







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania i przygotowania leku do stosowania
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMERY POZWOLEŃ NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Everolimus Sandoz, 2,5 mg, tabletki Everolimus Sandoz, 5 mg, tabletki Everolimus Sandoz, 10 mg, tabletki
Everolimus Sandoz, 2,5 mg
Każda tabletka zawiera 2,5 mg ewerolimusu (Everolimusum).
Substancja pomocnicza o znanym działaniu Każda tabletka zawiera 74,2 mg laktozy.
Everolimus Sandoz, 5 mg
Każda tabletka zawiera 5 mg ewerolimusu (Everolimusum).
Substancja pomocnicza o znanym działaniu Każda tabletka zawiera 148,4 mg laktozy.
Everolimus Sandoz, 10 mg
Każda tabletka zawiera 10 mg ewerolimusu (Everolimusum).
Substancja pomocnicza o znanym działaniu Każda tabletka zawiera 296,8 mg laktozy.
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Tabletka
Everolimus Sandoz, 2,5 mg
Białe do jasnożółtych, podłużne tabletki o przybliżonych wymiarach 10,1 x 4,1 mm, ze ściętą krawędzią, bez linii podziału, z oznakowaniem „LCL” po jednej stronie i „NVR” po drugiej stronie.
Everolimus Sandoz, 5 mg
Białe do jasnożółtych, podłużne tabletki o przybliżonych wymiarach 12,1 x 4,9 mm, ze ściętą krawędzią, bez linii podziału, z oznakowaniem „5” po jednej stronie i „NVR” po drugiej stronie.
Everolimus Sandoz, 10 mg
Białe do jasnożółtych, podłużne tabletki o przybliżonych wymiarach 15,1 x 6,0 mm, ze ściętą krawędzią, bez linii podziału, z oznakowaniem „UHE” po jednej stronie i „NVR” po drugiej stronie.
Nowotwory neuroendokrynne trzustki
Produkt leczniczy Everolimus Sandoz jest wskazany w leczeniu dorosłych pacjentów z wysoko lub średnio zróżnicowanymi nowotworami neuroendokrynnymi trzustki nieoperacyjnymi lub
z przerzutami, z chorobą o przebiegu postępującym.
Leczenie produktem Everolimus Sandoz powinien rozpoczynać i nadzorować lekarz z doświadczeniem w leczeniu przeciwnowotworowym.
Dawkowanie
W celu stosowania w różnych schematach dawkowania dostępne są tabletki produktu Everolimus Sandoz o mocy 2,5 mg, 5 mg i 10 mg.
Zalecana dawka ewerolimusu wynosi 10 mg jeden raz na dobę. Leczenie należy kontynuować tak długo, jak widoczne jest korzystne działanie kliniczne lub do czasu wystąpienia niemożliwych do zaakceptowania przez pacjenta objawów toksyczności.
Jeśli pacjent pominie dawkę produktu leczniczego, nie powinien przyjmować dodatkowej dawki, lecz przyjąć następną tabletkę o ustalonej porze.
Modyfikacja dawki ze względu na działania niepożądane
Postępowanie w przypadku poważnych i (lub) niemożliwych do zaakceptowania działań niepożądanych może wymagać zmniejszenia dawki i (lub) czasowego odstawienia produktu Everolimus Sandoz. W razie działań niepożądanych 1. stopnia modyfikacja dawki nie jest zazwyczaj konieczna. Jeśli zmniejszenie dawki jest konieczne, zalecana dawka wynosi 5 mg na dobę i nie może być mniejsza niż 5 mg na dobę.
W tabeli 1 przedstawiono zalecenia dotyczące modyfikacji dawki w przypadku różnych działań niepożądanych (patrz także punkt 4.4).
Tabela 1 Zalecenia dotyczące modyfikacji dawki produktu Everolimus Sandoz
Działanie niepożądane | Nasilenie1 | Modyfikacja dawki produktu Everolimus Sandoz |
Nieinfekcyjne zapalenie płuc | Stopień 2. | Rozważyć przerwanie leczenia do czasu ustąpienia objawów do stopnia ≤1. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. Jeśli poprawa nie nastąpi w ciągu 4 tygodni, przerwać leczenie. |
Stopień 3. | Przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Rozważyć ponowne rozpoczęcie leczenia z zastosowaniem dawki 5 mg na dobę. W razie nawrotu toksyczności stopnia 3., rozważyć przerwanie leczenia. | |
Stopień 4. | Przerwać leczenie. | |
Zapalenie błony śluzowej jamy ustnej | Stopień 2. | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Wznowić leczenie stosując tę samą dawkę. W razie nawrotu zapalenia jamy ustnej stopnia 2. przerwać podawanie do czasu ustąpienia objawów do stopnia ≤1. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. |
Stopień 3. | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. | |
Stopień 4. | Przerwać leczenie. |
Inne niehematologiczne objawy toksyczności (z wyłączeniem zaburzeń metabolicznych) | Stopień 2. | Modyfikacja dawki nie jest konieczna, jeśli pacjent jest w stanie tolerować objawy toksyczności. Jeśli toksyczność jest niemożliwa do zniesienia, czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Ponownie rozpocząć leczenie stosując tę samą dawkę. W razie nawrotu toksyczności stopnia 2., przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. |
Stopień 3. | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1. Rozważyć ponowne rozpoczęcie leczenia z zastosowaniem dawki 5 mg na dobę. W razie nawrotu toksyczności stopnia 3., rozważyć przerwanie leczenia. | |
Stopień 4. | Przerwać leczenie. | |
Zaburzenia metaboliczne (np. hiperglikemia, dyslipidemia) | Stopień 2. | Modyfikacja dawki nie jest konieczna. |
Stopień 3. | Czasowo przerwać podawanie. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. | |
Stopień 4. | Przerwać leczenie. | |
Małopłytkowość | Stopień 2. (<75, ≥50 x 109/l) | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1 (≥75 x 109/l). Ponownie rozpocząć leczenie stosując tę samą dawkę. |
Stopień 3. & 4. (<50 x 109/l) | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤1 (≥75 x 109/l). Ponownie rozpocząć leczenie stosując dawkę 5 mg/dobę. | |
Neutropenia | Stopień 2. (≥1 x 109/l) | Modyfikacja dawki nie jest konieczna. |
Stopień 3. (<1, ≥0,5 x 109/l) | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤2 (≥1 x 109/l). Ponownie rozpocząć leczenie stosując tę samą dawkę. | |
Stopień 4. (<0,5 x 109/l) | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤2 (≥1 x 109/l). Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. | |
Gorączka neutropeniczna | Stopień 3. | Czasowo przerwać leczenie do czasu ustąpienia objawów do stopnia ≤2 (≥1,25 x 109/l) i braku gorączki. Ponownie rozpocząć leczenie stosując dawkę 5 mg na dobę. |
Stopień 4. | Przerwać leczenie. | |
1 Stopniowanie nasilenia objawów wg Powszechnych kryteriów terminologicznych dla zdarzeń niepożądanych (ang. Common Terminology Criteria for Adverse Events, CTCAE) Narodowego Instytutu Raka (ang. National Cancer Institute, NCI), wersja 3.0 | ||
Szczególne grupy pacjentów
Osoby w podeszłym wieku (≥65 lat)
Modyfikacja dawki nie jest konieczna (patrz punkt 5.2).
Zaburzenia czynności nerek
Modyfikacja dawki nie jest konieczna (patrz punkt 5.2).
Zaburzenia czynności wątroby
Lekkie zaburzenia czynności wątroby (klasa A w skali Childa-Pugha) – zalecana dawka dobowa wynosi 7,5 mg.
Umiarkowane zaburzenia czynności wątroby (klasa B w skali Childa-Pugha) – zalecana dawka dobowa wynosi 5 mg.
Poważne zaburzenia czynności wątroby (klasa C w skali Childa-Pugha) – stosowanie produktu
Everolimus Sandoz zaleca się tylko wtedy, gdy pożądana korzyść przewyższa ryzyko. W takim przypadku nie wolno stosować dawki większej niż 2,5 mg na dobę.
Dawkę należy zmodyfikować, jeśli czynność wątroby pacjenta (w skali Childa-Pugha) zmieni się w trakcie leczenia (patrz także punkty 4.4 i 5.2).
Dzieci i młodzież
Nie ustalono bezpieczeństwa stosowania ani skuteczności produktu Everolimus Sandoz u dzieci w wieku od 0 do 18 lat. Brak danych.
Sposób podawania
Produkt leczniczy Everolimus Sandoz należy podawać doustnie raz na dobę, o tej samej porze dnia, konsekwentnie albo z posiłkiem, albo bez posiłku (patrz punkt 5.2). Tabletki należy połykać w całości, popijając szklanką wody. Tabletek nie należy żuć ani rozkruszać.
Nadwrażliwość na substancję czynną, inne pochodne rapamycyny lub na którąkolwiek substancję pomocniczą podaną w punkcie 6.1.
Nieinfekcyjne zapalenie płuc
Nieinfekcyjne zapalenie płuc jest efektem dotyczącym całej grupy pochodnych rapamycyny, w tym ewerolimusu. U pacjentów przyjmujących ewerolimus często zgłaszano występowanie nieinfekcyjnego zapalenia płuc (w tym śródmiąższowej choroby płuc), patrz punkt 4.8. Niektóre przypadki były ciężkie i rzadko prowadziły do zgonu. Rozpoznanie nieinfekcyjnego zapalenia płuc należy rozważyć u pacjentów z niespecyficznymi przedmiotowymi i podmiotowymi objawami ze strony układu oddechowego, takimi jak niedotlenienie, wysięk do opłucnej, kaszel lub duszność oraz u pacjentów, i u których wykluczono odpowiednimi metodami diagnostycznymi zakaźne, nowotworowe lub inne, niemedyczne przyczyny występujących objawów. W diagnostyce różnicowej nieinfekcyjnego zapalenia płuc należy wykluczyć zakażenia oportunistyczne, takie jak zapalenie płuc wywołane przez Pneumocystis jirovecii (carinii) (PJP, PCP) (patrz niżej „Zakażenia”). Pacjentów należy poinformować o konieczności natychmiastowego zgłaszania nowych lub pogorszeniu istniejących objawów ze strony układu oddechowego.
U pacjentów, u których wystąpią zmiany w obrazie radiologicznym wskazujące na nieinfekcyjne zapalenie płuc z nielicznymi objawami lub bez objawów, można dalej stosować produkt Everolimus Sandoz bez modyfikacji dawki. Jeśli nasilenie objawów jest umiarkowane (2. stopnia) lub ciężkie (3. stopnia), wskazane może być podawanie kortykosteroidów aż do momentu ustąpienia objawów klinicznych.
U pacjentów, u których konieczne jest podanie kortykosteroidów w leczeniu nieinfekcyjnego zapalenia płuc, należy rozważyć profilaktykę PJP, PCP.
Zakażenia
Ewerolimus hamuje czynność układu odpornościowego i może powodować podatność pacjentów na zakażenia bakteryjne, grzybicze, wirusowe lub, pierwotniakowe, w tym zakażenia patogenami oportunistycznymi (patrz punkt 4.8). U pacjentów przyjmujących ewerolimus opisywano zakażenia miejscowe i uogólnione, w tym infekcyjne zapalenie płuc, inne zakażenia bakteryjne, inwazyjne grzybice, takie jak aspergiloza, kandydoza lub PJP, PCP oraz zakażenia wirusowe, w tym reaktywację wirusa zapalenia wątroby typu B. Niektóre z tych zakażeń były poważne (np. prowadzące do posocznicy, niewydolności oddechowej lub niewydolności nerek) i sporadycznie prowadziły do zgonu.
Lekarze i pacjenci powinni być świadomi zwiększonego ryzyka zakażeń podczas leczenia produktem Everolimus Sandoz. Przed rozpoczęciem stosowania tego produktu leczniczego należy odpowiednio
leczyć istniejące zakażenia i doprowadzić do ich pełnego ustąpienia. Podczas terapii ewerolimusem należy kontrolować, czy u pacjenta nie występują objawy zakażenia, a w razie jego stwierdzenia niezwłocznie zastosować odpowiednie leczenie i rozważyć przerwanie lub zakończenie podawania produktu Everolimus Sandoz.
W razie stwierdzenia inwazyjnego zakażenia grzybiczego należy natychmiast odstawić definitywnie produkt Everolimus Sandoz i zastosować u pacjenta odpowiednie leczenie przeciwgrzybicze.
U pacjentów otrzymujących ewerolimus opisywano przypadki PJP, PCP, z których część zakończyła się zgonem. Występowanie PJP/PCP może wiązać się z jednoczesnym stosowaniem kortykosteroidów lub innych leków immunosupresyjnych. Jeśli konieczne jest jednoczesne stosowanie ewerolimusu i kortykosteroidów lub innych leków immunosupresyjnych, należy rozważyć profilaktykę PJP, PCP.
Reakcje nadwrażliwości
Podczas stosowania ewerolimusu obserwowano reakcje nadwrażliwości między innymi z takimi objawami, jak anafilaksja, duszność, zaczerwienienie skóry, ból w klatce piersiowej lub obrzęk naczynioruchowy (np. obrzęk dróg oddechowych lub języka, z zaburzeniami oddechowymi lub bez nich), patrz punkt 4.3.
Jednoczesne stosowanie z inhibitorami konwertazy angiotensyny (ACE)
U pacjentów przyjmujących jednocześnie inhibitor ACE (np. ramipryl) może istnieć zwiększone ryzyko obrzęku naczynioruchowego (np. obrzęk dróg oddechowych lub języka, z zaburzeniami oddechowymi lub bez nich), patrz punkt 4.5.
Zapalenie błony śluzowej jamy ustnej
Najczęściej zgłaszanym działaniem niepożądanym u pacjentów leczonych ewerolimusem jest zapalenie błony śluzowej jamy ustnej (w tym owrzodzenie jamy ustnej), patrz punkt 4.8. Zapalenie błony śluzowej jamy ustnej występuje najczęściej w ciągu pierwszych 8 tygodni leczenia.
Postępowanie w przypadku zapalenia błony śluzowej jamy ustnej może obejmować zapobiegawcze
i (lub) lecznicze stosowanie środków o działaniu miejscowym, takich jak płyn do płukania jamy ustnej z bezalkoholowym doustnym roztworem kortykosteroidu. Należy jednak unikać stosowania produktów zawierających alkohol, nadtlenek wodoru, jod i wyciągi z tymianku, gdyż mogą one zaostrzać objawy. Zaleca się monitorowanie pod kątem zakażenia grzybiczego i jego leczenia, zwłaszcza u pacjentów otrzymujących produkty lecznicze zawierające steroidy. Nie należy stosować leków przeciwgrzybiczych bez rozpoznania zakażenia grzybiczego (patrz punkt 4.5).
Niewydolność nerek
U pacjentów leczonych ewerolimusem obserwowano przypadki niewydolności nerek (w tym ostrej niewydolności nerek), niektóre zakończone zgonem (patrz punkt 4.8). Zaleca się kontrolowanie czynności nerek, zwłaszcza u pacjentów z dodatkowymi czynnikami ryzyka, które mogą spowodować dalsze zaburzenia czynności nerek.
Badania laboratoryjne
Czynność nerek
Zgłaszano zwiększenie stężenia kreatyniny (zazwyczaj niewielkie) i białkomocz (patrz punkt 4.8). Przed rozpoczęciem stosowania ewerolimusu i okresowo w trakcie leczenia zaleca się kontrolowanie czynności nerek, w tym oznaczenia stężenia azotu mocznikowego (BUN), białka w moczu lub kreatyniny w surowicy.
Stężenie glukozy we krwi
Zgłaszano przypadki hiperglikemii (patrz punkt 4.8). Przed rozpoczęciem stosowania ewerolimusu i okresowo w trakcie leczenia zaleca się kontrolowanie stężenia glukozy (na czczo) w surowicy.
Częstsze kontrole zaleca się w przypadku jednoczesnego stosowania ewerolimusu z innymi produktami leczniczymi, które mogą spowodować hiperglikemię. Jeśli to możliwe, przed rozpoczęciem stosowania ewerolimusu należy uzyskać optymalną kontrolę glikemii.
Stężenie lipidów we krwi
Zgłaszano przypadki dyslipidemii (w tym hipercholesterolemię i hipertrigliceridemię). Przed rozpoczęciem stosowania ewerolimusu i okresowo w trakcie leczenia zaleca się kontrolowanie stężenia cholesterolu i triglicerydów we krwi, a także wdrożenie odpowiedniego leczenia.
Wskaźniki hematologiczne
Zgłaszano przypadki zmniejszenia stężenia hemoglobiny, liczby limfocytów, neutrofilów i płytek krwi (patrz punkt 4.8). Przed rozpoczęciem stosowania ewerolimusu i okresowo w trakcie leczenia zaleca się kontrolowanie pełnej morfologii krwi.
Hormonalnie czynne rakowiaki
W randomizowanym, wieloośrodkowym badaniu z podwójnie ślepą próbą u pacjentów z hormonalnie czynnymi rakowiakami ewerolimus razem z oktreotydem w postaci depot porównywano z placebo
w skojarzeniu z oktreotydem w postaci depot. W badaniu nie uzyskano pierwszorzędowego punktu końcowego skuteczności (przeżycie wolne od progresji choroby, ang. progression free survival, PFS), a etapowa analiza przeżycia całkowitego (ang. overall survival, OS) wykazała liczbową przewagę na korzyść grupy otrzymującej placebo razem z oktreotydem w postaci depot. Dlatego nie ustalono bezpieczeństwa stosowania i skuteczności ewerolimusu u pacjentów z hormonalnie czynnymi rakowiakami.
Interakcje
Należy unikać jednoczesnego stosowania ewerolimusu z inhibitorami i induktorami CYP3A4 i (lub) pompy wielolekowej glikoproteiny P (PgP). Jeśli leczenie skojarzone z umiarkowanym inhibitorem lub induktorem CYP3A4 i (lub) PgP jest konieczne, należy brać pod uwagę modyfikację dawki ewerolimusu na podstawie przewidywanej wartości AUC (patrz punkt 4.5).
Jednoczesne stosowanie z silnymi inhibitorami CYP3A4 powodowało znaczne zwiększenie stężenia ewerolimusu w osoczu (patrz punkt 4.5). Aktualne dane są niewystarczające do określenia zaleceń dotyczących dawkowania w takiej sytuacji. Dlatego nie zaleca się stosowania ewerolimusu z silnymi inhibitorami CYP3A4.
Ze względu na możliwość wystąpienia interakcji należy zachować ostrożność podczas jednoczesnego stosowania ewerolimusu i doustnych substratów CYP3A4 o wąskim indeksie terapeutycznym. Jeśli ewerolimus jest przyjmowany z doustnymi substratami CYP3A4 o wąskim indeksie terapeutycznym (np. pimozyd, terfenadyna, astemizol, cyzapryd, chinidyna lub pochodne alkaloidów sporyszu), należy kontrolować, czy u pacjenta nie występują działania niepożądane opisane w ChPL substratów CYP3A4 podawanych doustnie (patrz punkt 4.5).
Zaburzenia czynności wątroby
Ekspozycja na ewerolimus byłą zwiększona u pacjentów z lekkimi (klasa A w skali Childa-Pugha), umiarkowanymi (klasa B w skali Childa-Pugha) i ciężkimi (klasa C w skali Childa-Pugha) zaburzeniami czynności wątroby (patrz punkt 5.2).
Stosowanie ewerolimusu u pacjentów z ciężkimi zaburzeniami czynności wątroby (klasa C w skali Childa-Pugha) zaleca się tylko wtedy, gdy możliwe korzyści przewyższają ryzyko (patrz punkty 4.2 i 5.2).
Nie są obecnie dostępne żadne dane kliniczne dotyczące bezpieczeństwa lub skuteczności, które pozwoliłyby na określenie modyfikacji dawkowania w przypadku wystąpienia działań niepożądanych u pacjentów z zaburzeniami czynności wątroby.
Szczepienia
Podczas leczenia ewerolimusem należy unikać stosowania szczepionek zawierających żywe drobnoustroje (patrz punkt 4.5).
Produkt Everolimus Sandoz zawiera laktozę
Ten produkt leczniczy nie powinien być stosowany u pacjentów z rzadko występującą dziedziczną
nietolerancją galaktozy, brakiem laktazy lub zespołem złego wchłaniania glukozy-galaktozy.
Zaburzenia gojenia się ran
Zaburzenia gojenia się ran są efektem dotyczącym całej grupy pochodnych rapamycyny, w tym ewerolimusu. Dlatego należy zachować ostrożność, jeśli ewerolimus stosowany jest w okresie okołooperacyjnym.
Ewerolimus jest substratem izoenzymu CYP3A4 oraz substratem i umiarkowanym inhibitorem glikoproteiny P (PgP). Dlatego na wchłanianie, a następnie eliminację ewerolimusu mogą wpływać produkty lecznicze, które oddziałują z CYP3A4 i (lub) glikoproteiną P. W warunkach in vitro ewerolimus jest kompetycyjnym inhibitorem CYP3A4 oraz mieszanym inhibitorem CYP2D6.
Znane i teoretycznie możliwe interakcje z wybranymi inhibitorami i induktorami CYP3A4 i PgP przedstawiono niżej w tabeli 2.
Inhibitory CYP3A4 i PgP zwiększające stężenie ewerolimusu
Substancje, które są inhibitorami CYP3A4 lub PgP, mogą zwiększyć stężenie ewerolimusu we krwi poprzez zmniejszenie jego metabolizmu lub usuwania go z komórek jelita.
Induktory CYP3A4 i PgP zmniejszające stężenie ewerolimusu
Substancje, które są induktorami CYP3A4 lub PgP, mogą zmniejszyć stężenie ewerolimusu we krwi zwiększając jego metabolizm lub usuwanie go z komórek jelita.
Tabela 2 Wpływ innych substancji czynnych na ewerolimus
Substancja czynna w zależności od interakcji | Interakcja – zmiana stosunku średnich geometrycznych AUC/Cmax ewerolimusu (obserwowany zakres) | Zalecenia dotyczące leczenia skojarzonego |
Silne inhibitory CYP3A4/PgP | ||
Ketokonazol | AUC ↑ 15,3-krotne | Nie zaleca się jednoczesnego leczenia produktem |
(zakres 11,2-22,5) | Everolimus Sandoz i silnymi inhibitorami. | |
Cmax ↑ 4,1-krotne | ||
(zakres 2,6-7,0) | ||
Itrakonazol, | Nie badano. Spodziewane jest | |
pozakonazol, | znaczne zwiększenie stężenia | |
worykonazol | ewerolimusu. | |
Telitromycyna, | ||
klarytromycyna | ||
Nefazodon | ||
Rytonawir, | ||
atazanawir, | ||
sakwinawir, | ||
darunawir, | ||
indynawir, | ||
nelfinawir | ||
Umiarkowane inhibitory CYP3A4/PgP | ||
Erytromycyna | AUC ↑ 4,4-krotne | Jeśli konieczne jest jednoczesne podawanie ewerolimusu i umiarkowanych inhibitorów CYP3A4 lub inhibitorów PgP, należy zachować ostrożność. Jeśli pacjent wymaga jednoczesnego stosowania umiarkowanego inhibitora CYP3A4 lub PgP, można rozważyć zmniejszenie dawki dobowej ewerolimusu |
(zakres 2,0-12,6) | ||
Cmax ↑ 2,0-krotne | ||
(zakres 0,9-3,5) | ||
Imatynib | AUC ↑ 3,7-krotne | |
Cmax ↑ 2,2-krotnie | ||
Werapamil | AUC ↑ 3,5-krotne (zakres 2,2-6,3) Cmax ↑ 2,3-krotne (zakres 1,3-3,8) | do 5 mg lub 2,5 mg, chociaż brak danych klinicznych z zastosowaniem takiej modyfikacji dawki. Ze względu na zmienność międzyosobniczą zalecana modyfikacja dawki może nie być optymalna dla wszystkich pacjentów, dlatego zaleca się ścisłe monitorowanie działań niepożądanych. Jeśli stosowanie umiarkowanego inhibitora jest przerywane, należy rozważyć zastosowanie okresu wymywania trwającego co najmniej 2 do 3 dni (średni czas eliminacji większości stosowanych umiarkowanych inhibitorów) przed przywróceniem dawki produktu Everolimus Sandoz do wartości sprzed leczenia skojarzonego. |
Cyklosporyna doustnie | AUC ↑ 2,7-krotne (zakres 1,5-4,7) Cmax ↑ 1,8-krotne (zakres 1,3-2,6) | |
Flukonazol | Nie badano. Spodziewane jest zwiększenie ekspozycji. | |
Diltiazem | ||
Dronedaron | Nie badano. Spodziewane jest zwiększenie ekspozycji. | |
Amprenawir, fosamprenawir | Nie badano. Spodziewane jest zwiększenie ekspozycji. | |
Sok grejpfrutowy lub inne pokarmy wpływające na CYP3A4/PgP | Nie badano. Spodziewane jest zwiększenie ekspozycji (znaczne zróżnicowanie wpływu). | Należy unikać jednoczesnego przyjmowania. |
Substancja czynna w zależności od interakcji | Interakcja – zmiana stosunku średnich geometrycznych AUC/Cmax ewerolimusu (obserwowany zakres) | Zalecenia dotyczące leczenia skojarzonego |
Silne i umiarkowane induktory CYP3A4 | ||
Ryfampicyna | AUC ↓ 63% (zakres 0-80%) Cmax ↓ 58% (zakres 10-70%) | Należy unikać jednoczesnego podawania ewerolimusu i silnych induktorów CYP3A4. Jeśli pacjent wymaga jednoczesnego stosowania silnego induktora CYP3A4, należy rozważyć stopniowe zwiększanie dawki dobowej produktu Everolimus Sandoz z 10 mg nawet do 20 mg co 5 mg lub mniej, podawanej w 4. oraz 8. dniu po rozpoczęciu podawania induktora. Przewiduje się, że taka dawka produktu Everolimus Sandoz spowoduje dostosowanie wartości AUC do zakresu obserwowanego bez podawania induktorów. Jednak brak danych klinicznych z zastosowaniem takiej modyfikacji dawki. Jeśli stosowanie induktora jest przerywane, należy rozważyć zastosowanie okresu wymywania trwającego co najmniej 3 do 5 dni (uzasadniony czas odwrócenia indukcji enzymów) przed przywróceniem dawki produktu Everolimus Sandoz do wartości sprzed leczenia skojarzonego. |
Deksametazon | Nie badano. Spodziewane jest zmniejszenie ekspozycji. | |
Karbamazepina, fenobarbital, fenytoina | Nie badano. Spodziewane jest zmniejszenie ekspozycji. | |
Efawirenz, newirapina | Nie badano. Spodziewane jest zmniejszenie ekspozycji. | |
Ziele dziurawca (Hypericum perforatum) | Nie badano. Spodziewane jest znaczne zmniejszenie ekspozycji. | Podczas leczenia ewerolimusem nie należy stosować preparatów zawierających ziele dziurawca. |
Leki, których stężenie w osoczu może być zmienione przez ewerolimus
Wyniki badań in vitro wskazują, że zahamowanie aktywności PgP, CYP3A4 i CYP2D6 przy stężeniach ogólnoustrojowych ewerolimusu uzyskanych po podaniu doustnych dawek 10 mg jest mało prawdopodobne. Nie można jednak wykluczyć zahamowania aktywności CYP3A4 i PgP w jelicie.
Badanie interakcji u zdrowych osób wykazało, że jednoczesne podanie doustnej dawki midazolamu (czułego znacznika dla substratu CYP3A4) i ewerolimusu spowodowało zwiększenie wartości Cmax oraz AUC0-inf dla midazolamu odpowiednio o 25% i 30%. Działanie to jest prawdopodobnie spowodowane zahamowaniem przez ewerolimus aktywności jelitowego CYP3A4. Dlatego
ewerolimus może wpływać na biodostępność podawanych doustnie substratów CYP3A4. Nie należy jednak spodziewać się klinicznie istotnego wpływu na ekspozycję na substraty CYP3A4 podawane ogólnie (patrz punkt 4.4).
Jednoczesne podawanie ewerolimusu z oktreotydem w postaci depot powodowało zwiększenie wartości Cmin oktreotydu przy stosunku średnich geometrycznych (ewerolimus/placebo) wynoszących 1,47. Nie udało się ustalić, czy ma to istotny kliniczny wpływ na skuteczność odpowiedzi na leczenie ewerolimusem pacjentów z zaawansowanymi nowotworami neuroendokrynnymi.
Jednoczesne stosowanie inhibitorów konwertazy angiotensyny (ACE)
U pacjentów przyjmujących jednocześnie inhibitor ACE (np. ramipryl) może istnieć zwiększone ryzyko obrzęku naczynioruchowego (patrz punkt 4.4).
Szczepienia
Ewerolimus może wpływać na odpowiedź immunologiczną na szczepienie, dlatego podczas stosowania produktu Everolimus Sandoz szczepienie może być mniej skuteczne. Należy unikać stosowania żywych szczepionek w czasie leczenia produktem Everolimus Sandoz (patrz punkt 4.4). Przykładami żywych szczepionek są: donosowa szczepionka przeciw grypie, szczepionki przeciw odrze, nagminnemu zapaleniu przyusznic, różyczce, doustna szczepionka przeciw polio, szczepionka przeciw gruźlicy (Bacillus Calmette-Guérin, BCG), przeciw żółtej febrze, ospie wietrznej oraz szczepionki przeciw durowi brzusznemu TY21.
Kobiety w wieku rozrodczym/antykoncepcja u kobiet i mężczyzn
Kobiety w wieku rozrodczym muszą stosować w trakcie stosowania ewerolimusu i do 8 tygodni po zakończeniu leczenia wysoce skuteczną metodę zapobiegania ciąży (tj. niezawierająca estrogenów hormonalna metoda w postaci doustnej, we wstrzyknięciu lub implancie, środki antykoncepcyjne zawierające progesteron, histerektomia, podwiązanie jajowodów, całkowita abstynencja, antykoncepcja barierowa, wkładka wewnątrzmaciczna i (lub) sterylizacja [kobiety/mężczyzny]).
Nie ma przeciwwskazań do poczęcia dziecka przez mężczyznę leczonego ewerolimusem.
Ciąża
Brak odpowiednich danych dotyczących stosowania ewerolimusu u kobiet w ciąży. Badania na zwierzętach wykazały toksyczny wpływ na reprodukcję, w tym na zarodki i płody (patrz punkt 5.3). Potencjalne ryzyko dla ludzi nie jest znane.
Nie zaleca się stosowania ewerolimusu w czasie ciąży ani u kobiet w wieku rozrodczym, które nie stosują antykoncepcji.
Karmienie piersią
Nie wiadomo czy ewerolimus przenika do mleka kobiecego. W badaniach na zwierzętach wykazano, że u szczurów ewerolimus i (lub) jego metabolity łatwo przenikały do mleka samic w okresie laktacji (patrz punkt 5.3). Dlatego kobiety przyjmujące ewerolimus nie powinny karmić piersią w trakcie leczenia i przez 2 tygodnie po przyjęciu ostatniej dawki.
Płodność
Nie wiadomo, czy ewerolimus może powodować niepłodność u kobiet i mężczyzn, jednak u kobiet obserwowano brak miesiączkowania (wtórny brak miesiączki i inne zaburzenia miesiączkowania) i związane z tym zaburzenia równowagi między hormonem luteinizującym (LH) a hormonem folikulotropowym (FSH). Wyniki badań nieklinicznych wskazują, że leczenie ewerolimusem może zmniejszać płodność mężczyzn i kobiet (patrz punkt 5.3).
Produkt Everolimus Sandoz może mieć niewielki lub umiarkowany wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn. Pacjentom należy zalecić ostrożność podczas wykonywania tych
czynności, jeśli podczas stosowania tego produktu leczniczego odczuwają zmęczenie.
Podsumowanie profilu bezpieczeństwa
Profil bezpieczeństwa ustalono na podstawie połączonych danych od 2879 pacjentów otrzymujących ewerolimus w ramach 11 badań klinicznych (5 randomizowanych, kontrolowanych placebo badań fazy III z podwójnie ślepą próbą oraz 6 otwartych badań fazy I i II) dotyczących zatwierdzonych wskazań.
Połączone dane wskazują, że najczęściej notowanymi działaniami niepożądanymi (występującymi z częstością ≥1/10) były (w malejącej kolejności): zapalenie błony śluzowej jamy ustnej, wysypka,
zmęczenie, biegunka, zakażenia, nudności, zmniejszenie apetytu, niedokrwistość, zaburzenia smaku, nieinfekcyjne zapalenie płuc, obrzęki obwodowe, hiperglikemia, astenia, świąd, zmniejszenie masy ciała, hipercholesterolemia, krwawienie z nosa, kaszel i ból głowy.
Najczęstszymi działaniami niepożądanymi 3.-4. stopnia (występującymi z częstością ≥1/100 do <1/10) były: zapalenie błony śluzowej jamy ustnej, niedokrwistość, hiperglikemia, zakażenia, zmęczenie, biegunka, nieinfekcyjne zapalenie płuc, astenia, małopłytkowość, neutropenia, duszność, białkomocz, limfopenia, krwotok, hipofosfatemia, wysypka, nadciśnienie tętnicze, infekcyjne zapalenie płuc, zwiększenie aktywności aminotransferazy alaninowej (AlAT), zwiększenie aktywności aminotransferazy asparaginianowej (AspAT) i cukrzyca (wg CTCAE, wersja 3.0 i 4.03).
Tabelaryczne zestawienie działań niepożądanych
Tabela 3 przedstawia częstości działań niepożądanych odnotowanych w analizie połączonych danych w populacji ocenianej pod względem bezpieczeństwa. Działania niepożądane wymieniono zgodnie
z klasyfikacją układów i narządów MedDRA, a częstości zdefiniowano następująco: bardzo często (≥1/10); często (≥1/100 do <1/10); niezbyt często (≥1/1000 do <1/100); rzadko (≥1/10 000 do
<1/1000); bardzo rzadko (<1/10 000). W obrębie każdej z grup o określonej częstości działania niepożądane wymieniono zgodnie ze zmniejszającym się nasileniem.
Tabela 3 Działania niepożądane zgłaszane w badaniach klinicznych
Zakażenia i zarażenia pasożytnicze | |
Bardzo często | Zakażeniaa,* |
Zaburzenia krwi i układu chłonnego | |
Bardzo często | Niedokrwistość |
Często | Małopłytkowość, neutropenia, leukopenia, limfopenia |
Niezbyt często | Pancytopenia |
Rzadko | Aplazja czysto czerwonokrwinkowa |
Zaburzenia układu immunologicznego | |
Niezbyt często | Nadwrażliwość |
Zaburzenia metabolizmu i odżywiania | |
Bardzo często | Zmniejszenie apetytu, hiperglikemia, hipercholesterolemia |
Często | Hipertriglicerydemia, hipofosfatemia, cukrzyca, hiperlipidemia, hipokaliemia, odwodnienie, hipokalcemia |
Zaburzenia psychiczne | |
Często | Bezsenność |
Zaburzenia układu nerwowego | |
Bardzo często | Zaburzenia smaku, ból głowy |
Niezbyt często | Brak odczuwania smaku |
Zaburzenia oka | |
Często | Obrzęk powieki |
Niezbyt często | Zapalenie spojówek |
Zaburzenia serca | |
Niezbyt często | Zastoinowa niewydolność serca |
Zaburzenia naczyniowe | |
Często | Krwotokb, nadciśnienie tętnicze |
Niezbyt często | Zaczerwienienie skóry, zakrzepica żył głębokich |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | |
Bardzo często | Nieinfekcyjne zapalenie płucc, krwawienie z nosa, kaszel |
Często | Duszność |
Niezbyt często | Krwioplucie, zator płucny |
Rzadko | Zespół ostrej niewydolności oddechowej |
Zaburzenia żołądka i jelit | |
Bardzo często | Zapalenie jamy ustnejd, biegunka, nudności |
Często | Wymioty, suchość w jamie ustnej, ból brzucha, zapalenie błony śluzowej, ból jamy ustnej, niestrawność, zaburzenia połykania |
Zaburzenia wątroby i dróg żółciowych | |
Często | Zwiększenie aktywności aminotransferazy asparaginianowej, zwiększenie aktywności aminotransferazy alaninowej |
Zaburzenia skóry i tkanki podskórnej | |
Bardzo często | Wysypka, świąd |
Często | Suchość skóry, zaburzenia dotyczące paznokci, lekkie łysienie, trądzik, rumień, łamliwość paznokci, erytrodyzestezja dłoniowo-podeszwowa, złuszczanie się skóry, zmiany skórne |
Rzadko | Obrzęk naczynioruchowy |
Zaburzenia mięśniowo-szkieletowe i tkanki łącznej | |
Często | Ból stawów |
Zaburzenia nerek i dróg moczowych | |
Często | Białkomocz*, zwiększenie stężenia kreatyniny we krwi, niewydolność nerek* |
Niezbyt często | Zwiększone wydalanie moczu w ciągu dnia, ostra niewydolność nerek* |
Zaburzenia układu rozrodczego i piersi | |
Często | Nieregularne miesiączkowaniee |
Niezbyt często | Brak miesiączkie |
Zaburzenia ogólne i stany w miejscu podania | |
Bardzo często | Zmęczenie, astenia, obrzęki obwodowe |
Często | Gorączka |
Niezbyt często | Ból w klatce piersiowej pochodzenia pozasercowego, zaburzenia gojenia się ran |
Badania diagnostyczne | |
Bardzo często | Zmniejszenie masy ciała |
* Patrz także niżej “Opis wybranych działań niepożądanych”
a Obejmuje wszystkie reakcje w obrębie klasy „Zakażenia i zarażenia pasożytnicze”, w tym (często) nieinfekcyjne zapalenie płuc, zakażenia dróg moczowych; (niezbyt często) zapalenie oskrzeli, półpasiec, posocznicę, ropnie i pojedyncze przypadki zakażeń oportunistycznych [np. aspergiloza, kandydoza, PJP, PCP i zapalenie wątroby typu B (patrz także punkt 4.4)] oraz (rzadko) wirusowe zapalenie mięśnia sercowego
b Obejmuje inne, niewymienione pojedynczo krwawieniem z różnych miejsc
c Obejmuje (bardzo często) nieinfekcyjne zapalenie płuc, (często) śródmiąższową chorobę płuc, nacieki w płucach i (rzadko) krwotok do pęcherzyków płucnych, objawy działania toksycznego na płuca i zapalenie pęcherzyków płucnych
d Obejmuje (bardzo często) zapalenie jamy ustnej, (często) aftowe zapalenie jamy ustnej, owrzodzenie jamy ustnej i języka oraz (niezbyt często) ból języka, zapalenie języka
e Częstość na podstawie połączonych danych od kobiet w wieku od 10 do 55 lat
Opis wybranych działań niepożądanych
Wyniki badań klinicznych i spontaniczne zgłoszenia po wprowadzeniu ewerolimusu do obrotu wskazują, że stosowanie ewerolimusu wiąże się z ciężkimi przypadkami reaktywacji wirusowego zapalenia wątroby typu B, włącznie z przypadkami zgonu. Reaktywacja zakażenia jest spodziewanym działaniem w okresach immunosupresji.
Wyniki badań klinicznych i spontaniczne zgłoszenia po wprowadzeniu ewerolimusu do obrotu wskazują, że stosowanie ewerolimusu wiąże się z występowaniem niewydolności nerek (również zakończonej zgonem) i białkomoczu. Zaleca się kontrolowanie czynności nerek (patrz punkt 4.4).
Wyniki badań klinicznych i spontaniczne zgłoszenia po wprowadzeniu ewerolimusu do obrotu wskazują, że stosowanie ewerolimusu wiąże się z przypadkami braku miesiączki (wtórny brak miesiączki i inne zaburzenia miesiączkowania).
Wyniki badań klinicznych i spontaniczne zgłoszenia po wprowadzeniu ewerolimusu do obrotu wskazują, że stosowanie ewerolimusu wiąże się z występowaniem PJP, PCP, również z przypadkami zgonu (patrz punkt 4.4).
Osoby w podeszłym wieku
W połączonej populacji ocenianej pod względem bezpieczeństwa 37% pacjentów otrzymujących ewerolimus miało co najmniej 65 lat. U większej liczby pacjentów w wieku ≥65 lat wystąpiły działania niepożądane prowadzące do przerwania leczenia (20% vs. 13%). Najczęstszymi z takich działań były: nieinfekcyjne zapalenie płuc (w tym choroba śródmiąższowa płuc), zapalenie jamy ustnej, zmęczenie i duszność.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departamentu Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych,
Wyrobów Medycznych i Produktów Biobójczych: Al. Jerozolimskie 181C, 02-222 Warszawa tel.: + 48 22 49 21 301, fax: +48 22 49 21 309, strona internetowa: https://smz.ezdrowie.gov.pl Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu.
Doświadczenie dotyczące przedawkowania ewerolimusu u ludzi jest bardzo ograniczone. Pojedyncze dawki do 70 mg były podawane z możliwą do zaakceptowania ostrą tolerancją. W każdym przypadku przedawkowania należy zastosować ogólne leczenie podtrzymujące.
Grupa farmakoterapeutyczna: leki przeciwnowotworowe, inne leki przeciwnowotworowe, inhibitory kinazy białkowej.
Kod ATC: L01XE10
Mechanizm działania
Ewerolimus jest wybiórczym inhibitorem ssaczego białka docelowego dla rapamycyny mTOR (ang. mammalian target of rapamycin). mTOR jest kluczową kinazą serynowo-treoninową, której aktywność jest nasilona w wielu ludzkich nowotworach złośliwych. Ewerolimus wiąże się
z międzykomórkowym białkiem FKBP-12, tworząc kompleks, który hamuje aktywność kompleksu 1 kinazy mTOR (mTORC1). Hamowanie szlaku przekazywania sygnałów mTORC1 zaburza translację i syntezę białek przez zmniejszenie aktywności rybosomalnej kinazy S6 (S6K1) i 4EBP1 (białka wiążącego eukariotyczny czynnik elongacyjny 4E), które regulują aktywność białek włączonych w cykl komórkowy, angiogenezę i glikolizę. Uważa się, że substrat S6K1 fosforyluje domenę aktywacyjną 1 receptora estrogenowego, odpowiedzialną za aktywację receptora niezależną od ligandu. Ewerolimus zmniejsza stężenie czynnika wzrostu śródbłonka naczyniowego (VEGF), który nasila procesy angiogenezy guza. Ewerolimus jest silnym inhibitorem wzrostu i namnażania komórek guza, komórek śródbłonka, fibroblastów i komórek mięśni gładkich naczyń krwionośnych.
Wykazano również, że ewerolimus hamuje glikolizę guzów litych in vitro i in vivo. Skuteczność kliniczna i bezpieczeństwo stosowania
Zaawansowane nowotwory neuroendokrynne trzustki (pNET)
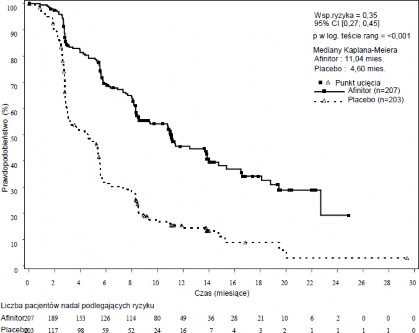
Wyniki wieloośrodkowego, randomizowanego badania III fazy z podwójnie ślepą próbą RADIANT-3 (CRAD001C2324) porównującego ewerolimus plus najlepsze leczenie wspomagające (ang. best supportive care, BSC) z placebo plus BSC u pacjentów z zaawansowanymi nowotworami neuroendokrynnymi trzustki, wykazały statystycznie istotną przewagę korzyści klinicznej
z zastosowania ewerolimusu nad placebo w postaci 2,4-krotnego zwiększenia mediany przeżycia wolnego od progresji choroby (PFS): 11,04 miesiąca wobec 4,6 miesiąca, HR 0,35; 95% CI; 0,27,
0,45; p<0,0001 (patrz tabela 3 i rycina 1).
Do badania RADIANT-3 włączono pacjentów z wysoko lub średnio zróżnicowanymi nowotworami neuroendokrynnymi trzustki, u których wystąpiła progresja choroby w ciągu poprzednich 12 miesięcy. Dozwolone było stosowanie analogów somatostatyny, jako elementu BSC.
Pierwszorzędowym punktem końcowym badania był PFS, oceniony za pomocą kryteriów RECIST. Po udokumentowanej radiologicznie progresji choroby badacz mógł ujawnić, którą kombinację leków otrzymywał pacjent. Pacjenci, których przydzielono losowo do grupy przyjmującej placebo, mogli następnie w otwartej fazie badania otrzymywać ewerolimus.
Do drugorzędowych punktów końcowych należały: bezpieczeństwo stosowania, wskaźnik obiektywnej odpowiedzi na leczenie, czas trwania odpowiedzi na leczenie i przeżycie całkowite (OS).
Łącznie 410 pacjentów przydzielono losowo w stosunku 1:1 do grupy otrzymującej ewerolimus
w dawce 10 mg/dobę (n=207) lub placebo (n=203). Grupy były dobrze zrównoważone w odniesieniu do charakterystyki demograficznej (mediana wieku: 58 lat, mężczyźni: 55%, pacjenci rasy białej: 78,5%). Wcześniejsze leczenie systemowe otrzymało 58% pacjentów w obu ramionach badania.
Mediana czasu trwania zaślepionego leczenia w badaniu wyniosła 37,8 tygodnia (zakres 1,1-129,9 tygodnia) w grupie otrzymującej ewerolimus i 16,1 tygodnia (zakres 0,4-147 tygodni) w grupie placebo.
Po wystąpieniu progresji choroby lub po rozkodowaniu badania 172 z 203 pacjentów (84,7%) początkowo przydzielonych do grupy placebo zmieniło leczenie w fazie otwartej badania na ewerolimus. Mediana czasu trwania leczenia otwartego wyniosła 47,7 tygodnia u wszystkich pacjentów; 67,1 tygodnia u 53 pacjentów zrandomizowanych do grupy ewerolimusu, którzy przeszli na otwarte leczenie ewerolimusem oraz 44,1 tygodnia u 172 pacjentów zrandomizowanych do grupy placebo, którzy przeszli do grupy otwartego leczenia ewerolimusem.
Tabela 4 Wyniki dotyczące skuteczności w badaniu RADIANT-3
Populacja | Ewerolimus n=207 | Placebo n=203 | Współczynnik ryzyka (95% CI) | Wartość p |
Mediana przeżycia bez progresji choroby (miesiące) (95% CI) | ||||
Ocena badania radiologicznego | 11,04 | 4,60 | 0,35 | <0,0001 |
dokonana przez badacza | (8,41, 13,86) | (3,06, 5,39) | (0,27, 0,45) | |
Niezależna ocena radiologiczna | 13,67 | 5,68 | 0,38 | <0,0001 |
(11,17, 18,79) | (5,39, 8,31) | (0,28, 0,51) | ||
Mediana całkowitego przeżycia (miesiące) (95% CI) | ||||
Mediana całkowitego przeżycia | 44,02 | 37,68 | 0,94 | 0,300 |
(35,61, 51,75) | (29,14, 45,77) | (0,73, 1,20) | ||
Rycina 1 Krzywe Kaplana-Meiera dotyczące przeżycia bez progresji choroby w badaniu RADIANT-3 (ocena wyniku badania radiologicznego dokonana przez badacza)
Ewerolimus
Ewerolimus (n=207) Placebo (n=203)
Ewerolimus
Placebo
Dzieci i młodzież
Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań ewerolimusu we wszystkich podgrupach populacji dzieci i młodzieży z nowotworami neuroendokrynnymi trzustki (stosowanie u dzieci i młodzieży, patrz punkt 4.2).
Wchłanianie
U pacjentów z zaawansowanymi guzami litymi ewerolimus podawany w dawce dobowej 5 mg
i 10 mg na czczo lub z lekką beztłuszczową przekąską osiąga maksymalne stężenie w osoczu (Cmax) średnio (mediana) po 1 godzinie. Wartość Cmax koreluje z dawką od 5 do 10 mg. Ewerolimus jest substratem i umiarkowanych inhibitorem PgP.
Wpływ pokarmu
U zdrowych osób bogatotłuszczowy posiłek zmniejszał wartości ogólnoustrojową ekspozycję na ewerolimus (mierzoną jako AUC) o 22%, a maksymalne stężenie w osoczu Cmax o 54%. Po spożyciu posiłku z niewielką zawartością tłuszczu oba wskaźniki były mniejsze, odpowiednio o 32% i 42%. Jednak pokarm nie ma wyraźnego wpływu na profil zależności stężenia od czasu w fazie po wchłanianiu.
Dystrybucja
Stosunek dystrybucji pełna krew/osocze dla ewerolimusu zależy od stężenia i w zakresie od 5 do 5000 ng/ml ma wartość od 17% do 73%. U pacjentów z chorobą nowotworową otrzymujących dawkę 10 mg na dobę około 20% stężenia ewerolimusu w pełnej krwi ogranicza się do osocza. U zdrowych osób i u pacjentów z umiarkowanymi zaburzeniami czynności wątroby ewerolimus wiąże się
z białkami osocza w około 74%. U pacjentów z zaawansowanymi guzami litymi wartość Vd w kompartmencie centralnym wyniosła 191 l, a w kompartmencie obwodowym 517 l.
Metabolizm
Ewerolimus jest substratem dla CYP3A4 i glikoproteiny P. Po podaniu doustnym jest on głównym składnikiem krążącym we krwi człowieka. W ludzkiej krwi wykryto sześć głównych metabolitów ewerolimusu, w tym trzy metabolity utworzone w wyniku wprowadzenia do cząsteczki jednej grupy hydroksylowej, dwa produkty hydrolizy o otwartym pierścieniu oraz związek powstały ze sprzężenia
ewerolimusu z fosfatydylocholiną. Metabolity te zidentyfikowano również u zwierząt poddanych badaniom toksyczności i wykazywały one 100-krotnie mniejszą aktywność niż sam ewerolimus. Dlatego uważa się, że substancja macierzysta ma główny udział w ogólnej aktywności farmakologicznej ewerolimusu.
Wydalanie
Średni klirens ewerolimusu po podaniu doustnym (CL/F) u pacjentów z zaawansowanymi guzami litymi otrzymujących dawkę 10 mg na dobę miał wartość 24,5 l/godzinę. Średni okres półtrwania w fazie eliminacji wynosi około 30 godzin.
Nie przeprowadzono szczególnych badań wydalania u pacjentów z chorobą nowotworową, ale dostępne są dane z badań u biorców przeszczepów. Po podaniu pojedynczej dawki ewerolimusu znakowanego radioaktywnie pacjentom po przeszczepieniu otrzymującym cyklosporynę, 80% radioaktywności stwierdzano w kale, a 5% wydalana była w moczu. Związku macierzystego nie stwierdzono ani w moczu, ani w kale.
Farmakokinetyka w stanie stacjonarnym
Po podaniu ewerolimusu pacjentom z zaawansowanymi guzami litymi wartość AUC0-τ w stanie stacjonarnym była proporcjonalna do dawki w zakresie dawek dobowych od 5 do 10 mg. Stan stacjonarny uzyskano w ciągu 2 tygodni. Wartość Cmax koreluje z dawką od 5 do 10 mg, a tmax występuje po 1 do 2 godzin od podania dawki. Stwierdzono istotną korelację między AUC0-τ
a minimalnym stężeniem przed podaniem kolejnej dawki w stanie stacjonarnym. Szczególne grupy pacjentów
Zaburzenia czynności wątroby
Bezpieczeństwo stosowania, tolerancję i farmakokinetykę ewerolimusu oceniano w dwóch badaniach z zastosowaniem dawki pojedynczej ewerolimusu w tabletkach u 8 i 34 osób z zaburzeniami czynności wątroby i porównano z osobami z prawidłową czynnością wątroby.
W pierwszym badaniu średnia wartość AUC u 8 osób z umiarkowanymi zaburzeniami czynności wątroby (klasa B w skali Childa-Pugha) była dwukrotnie większa niż u 8 osób z prawidłową czynnością wątroby.
W drugim badaniu stwierdzono, że w porównaniu z osobami z prawidłową czynnością wątroby,
u 34 osób z różnego stopnia zaburzeniami czynności wątroby ekspozycja (tj. AUC0-inf) była większa 1,6-, 3,3- i 3,6-krotnie w przypadku odpowiednio lekkich (klasa A w skali Childa-Pugha), umiarkowanych (klasa B w skali Childa-Pugha) lub poważnych (klasa C w skali Childa-Pugha) zaburzeń czynności wątroby.
Symulacje farmakokinetyki po podaniu dawek wielokrotnych potwierdzają zalecenia dotyczące dawkowania u osób z zaburzeniami czynności wątroby na podstawie ich statusu w skali Childa-Pugha.
Wyniki dwóch badań wskazują, że u pacjentów z zaburzeniami czynności wątroby zalecana jest modyfikacja dawki ewerolimusu (patrz punkty 4.2 i 4.4).
Zaburzenia czynności nerek
Populacyjna analiza farmakokinetyki obejmująca 170 pacjentów z zaawansowanymi guzami litymi nie wykazała znaczącego wpływu klirensu kreatyniny (w zakresie 25-178 ml/min) na klirens ewerolimusu po podaniu doustnym (CL/F). Zaburzenia czynności przeszczepionej nerki (Clkr 11-107 ml/min) nie wpływały na farmakokinetykę ewerolimusu u biorców przeszczepu.
Pacjenci w podeszłym wieku
W populacyjnej ocenie farmakokinetyki u pacjentów z chorobą nowotworową nie stwierdzono znaczącego wpływu wieku (27-85 lat) na klirens ewerolimusu po podaniu doustnym.
Grupy etniczne
Klirens ewerolimusu po podaniu doustnym (CL/F) u pacjentów pochodzenia japońskiego
i kaukaskiego z chorobą nowotworową oraz zbliżoną czynnością wątroby jest podobny. Populacyjna analiza farmakokinetyki wykazała, że wartość CL/F jest średnio o 20% większy u biorców przeszczepu rasy czarnej.
Niekliniczny profil bezpieczeństwa ewerolimusu oceniano u myszy, szczurów, świń miniaturowych, małp i królików. Głównymi narządami docelowymi był żeński i męski układ rozrodczy (zwyrodnienie kanalików plemnikotwórczych, zmniejszona zawartość nasienia w najądrzach i zanik macicy) u kilku gatunków zwierząt; płuca (zwiększenie liczby makrofagów pęcherzykowych u szczura i myszy); trzustka (degranulacja i wakuolizacja komórek egzokrynnych odpowiednio u małp i świń miniaturowych oraz degeneracja komórek wysp trzustkowych u małp) i oczy (zmętnienia przednich szwów soczewki tylko u szczura). Odnotowano niewielkie zmiany w nerkach u szczura (nasilenie związanego z wiekiem spichrzania lipofuscyn w nabłonku kanalików nerkowych) i myszy (nasilenie współistniejących zmian chorobowych). Nie stwierdzono zmian wskazujących na toksyczne działanie ewerolimusu na nerki małp lub świń miniaturowych.
Okazało się, że podawanie ewerolimusu nasila samoistnie występujące choroby towarzyszące (przewlekłe zapalenie mięśnia sercowego u szczurów, zakażenie wirusem Coxsackie w osoczu i sercu u małp, zakażenia ziarniakami przewodu pokarmowego świń miniaturowych, zmiany skórne u myszy i małp). Obserwowano to zazwyczaj przy ekspozycji ogólnoustrojowej na ewerolimus zapewniającej stężenia terapeutyczne lub większe, z wyjątkiem zmian u szczurów, które występowały przy ekspozycji mniejszej niż terapeutyczna ze względu na dużą dystrybucję do tkanek.
W badaniu płodności samców szczura dawki 0,5 mg/kg mc. i większe wpływały na morfologię jąder, a dawka 5 mg/kg mc. powodowała zmniejszenie ruchliwości nasienia, liczby plemników i stężenia testosteronu w osoczu, zmniejszając w ten sposób płodność. Wykazano, że zmiany te są odwracalne.
W badaniu wpływu na reprodukcję ewerolimus nie zaburzał płodności samic badanych zwierząt.
Jednak w doustnych dawkach ≥0,1 mg/kg mc. (co odpowiada około 4% wartości AUC0-24h
u pacjentów otrzymujących dawkę dobową 10 mg) powodował częstszą utratę zarodków przed zagnieżdżeniem.
Ewerolimus przenika przez łożysko i działa toksycznie na płód. U szczurów ewerolimus wykazywał szkodliwe działanie na zarodek i (lub) płód przy układowej ekspozycji poniżej wartości terapeutycznych, co objawiało się śmiertelnością i zmniejszoną masą ciała płodów. W dawkach
0,3 mg/kg mc. i 0,9 mg/kg mc. powodował zwiększenie częstości zmian w obrębie kośćca i wad rozwojowych (np. rozszczep mostka). U królików działanie embriotoksyczne prowadziło do zwiększenia późnych resorpcji.
Odpowiednie punkty końcowe w badaniach genotoksyczności nie wskazały na klastogenne lub mutagenne działanie ewerolimusu. Skutki podawania ewerolimusu przez okres do 2 lat nie wskazują na potencjał rakotwórczy u myszy i szczurów otrzymujących nawet największe dawki ewerolimusu, odpowiednio 3,9- i 0,2-krotnie większe od szacowanej ekspozycji podczas leczenia u ludzi.
Butylohydroksytoluen (E 321) Magnezu stearynian
Laktoza
Hypromeloza (3 mPa.s) Krospowidon (Typ A)
Nie dotyczy.
3 lata
Nie przechowywać w temperaturze powyżej 30°C.
Przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem i wilgocią.
Tabletki są pakowane w blistry z folii Aluminium/PA/Aluminium/PVC i umieszczane w tekturowym pudełku.
Wielkości opakowań:
Blistry: 10, 30, 90 tabletek.
Blistry jednodawkowe: 10x1, 30x1, 90x1 tabletek.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami.
Sandoz GmbH Biochemiestrasse 10
6250 Kundl, Austria
Everolimus Sandoz, 2,5 mg Pozwolenie nr 25192 Everolimus Sandoz, 5 mg Pozwolenie nr 25193 Everolimus Sandoz, 10 mg Pozwolenie nr 25194
Data wydania pierwszego pozwolenia: 15.03.2019
11.02.2021 r.








