







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Preato, 25 mg, tabletki Preato, 50 mg, tabletki Preato, 75 mg, tabletki Preato, 100 mg, tabletki Preato, 150 mg, tabletki Preato, 200 mg, tabletki Preato, 225 mg, tabletki Preato, 300 mg, tabletki
Preato,25 mg, tabletki
Każda tabletka zawiera 25 mg pregabaliny.
Preato, 50 mg, tabletki
Każda tabletka zawiera 50 mg pregabaliny.
Preato, 75 mg, tabletki
Każda tabletka zawiera 75 mg pregabaliny.
Preato, 100 mg, tabletki
Każda tabletka zawiera 100 mg pregabaliny.
Preato, 150 mg, tabletki
Każda tabletka zawiera 150 mg pregabaliny.
Preato, 200 mg, tabletki
Każda tabletka zawiera 200 mg pregabaliny.
Preato, 225 mg, tabletki
Każda tabletka zawiera 225 mg pregabaliny.
Preato, 300 mg, tabletki
Każda tabletka zawiera 300 mg pregabaliny.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletki
Preato, 25 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “I“ z jednej strony, o średnicy 5 mm
Preato, 50 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “M1“ z jednej strony, o średnicy 7 mm
Preato, 75 mg, tabletki
Białe, okragłe, obustronnie wypukłe tabletki z wytłoczonym napisem “I1“ z jednej strony, o średnicy 8 mm
Preato,100 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “M2“ z jednej strony, o średnicy 9 mm
Preato, 150 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “I2“ z jednej strony, o średnicy 10 mm
Preato, 200 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “M3“ z jednej strony, o średnicy 12 mm
Preato, 225 mg, tabletki
Białe, okrągłe, obustronnie wypukłe tabletki z wytłoczonym napisem “M7“ z jednej strony, o średnicy 12 mm
Preato, 300 mg, tabletki
Białe, podłużne, obustronnie wypukłe tabletki z wytłoczonym napisem "I3" z jednej strony, o średnicy dłuższej 20 mm i średnicy krótszej 8 mm
Tabletkę można podzielić na równe dawki.
Ból neuropatyczny
Produkt leczniczy Preato jest wskazany w leczeniu bólu neuropatycznego pochodzenia obwodowego i ośrodkowego u osób dorosłych.
Padaczka
Produkt leczniczy Preato jest wskazany u osób dorosłych w leczeniu skojarzonym napadów częściowych, które są lub nie są wtórnie uogólnione.
Uogólnione zaburzenia lękowe
Produkt leczniczy Preato jest wskazany w leczeniu uogólnionych zaburzeń lękowych (Generalised Anxiety Disorder – GAD) u osób dorosłych.
Dawkowanie
Dawka wynosi od 150 mg do 600 mg na dobę, podawana w dwóch lub trzech dawkach podzielonych.
Ból neuropatyczny
Leczenie pregabaliną można rozpocząć od dawki 150 mg na dobę, podawanej w dwóch lub trzech dawkach podzielonych. W zależności od indywidualnej reakcji pacjenta i tolerancji leczenia, po
3-7 dniach dawkę można zwiększyć do 300 mg na dobę, a następnie w zależności od potrzeby, po kolejnych 7 dniach do maksymalnej dawki 600 mg na dobę.
Padaczka
Leczenie pregabaliną można rozpocząć od dawki 150 mg na dobę, podawanej w dwóch lub trzech dawkach podzielonych. W zależności od indywidualnej reakcji pacjenta i tolerancji leczenia, dawkę można zwiększyć po 1 tygodniu leczenia do 300 mg na dobę. Dawkę maksymalną 600 mg na dobę można wprowadzić po kolejnym tygodniu.
Uogólnione zaburzenia lękowe
Dawka wynosi od 150 do 600 mg na dobę podawana w dwóch lub trzech dawkach podzielonych. Należy regularnie oceniać konieczność dalszego leczenia.
Leczenie pregabaliną można rozpocząć od dawki 150 mg na dobę. W zależności od indywidualnej odpowiedzi pacjenta oraz tolerancji, dawkę tę po 1 tygodniu leczenia można zwiększyć do 300 mg na dobę. Po upływie kolejnego tygodnia dawkę można zwiększyć do 450 mg na dobę. Po upływie jeszcze jednego tygodnia można wprowadzić dawkę maksymalną 600 mg na dobę.
Przerwanie leczenia pregabaliną
Zgodnie z obecną praktyką kliniczną w razie konieczności przerwania leczenia pregabaliną należy dokonywać tego stopniowo przez okres co najmniej 1 tygodnia niezależnie od wskazania (patrz punkty 4.4 i 4.8).
Zaburzenia czynności nerek
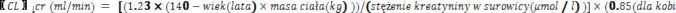
Pregabalina jest usuwana z krążenia ogólnego głównie przez wydalanie nerkowe w postaci niezmienionej. Ze względu na fakt, że klirens pregabaliny jest wprost proporcjonalny do klirensu kreatyniny (patrz punkt 5.2), redukcja dawki u pacjentów z zaburzeniami czynności nerek powinna być przeprowadzona indywidualnie, zgodnie z klirensem kreatyniny (CLcr), jak pokazano w Tabeli 1, przy użyciu następującego wzoru:
Pregabalina jest skutecznie usuwana z osocza w trakcie hemodializy (50% leku w ciągu 4 godzin). U pacjentów dializowanych dobowa dawka pregabaliny powinna być dostosowana do czynności nerek. Oprócz dawki dobowej, pacjent powinien otrzymywać dodatkową dawkę bezpośrednio po każdym czterogodzinnym zabiegu hemodializy (patrz Tabela 1).
Tabela 1. Dostosowanie dawki pregabaliny w zależności od czynności nerek
Klirens kreatyniny (CLcr) (ml/min) | Całkowita dawka dobowa pregabaliny * | Schemat dawkowania | |
Dawka początkowa (mg/dobę) | Dawka maksymalna (mg/dobę) | ||
60 | 150 | 600 | BID lub TID |
30 - 60 | 75 | 300 | BID lub TID |
15 -30 | 25– 50 | 150 | Raz na dobę lub BID |
< 15 | 25 | 75 | Raz na dobę |
Dodatkowa dawka pregabaliny podawana po zabiegu hemodializy (mg) | |||
25 | 100 | Pojedyncza dawka+ | |
TID = 3 razy na dobę
BID = 2 razy na dobę
*Całkowita dawka dobowa (mg/dobę) powinna być podzielona według schematu dawkowania
+ Dawka uzupełniająca podawana jednorazowo Zaburzenia czynności wątroby
U pacjentów z zaburzoną czynnością wątroby nie jest wymagana modyfikacja dawki (patrz punkt 5.2).
Dzieci i młodzież
Nie określono bezpieczeństwa stosowania ani skuteczności produktu leczniczego Preato u dzieci w wieku poniżej 12 lat i młodzieży (12–17 lat). Dostępne obecnie dane przedstawiono w punktach 4.8, 5.1 i 5.2, nie można jednak sformułować żadnych zaleceń dotyczących dawkowania.
Pacjenci w podeszłym wieku
U pacjentów w podeszłym wieku może być konieczne zmniejszenie dawki pregabaliny, ze względu na zmniejszoną czynność nerek (patrz punkt 5.2).
Sposób podawania
Produkt leczniczy Preato może być przyjmowany podczas posiłku lub niezależnie od niego. Produkt leczniczy Preato jest przeznaczony wyłącznie do przyjmowania doustnego.
Nadwrażliwość na substancję czynną lub na którąkolwiek substancji pomocniczych wymienionych w punkcie 6.1.
Pacjenci z cukrzycą
Zgodnie z aktualną praktyką kliniczną pacjenci z cukrzycą, którzy przybierają na wadze podczas leczenia pregabaliną, mogą wymagać dostosowania dawek hipoglikemizujących produktów leczniczych.
Reakcje nadwrażliwości
Po wprowadzeniu leku do obrotu odnotowano występowanie reakcji nadwrażliwości, w tym przypadków obrzęku naczynioruchowego. Pregabalinę należy odstawić natychmiast po wystąpieniu objawów obrzęku naczynioruchowego, takich jak obrzęk twarzy, obrzęk ust lub obrzęk w obrębie górnych dróg oddechowych.
Zawroty głowy, senność, utrata przytomności, splątanie i zaburzenia psychiczne
Leczenie pregabaliną było związane z występowaniem zawrotów głowy i senności, które mogą zwiększać ryzyko wystąpienia przypadkowego zranienia (upadku) u osób w podeszłym wieku. Po wprowadzeniu leku do obrotu zgłaszano również przypadki utraty przytomności, splątania i zaburzeń psychicznych. Dlatego należy zalecić pacjentom zachowanie ostrożności do chwili ustalenia, jakie potencjalne działania wywiera na nich stosowany produkt leczniczy.
Objawy związane z narządem wzroku
W kontrolowanych badaniach większy odsetek osób, u których wystąpiło niewyraźne widzenie, stwierdzono wśród pacjentów leczonych pregabaliną niż wśród pacjentów otrzymujących placebo. Objaw ten ustąpił w większości przypadków w miarę kontynuacji leczenia. W badaniach klinicznych, w których wykonywano badania okulistyczne, częstość występowania zmniejszenia ostrości widzenia i zmian w polu widzenia była większa u pacjentów leczonych pregabaliną niż u pacjentów otrzymujących placebo; z kolei częstość występowania zmian w badaniach dna oka była większa u pacjentów otrzymujących placebo (patrz punkt 5.1).
Po wprowadzeniu leku do obrotu donoszono również o występowaniu działań niepożądanych ze strony narządu wzroku, w tym o utracie wzroku, niewyraźnym widzeniu lub innych zmianach ostrości widzenia, z których wiele było przemijających. Przerwanie stosowania pregabaliny może doprowadzić do ustąpienia tych objawów lub ich zmniejszenia.
Niewydolność nerek
Zgłaszano przypadki niewydolności nerek, ale niekiedy to działanie niepożądane ustępowało po przerwaniu stosowania pregabaliny.
Odstawienie jednocześnie stosowanych leków przeciwpadaczkowych
Brak wystarczających danych odnośnie odstawienia jednocześnie stosowanych leków przeciwpadaczkowych i zastosowania monoterapii pregabaliną po opanowaniu napadów przy użyciu pregabaliny, wprowadzonej jako leczenie wspomagające.
Objawy odstawienia
U niektórych pacjentów obserwowano objawy odstawienia po przerwaniu krótko- i długotrwałego leczenia pregabaliną. Wystąpiły następujące zdarzenia: bezsenność, ból głowy, nudności, lęk, biegunka, objawy grypopodobne, nerwowość, depresja, ból, drgawki, nadmierrne pocenie się i zawroty głowy, sugerujące uzależnienie fizyczne. Przed rozpoczęciem leczenia należy poinformować pacjenta o możliwości wystąpienia tych stanów.
Podczas stosowania pregabaliny lub krótko po zaprzestaniu leczenia mogą wystąpić drgawki, w tym stan padaczkowy oraz napady padaczkowe typu grand mal.
Odnośnie przerwania długotrwałego leczenia pregabaliną, dane wskazują, że częstość występowania i nasilenie objawów odstawienia mogą być zależne od dawki.
Zastoinowa niewydolność serca
Po wprowadzeniu pregabaliny do obrotu opisywano przypadki zastoinowej niewydolności serca u niektórych pacjentów otrzymujących pregabalinę. Reakcje te obserwowano głównie u pacjentów w podeszłym wieku z zaburzeniami ze strony układu sercowo-naczyniowego, podczas terapii pregabaliną wskazaną w bólu neuropatycznym. Pregabalinę należy ostrożnie stosować u takich pacjentów. Przerwanie stosowania pregabaliny może spowodować wycofanie się objawów.
Leczenie ośrodkowego bólu neuropatycznego wywołanego urazem rdzenia kręgowego
W leczeniu ośrodkowego bólu neuropatycznego wywołanego urazem rdzenia kręgowego stwierdzono zwiększoną ogólną częstość występowania działań niepożądanych oraz działań niepożądanych ze strony ośrodkowego układu nerwowego, zwłaszcza senności. Powodem może być działanie addytywne innych, jednocześnie stosowanych produktów leczniczych (np. leków przeciwspastycznych) niezbędnych w leczeniu tego stanu. Fakt ten należy brać pod uwagę w przypadku zapisywania pregabaliny takim pacjentom.
Depresja oddechowa
Zgłaszano przypadki ciężkiej depresji oddechowej związane ze stosowaniem pregabaliny. Pacjenci z upośledzeniem czynności układu oddechowego, chorobami układu oddechowego lub chorobami neurologicznymi, zaburzeniami czynności nerek, stosujący jednocześnie leki działające depresyjnie na OUN oraz w podeszłym wieku mogą być bardziej narażeni na wystąpienie tego ciężkiego działania niepożądanego. U tych pacjentów może być konieczne dostosowanie dawki (patrz punkt 4.2).
Myśli i zachowania samobójcze
Odnotowano występowanie myśli i zachowań samobójczych u pacjentów przyjmujących leki przeciwpadaczkowe z różnych wskazań. Metaanaliza randomizowanych, kontrolowanych placebo badań oceniających leki przeciwpadaczkowe również wykazała niewielki wzrost ryzyka wystąpienia myśli i zachowań samobójczych. Mechanizm leżący u podstaw tego zjawiska nie jest znany, dostępne dane nie wykluczają możliwości wzrostu ryzyka pod wpływem pregabaliny.
Z tego względu należy obserwować pacjentów pod kątem oznak myśli i zachowań samobójczych, a także rozważyć odpowiednie leczenie. Powinno się doradzać pacjentom (i ich opiekunom), aby zwracali się o pomoc medyczną, jeśli pojawią się oznaki myśli lub zachowań samobójczych.
Osłabienie czynności dolnego odcinka przewodu pokarmowego
Po wprowadzeniu produktu do obrotu zgłaszano przypadki działań niepożądanych związanych z osłabieniem czynności dolnego odcinka przewodu pokarmowego (np. niedrożność jelita, porażenna niedrożność jelit, zaparcia), kiedy stosowano pregabalinę z produktami, które mogą potencjalnie wywoływać zaparcie, takimi jak opioidowe leki przeciwbólowe. Jeśli pregabalina ma być stosowana jednocześnie z opioidami, należy rozważyć podjęcie środków przeciwdziałających zaparciom (szczególnie u kobiet i pacjentów w podeszłym wieku).
Niewłaściwe stosowanie, nadużywanie oraz uzależnienie od leku
Zgłaszano przypadki niewłaściwego stosowania, nadużywania oraz uzależnienia od leku. Należy zachować ostrożność w przypadku pacjentów, którzy w przeszłości nadużywali leków, oraz obserwować tych pacjentów w kierunku objawów niewłaściwego stosowania, nadużywania oraz uzależnienia od pregabaliny (zgłaszano przypadki rozwoju tolerancji na lek, zwiększania dawki, poszukiwania środka uzależniającego).
Encefalopatia
Zgłaszano przypadki encefalopatii, głównie u pacjentów z chorobami współistniejącymi, które mogą przyspieszyć wystąpienie encefalopatii.
Ciężkie niepożądane reakcje skórne
W związku z leczeniem pregabaliną rzadko notowano ciężkie niepożądane reakcje skórne, w tym zespół Stevensa-Johnsona (SJS) i toksyczne martwicze oddzielanie się naskórka (TEN), mogące zagrażać życiu lub powodować zgon. W momencie przepisywania leku należy pacjenta poinformować o objawach przedmiotowych i podmiotowych oraz ściśle obserwować, czy nie występują u niego reakcje skórne. W razie pojawienia się objawów przedmiotowych i podmiotowych wskazujących na występowanie tych reakcji, należy natychmiast przerwać stosowanie pregabaliny i rozważyć alternatywną metodę leczenia (stosownie do przypadku).
Pregabalina jest wydalana z organizmu głównie w postaci niezmienionej w moczu, tak więc jej metabolizm u ludzi nie ma istotnego znaczenia (< 2% dawki jest usuwane w moczu w postaci
metabolitów), nie hamuje metabolizmu leków in vitro, nie wiąże się z białkami osocza i jest mało prawdopodobne, że wywołuje lub podlega interakcjom farmakokinetycznym.
Badania in vivo i analiza farmakokinetyczna populacji
Odpowiednio, w badaniach in vivo nie obserwowano istotnych klinicznie interakcji farmakokinetycznych pomiędzy pregabaliną a fenytoiną, karbamazepiną, kwasem walproinowym, lamotryginą, gabapentyną, lorazepamem, oksykodonem lub etanolem. Analiza farmakokinetyczna populacji wykazała, że doustne leki przeciwcukrzycowe, diuretyki, insulina, fenobarbital, tiagabina i topiramat nie miały istotnego klinicznie wpływu na klirens pregabaliny.
Doustne leki antykoncepcyjne, noretysteron i (lub) etynyloestradiol
Jednoczesne stosowanie pregabaliny i doustnych leków antykoncepcyjnych noretysteronu i (lub) etynyloestradiolu nie wpływa na farmakokinetykę żadnego z leków w stanie stacjonarnym.
Produkty lecznicze działające na ośrodkowy układ nerwowy
Pregabalina może nasilać działanie etanolu i lorazepamu. W kontrolowanych badaniach klinicznych wielokrotne podawanie doustnych dawek pregabaliny wraz z oksykodonem, lorazepamem lub etanolem nie wywierało istotnego klinicznie wpływu na oddychanie. Po wprowadzeniu produktu do obrotu zgłaszano przypadki niewydolności oddechowej i śpiączki u pacjentów, którzy przyjmowali pregabalinę wraz z innymi produktami leczniczymi działającymi hamująco na ośrodkowy układ nerwowy (OUN). Pregabalina działa prawdopodobnie addytywnie do oksykodonu, jeśli chodzi o zaburzenia funkcji poznawczych i motoryki dużej.
Interakcje u pacjentów w wieku podeszłym
Nie prowadzono specyficznych badań dotyczących farmakodynamicznych interakcji u ochotników w wieku podeszłym. Interakcje z innymi lekami badano wyłącznie u osób dorosłych.
Kobiety w wieku rozrodczym / antykoncepcja u mężczyzn i kobiet
Ponieważ potencjalne zagrożenia dla ludzi nie są znane, kobiety w wieku rozrodczym muszą stosować skuteczną metodę antykoncepcji.
Ciąża
Nie ma odpowiednich danych dotyczących stosowania pregabaliny u kobiet w ciąży.
Badania na zwierzętach wykazały toksyczny wpływ na rozród (patrz punkt 5.3). Potencjalne ryzyko u ludzi nie jest znane.
Produktu leczniczego Preato nie należy stosować w okresie ciąży dopóki nie jest to konieczne (tzn. w przypadku gdy oczekiwane korzyści dla matki znacznie przewyższają ryzyko dla płodu).
Karmienie piersią
Pregabalina przenika do mleka ludzkiego (patrz punkt 5.2). Wpływ pregabaliny na organizm noworodków/dzieci jest nieznany. Należy podjąć decyzję, czy przerwać karmienie piersią, czy przerwać podawanie pregabaliny, biorąc pod uwagę korzyści z karmienia piersią dla dziecka
i korzyści z leczenia dla matki.
Płodność
Brak danych z badań klinicznych na temat wpływu pregabaliny na płodność kobiet.
W badaniu klinicznym oceniającym wpływ pregabaliny na ruchliwość plemników, zdrowym mężczyznom podawano pregabalinę w dawce 600 mg/dobę. Po 3 miesiącach przyjmowania leku nie stwierdzono żadnego wpływu na ruchliwość plemników.
W badaniu wpływu na płodność u samic szczura wykazano niekorzystny wpływ na rozród. Badania wpływu na płodność u samców szczura wykazały niekorzystny wpływ na rozród i rozwój. Znaczenie kliniczne poczynionych obserwacji nie jest znane (patrz punkt 5.3).
Produkt leczniczy Preato może wywierać niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Pregabalina może wywoływać zawroty głowy i senność, i z tego powodu może mieć wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn. Pacjentom zaleca się, aby nie prowadzili pojazdów, nie obsługiwali urządzeń mechanicznych ani nie wykonywali potencjalnie niebezpiecznych czynności do czasu ustalenia, czy produkt leczniczy wpływa u nich na zdolność wykonywania tychże czynności.
Program badań klinicznych z pregabaliną obejmował ponad 8900 pacjentów przyjmujących lek, ponad 5600 z nich brało udział w badaniach kontrolowanych placebo metodą podwójnie ślepej próby. Do najczęściej odnotowywanych działań niepożądanych należały zawroty głowy i senność. Działania niepożądane były zwykle łagodnie lub umiarkowanie nasilone. We wszystkich badaniach kontrolowanych odsetek przypadków odstawienia leku z powodu działań niepożądanych wyniósł 12% w grupie pacjentów otrzymujących pregabalinę i 5% w grupie pacjentów otrzymujących placebo. Do najczęstszych działań niepożądanych powodujących przerwanie leczenia należały zawroty głowy i senność.
W tabeli 2, poniżej, przedstawiono wszystkie działania niepożądane, które wystąpiły z częstością większą niż w grupie placebo i u więcej niż jednego pacjenta według klasy i częstości ich występowania [bardzo często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1 000 do <1/100), rzadko (≥1/10 000 do <1/1 000), bardzo rzadko (<1/10 000), częstość nieznana (częstość nie może być określona na podstawie dostępnych danych)]. Działania niepożądane w każdej kategorii częstości występowania uporządkowano według malejącej ciężkości tych działań.
Przedstawione poniżej działania niepożądane mogą mieć również związek z chorobą podstawową i (lub) z jednoczesnym stosowaniem innych produktów leczniczych.
W leczeniu ośrodkowego bólu neuropatycznego wywołanego urazem rdzenia kręgowego stwierdzono zwiększoną ogólną częstość występowania działań niepożądanych oraz działań niepożądanych ze strony OUN, zwłaszcza senności (patrz punkt 4.4).
W poniższej tabeli pismem pochyłym przedstawiono dodatkowe reakcje pochodzące z danych zebranych po wprowadzeniu produktu do obrotu.
Tabela 2. Działania niepożądane pregabaliny
Klasyfikacja układów i nrządów | Działania niepożądane |
Zakażenia i zarażenia pasożytnicze | |
Często | Zapalenie jamy nosowo-gardłowej |
Zaburzenia krwi i układu chłonnego | |
Niezbyt często | Neutropenia |
Zaburzenia układu immunologicznego | |
Niezbyt często | Nadwrażliwość |
Rzadko | Obrzęk naczyniowo-ruchowy, reakcje alergiczne |
Zaburzenia metabolizmu i odżywiania | |
Często | Zwiększenie apetytu |
Niezbyt często | Anoreksja, hipoglikemia |
Zaburzenia psychiczne | |
Często | Nastrój euforyczny, splątanie, drażliwość, dezorientacja, bezsenność, zmniejszone libido |
Niezbyt często | Omamy, napady paniki, niepokój ruchowy, pobudzenie, depresja, nastrój depresyjny, podwyższony nastrój, agresja, zmiany nastroju, depersonalizacja, trudności ze znalezieniem odpowiednich słów, niezwykłe sny, zwiększone libido, anorgazmia, apatia |
Rzadko | Brak zahamowań |
Zaburzenia układu nerwowego | |
Bardzo często | Zawroty głowy, senność, bóle głowy |
Często | Ataksja, zaburzenia koordynacji, drżenia, zaburzenia mowy, amnezja, zaburzenia pamięci, trudności w skupieniu uwagi, parestezje, niedoczulica, uspokojenie, zaburzenia równowagi, letarg |
Niezbyt często | Omdlenie, stupor, drgawki kloniczne mięśni, utrata przytomności, nadreaktywność psychoruchowa, dyskineza, ułożeniowe zawroty głowy, drżenie zamiarowe, oczopląs, zaburzenia poznawcze, zaburzenia psychiczne, zaburzenia mowy, osłabienie odruchów, przeczulica, uczucie pieczenia, brak smaku, złe samopoczucie |
Rzadko | Drgawki, zmieniony węch, hipokineza, dysgrafia, zespół parkinsonowski |
Zaburzenia oka | |
Często | Nieostre widzenie, podwójne widzenie |
Niezbyt często | Utrata obwodowej części pola widzenia, zaburzenia widzenia, obrzęk oka, zaburzenia pola widzenia, zmniejszenie ostrości widzenia, ból oka, niedowidzenie, wrażenie błysków, uczucie suchości w oku, zwiększone wydzielanie łez, podrażnienie oka |
Rzadko | Utrata wzroku, zapalenie rogówki, wrażenie drgania obrazu widzianego, zmienione wrażenie głębi obrazu widzianego, rozszerzenie źrenic, zez, jaskrawe |
widzenie/olśnienie | |
Zaburzenia ucha i błędnika | |
Często | Zawroty głowy |
Niezbyt często | Przeczulica słuchowa |
Zaburzenia serca | |
Niezbyt często | Tachykardia, blok przedsionkowo- komorowy pierwszego stopnia, bradykardia zatokowa, zastoinowa niewydolność serca |
Rzadko | Wydłużenie odstępu QT, tachykardia zatokowa, arytmia zatokowa |
Zaburzenia naczyniowe | |
Niezbyt często | Niedociśnienie, nadciśnienie tętnicze, nagłe uderzenia gorąca, zaczerwienienie, uczucie zimna w kończynach |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Niezbyt często | Duszność, krwawienie z nosa, kaszel, uczucie zatkanego nosa, zapalenie błony śluzowej nosa, chrapanie, suchość śluzówki nosa |
Rzadko | Obrzęk płuc, uczucie ucisku w gardle |
Częstość nieznana | Depresja oddechowa |
Zaburzenia żołądka i jelit | |
Często | Wymioty, nudności, zaparcie, biegunka, wzdęcia, uczucie rozdęcia brzucha, suchość błony śluzowej jamy ustnej |
Niezbyt często | Refluks żołądkowo-przełykowy, nadmierne wydzielanie śliny, niedoczulica w okolicy ust |
Rzadko | Wodobrzusze, zapalenie trzustki, obrzęk języka, zaburzenia połykania |
Zaburzenia wątroby i dróg żółciowych | |
Często | Zwiększona aktywność enzymów wątrobowych* |
Rzadko | Żółtaczka |
Bardzo rzadko | Niewydolność wątroby, zapalenie wątroby |
Zaburzenia skóry i tkanki podskórnej | |
Niezbyt często | Wysypka grudkowa, pokrzywka, nadmierne pocenie się, świąd |
Rzadko | Zespół Stevensa-Johnsona, zimne poty, toksyczne martwicze oddzielanie się naskórka |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często | Kurcze mięśni, bóle stawów, bóle pleców, bóle kończyn, kręcz szyi |
Niezbyt często | Obrzęk stawów, bóle mięśni, drganie mięśniowe, bóle szyi, sztywność mięśni |
Rzadko | Rabdomioliza |
Zaburzenia nerek i dróg moczowych | |
Niezbyt często | Nietrzymanie moczu, dyzuria |
Rzadko | Niewydolność nerek, skąpomocz, retencja moczu |
Zaburzenia układu rozrodczego i piersi | |
Często | Zaburzenia erekcji |
Niezbyt często | Zaburzenia czynności seksualnych, opóźnienie ejakulacji, bolesne miesiączkowanie, bóle piersi |
Rzadko | Brak miesiączki, wyciek z brodawki sutkowej, powiększenie piersi, ginekomastia |
Zaburzenia ogólne i stany w miejscu podania | |
Często | Obrzęki obwodowe, obrzęki, zaburzenia chodu, upadki, uczucie podobne do występującego po spożyciu alkoholu, zmienione samopoczucie, zmęczenie |
Niezbyt często | Uogólniony obrzęk tkanki podskórnej, obrzęk twarzy, ucisk w klatce piersiowej, ból, gorączka, pragnienie, dreszcze, osłabienie |
Badania diagnostyczne | |
Często | Zwiększenie masy ciała |
Niezbyt często | Zwiększenie aktywności fosfokinazy kreatynowej, zwiększenie stężenia glukozy we krwi, zmniejszenie liczby płytek krwi we krwi, zwiększenie stężenia kreatyniny we krwi, zmniejszenie stężenia potasu we krwi, zmniejszenie masy ciała |
Rzadko | Zmniejszenie liczby krwinek białych we krwi |
* Zwiększenie aktywności aminotransferazy alaninowej (AlAT) i aminotransferazy asparaginianowej (AspAT).
U niektórych pacjentów obserwowano objawy odstawienia po przerwaniu krótko- i długotrwałego leczenia pregabaliną. Wystąpiły następujące objawy: bezsenność, bóle głowy, nudności, lęk, biegunka, zespół grypopodobny, drgawki, nerwowość, depresja, ból, nadmierne pocenie się i zawroty głowy, sugerujące uzależnienie fizyczne. Przed rozpoczęciem leczenia należy poinformować pacjenta o możliwości wystąpienia tych stanów.
Odnośnie przerwania długotrwałego leczenia pregabaliną, dane wskazują, że częstość występowania i nasilenie objawów odstawienia mogą być zależne od dawki.
Dzieci i młodzież
Profil bezpieczeństwa stosowania pregabaliny obserwowany w trzech badaniach pediatrycznych z udziałem pacjentów z napadami padaczkowymi częściowymi wtórnie uogólnionymi lub bez wtórnego uogólnienia (12-tygodniowe badanie skuteczności i bezpieczeństwa stosowania u pacjentów z napadami częściowymi, n=295; badanie farmakokinetyki i tolerancji, n=65 i trwająca rok kontynuacja badania, prowadzona metodą otwartej próby, mająca na celu ocenę bezpieczeństwa, n=54) był podobny do występującego w badaniach u dorosłych pacjentów z padaczką. W 12-tygodniowym badaniu u pacjentów leczonych pregabaliną najczęściej występującymi zdarzeniami niepożądanymi były: senność, gorączka, zakażenie górnych dróg
oddechowych, zwiększenie łaknienia, zwiększenie masy ciała i zapalenie błony śluzowej nosa i gardła (patrz punkty 4.2, 5.1 i 5.2).
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych
Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych Al. Jerozolimskie 181C, 02-222 Warszawa,
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Po wprowadzeniu produktu do obrotu do najczęściej obserwowanych działań niepożądanych występujących po przedawkowaniu pregabaliny należały: senność, splątanie, pobudzenie i niepokój. Zgłaszano także napady padaczkowe.
Rzadko zgłaszano przypadki śpiączki.
Leczenie przedawkowania pregabaliny powinno polegać na ogólnym leczeniu podtrzymującym i, w razie potrzeby, zastosowaniu hemodializy (patrz punkt 4.2, Tabela 1).
Grupa farmakoterapeutyczna: leki przeciwpadaczkowe, inne leki przeciwpadaczkowe, kod ATC: N03AX16.
Aktywna substancja pregabalina to pochodna kwasu gamma-aminomasłowego (GABA)[kwas(S)-3- (aminometylo)-5-metyloheksanowy].
Mechanizm działania
Pregabalina wiąże się z pomocniczą podjednostką (białko α2-δ) otwieranego poprzez zmianę napięcia błonowego kanału wapniowego w ośrodkowym układzie nerwowym.
Skuteczność kliniczna i bezpieczeństwo stosowania
Ból neuropatyczny
W badaniach wykazano skuteczność leku w neuropatii cukrzycowej, neuralgii po przebytym półpaścu i urazie rdzenia kręgowego. Nie przeprowadzono badań na innych modelach bólu neuropatycznego.
Pregabalina była oceniana w 10 kontrolowanych badaniach klinicznych trwających do 13 tygodni z zastosowaniem dawkowania dwa razy na dobę (BID) i do 8 tygodni z zastosowaniem dawkowania trzy razy na dobę (TID). Ogólnie profile bezpieczeństwa stosowania i skuteczności dla schematów dawkowania BID i TID były podobne.
W badaniach klinicznych trwających do 12 tygodni w bólu neuropatycznym pochodzenia obwodowego i ośrodkowego, zmniejszenie nasilenia bólu obserwowano w ciągu pierwszego tygodnia i utrzymywało się przez cały okres leczenia.
W kontrolowanych badaniach klinicznych w obwodowym bólu neuropatycznym 35% pacjentów leczonych pregabaliną i 18% pacjentów przyjmujących placebo wykazało poprawę w wynikach skali bólu. Wśród pacjentów nieodczuwających senności poprawę taką obserwowano u 33% pacjentów przyjmujących pregabalinę i 18% pacjentów przyjmujących placebo. Wśród pacjentów, którzy odczuwali senność, poprawę obserwowano u 48% pacjentów leczonych pregabaliną i u 16% przyjmujących placebo.
W kontrolowanym badaniu klinicznym w ośrodkowym bólu neuropatycznym u 22% pacjentów leczonych pregabaliną i u 7% pacjentów otrzymujących placebo stwierdzono poprawę w punktowej ocenie nasilenia bólu o 50%.
Epilepsja
Terapia skojarzona
Pregabalina była oceniana w 3 kontrolowanych badaniach klinicznych, trwających 12 tygodni, w których podawano lek według schematów dawkowania BID lub TID. Ogólnie, profile bezpieczeństwa stosowania i skuteczności dla schematów dawkowania BID i TID były podobne.
Zmniejszenie częstości napadów padaczkowych obserwowano przed końcem pierwszego tygodnia leczenia.
Dzieci i młodzież
Nie określono skuteczności ani bezpieczeństwa stosowania pregabaliny w terapii skojarzonej w leczeniu padaczki u dzieci w wieku poniżej 12 lat i młodzieży. Działania niepożądane obserwowane w badaniu farmakokinetyki i tolerancji, obejmującym pacjentów z napadami padaczkowymi częściowymi w wieku od 3 miesięcy do 16 lat (n=65) były podobne do występujących u dorosłych.
Wyniki 12-tygodniowego badania kontrolowanego placebo z udziałem 295 pacjentów pediatrycznych w wieku od 4 do 16 lat prowadzonego w celu oceny skuteczności i bezpieczeństwa stosowania pregabaliny jako terapii wspomagającej w leczeniu napadów częściowych oraz trwającego 1 rok otwartego badania bezpieczeństwa obejmującego 54- osobową grupę dzieci i młodzieży z padaczką, w wieku od 3 miesięcy do 16 lat, wykazały częstsze występowanie działań niepożądanych, takich jak gorączka i zakażenia górnych dróg oddechowych, niż w badaniach u dorosłych pacjentów z padaczką (patrz punkty 4.2, 4.8 i 5.2).
W trwającym 12 tygodni badaniu kontrolowanym placebo, pacjenci pediatryczni zostali przypisani do jednej z trzech grup: grupy otrzymującej pregabalinę w dawce 2,5 mg/kg mc./dobę (maksymalnie 150 mg/dobę), grupy otrzymującej pregabalinę w dawce 10 mg/kg mc./dobę (maksymalnie 600 mg/dobę) lub do grupy placebo. Odsetek pacjentów, u których odnotowano co najmniej 50% zmniejszenie liczby napadów częściowych w porównaniu do wartości wyjściowych, wynosił 40,6% w grupie pacjentów leczonych pregabaliną w dawce 10 mg/kg mc./dobę (p = 0,0068 versus placebo), 29,1% w grupie pacjentów leczonych pregabaliną w dawce 2,5 mg/kg mc./dobę (p = 0,2600 versus placebo) oraz 22,6% w grupie pacjentów otrzymujących placebo.
Monoterapia (pacjenci nowo zdiagnozowani)
Pregabalina była oceniana w 1 kontrolowanym badaniu klinicznym, trwającym 56 tygodni, w którym lek podawano według schematu dawkowania BID. Nie wykazano równoważności pregabaliny względem lamotryginy w odniesieniu do punktu końcowego zdefiniowanego jako 6-miesięczny okres wolny od napadów drgawek. Pregabalina i lamotrygina były podobnie bezpieczne i dobrze tolerowane.
Uogólnione zaburzenia lękowe
Pregabalina była oceniana w 6 kontrolowanych badaniach trwających 4–6 tygodni, w 8- tygodniowym badaniu z udziałem pacjentów w podeszłym wieku, a także w długookresowym, dotyczącym zapobiegania nawrotom objawów zaburzenia badaniu z 6-miesięczną fazą prowadzoną metodą podwójnie ślepej próby.
Zmniejszenie nasilenia objawów GAD określane za pomocą skali oceny lęku Hamiltona (Hamilton Anxiety Rating Scale – HAM-A) zaobserwowano w pierwszym tygodniu leczenia.
W kontrolowanych badaniach klinicznych (trwających 4–8 tygodni) poprawa o co najmniej 50% całkowitej punktacji w skali HAM-A od momentu rozpoczęcia badania do chwili osiągnięcia punktu końcowego obserwowana była u 52% pacjentów leczonych pregabaliną i u 38% pacjentów otrzymujących placebo.
W kontrolowanych badaniach niewyraźne widzenie stwierdzono u większego odsetka pacjentów leczonych pregabaliną niż u pacjentów otrzymujących placebo. Objaw ten ustąpił w większości przypadków w miarę kontynuacji leczenia. W kontrolowanych badaniach klinicznych u ponad 3600 pacjentów przeprowadzono badania okulistyczne (obejmujące badania ostrości widzenia, formalne badania pola widzenia i badania dna oka po rozszerzeniu źrenicy). W tej grupie stwierdzono zmniejszenie ostrości widzenia u 6,5% pacjentów leczonych pregabaliną i u 4,8% pacjentów otrzymujących placebo. Zmiany pola widzenia wykryto u 12,4% pacjentów leczonych pregabaliną i u 11,7% pacjentów otrzymujących placebo. Zmiany w badaniach dna oka stwierdzono u 1,7% pacjentów leczonych pregabaliną i u 2,1% pacjentów otrzymujących placebo.
Farmakokinetyka pregabaliny w stanie stacjonarnym jest podobna u zdrowych ochotników, pacjentów z padaczką otrzymujących leki przeciwpadaczkowe oraz pacjentów z bólem przewlekłym.
Wchłanianie
Pregabalina jest szybko wchłaniana po podaniu na czczo i osiąga maksymalne stężenia w osoczu w ciągu 1 godziny po podaniu dawki pojedynczej i dawek wielokrotnych. Biodostępność pregabaliny po podaniu doustnym wynosi ≥90% i jest niezależna od dawki. Po podaniu dawek wielokrotnych stan stacjonarny zostaje osiągnięty po 24 do 48 godzinach. Szybkość wchłaniania pregabaliny jest zmniejszona, jeśli lek podawany jest wraz z pokarmem, co powoduje zmniejszenie Cmax o około 25- 30% i opóźnienie tmax o około 2,5 godziny. Jakkolwiek, podawanie pregabaliny wraz z pokarmem nie ma istotnego klinicznie wpływu na stopień wchłaniania pregabaliny.
Dystrybucja
W badaniach przedklinicznych pregabalina przenikała przez barierę krew-mózg u myszy, szczurów i małp. Pregabalina przenika przez łożysko u szczurów i jest obecna w mleku karmiących szczurów. U ludzi pozorna objętość dystrybucji pregabaliny po podaniu doustnym wynosi około 0,56 l/kg. Pregabalina nie wiąże się z białkami osocza.
Metabolizm
W organizmie ludzkim pregabalina podlega metabolizmowi w nieistotnym stopniu. Po podaniu dawki pregabaliny znaczonej radioaktywnie, około 98% radioaktywności wykrywano w moczu w postaci niezmienionej pregabaliny. N-metylowa pochodna pregabaliny, główny metabolit obecny w moczu, stanowiła 0,9% dawki. W badaniach przedklinicznych nie obserwowano przechodzenia form racemicznych pregabaliny z S-enancjomeru do R-enancjomeru.
Eliminacja
Pregabalina jest wydalana z krążenia ogólnego głównie przez nerki w postaci niezmienionej. Średni okres półtrwania w fazie eliminacji pregabaliny wynosi 6,3 godziny. Klirensy: osoczowy i nerkowy pregabaliny są wprost proporcjonalne do klirensu kreatyniny (patrz punkt 5.2 Zaburzenie czynności nerek).
Wymagane jest dostosowanie dawki u pacjentów z zaburzeniem czynności nerek lub u pacjentów hemodializowanych (patrz punkt 4.2 Tabela 1).
Liniowość lub nieliniowość
W zalecanym zakresie dawek dobowych farmakokinetyka pregabaliny jest liniowa. Międzyosobnicze różnice w farmakokinetyce leku są małe (<20%). Farmakokinetyka po podaniu dawek wielokrotnych jest przewidywalna i można ją określić na podstawie danych uzyskanych po podaniu dawki jednorazowej. Dlatego nie ma potrzeby rutynowego monitorowania stężenia pregabaliny w osoczu.
Płeć
Badania kliniczne wskazują, że płeć nie wpływa w sposób istotny klinicznie na stężenia pregabaliny w osoczu.
Zaburzenie czynności nerek
Klirens pregabaliny jest wprost proporcjonalny do klirensu kreatyniny. Dodatkowo, pregabalina jest skutecznie usuwana z osocza podczas zabiegu hemodializy (po 4 godzinnym zabiegu hemodializy stężenie pregabaliny w osoczu zostaje zmniejszone o około 50%). Ze względu na to, że główną drogą eliminacji leku jest eliminacja przez nerki, u pacjentów z zaburzoną czynnością nerek konieczne jest zmniejszenie dawki, po zabiegu hemodializy zaś należy podać dawkę dodatkową (patrz punkt 4.2 Tabela 1).
Zaburzenie czynności wątroby
Nie przeprowadzono specjalnych badań dotyczących farmakokinetyki u osób z zaburzeniami czynności wątroby. Ze względu na to, że pregabalina nie ulega istotnemu metabolizmowi i jest wydalana głównie w postaci niezmienionej w moczu, zaburzenia czynności wątroby nie powinny istotnie zmieniać stężenia pregabaliny w osoczu.
Dzieci i młodzież
Farmakokinetykę pregabaliny stosowanej w dawkach 2,5, 5, 10 i 15 mg/kg mc./dobę oceniano u dzieci i młodzieży z padaczką (grupy wiekowe: od 1 do 23 miesięcy, od 2 do 6 lat, od 7 do 11 lat, od 12 do 16 lat) w badaniu farmakokinetyki i tolerancji.
Czas do osiągnięcia maksymalnego stężenia pregabaliny w osoczu po podaniu doustnym na czczo u dzieci i młodzieży był na ogół podobny we wszystkich grupach wiekowych i wynosił od 0,5 godziny do 2 godzin od podania dawki.
Wartości Cmax i AUC dla pregabaliny wzrastały liniowo wraz ze zwiększaną dawką w każdej grupie wiekowej. Wartość AUC była niższa o 30% u dzieci i młodzieży o masie ciała poniżej 30 kg, ponieważ u tych pacjentów klirens skorygowany względem masy ciała był o 43%
większy niż u pacjentów, których masa ciała wynosiła ≥30 kg.
Czas półtrwania pregabaliny w końcowej fazie eliminacji wynosił średnio od około 3 do 4 godzin u dzieci w wieku do 6 lat i od 4 do 6 godzin u pacjentów w wieku 7 lat i starszych.
Analiza farmakokinetyczna populacji wykazała, że klirens kreatyniny był istotną współzmienną wpływającą na klirens pregabaliny po podaniu doustnym, masa ciała była istotną współzmienną wpływającą na pozorną objętość dystrybucji po podaniu doustnym, a zależności te były podobne u dzieci i młodzieży oraz u pacjentów dorosłych.
Nie ustalono farmakokinetyki pregabaliny u pacjentów w wieku poniżej 3 miesięcy (patrz punkty 4.2, 4.8 i 5.1).
Osoby w podeszłym wieku
Klirens pregabaliny zmniejsza się wraz z wiekiem. Zmniejszenie klirensu pregabaliny po podaniu doustnym jest zgodne ze zmniejszeniem klirensu kreatyniny, co jest typowe dla osób starszych. Może być zatem wymagane zmniejszenie dawki pregabaliny u pacjentów, u których doszło do związanego z wiekiem zaburzenia czynności nerek (patrz punkt 4.2 Tabela 1).
Kobiety karmiące piersią
Farmakokinetykę pregabaliny w dawce 150 mg podawanej co 12 godzin (300 mg na dobę) oceniano u 10 kobiet w okresie laktacji, będących co najmniej 12 tygodni po porodzie. Laktacja miała niewielki (lub żaden) wpływ na farmakokinetykę pregabaliny. Pregabalina przenikała do mleka matki, a jej średnie stężenie w stanie stacjonarnym wynosiło około 76% stężenia w osoczu matki. Szacunkowa dawka przyjmowana przez dziecko z mlekiem matki (zakładając średnie dzienne spożycie mleka 150 ml/kg mc./dobę) w przypadku kobiet otrzymujących pregabalinę w dawce 300 mg/dobę wynosiłaby 0,31 mg/kg mc./dobę, a przy maksymalnej dawce 600 mg/dobę — 0,62 mg/kg mc./dobę. Te szacunkowe dawki stanowią około 7% łącznej dawki dobowej przyjętej przez matkę, w przeliczeniu na mg/kg mc.
W konwencjonalnych farmakologicznych badaniach bezpieczeństwa prowadzonych na zwierzętach pregabalina była dobrze tolerowana w zakresie dawek stosowanych klinicznie. W badaniach toksyczności z zastosowaniem dawek wielokrotnych, przeprowadzonych u szczurów i małp obserwowano wpływ na ośrodkowy układ nerwowy w postaci zmniejszenia aktywności, nadmiernej aktywności i ataksji. Po długotrwałej ekspozycji na pregabalinę w dawkach 5-krotnie lub więcej przekraczających średnią ekspozycję u ludzi po zastosowaniu maksymalnych dawek klinicznych, u starszych szczurów albinotycznych często obserwowano zwiększoną częstość występowania zaniku siatkówki.
Pregabalina nie była teratogenna u myszy, szczurów lub królików. Toksyczność względem płodów u szczurów i królików obserwowano jedynie po zastosowaniu dawek istotnie większych od tych stosowanych u ludzi. W badaniach dotyczących toksyczności prenatalnej i pourodzeniowej pregabalina wywoływała zaburzenia rozwojowe u młodych szczurów przy ekspozycji >2 razy większej od maksymalnej, zalecanej ekspozycji u człowieka.
Niekorzystny wpływ na płodność samców i samic szczura stwierdzano wyłącznie po ekspozycji znacznie przekraczającej ekspozycję leczniczą. Niekorzystny wpływ na męskie narządy rozrodcze i parametry nasienia był przemijający, i objawiał się wyłącznie po ekspozycji znacznie przekraczającej ekspozycję leczniczą lub był związany z samoistnymi procesami zwyrodnieniowymi w męskich narządach rozrodczych u szczurów. W związku z tym opisane działania uznano za bez znaczenia klinicznego bądź mające minimalne znaczenie kliniczne.
W serii testów przeprowadzonych in vitro i in vivo pregabalina nie była genotoksyczna.
U szczurów i myszy przeprowadzono dwuletnie badania karcynogenności pregabaliny. Nie obserwowano występowania guzów u szczurów przy ekspozycji 24-krotnie przekraczającej średnią ekspozycję u ludzi po zastosowaniu maksymalnej zalecanej dawki dobowej 600 mg. U myszy nie obserwowano zwiększenia częstości występowania guzów przy ekspozycji porównywalnej do średniej ekspozycji u ludzi, zaś przy większych ekspozycjach obserwowano zwiększenie częstości występowania naczyniakomięsaka krwionośnego (haemangiosarcoma). Spośród niegenotoksycznych mechanizmów powstawania guzów u myszy wywołanych przez pregabalinę wymienia się zmiany dotyczące płytek krwi i związaną z nimi proliferację komórek śródbłonka. W obserwacji krótkoterminowej i ograniczonej długoterminowej obserwacji klinicznej zmiany płytek krwi nie występowały u szczurów ani u ludzi. Nie ma dowodów, na podstawie których można by wnioskować o ryzyku u ludzi.
Rodzaje toksyczności nie różniły się jakościowo w grupie młodych szczurów i szczurów dorosłych, jakkolwiek młode szczury są bardziej wrażliwe na działanie leku. Po zastosowaniu dawek terapeutycznych obserwowano objawy kliniczne dotyczące OUN - nadreaktywność
i bruksizm, a także zmiany we wzrastaniu (przejściowe zahamowanie tempa zwiększenia masy ciała). Wpływ na cykl rozrodczy obserwowano po dawkach 5-krotnie przekraczających ekspozycję u ludzi. U młodych szczurów 1-2 tygodnia po ekspozycji na dawkę ponad dwukrotnie przekraczającą ekspozycję u ludzi obserwowano osłabienie reakcji na dźwięk. Dziewięć tygodni po ekspozycji efekt ten nie był widoczny.
Celuloza mikrokrystaliczna Magnezu stearynian
Nie dotyczy.
3 lata.
Brak specjalnych zaleceń dotyczących przechowywania produktu leczniczego.
Blistry z folii PA/Aluminium /PVC/Aluminium zawierające 14, 28, 56, 84 tabletek w tekturowym pudełku.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Bez specjalnych wymagań dotyczących usuwania.
neuraxpharm Arzneimittel GmbH
Elisabeth-Selbert-Straße 23
40764 Langenfeld Niemcy
25293, 25294, 25295, 25296, 25297, 25298, 25299, 25300
19/04/2019
22.03.2022








