Spis treści:
- NAZWA WETERYNARYJNEGO PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- DANE KLINICZNE
- DANE FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- NAZWA PODMIOTU ODPOWIEDZIALNEGO
- NUMER(-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA OSTATNIEJ AKTUALIZACJI CHARAKTERYSTYKI PRODUKTU LECZNICZEGO WETERYNARYJNEGO
- KLASYFIKACJA WETERYNARYJNYCH PRODUKTÓW LECZNICZYCH
CHARAKTERYSTYKA WETERYNARYJNEGO PRODUKTU LECZNICZEGO
NAZWA WETERYNARYJNEGO PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
DANE KLINICZNE
Docelowe gatunki zwierząt
Wskazania lecznicze dla każdego z docelowych gatunków zwierząt
Przeciwwskazania
Specjalne ostrzeżenia
Specjalne środki ostrożności dotyczące stosowania
Zdarzenia niepożądane
Stosowanie w ciąży, podczas laktacji lub w okresie nieśności
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Droga podania i dawkowanie
Objawy przedawkowania (oraz sposób postępowania przy udzielaniu natychmiastowej
Szczególne ograniczenia dotyczące stosowania i specjalne warunki stosowania, w tym ograniczenia dotyczące stosowania przeciwdrobnoustrojowych i przeciwpasożytniczych weterynaryjnych produktów leczniczych w celu ograniczenia ryzyka rozwoju oporności
Okresy karencji
DANE FARMAKOLOGICZNE
Kod ATCvet: QN04BC02
Dane farmakodynamiczne
Dane farmakokinetyczne
DANE FARMACEUTYCZNE
Główne niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności przy przechowywaniu
Rodzaj i skład opakowania bezpośredniego
Specjalne środki ostrożności dotyczące usuwania niezużytych weterynaryjnych produktów leczniczych lub pochodzących z nich odpadów
NAZWA PODMIOTU ODPOWIEDZIALNEGO
NUMER(-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA OSTATNIEJ AKTUALIZACJI CHARAKTERYSTYKI PRODUKTU LECZNICZEGO WETERYNARYJNEGO
KLASYFIKACJA WETERYNARYJNYCH PRODUKTÓW LECZNICZYCH
Prasequin vet. 1 mg tabletki dla koni
Prasequine 1 mg tablets for horses (AT, BE, DE, EE, FR, HU, LT, LV, NL, PT, SK, UK-NI) Pras-equine 1 mg tablets for horses (CZ)
Każda tabletka zawiera:
Substancja czynna:
Pergolid 1,0 mg
(co odpowiada 1,31 mg pergolidu mezylanu)
Substancje pomocnicze:
Skład jakościowy substancji pomocniczych i pozostałych składników |
Laktoza jednowodna |
Kroskarmeloza sodowa |
Powidon |
Magnezu stearynian |
Żelaza tlenek żółty (E172) |
Tabletka;
Prawie biała, okrągła i wypukła tabletka z linią podziału w kształcie krzyża po jednej stronie; tabletki
można podzielić na 2 lub 4 równe części.
Konie (nieprzeznaczone do spożycia przez ludzi)
Leczenie objawowe symptomów klinicznych związanych z dysfunkcją części pośredniej przysadki
mózgowej (ang. Pituitary Pars Intermedia Dysfunction, PPID; zespół Cushinga koni).
Nie stosować u koni ze znaną nadwrażliwością na pergolidu mezylan lub inne pochodne sporyszu, lub
na dowolną substancję pomocniczą.
Nie stosować u koni w wieku poniżej 2 lat.
W celu rozpoznania PPID należy wykonać odpowiednie endokrynologiczne badania laboratoryjne
oraz ocenę objawów klinicznych.
Specjalne środki ostrożności dotyczące bezpiecznego stosowania u docelowych gatunków zwierząt: Ponieważ większość przypadków PPID rozpoznaje się u starszych koni, często równocześnie występują też inne procesy patologiczne. Informacje dotyczące monitorowania i częstości badań, patrz punkt 3.9.
Specjalne środki ostrożności dla osób podających weterynaryjny produkt leczniczy zwierzętom:
Ten weterynaryjny produkt leczniczy może powodować podrażnienie oczu, drażniący zapach lub ból głowy po dzieleniu tabletek. Unikać kontaktu z oczami i wdychania podczas postępowaniu
z tabletkami.
Zminimalizuj ryzyko narażenia podczas dzielenia lub rozpuszczania tabletek, np. tabletek nie należy rozgniatać. W przypadku kontaktu ze skórą, zmyć narażoną skórę wodą. W razie narażenia oczu niezwłocznie spłukać dotknięte oko wodą i uzyskać poradę lekarską. W przypadku podrażnienia nosa wyjść na świeże powietrze i uzyskać pomoc lekarską w przypadku wystąpienia trudności
z oddychaniem.
Ten weterynaryjny produkt leczniczy może powodować reakcje nadwrażliwości (alergiczne). Osoby o znanej nadwrażliwości na pergolid lub inne pochodne sporyszu powinny unikać kontaktu
z weterynaryjnym produktem leczniczym.
Ten weterynaryjny produkt leczniczy może wywoływać zdarzenia niepożądane na skutek obniżonego poziomu prolaktyny, co stanowi szczególne ryzyko dla kobiet w ciąży i karmiących piersią. Kobiety
w ciąży i karmiące piersią powinny unikać kontaktu produktu ze skórą oraz przenoszenia go z dłoni do ust przez noszenie rękawiczek podczas podawania.
Przypadkowe połknięcie, w szczególności przez dzieci, może powodować zdarzenia niepożądane, takie jak wymioty, zawroty głowy, letarg lub obniżenie ciśnienia tętniczego krwi. Aby uniknąć przypadkowego połknięcia, starannie przechowywać weterynaryjny produkt leczniczy w miejscu niewidocznym i niedostępnym dla dzieci. Części tabletek należy umieścić z powrotem w otwartym gnieździe blistra. Blistry należy umieszczać z powrotem w opakowaniu zewnętrznym i przechowywać w bezpiecznym miejscu. Po przypadkowym połknięciu należy niezwłocznie zasięgnąć porady lekarza oraz przedstawić lekarzowi ulotkę informacyjną lub opakowanie.
Nie jeść, nie pić ani nie palić podczas stosowania tego weterynaryjny produktu leczniczego. Po użyciu umyć ręce.
Specjalne środki ostrożności dotyczące ochrony środowiska:
Nie dotyczy.
Konie:
Rzadko (1 do 10 zwierząt/10 000 leczonych zwierząt): | Brak apetytu, przejściowy brak łaknienia i apatia, łagodne objawy ze strony ośrodkowego układu nerwowego (np. łagodna osowiałość i łagodna ataksja), biegunka i kolka. |
Bardzo rzadko (< 1 zwierzę/10 000 leczonych zwierząt, włączając pojedyncze raporty): | Potliwość. |
Zgłaszanie zdarzeń niepożądanych jest istotne, ponieważ umożliwia ciągłe monitorowanie
bezpieczeństwa stosowania weterynaryjnego produktu leczniczego. Zgłoszenia najlepiej przesłać za
pośrednictwem lekarza weterynarii do właściwych organów krajowych lub do podmiotu
odpowiedzialnego za pośrednictwem krajowego systemu zgłaszania. Właściwe dane kontaktowe znajdują się w punkcie 16 ulotki informacyjnej.
Ciąża:
Bezpieczeństwo weterynaryjnego produktu leczniczego stosowanego u klaczy w ciąży nie zostało określone. Badania laboratoryjne na myszach i królikach nie dały żadnych dowodów działania teratogennego. Stwierdzono ograniczenie płodności u myszy przy dawce 5,6 mg/kg masy ciała na dobę. Stosować jedynie po dokonaniu przez lekarza weterynarii oceny stosunku korzyści do ryzyka.
Laktacja:
Nie zaleca się stosowania u koni w okresie laktacji, u których to nie wykazano bezpieczeństwa stosowania tego weterynaryjnego produktu leczniczego. U myszy obniżona masa ciała i wskaźniki przeżywalności potomstwa przypisano farmakologicznemu hamowaniu wydzielania prolaktyny prowadzącemu do braku laktacji.
Należy zachować środki ostrożności w przypadku, gdy produkt leczniczy weterynaryjny podawany jest w skojarzeniu z innymi lekami silnie wiążącymi się z białkami osocza. Nie podawać równocześnie z antagonistami dopaminy, takimi jak neuroleptyki (fenotiazyny – np. acepromazyna) domperidon lub metoklopramid, ponieważ mogą one ograniczać skuteczność pergolidu
Podanie doustne, raz dziennie.
W celu ułatwienia podania należy umieścić wymaganą dzienną dawkę produktu w małej ilości wody i (lub) zmieszać z melasą lub innym słodzikiem, a następnie mieszać do rozpuszczenia. W takim przypadku rozpuszczone tabletki należy podać za pomocą strzykawki. Całą objętość należy podać natychmiast. Nie należy rozgniatać tabletek, patrz punkt 3.5. Po podzieleniu tabletek pozostałą część tabletki należy podać przy następnym podaniu.
Dawka początkowa
Dawka początkowa wynosi ok. 2 μg pergolidu/kg (zakres dawki: od 1,7 do 2,5 µg/kg; patrz tabela poniżej). Dawkę podtrzymującą (od 0,6 do 10 µg pergolidu/kg masy ciała, średnio 2 µg pergolidu/kg masy ciała) należy dostosować odpowiednio do indywidualnej odpowiedzi, stwierdzonej na podstawie monitorowania zwierzęcia (patrz poniżej).
Zaleca się następujące dawki początkowe:
Masa ciała konia kg | Liczba tabletek | Dawka początkowa mg/koni | Zakres dawkowania μg/kg |
200–300 | ½ | 0,50 | 1,7–2,5 |
301–400 | ¾ | 0,75 | 1,9–2,5 |
401–600 | 1 | 1,00 | 1,7–2,5 |
601–850 | 1 ½ | 1,50 | 1,8–2,5 |
851–1000 | 2 | 2,00 | 2,0–2,4 |
Dawka podtrzymująca
W przypadku tej choroby przewiduje się leczenie przez całe życia.
U większości koni reakcja na leczenie i stabilizacja następuje przy średniej dawce wynoszącej 2 µg pergolidu/kg masy ciała. Poprawy klinicznej po zastosowaniu pergolidu należy się spodziewać
w ciągu 6 do 12 tygodni. Odpowiedź kliniczna u koni może wystąpić przy niższych lub zmiennych dawkach, w związku z czym zaleca się stopniowe dostosowanie dawki do najniższej dawki skutecznej na podstawie odpowiedzi na leczenie, niezależnie od tego, czy jest to skuteczność, czy objawy
nietolerancji. W przypadku niektórych koni konieczna może być dawka sięgająca 10 µg pergolidu/kg masy ciała dziennie. W takich rzadkich przypadkach zalecane jest odpowiednie dodatkowe monitorowanie zwierzęcia.
W celu dostosowania dawki i monitorowania leczenia po początkowym rozpoznaniu należy powtarzać badania endokrynologiczne w odstępach 4–6 tygodni do czasu stabilizacji lub złagodzenia objawów klinicznych i (lub) poprawy wyników badań diagnostycznych.
Jeśli objawy kliniczne lub wyniki badań diagnostycznych nie ulegną poprawie po pierwszym okresie wynoszącym 4–6 tygodni, całkowitą dawkę dzienną można zwiększyć o 0.25 - 0,50 mg.
W przypadku, gdy objawy kliniczne uległy jedynie złagodzeniu, lekarz weterynarii może kontynuować dostosowywanie dawki aż do całkowitego ustabilizowania pacjenta, bądź też
zrezygnować z tego – w zależności od indywidualnej odpowiedzi na leczenie lub tolerancji produktu.
W przypadku, gdy objawy kliniczne nie są odpowiednio kontrolowane (na podstawie oceny klinicznej i (lub) testów diagnostycznych), zaleca się zwiększanie całkowitej dawki dobowej o 0.25 - 0,50 mg (jeśli lek jest tolerowany przy tej dawce) co 4 do 6 tygodni do czasu stabilizacji. W razie wystąpienia objawów nietolerancji dawki należy wstrzymać leczenie na 2–3 dni, a następnie wznowić, podając połowę poprzedniej dawki. Następnie można kontynuować zwiększanie całkowitej dawki dobowej
o 0.25 - 0,5 mg co 2–4 tygodnie do uzyskania pożądanego efektu klinicznego. W przypadku pominięcia dawki następną zaplanowaną dawkę należy podać zgodnie z zaleceniami.
Po uzyskaniu stabilizacji należy regularnie co 6 miesięcy powtarzać ocenę stanu klinicznego i badania diagnostyczne w celu monitorowania leczenia i dawki. W przypadku braku widocznej odpowiedzi na leczenie należy zweryfikować rozpoznanie i (lub) plan leczenia.
Tabletki można dzielić na 2 lub 4 równe części w celu zapewnienia dokładnego dawkowania. Położyć tabletkę na płaskiej powierzchni stroną z linią podziału do góry i wypukłą (zaokrągloną) stroną w dół.
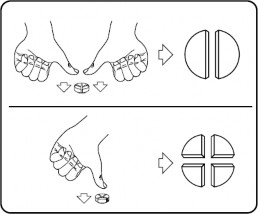
2 równe części: nacisnąć w dół kciukami po obu stronach tabletki. 4 równe części: nacisnąć w dół kciukiem pośrodku tabletki.
pomocy i odtrutki, w stosownych przypadkach)
Brak dostępnych informacji
Nie dotyczy.
Produkt niedopuszczony do stosowania u koni przeznaczonych do spożycia przez ludzi.
Koń musi zostać zadeklarowany jako zwierzę nieprzeznaczone do spożycia przez ludzi w ramach przepisów krajowych dotyczących paszportów dla koni.
Produkt niedopuszczony do stosowania u klaczy produkujących mleko do spożycia przez ludzi.
Pergolid jest syntetyczną pochodną sporyszu i wykazuje działanie właściwe silnemu, długotrwałemu agoniście receptora dla dopaminy. Badania farmakologiczne in vitro oraz in vivo dowiodły aktywności pergolidu jako wybiórczego agonisty dopaminy, z bardzo niewielkim działaniem lub jego brakiem
w dawkach terapeutycznych na ścieżki norepinefryny, epinefryny lub serotoniny. Podobnie jak
w przypadku innych agonistów dopaminy, pergolid hamuje uwalnianie prolaktyny. U koni cierpiących na dysfunkcję części pośredniej przysadki mózgowej (PPID) pergolid wykazuje działanie terapeutyczne poprzez stymulację receptorów dla dopaminy. Ponadto wykazano, że u koni z PPID pergolid obniża w osoczu poziom ACTH, MSH i innych peptydów proopiomelanokortynowych.
Dane farmakokinetyczne dotyczące koni są dostępne dla dawek 2, 4 i 10 μg pergolidu/kg masy ciała. Wykazano, że pergolid jest szybko wchłaniany, z krótkim czasem do osiągnięcia stężenia maksymalnego.
Stężenia maksymalne (Cmax) po podaniu dawki 10 μg/kg były niskie i zmienne, ze średnią ok. 4 ng/ml, a średni końcowy okres półtrwania (T ½) wynosił ok. 6 godzin. Mediana czasu stężenia maksymalnego (Tmax) wynosiła ok. 0,4 godziny, a powierzchnia pod krzywą (AUC) wynosiła ok. 14 ng x godz./ml.
W bardziej czułym teście analitycznym stężenia w osoczu po podaniu dawki 2 μg pergolidu/kg były bardzo niskie i zmienne, ze stężeniami maksymalnymi w zakresie od 0,138 do 0,551 ng/ml. Stężenia maksymalne wystąpiły po 1,25 +/– 0,5 godz. (Tmax). Stężenia w osoczu u większości koni były mierzalne tylko przez 6 godzin po podaniu. U jednego konia mierzalne stężenia odnotowywano przez 24 godziny. Końcowe okresy półtrwania nie zostały wyliczone, ponieważ w przypadku większości koni nie było pełnej jasności dotyczącej krzywej stężenia w osoczu w czasie.
Stężenia maksymalne (Cmax) po podaniu dawki 4 μg/kg były niskie i zmienne, w zakresie od 0,4 do 4,2 ng/ml, ze średnią 1,8 ng/ml, a średni końcowy okres półtrwania (T ½) wynosił ok. 5 godzin.
Mediana czasu stężenia maksymalnego (Tmax) wynosiła ok. 0,6 godz., a AUCt ok. 3,4 ng x godz./ml.
Pergolidu mezylan wiąże się w około 90% z białkami osocza u ludzi i zwierząt laboratoryjnych. Wydalanie zachodzi przez nerki.
Nie dotyczy.
Okres ważności weterynaryjnego produktu leczniczego zapakowanego do sprzedaży: 2 lata
Brak specjalnych środków ostrożności dotyczących przechowywania weterynaryjnego produktu leczniczego.
Blister OPA/Aluminium/PVC-Aluminium, zawierający 7 lub 10 tabletek. Pudełko tekturowe zawierające 60, 91, 100, 160 lub 240 tabletek.
Niektóre wielkości opakowań mogą nie być dostępne w obrocie.
Leków nie należy usuwać do kanalizacji ani wyrzucać do śmieci.
Należy skorzystać z krajowego systemu odbioru odpadów w celu usunięcia niewykorzystanego weterynaryjnego produktu leczniczego lub materiałów odpadowych pochodzących z jego
zastosowania w sposób zgodny z obowiązującymi przepisami oraz krajowymi systemami odbioru
odpadów dotyczącymi danego weterynaryjnego produktu leczniczego.
CP-Pharma Handelsgesellschaft mbH
Pozwolenie nr
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: {DD/MM/RRRR}
{DD/MM/RRRR}
Wydawany na receptę weterynaryjną
Szczegółowe informacje dotyczące powyższego weterynaryjnego produktu leczniczego są dostępne
w unijnej bazie danych produktów (https://medicines.health.europa.eu/veterinary).