieczne zmniejszanie podawanej dawki u pacjentów z zaburzeniami czynności nerek, w tym z ciężkimi zaburzeniami czynności nerek (patrz punkt 4.2). Jednakże, brak jest danych klinicznych u pacjentów z rakiem gruczołu krokowego i ciężkimi zaburzeniami czynności nerek. Należy zachować ostrożność u tych pacjentów.
We wszystkich badaniach toksyczności u zwierząt stwierdzano znaczne zmniejszenie stężeń krążącego testosteronu. Skutkiem tego, występowało zmniejszenie masy narządów oraz zmiany morfologiczne i (lub) histopatologiczne w narządach rozrodczych, nadnerczach, przysadce i sutkach. Wszystkie zmiany były całkowicie lub częściowo odwracalne. Zmiany w narządach rozrodczych oraz narządach wrażliwych na androgeny są zgodne z farmakologią abirateronu. Wszystkie związane
z leczeniem zmiany hormonalne były odwracalne lub ustępowały po okresie 4 tygodni.
W badaniach nad płodnością, zarówno u samców jak i samic szczurów, abirateronu octan zmniejszał płodność, co było całkowicie odwracalne w ciągu 4 do 16 tygodni od przerwania podawania abirateronu octanu.
W badaniu toksycznego wpływu na rozwój u szczurów, abirateronu octan wpływał na ciążę, m.in. skutkował zmniejszeniem masy płodu i przeżycia. Stwierdzano wpływ na zewnętrzne narządy płciowe, chociaż abirateronu octan nie był teratogenny.
W tych badaniach płodności i toksycznego wpływu na rozwój przeprowadzonych na szczurach,
wszystkie działania były związane z farmakologicznym działaniem abirateronu.
Oprócz zmian w narządach rozrodczych, stwierdzonych we wszystkich badaniach toksyczności u zwierząt, nie ujawniono szczególnego zagrożenia dla człowieka w oparciu o dane niekliniczne, wynikające z konwencjonalnych badań farmakologicznych dotyczących bezpieczeństwa, badań
toksyczności po podaniu wielokrotnym, genotoksyczności i rakotwórczości. Abirateronu octan nie wykazywał działania rakotwórczego w 6-miesięcznym badaniu u transgenicznych myszy (Tg.rasH2). W 24-miesięcznym badaniu rakotwórczości u szczurów, abirateronu octan zwiększał częstość występowania nowotworów komórek interstycjalnych w jądrach. To odkrycie uważa się za związane z działaniem farmakologicznym abirateronu i specyficzne dla szczurów. Abirateronu octan nie był rakotwórczy u samic szczurów.
Ocena ryzyka środowiskowego (ang. Environmental risk assessment, ERA)
Substancja czynna, abirateronu octan stanowi zagrożenie dla środowiska wodnego, w szczególności
dla ryb.
Rdzeń tabletki
Laktoza jednowodna Kroskarmeloza sodowa Hypromeloza 2910 (15mPas) Sodu laurylosiarczan
Celuloza mikrokrystaliczna (silikonowana) Krzemionka koloidalna bezwodna Magnezu stearynian.
Otoczka tabletki
Żelaza tlenek czarny (E 172) Żelaza tlenek czerwony (E 172) Makrogol 3350
Alkohol poliwinylowy Talk
Tytanu dwutlenek (E 171).
Nie dotyczy.
2 lata
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Każde pudełko tekturowe zawiera 56 tabletek powlekanych w 4 przezroczystych blistrach z folii PVC/PE/PVDC/Aluminium po 14 tabletek powlekanych każdy.
Każde pudełko tekturowe zawiera 60 tabletek powlekanych w 5 przezroczystych blistrach z folii PVC/PE/PVDC/Aluminium po 12 tabletek powlekanych każdy.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie
z lokalnymi przepisami. Ten produkt leczniczy może stanowić zagrożenie dla środowiska wodnego
(patrz punkt 5.3).
DOPUSZCZENIE DO OBROTU
Pharmascience International Limited Lampousas 1
1095 Nikozja Cypr
27116
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 10-06-2022
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
30-11-2022








