







Spis treści:
- NAZWA PRODUKTU LECZNICZEGO
- SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- POSTAĆ FARMACEUTYCZNA
- SZCZEGÓŁOWE DANE KLINICZNE
- WŁAŚCIWOŚCI FARMAKOLOGICZNE
- DANE FARMACEUTYCZNE
- PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
- NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
- DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
- DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO
NAZWA PRODUKTU LECZNICZEGO
SKŁAD JAKOŚCIOWY I ILOŚCIOWY
POSTAĆ FARMACEUTYCZNA
SZCZEGÓŁOWE DANE KLINICZNE
Wskazania do stosowania
w monoterapii do leczenia raka piersi z obecnymi receptorami estrogenowymi, miejscowo zaawansowanego lub z przerzutami, u kobiet po menopauzie:
wcześniej nieleczonych terapią hormonalną lub
z nawrotem choroby podczas lub po zakończeniu leczenia uzupełniającego lekiem
z grupy antyestrogenów lub, gdy nastąpiła progresja choroby podczas leczenia lekiem z grupy antyestrogenów.
w skojarzeniu z palbocyklibem w leczeniu miejscowo zaawansowanego lub rozsianego raka piersi z obecnością receptorów hormonalnych (ang. hormone receptor, HR), bez nadmiernej ekspresji receptora ludzkiego naskórkowego czynnika wzrostu 2 (ang. human epidermal growth factor receptor 2, HER2) u kobiet, które wcześniej otrzymały leczenie hormonalne (patrz punkt 5.1).
U kobiet przed menopauzą i w okresie okołomenopauzalnym leczenie skojarzone z palbocyklibem należy stosować jednocześnie z agonistą hormonu uwalniającego hormon luteinizujący (ang. luteinizing hormone releasing hormone, LHRH).
Dawkowanie i sposób podawania
Przeciwwskazania
Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania
Interakcje z innymi produktami leczniczymi i inne rodzaje interakcji
Wpływ na płodność, ciążę i laktację
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn
Działania niepożądane
Obejmuje działania niepożądane, dla których nie można określić dokładnego udziału fulwestrantu ze względu na chorobę podstawową.
Termin “odczyny w miejscu podania” nie obejmuje terminów krwotok z miejsca podania, krwiak w miejscu podania, rwa kulszowa, nerwoból, neuropatia obwodowa.
Działania nie obserwowano w trakcie dużych badań klinicznych (CONFIRM, FINDER 1, FINDER 2, NEWEST). Częstość obliczono przyjmując górną granicę 95% przedziału ufności dla estymacji w punkcie. Uzyskano wartość 3/560 (gdzie 560 to liczba pacjentów w dużych badaniach klinicznych), co odpowiada kategorii „niezbyt często”.
Obejmuje: ból stawów, rzadziej bóle mięśniowo-szkieletowe, ból mięśni i ból kończyn. e Różnice w częstości między zbiorczą analizą bezpieczeństwa a badaniem FALCON.
Preferowana terminologia (PT) podana zgodnie z MedDRA 17.1.
Zakażenia obejmują wszystkie PT, które należą do zakażeń i zarażeń pasożytniczych w klasyfikacji układów i narządów.
Neutropenia obejmuje następujące PT: neutropenia, zmniejszona liczba neutrofilów.
Leukopenia obejmuje następujące PT: leukopenia, zmniejszona liczba krwinek białych.
Niedokrwistość obejmuje następujące PT: niedokrwistość, zmniejszenie stężenia hemoglobiny, zmniejszenie wartości hematokrytu.
Małopłytkowość obejmuje następujące PT: małopłytkowość, zmniejszona liczba płytek krwi. g Zapalenie jamy ustnej obejmuje następujące PT: aftowe zapalenie jamy ustnej, zapalenie
Przedawkowanie
WŁAŚCIWOŚCI FARMAKOLOGICZNE
Właściwości farmakodynamiczne
Fulwestrant jest wskazany u pacjentek, u których nastąpił nawrót lub progresja choroby podczas leczenia antyestrogenowego. Wyniki w podgrupie inhibitorów aromatazy nie są rozstrzygające.
Wartość OS (przeżycie całkowite) dla końcowej analizy przeżycia przy 75% dojrzałości wyników.
Nominalna wartość p bez korekty poziomu istotności dla testów wielokrotnych pomiędzy wstępną analizą całkowitego przeżycia przy 50% dojrzałości wyników i zaktualizowaną analizą przeżycia przy 75% dojrzałości wyników.
ORR oceniano u pacjentek, które na wstępie zakwalifikowano do grupy podlegającej ocenie odpowiedzi na leczenie (tzn. do grupy pacjentek, u których na początku badania możliwa była ocena wielkości zmian nowotworowych: 240 pacjentek w grupie otrzymującej fulwestrant
w dawce 500 mg i 261 pacjentek w grupie otrzymującej fulwestrant w dawce 250 mg).
Pacjentki z odpowiedzią obiektywną całkowitą, częściową lub stabilną chorobą ≥24 tygodni. PFS: przeżycie wolne od progresji choroby; ORR: odsetek odpowiedzi obiektywnych; OR: odpowiedź obiektywna; CRB: odsetek korzyści klinicznych; CB: korzyść kliniczna; OS: całkowite przeżycie;
mg. Stwierdzono, że fulwestrant w comiesięcznej dawce 250 mg jest co najmniej tak skuteczny, jak anastrozol w odniesieniu do przeżycia wolnego od progresji choroby, obiektywnej odpowiedzi na leczenie i czasu do zgonu. Nie stwierdzono istotnej statystycznie różnicy między obiema grupami
w odniesieniu do żadnego z punktów końcowych badania. Pierwszorzędowym punktem końcowym badania był czas wolny od progresji choroby. Łączna analiza wyników obu badań wykazała, że progresja choroby wystąpiła u 83% pacjentek otrzymujących fulwestrant i u 85% pacjentek otrzymujących anastrozol. W tej analizie współczynnik ryzyka dla porównania fulwestrantu w dawce 250 mg z anastrozolem w odniesieniu do czasu wolnego od progresji choroby wyniósł 0,95 (95% CI 0,82 do 1,10). Odsetek obiektywnych odpowiedzi dla fulwestrantu 250 mg wynosił 19,2%, w porównaniu z 16,5% dla anastrozolu. Mediana czasu do wystąpienia zgonu wynosiła 27,4 miesiąca
u pacjentek otrzymujących fulwestrant i 27,6 miesiąca u pacjentek leczonych anastrozolem. Współczynnik ryzyka dla porównania fulwestrantu 250 mg z anastrozolem pod względem czasu do wystąpienia zgonu wynosił 1,01 (95% CI 0,86 do 1,19).
Leczenie skojarzone palbocyklibem
Przeprowadzono międzynarodowe, randomizowane, podwójnie zaślepione, wieloośrodkowe badanie III fazy w grupach równoległych, porównujące leczenie fulwestrantem 500 mg w skojarzeniu
z palbocyklibem w dawce 125 mg z leczeniem fulwestrantem 500 mg w skojarzeniu z placebo u kobiet z hormonozależnym rakiem piersi, bez nadmiernej ekspresji HER2, miejscowo zaawansowanym, niekwalifikującym się do leczenia chirurgicznego ani radioterapii z intencją
wyleczenia, lub z rozsianym rakiem piersi, niezależnie od statusu menopauzy, u których doszło do
progresji choroby po wcześniejszej terapii hormonalnej w leczeniu (neo) adjuwantowym lub w chorobie rozsianej.
Łącznie 521 kobiet w okresie przed-/około- i pomenopauzalnym, u których doszło do progresji choroby w trakcie lub w ciągu 12 miesięcy od zakończenia uzupełniającej terapii hormonalnej, lub podczas albo w ciągu 1 miesiąca od wcześniejszej terapii hormonalnej z powodu choroby zaawansowanej, zostało losowo przydzielonych w stosunku 2:1 do grupy otrzymującej fulwestrant w skojarzeniu z palbocyklibem lub do grupy otrzymującej fulwestrant w skojarzeniu z placebo.
Randomizację stratyfikowano według udokumentowanej wrażliwości na wcześniejszą terapię hormonalną, okresu menopauzy w chwili przystąpienia do badania (okres przed-/okołomenopauzalny w porównaniu z okresem pomenopauzalnym) i obecności przerzutów do narządów miąższowych.
Kobiety w okresie przed-/okołomenopauzalnym otrzymywały agonistę LHRH, goserelinę. Pacjentki z chorobą zaawansowaną/rozsianą, objawową, z zajęciem narządów miąższowych, u których istniało ryzyko wystąpienia w krótkim czasie powikłań zagrażających życiu (w tym pacjentki z masywnymi niekontrolowanymi wysiękami [opłucnowymi, osierdziowymi, otrzewnowymi], zapaleniem naczyń chłonnych płuc i zajęciem ponad 50% miąższu wątroby) nie spełniały kryteriów włączenia do tego badania.
Pacjentki kontynuowały przydzielone leczenie do chwili wystąpienia obiektywnej progresji choroby, nasilenia objawów, wystąpienia niemożliwych do zaakceptowania objawów toksyczności, zgonu lub wycofania zgody na udział w badaniu, w zależności od tego, które z tych zdarzeń wystąpiło jako pierwsze. Zmiana grup leczenia nie była dozwolona.
Pacjentki z grupy otrzymującej fulwestrant w skojarzeniu z palbocyklibem i pacjentki z grupy otrzymującej fulwestrant w skojarzeniu z placebo były dobrze dobrane pod względem wyjściowych danych demograficznych i cech prognostycznych. Mediana wieku pacjentek włączonych do tego badania wyniosła 57 lat (zakres 29, 88). W każdej grupie badanej większość stanowiły kobiety rasy białej, z udokumentowaną wrażliwością na wcześniejszą terapię hormonalną i w okresie pomenopauzalnym. Około 20% pacjentek stanowiły kobiety w okresie przed-/okołomenopauzalnym. Wszystkie pacjentki otrzymały wcześniej leczenie systemowe i większość pacjentek z każdej grupy terapeutycznej była poddana wcześniej chemioterapii z powodu pierwotnego rozpoznania. U ponad połowy (62%) stan sprawności wg ECOG wyniósł 0, u 60% występowały przerzuty do narządów miąższowych, a 60% otrzymało wcześniej więcej niż 1 linię hormonoterapii z powodu pierwotnego rozpoznania.
Pierwszorzędowym punktem końcowym w badaniu było PFS oceniane przez badacza według kryteriów RECIST 1.1. Wspomagające analizy PFS opierały się na niezależnej, centralnej ocenie radiologicznej. Drugorzędowe punkty końcowe obejmowały OR, CBR, OS, bezpieczeństwo stosowania i czas do pogorszenia (ang. time-to-deterioration, TTD) punktu końcowego dotyczącego bólu.
W badaniu osiągnięto pierwszorzędowy punkt końcowy, uzyskano wydłużenie PFS wg oceny badacza w analizie etapowej przeprowadzonej po wystąpieniu 82% planowanych zdarzeń PFS; wyniki przekroczyły predefiniowaną granicę skuteczności Haybittle-Peto (α=0,00135) wykazując statystycznie znamienne wydłużenie PFS i klinicznie znaczący efekt leczenia. Bardziej dojrzałą aktualizację danych dotyczących skuteczności przedstawiono w Tabeli 5.
Po obserwacji o medianie czasu trwania wynoszącej 45 miesięcy, przeprowadzono końcową analizę OS na podstawie 310 zdarzeń (60% zrandomizowanych pacjentek). Zaobserwowano różnicę
w medianie OS wynoszącą 6,9 miesiąca między grupą otrzymującą palbocyklib w skojarzeniu z fulwestrantem a grupą otrzymująca placebo w skojarzeniu z fulwestrantem; ten wynik nie był
statystycznie znamienny przy określonym wcześniej poziomie istotności wynoszącym 0,0235 (test 1- stronny). W grupie leczonej placebo w skojarzeniu z fulwestrantem 15,5% zradnomizowanych pacjentek otrzymało następnie palbocyklib lub inne inhibitory CDK w ramach kolejnych linii leczenia po progresji choroby.
Wyniki dotyczące danych o PFS według oceny badaczy i końcowych danych o OS w badaniu PALOMA3 przedstawiono w Tabeli 5. Odpowiednie wykresy Kaplana-Meiera pokazano na Rycinach 2 i 3.
Tabela 5. Wyniki dotyczące skuteczności – badanie PALOMA3 (ocena badacza, populacja zgodna z zaplanowanym leczeniem)
Aktualizacja analizy
(data odcięcia danych: 23 października 2015 r.)
Fulwestrant + palbocyklib (N=347)
Fulwestrant + placebo (N=174)
Przeżycie wolne od progresji
choroby
Mediana [miesiące (95% CI)]
11,2 (9,5; 12,9)
4,6 (3,5; 5,6)
Współczynnik ryzyka (95% CI) i wartość p
0,497 (0,398; 0,620), p <0,000001
Drugorzędowe punkty końcowe*
OR [% (95% CI)]
26,2 (21,7; 31,2)
13,8 (9,0; 19,8)
OR (zmiany mierzalne) [% (95% CI)]
33,7 (28,1; 39,7)
17,4 (11,5; 24,8)
CBR [% (95% CI)]
68,0 (62,8; 72,9)
39,7 (32,3; 47,3)
Przeżycie całkowite (OS) w analizie końcowej
(data odcięcia danych: 13 kwietnia 2018r.)
Liczba zdarzeń (%)
201 (57,9)
109 (62,6)
Mediana [miesiące (95% CI)]
34,9 (28,8; 40,0)
28,0 (23,6; 34,6)
Współczynnik ryzyka (95% CI) i wartość p†
0,814 (0,644, 1,029) p=0,0429†
CBR=korzyść kliniczna; CI=przedział ufności; N=liczba pacjentek; OR=odpowiedź obiektywna Wyniki dotyczące drugorzędowych punktów końcowych opierają się na potwierdzonych i niepotwierdzonych odpowiedziach według RECIST 1.1.
*Statystycznie nieistotne.
† 1-stronna wartość p w logarytmicznym teście rang ze stratyfikacją uwzględniającą obecność przerzutów do narządów trzewnych i wrażliwość na wcześniejszą terapię hormonalną według randomizacji.
Rycina 2 Wykres Kaplana-Meiera dla przeżycia wolnego od progresji choroby (ocena badacza, populacja zgodna z zaplanowanym leczeniem) – badanie PALOMA3 (data odcięcia danych: 23 października 2015r.)
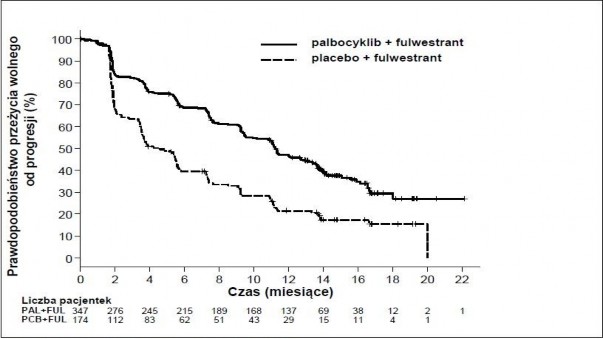
FUL=fulwestrant; PAL=palbocyklib; PCB=placebo
Redukcja ryzyka progresji choroby lub zgonu w grupie otrzymującej fulwestrant w skojarzeniu
z palbocyklibem obserwowano we wszystkich poszczególnych podgrupach pacjentek definiowanych w oparciu o czynniki stratyfikacji i charakterystykę wyjściową. Redukcję ryzyka wykazano u kobiet w okresie przed-/okołomenopauzalnym (HR=0,46 [95% CI: 0,28; 0,75]), u kobiet w okresie pomenopauzalnym (HR=0,52 [95% CI: 0,40; 0,66]), u pacjentek z lokalizacją przerzutów w narządach miąższowych (HR=0,50 [95% CI: 0,38; 0,65]) i lokalizacją przerzutów poza narządami miąższowymi (HR=0,48 [95% CI: 0,33; 0,71]). Korzyści również obserwowano niezależnie od liczby linii wcześniejszych terapii z powodu choroby rozsianej, niezależnie od tego, czy było to 0 (HR=0,59 [95% CI: 0,37; 0,93]), 1 (HR=0,46 [95% CI: 0,32; 0,64]), 2 (HR=0,48 [95% CI: 0,30; 0,76]) lub ≥3 linie
leczenia (HR=0,59 [95% CI: 0,28; 1,22]).
Rycina 3. Wykres Kaplana-Meiera dla przeżycia całkowitego (populacja zgodna z zaplanowanym leczeniem) – badanie PALOMA3 (data odcięcia danych: 13 kwietnia 2018r.)
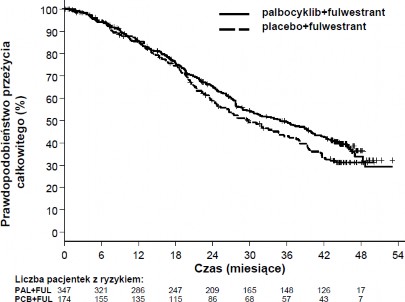
FUL=fulwestrant; PAL=palbocyklib; PCB=placebo
Dodatkowe miary skuteczności (OR i TTR) oceniane w podgrupach pacjentek z zajęciem narządów miąższowych lub bez zostały przedstawione w Tabeli 6.
Tabela 6 Wyniki dotyczące skuteczności u pacjentek z zajęciem narządów miąższowych lub bez, w badaniu PALOMA3 (populacja zgodna z zaplanowanym leczeniem)
Przerzuty do narządów miąższowych
Brak przerzutów do narządów
miąższowych
Fulwestrant
+palbocyklib (N=206)
Fulwestrant+placebo (N=105)
Fulwestrant
+palbocyklib (N=141)
Fulwestrant+placebo (N=69)
OR [%
(95% CI)]
35,0
13,3
13,5
14,5
(28,5; 41,9)
(7,5; 21,4)
(8,3; 20,2)
(7,2; 25,0)
TTR*,
Mediana
3,8
5,4
3,7
3,6
[miesiące
(3,5; 16,7)
(3,5; 16,7)
(1,9; 13,7)
(3,4; 3,7)
(zakres)]
*Wyniki dotyczące odpowiedzi na leczenie na podstawie potwierdzonych i niepotwierdzonych odpowiedzi.
N=liczba pacjentek; CI=przedział ufności; OR= odpowiedź obiektywna; TTR=czas do pierwszej odpowiedzi ze strony guza.
Objawy zgłaszane przez pacjentki były oceniane za pomocą kwestionariusza jakości życia (QLQ)-C30 Europejskiej Organizacji na rzecz Badań i Leczenia Raka (EORTC) i jego modułu dotyczącego raka piersi (EORTC QLQ-BR23). Łącznie 335 pacjentek z grupy otrzymującej fulwestrant w skojarzeniu
z palbocyklibem i 166 pacjentek w grupie otrzymującej fulwestrant w skojarzeniu z placebo wypełniło kwestionariusz przy rozpoczęciu badania i przynajmniej 1 raz na wizycie podczas udziału w badaniu.
Czas do pogorszenia objawów został wcześniej określony jako czas między wynikiem wyjściowym
a pierwszym wystąpieniem zwiększenia o ≥10 punktów w wynikach dotyczących objawów bólowych
względem stanu wyjściowego. Dołączenie palbocyklibu do leczenia fulwestrantem miało korzystny wpływ na objawy przez istotne wydłużenie czasu do nasilenia objawów bólowych w porównaniu z leczeniem fulwestrantem w skojarzeniu z placebo (mediana 8,0 miesięcy w porównaniu z
2,8 miesiąca; HR=0,64 [95% CI: 0,49; 0,85]; p<0,001).
Wpływ na endometrium w okresie po menopauzie
Na podstawie wyników badań nieklinicznych nie można sądzić, że fulwestrant wpływa pobudzająco na błonę śluzową macicy po menopauzie (patrz punkt 5.3). W 2-tygodniowym badaniu klinicznym z udziałem zdrowych ochotniczek po menopauzie stwierdzono, że w porównaniu z placebo leczenie wstępne fulwestrantem w dawce 250 mg powoduje istotne osłabienie działania pobudzającego na błonę śluzową macicy etynyloestradiolu podawanego w dawce 20 mikrogramów. Działanie pobudzające oceniano, mierząc grubość endometrium ultrasonograficznie.
Leczenie neoadjuntowe u pacjentek z rakiem piersi trwające do 16 tygodni, zarówno fulwestrantem w dawce 500 mg jak i fulwestrantem w dawce 250 mg nie spowodowało znaczącej klinicznie zmiany grubości błony śluzowej macicy, co wskazuje na brak działania agonistycznego. Nie ma dowodów na niekorzystny wpływ na błonę śluzową macicy u badanych pacjentek z rakiem piersi. Nie ma danych dotyczących wpływu na budowę błony śluzowej macicy.
W dwóch krótkotrwałych badaniach klinicznych (trwających 1 i 12 tygodni) przeprowadzonych
u pacjentek w okresie przed menopauzą z łagodnymi chorobami ginekologicznymi nie stwierdzono znaczącej różnicy grubości endometrium (mierzonego ultrasonograficznie) między grupą otrzymującą fulwestrant i placebo.
Wpływ na kości
Nie ma danych dotyczących odległych skutków działania fulwestrantu na kości. Leczenie neoadjuntowe u pacjentek z rakiem piersi trwające do 16 tygodni, zarówno fulwestrantem w dawce 500 mg jak i w dawce 250 mg nie spowodowało znaczącej klinicznie zmiany w stężeniach markerów metabolizmu kości w surowicy.
Dzieci i młodzież
Fulwestrant nie jest wskazany do stosowania u dzieci. Europejska Agencja Leków uchyliła obowiązek dołączania wyników badań referencyjnego produktu leczniczego zawierającego fulwestrant we wszystkich podgrupach populacji dzieci i młodzieży we wskazaniu rak piersi (stosowanie u dzieci
i młodzieży, patrz punkt 4.2).
W otwartym badaniu klinicznym II fazy oceniano bezpieczeństwo stosowania, skuteczność oraz parametry farmakokinetyczne fulwestrantu, wśród 30 dziewcząt w wieku od 1 do 8 lat
z przedwczesnym dojrzewaniem płciowym (ang. Progressive Precocious Puberty) związanym z zespołem McCune Albrighta (MAS). Dzieci otrzymywały domięśniowo fulwestrant w dawce 4 mg/kg masy ciała co miesiąc. To 12-miesięczne badanie pozwoliło na ocenę wielu punktów
końcowych istotnych dla leczenia MAS i wykazało zmniejszenie częstości krwawień z dróg rodnych oraz zmniejszenie wskaźnika zaawansowania wieku kostnego. W stanie stacjonarnym wartości stężeń fulwestrantu ocenione w tym badaniu w osoczu u dzieci odpowiadały wartościom stwierdzanym u pacjentów dorosłych (patrz punkt 5.2). To małe badanie nie ujawniło żadnych nowych doniesień dotyczących bezpieczeństwa stosowania, ale dane z 5-letniej obserwacji nie są jeszcze dostępne.
Właściwości farmakokinetyczne
Przedkliniczne dane o bezpieczeństwie
DANE FARMACEUTYCZNE
Wykaz substancji pomocniczych
Niezgodności farmaceutyczne
Okres ważności
lata
Specjalne środki ostrożności podczas przechowywania
Rodzaj i zawartość opakowania
Specjalne środki ostrożności dotyczące usuwania
Należy wyjąć szklaną ampułkę z pojemnika i sprawdzić, czy nie jest uszkodzona.
Otworzyć opakowanie zewnętrzne igły z systemem osłaniającym (SafetyGlide).
Przed podaniem roztworów parenteralnych należy dokonać ich wizualnej oceny w celu wykrycia obecności cząstek stałych i zmiany barwy.
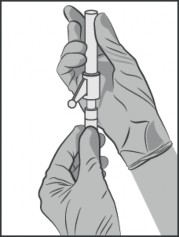
Trzymać strzykawkę pionowo w części prążkowanej (C). Drugą ręką chwycić nasadkę (A) i ostrożnie przechylać do przodu i do tyłu, aż nasadka rozłączy się i możliwe będzie jej ściągnięcie, nie przekręcać (patrz Rysunek 1).
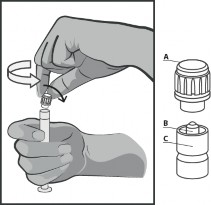
Rysunek 1.
Usunąć nasadkę (A) w pozycji pionowej ku górze. W celu zachowania sterylności nie dotykać końcówki strzykawki (B) (patrz Rysunek 2).
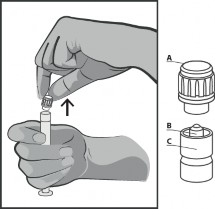
Rysunek 2.
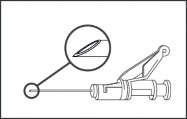
Dołączyć igłę z systemem osłaniającym do końcówki Luer-Lock i dokręcić w celu trwałego umocowania (patrz Rysunek 3).
Sprawdzić czy igła jest połączona z końcówką Luer przed przejściem do pozycji pionowej.
Przy dokręcaniu igły należy postępować tak, aby nie uszkodzić jej ostrego końca.
Igłę z nasadką należy zbliżyć do miejsca podania.
Zdjąć nasadkę z igły.
Usunąć nadmiar powietrza ze strzykawki.
Rysunek 3.
Lek należy podawać domięśniowo, powoli (1-2 minuty/wstrzyknięcie), w mięsień pośladkowy (miejsce na pośladku). Dla wygody osoby podającej, ścięcie igły znajduje się na tej samej powierzchni igły co dźwignia systemu osłaniającego igłę (patrz Rysunek 4).
Rysunek 4.
Natychmiast po podaniu leku należy uruchomić system osłaniający igłę przez popchnięcie do przodu jego dźwigni (patrz Rysunek 5).
PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU
NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU
DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU I DATA PRZEDŁUŻENIA POZWOLENIA
DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU CHARAKTERYSTYKI PRODUKTU LECZNICZEGO
Fulvestrant Zentiva, 250 mg/5 ml, roztwór do wstrzykiwań w ampułko-strzykawce
Każda ampułko-strzykawka o pojemności 5 ml zawiera 250 mg fulwestrantu. Substancje pomocnicze o znanym działaniu (w 5 ml):
Etanol 96% (alkohol), 500 mg
Alkohol benzylowy, 500 mg Benzylu benzoesan, 750 mg
Pełny wykaz substancji pomocniczych, patrz punkt 6.1.
Roztwór do wstrzykiwań w ampułko-strzykawce.
Przezroczysty, bezbarwny do żółtego, lepki roztwór, bez widocznych cząstek stałych.
Fulwestrant jest wskazany:
Dawkowanie
Dorosłe pacjentki (w tym pacjentki w podeszłym wieku)
Zalecana dawka leku to 500 mg, podawana w odstępach jednomiesięcznych z dodatkową dawką 500 mg po upływie 2 tygodni od podania pierwszej dawki.
Gdy fulwestrant jest stosowany w skojarzeniu z palbocyklibem, należy zapoznać się z Charakterystyką Produktu Leczniczego palbocyklibu.
Przed rozpoczęciem leczenia skojarzonego fulwestrantem z palbocyklibem i przez cały czas jego trwania pacjentki w wieku przed- i okołomenopauzalnym powinny otrzymywać leczenie agonistami LHRH zgodnie z lokalnie przyjętą praktyką kliniczną.
Szczególne grupy pacjentów
Zaburzenie czynności nerek
Nie ma konieczności zmiany dawki produktu u pacjentek z łagodnymi i umiarkowanymi zaburzeniami czynności nerek (klirens kreatyniny ≥30 ml/min). Nie badano skuteczności i bezpieczeństwa stosowania produktu u pacjentek z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny
<30 ml/min), dlatego zaleca się ostrożność podczas stosowania produktu w tej grupie pacjentek (patrz punkt 4.4).
Zaburzenie czynności wątroby
Nie ma konieczności zmiany dawki produktu leczniczego u pacjentek z łagodnymi i umiarkowanymi zaburzeniami czynności wątroby. Jednak w tej grupy pacjentek fulwestrant należy stosować ostrożnie ze względu na możliwość zwiększenia ekspozycji na fulwestrant. Brak danych dotyczących stosowania produktu u pacjentek z ciężkimi zaburzeniami czynności wątroby (patrz punkty 4.3, 4.4
i 5.2).
Dzieci i młodzież
Bezpieczeństwo stosowania i skuteczność działania fulwestrantu u dzieci i młodzieży w wieku do 18 lat nie zostały ustalone. Aktualnie dostępne dane są opisane w punkcie 5.1 i 5.2, ale niemożliwe jest ustalenie dawkowania.
Sposób podawania
Fulwestrant należy podawać powoli (czas jednego wstrzyknięcia 1 do 2 minut), domięśniowo w dwóch kolejnych wstrzyknięciach po 5 ml, każde w inny pośladek (miejsce na pośladku).
Należy zachować ostrożność podczas podawania fulwestrantu w górno-boczną okolicę pośladka ze względu na bliskość nerwu kulszowego.
Pełna instrukcja podawania patrz punkt 6.6.
Nadwrażliwość na substancje czynną lub na którąkolwiek substancję pomocniczą wymienioną w punkcie 6.1.
Ciąża i karmienie piersią (patrz punkt 4.6).
Ciężkie zaburzenie czynności wątroby (patrz punkty 4.4 i 5.2).
Fulwestrant należy stosować ostrożnie u pacjentek z łagodnymi i umiarkowanymi zaburzeniami czynności wątroby (patrz punkty 4.2, 4.3 i 5.2).
Fulwestrant należy stosować ostrożnie u pacjentek z ciężkimi zaburzeniami czynności nerek (klirens kreatyniny mniejszy niż 30 ml/min).
Ze względu na domięśniową drogę podania, fulwestrant należy stosować ostrożnie u pacjentek ze skazą krwotoczną, trombocytopenią lub u pacjentek stosujących leki przeciwzakrzepowe.
U pacjentek z zaawansowanym rakiem piersi często występują zaburzenia zatorowo-zakrzepowe. Zaburzenia te obserwowano także u pacjentek w badaniach klinicznych z zastosowaniem fulwestrantu (patrz punkt 4.8). Należy wziąć to pod uwagę przepisując fulwestrant pacjentkom z grupy ryzyka.
W związku ze wstrzyknięciem fulwestrantu zgłaszano reakcje w miejscu wstrzyknięcia takie jak rwa kulszowa, nerwoból, ból neuropatyczny i neuropatia obwodowa. Należy zachować ostrożność podczas podawania fulwestrantu w górno-boczną okolicę pośladka ze względu na bliskość nerwu kulszowego (patrz punkty 4.2 i 4.8).
Brak jest danych dotyczących odległych skutków działania fulwestrantu na kości. W związku z mechanizmem działania fulwestrantu istnieje potencjalne ryzyko wystąpienia osteoporozy.
Nie badano skuteczności i bezpieczeństwa stosowania fulwestrantu (podawanego w monoterapii lub w skojarzeniu z palbocyklibem) u pacjentek z masywnymi przerzutami do narządów miąższowych.
Gdy fulwestrant jest stosowany w skojarzeniu z palbocyklibem, należy zapoznać się także z Charakterystyką Produktu Leczniczego palbocyklibu.
Interakcje z testami przeciwciał estradiolu
Ze względu na podobieństwo struktury fulwestrantu i estradiolu, fulwestrant może wpływać na wyniki testów wykrywających poziom estradiolu a opartych na przeciwciałach oraz może powodować fałszywie dodatnie wyniki wskazujące na wysoki poziomu estradiolu.
Etanol
Ten produkt leczniczy zawiera 500 mg alkoholu (etanolu) w każdej ampułko-strzykawce, co jest równoważne 100 mg/ml (10% w/v). Ilość alkoholu w każdym wstrzyknięciu tego produktu leczniczego jest równoważna 13 ml piwa lub 5 ml wina.
Dawka 500 mg tego produktu leczniczego (2 ampułko-strzykawki) podana dorosłej kobiecie o masie ciała 70 kg spowoduje narażenie na etanol wynoszące 14,3 mg/kg mc., co może spowodować zwiększenie stężenia alkoholu we krwi (ang. blood alcohol concentration, BAC) o około
2,4 mg/100 ml (patrz załącznik 1 do raportu EMA/CHMP/43486/2018). Dla porównania, u osoby dorosłej pijącej kieliszek wina lub 500 ml piwa, stężenie alkoholu we krwi wyniesie prawdopodobnie około 50 mg/100 ml.
Jednoczesne podawanie z lekami zawierającymi, np. glikol propylenowy lub etanol może prowadzić do kumulacji etanolu i wywoływać działania niepożądane.
Alkohol benzylowy
Lek zawiera 500 mg alkoholu benzylowego w każdej ampułkostrzykawce. Alkohol benzylowy może powodować reakcje alergiczne.
Dzieci i młodzież
Fulwestrant nie jest zalecany do stosowania u dzieci i młodzieży ponieważ nie zostało ustalone bezpieczeństwo stosowania i skuteczność w tej grupie pacjentów (patrz punkt 5.1).
Wyniki badania klinicznego nad interakcją z midazolamem (substratem CYP3A4) wykazują, że fulwestrant nie wpływa hamująco na CYP3A4. W przeprowadzonych badaniach klinicznych nad interakcją z ryfampicyną (induktorem CYP3A4) i ketokonazolem (inhibitorem CYP3A4), nie stwierdzono istotnej klinicznie zmiany klirensu fulwestrantu. W związku z powyższym nie ma konieczności zmiany dawki fulwestrantu, jeśli jest on stosowany jednocześnie z innymi lekami
o działaniu hamującym lub pobudzającym aktywność CYP3A4.
Kobiety w wieku rozrodczym
Pacjentki w wieku rozrodczym należy poinformować o konieczności stosowania skutecznych metod antykoncepcji w trakcie leczenia.
Ciąża
Fulwestrant jest przeciwwskazany do stosowania w ciąży (patrz punkt 4.3). W badaniach na szczurach i królikach wykazano, że fulwestrant po podaniu pojedynczej dawki domięśniowej przenika przez łożysko. Badania na zwierzętach wykazały toksyczny wpływ na reprodukcję, w tym zwiększoną liczbę nieprawidłowości i zgonów płodów (patrz punkt 5.3). Jeśli podczas stosowania fulwestrantu zostanie stwierdzona ciąża, pacjentkę należy niezwłocznie poinformować o potencjalnym ryzyku uszkodzenia płodu i utraty ciąży.
Karmienie piersią
Należy przerwać karmienie piersią jeśli konieczne jest leczenie fulwestrantem. Fulwestrant przenika do mleka karmiących samic szczura. Nie wiadomo, czy fulwestrant przenika do mleka kobiet karmiących. W związku z możliwością wystąpienia ciężkich działań niepożądanych u karmionego piersią dziecka, którego matka jest leczona fulwestrantem, stosowanie leku w okresie karmienia piersią jest przeciwwskazane (patrz punkt 4.3).
Płodność
Nie badano wpływu stosowania fulwestrantu na płodność u ludzi.
Fulwestrant nie ma wpływu lub wywiera nieistotny wpływ na zdolność prowadzenia pojazdów
i obsługiwania maszyn. Jednak ze względu na częste występowanie astenii w trakcie stosowania fulwestrantu, pacjenci, u których występuje to działanie niepożądane, powinni zachować szczególną ostrożność podczas prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu.
Podsumowanie profilu bezpieczeństwa
Monoterapia
W tym punkcie przedstawiono informacje o wszystkich działaniach niepożądanych uzyskane podczas badań klinicznych, badań po wprowadzeniu fulwestrantu do obrotu oraz zgłoszeń spontanicznych. W zbiorczej grupie danych dotyczących stosowania fulwestrantu w monoterapii, do najczęściej opisywanych działań niepożądanych należą: odczyny w miejscu podania, astenia, nudności i zwiększenie aktywności enzymów wątrobowych (AlAT, AspAT, fosfataza alkaliczna).
W Tabeli 1, podane kategorie częstości występowania działań niepożądanych zostały zdefiniowane w oparciu o zbiorcze analizy bezpieczeństwa stosowania w grupie terapeutycznej przyjmującej fulwestrant 500 mg w badaniach porównujących fulwestrant 500 mg z fulwestrantem 250 mg
[CONFIRM (badanie D6997C00002), FINDER 1 (badanie D6997C00004), FINDER 2 (badanie
D6997C00006), NEWEST (badanie D6997C00003)] lub tylko z badania FALCON (badanie D699BC00001), w którym porównywano fulwestrant 500 mg z anastrozolem 1 mg. W przypadku różnic w częstości występowania między zbiorczą analizą bezpieczeństwa a badaniem FALCON, przedstawiono największą częstość występowania. Częstości wymienione w Tabeli 1 są podane w oparciu o wszystkie zgłoszone incydenty niezależnie od oceny związku przyczynowo-skutkowego przez badacza. Mediana czasu trwania leczenia fulwestrantem w dawce 500 mg dla zbiorczej grupy danych (w tym w badaniach wymienionych wyżej i w badaniu FALCON) wyniosła 6,5 miesiąca.
Tabelaryczne zestawienie działań niepożądanych
Działania niepożądane zostały podane zgodnie z częstością ich występowania i klasyfikacją układów i narządów (ang. System Organ Class, SOC). Częstość występowania określono następująco: bardzo
często (≥1/10), często (≥1/100 do <1/10), niezbyt często (≥1/1000 do <1/100). W obrębie każdej grupy o określonej częstości występowania działania niepożądane są wymienione zgodnie ze zmniejszającym się nasileniem.
Tabela 1 Działania niepożądane zgłaszane podczas stosowania fulwestrantu w monoterapii
Działania niepożądane według klasyfikacji układów i narządów oraz częstości występowania | ||
Zakażenia i zarażenia pasożytnicze | Często | Zakażenia dróg moczowych |
Zaburzenia krwi i układu chłonnego | Często | Zmniejszona liczba płytek krwie |
Zaburzenia układu immunologicznego | Bardzo często | Reakcje nadwrażliwoście |
Niezbyt często | Reakcje anafilaktyczne | |
Zaburzenia metabolizmu i odżywiania | Często | Jadłowstręta |
Zaburzenia układu nerwowego | Często | Ból głowy |
Zaburzenia naczyniowe | Bardzo często | Uderzenia gorącae |
Często | Żylna choroba zakrzepowo-zatorowaa | |
Zaburzenia żołądkowo-jelitowe | Bardzo często | Nudności |
Często | Wymioty, biegunka | |
Zaburzenia wątroby i dróg żółciowych | Bardzo często | Zwiększona aktywność enzymów wątrobowych (AlAT, AspAT, fosfataza zasadowa)a |
Często | Zwiększone stężenie bilirubinya | |
Niezbyt często | Niewydolność wątrobyc,f, zapalenie wątrobyf, zwiększona aktywność gamma-GTf | |
Zaburzenia skóry i tkanki podskórnej | Bardzo często | Wysypkae |
Zaburzenia mięśniowo- szkieletowe i tkanki łącznej | Bardzo często | Bóle stawów i bóle mięśniowo-szkieletowe |
Często | Ból pleców | |
Często | Krwotok z pochwye | |
Zaburzenia układu rozrodczego i piersi | Niezbyt często | Kandydoza pochwyf, obfite upławyf |
Zaburzenia ogólne i stany w miejscu podania | Bardzo często | Asteniaa, odczyny w miejscu podaniab |
Często | Neuropatia obwodowae, zapalenie nerwu kulszowegoe | |
Niezbyt często | Krwotok w miejscu podaniaf, krwiak w miejscu podaniaf, nerwobólc,f |
f Działania niepożądanego nie obserwowano w badaniu FALCON.
Opis wybranych działań niepożądanych
Podane niżej opisy opierają się na analizie bezpieczeństwa obejmującej 228 pacjentek, które otrzymały co najmniej jedną (1) dawkę fulwestrantu oraz 232 pacjentek, które otrzymały co najmniej jedną (1) dawkę anastrozolu w badaniu III fazy FALCON.
Ból stawów i bóle mięśniowo-szkieletowe
W badaniu FALCON liczba pacjentek, które zgłosiły zdarzenie niepożądane w postaci bólu stawów
i bólów mięśniowo-szkieletowych, wyniosła w grupie otrzymującej fulwestrant 65 (31,2%) i w grupie otrzymującej anastrozol 48 (24,1%). Spośród 65 pacjentek z grupy otrzymującej fulwestrant 40% (26/65) zgłaszało ból stawów i bóle mięśniowo-szkieletowe w pierwszym miesiącu leczenia, zaś 66,2% (43/65) pacjentek zgłaszało te bóle w pierwszych 3 miesiącach leczenia. Żadna z pacjentek nie zgłosiła zdarzeń stopnia ≥3 wg CTCAE (ang. Common Terminology Criteria for Adverse Events) lub wymagających zmniejszenia dawki, przerwania podawania dawki leku bądź zakończenia leczenia
z powodu tych zdarzeń niepożądanych.
Leczenie skojarzone z palbocyklibem
Ogólny profil bezpieczeństwa fulwestrantu stosowanego w skojarzeniu z palbocyklibem opiera się na danych uzyskanych od 517 pacjentek z miejscowo zaawansowanym lub rozsianym rakiem piersi
z obecnością receptorów hormonalnych, bez nadmiernej ekspresji HER2, uczestniczących
w randomizowanym badaniu PALOMA3 (patrz punkt 5.1). Najczęstszymi (≥20%) zdarzeniami niepożądanymi dowolnego stopnia nasilenia, które zgłaszały pacjentki otrzymujące fulwestrant w skojarzeniu z palbocyklibem były: neutropenia, leukopenia, zakażenia, uczucie zmęczenia, nudności, niedokrwistość, zapalenie jamy ustnej, biegunka, małopłytkowość i wymioty. Najczęstszymi (≥2%) zdarzeniami niepożądanymi o stopniu nasilenia ≥3 były: neutropenia, leukopenia, niedokrwistość, zakażenia, zwiększenie aktywności AspAT, małopłytkowość i uczucie zmęczenia.
W tabeli 2. przedstawiono działania niepożądane z badania PALOMA3.
Mediana czasu trwania ekspozycji na fulwestrant wyniosła 11,2 miesiąca w grupie otrzymującej fulwestrant z palbocyklibem oraz 4,8 miesiąca w grupie otrzymującej fulwestrant i placebo. Mediana czasu trwania ekspozycji na palbocyklib w grupie fulwestrant + palbocyklib, wyniosła 10,8 miesiąca.
Tabela 2 Działania niepożądane zgłaszane w badaniu PALOMA 3 (N=517)
Klasyfikacja układów i narządów Częstość Preferowany termina | Fulwestrant + palbocyklib (N=345) | Fulwestrant + placebo (N=172) | ||
Wszystkich stopni (n%) | Stopnia ≥3. (%) | Wszystkich stopnie(n%) | Stopnia ≥3. (%) | |
Zakażenia i zarażenia pasożytnicze | ||||
Bardzo często | ||||
Zakażeniab | 188 (54,5) | 19 (5,5) | 60 (34,9) | 6 (3,5) |
Zaburzenia krwi i układu chłonnego | ||||
Bardzo często | ||||
Neutropeniac | 290 (84,1) | 240 (69,6) | 6 (3,5) | 0 |
Leukopeniad | 207 (60,0) | 132 (38,3) | 9 (5,2) | 1 (0,6) |
Niedokrwistośće | 109 (31,6) | 15 (4,3) | 24 (14,0) | 4 (2,3) |
Małopłytkowośćf | 88 (25,5) | 10 (2,9) | 0 | 0 |
Niezbyt często | ||||
Gorączka neutropeniczna | 3 (0,9) | 3 (0,9) | 0 | 0 |
Zaburzenia metabolizmu i odżywiania | ||||
Bardzo często | ||||
Zmniejszony apetyt | 60 (17,4) | 4 (1,2) | 18 (10,5) | 1 (0,6) |
Zaburzenia układu nerwowego | ||||
Często | ||||
Zaburzenia smaku | 27 (7,8) | 0 | 6 (3,5) | 0 |
Zaburzenia oka | ||||
Często | ||||
Nasilone łzawienie | 25 (7,2) | 0 | 2 (1,2) | 0 |
Niewyraźne widzenie | 24 (7,0) | 0 | 3 (1,7) | 0 |
Suchość oka | 15 (4,3) | 0 | 3 (1,7) | 0 |
Zaburzenia układu oddechowego, klatki piersiowej i śródpiersia | ||||
Często | ||||
Krwawienie z nosa | 25 (7,2) | 0 | 4 (2,3) | 0 |
Zaburzenia żołądka i jelit | ||||
Bardzo często | ||||
Nudności | 124 (35,9) | 2 (0,6) | 53 (30,8) | 1 (0,6) |
Zapalenie jamy ustnejg | 104 (30,1) | 3 (0,9) | 24 (14,0) | 0 |
Biegunka | 94 (27,2) | 0 | 35 (20,3) | 2 (1,2) |
Wymioty | 75 (21,7) | 2 (0,6) | 28 (16,3) | 1 (0,6) |
Zaburzenia skóry i tkanki podskórnej | ||||
Bardzo często | ||||
Łysienie | 67 (19,4) | NA | 11 (6,4) | NA |
Wysypkah | 63 (18,3) | 3 (0,9) | 10 (5,8) | 0 |
Często | ||||
Suchość skóry | 28 (8,1) | 0 | 3 (1,7) | 0 |
Zaburzenia ogólne i stany w miejscu podania | ||||
Bardzo często | ||||
Zmęczenie | 152 (44,1) | 9 (2,6) | 54 (31,4) | 2 (1,2) |
Gorączka | 47 (13,6) | 1 (0,3) | 10 (5,8) | 0 |
Często | ||||
Astenia | 27 (7,8) | 1 (0,3) | 13 (7,6) | 2 (1,2) |
Badania diagnostyczne | ||||
Bardzo często | ||||
Zwiększenie aktywności AspAT | 40 (11,6) | 11 (3,2) | 13 (7,6) | 4 (2,3) |
Często | ||||
Zwiększenie aktywności AlAT | 30 (8,7) | 7 (2,0) | 10 (5,8) | 1 (0,6) |
AlAT=aminotransferaza alaninowa; AspAT=aminotransferaza asparaginianowa; N=liczba pacjentek,
NA=nie dotyczy
czerwieni warg, zapalenie języka, ból języka, owrzodzenie jamy ustnej, zapalenie błony śluzowej, ból jamy ustnej, uczucie dyskomfortu w jamie ustnej i gardle, ból jamy ustnej i gardła, zapalenie jamy ustnej.
h Wysypka obejmuje następujące PT: wysypka, wysypka grudkowo-plamkowa, wysypka ze świądem, wysypka z rumieniem, wysypka grudkowa, zapalenie skóry, trądzikopodobne zapalenie skóry, toksyczne wykwity skórne.
Opis wybranych działań niepożądanych
Neutropenia
U pacjentek otrzymujących fulwestrant w skojarzeniu z palbocyklibem w ramach badania PALOMA3 neutropenię dowolnego stopnia zgłaszano u 290 (84,1%) pacjentek, neutropenię stopnia 3. zgłoszono u 200 (58,0%) pacjentek, a neutropenię 4. stopnia zgłoszono u 40 (11,6%) pacjentek. W grupie otrzymującej fulwestrant i placebo (n=172) neutropenię dowolnego stopnia odnotowano u 6 (3,5%) pacjentek. Nie zgłoszono żadnego przypadku neutropenii 3. i 4. stopnia w grupie przyjmującej fulwestrant i placebo.
U pacjentek otrzymujących fulwestrant w skojarzeniu z palbocyklibem mediana czasu do wystąpienia pierwszego epizodu neutropenii dowolnego stopnia nasilenia wyniosła 15 dni (zakres: 13-512),
a mediana czasu trwania neutropenii stopnia ≥3 wyniosła 16 dni. Gorączkę neutropeniczną odnotowano u 3 (0,9%) pacjentek otrzymujących fulwestrant z palbocyklibem.
Zgłaszanie podejrzewanych działań niepożądanych
Po dopuszczeniu produktu leczniczego do obrotu istotne jest zgłaszanie podejrzewanych działań niepożądanych. Umożliwia to nieprzerwane monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Osoby należące do fachowego personelu medycznego powinny zgłaszać wszelkie podejrzewane działania niepożądane za pośrednictwem Departament Monitorowania Niepożądanych Działań Produktów Leczniczych Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych
Al. Jerozolimskie 181C 02-222 Warszawa
Tel.: + 48 22 49 21 301
Faks: + 48 22 49 21 309
Strona internetowa: https://smz.ezdrowie.gov.pl
Działania niepożądane można zgłaszać również podmiotowi odpowiedzialnemu lub przedstawicielowi podmiotu odpowiedzialnego w Polsce.
Istnieją pojedyncze doniesienia o przypadkach przedawkowania fulwestrantu u ludzi. W razie przedawkowania zaleca się podtrzymujące leczenie objawowe. Badania na zwierzętach nie wskazują,
aby stosowanie fulwestrantu w większych dawkach wywoływało objawy inne niż wynikające z bezpośredniego lub pośredniego działania antyestrogenowego (patrz punkt 5.3).
Grupa farmakoterapeutyczna: leki stosowane w terapii hormonalnej, antyestrogeny Kod ATC: L02BA03
Mechanizm działania i działania farmakodynamiczne
Fulwestrant jest kompetycyjnym antagonistą receptora estrogenowego (ER) o powinowactwie porównywalnym z estradiolem. Fulwestrant blokuje troficzne działanie estrogenów bez nawet częściowego działania agonistycznego (estrogenopodobnego). Mechanizm działania polega na zmniejszeniu ilości (ang. down-regulation) białka receptorowego receptora estrogenowego.
W badaniach klinicznych u kobiet po menopauzie z pierwotnym rakiem piersi wykazano, że
w porównaniu z placebo fulwestrant znacząco zmniejsza ilość (down-regulation) białka receptora estrogenowego w guzach zawierających receptory estrogenowe. Stwierdzono także znaczące zmniejszenie ekspresji receptora progesteronowego, co potwierdza brak wewnętrznej aktywności estrogenowej. Wykazano również, że w neoadjuwantowym leczeniu guzów piersi u kobiet po menopauzie fulwestrant w dawce 500 mg w większym stopniu ogranicza ekspresję receptora estrogenowego i markera proliferacji Ki67 niż fulwestrant w dawce 250 mg.
Bezpieczeństwo kliniczne i skuteczność w zaawansowanym raku piersi
Monoterapia
Badanie kliniczne III fazy ukończyło 736 kobiet po menopauzie z zaawansowanym rakiem piersi, z nawrotem w trakcie lub po zakończeniu hormonalnego leczenia uzupełniającego albo z progresją choroby po leczeniu hormonalnym nowotworu zaawansowanego. Badanie objęło 423 pacjentki, u
których nastąpił nawrót albo progresja w trakcie leczenia antyestrogenowego (podgrupa AE) oraz 313 pacjentek z nawrotem lub progresją w trakcie leczenia inhibitorem aromatazy (podgrupa AI). Celem badania było porównanie skuteczności i bezpieczeństwa stosowania fulwestrantu w dawce 500 mg (n=362) i w dawce 250 mg (n=374). Pierwszorzędowym punktem końcowym było przeżycie wolne od progresji choroby (PFS); najważniejsze drugorzędowe punkty końcowe oceniające skuteczność leczenia objęły odsetek odpowiedzi obiektywnych (ORR), wskaźnik korzyści klinicznej (CBR)
i przeżycie całkowite (OS). Wyniki badania CONFIRM dotyczące skuteczności leczenia podsumowano w tabeli 3.
Tabela 3 Podsumowanie wyników skuteczności leczenia w badaniu CONFIRM: pierwszorzędowy punkt końcowy (PFS) i najważniejsze drugorzędowe punkty końcowe.
Zmienna | Sposób oceny; porównanie leczenia | Fulwestrant 500 mg (N=362) | Fulwestrant 250 mg (N=374) | Porównanie grup (fulwestrant 500 mg/fulwestrant 250 mg) | ||
Współczynnik ryzyka | 95% CI | Wartość p | ||||
PFS | Mediana czasu przeżycia [miesiące] wg K-M; współczynnik ryzyka | |||||
Wszystkie pacjentki | 6,5 | 5,5 | 0,80 | 0,68, 0,94 | 0,006 | |
-Podgrupa AE (n=423) | 8,6 | 5,8 | 0,76 | 0,62, 0,94 | 0,013 | |
-Podgrupa AI (n=313)a | 5,4 | 4,1 | 0,85 | 0,67, 1,08 | 0,195 | |
OSb | Mediana czasu przeżycia [miesiące] wg K-M; współczynnik ryzyka | |||||
Wszystkie pacjentki | 26,4 | 22,3 | 0,81 | 0,69, 0,96 | 0,016c | |
-Podgrupa AE (n=423) | 30,6 | 23,9 | 0,79 | 0,63, 0,99 | 0,038c | |
-Podgrupa AI (n=313)a | 24,1 | 20,8 | 0,86 | 0,67, 1,11 | 0,241c | |
Zmienna | Sposób oceny; porównanie leczenia | Fulwestrant 500 mg (N=362) | Fulwestrant 250 mg (N=374) | Porównanie grup (fulwestrant 500 mg/fulwestrant 250 mg) | ||
Bezwzględna różnica w % | 95% CI | |||||
ORRd | % pacjentek z OR; bezwzględna różnica w % | |||||
Wszystkie pacjentki | 13,8 | 14,6 | -0,8 | -5,8, 6,3 | ||
-Podgrupa AE (n=296) | 19,1 | -1,0 | -8,2, 9,3 | |||
-Podgrupa AI (n=205)a | 8,3 | -1,0 | -5,5, 9,8 | |||
CBRe | % pacjentek z CB; bezwzględna różnica w % | |||||
Wszystkie pacjentki | 45,6 | 39,6 | 6,0 | -1,1, 13,3 | ||
-Podgrupa AE (n=423) | 45,1 | 7,3 | -2,2, 16,6 | |||
-Podgrupa AI (n=313)a | 32,3 | 3,9 | -6,1, 15,2 | |||
K-M: krzywe Kaplana-Meiera; CI: przedział ufności, IA: inhibitory aromatazy; AE: antyestrogeny.
Przeprowadzono randomizowane, wieloośrodkowe, podwójnie pozorowane badanie trzeciej fazy z podwójnie ślepą próbą, porównujące fulwestrant o mocy 500 mg z anastrozolem o mocy 1 mg u kobiet po menopauzie z ER-dodatnim i (lub) PgR-dodatnim miejscowo zaawansowanym lub
rozsianym rakiem piersi, które wcześniej nie otrzymywały żadnej terapii hormonalnej. Łącznie 462 pacjentki były sekwencyjnie losowo przydzielane w stosunku 1:1 do grupy otrzymującej 500 mg fulwestrantu lub do grupy otrzymującej 1 mg anastrozolu.
Randomizacja była stratyfikowana według stopnia zaawansowania choroby (miejscowo zaawansowana lub rozsiana), wcześniejszej chemioterapii z powodu zaawansowanej choroby i obecności zmian możliwych do oceny.
Pierwszorzędowym punktem końcowym dotyczącym skuteczności w tym badaniu było przeżycie wolne od progresji choroby (PFS) oceniane przez badacza według kryteriów oceny odpowiedzi na leczenie w guzach litych RECIST 1.1 (ang. Response Evaluation Criteria in Solid Tumours).
Najważniejsze drugorzędowe punkty końcowe oceny skuteczności obejmowały przeżycie całkowite (OS) i odsetek odpowiedzi obiektywnych (ORR).
Mediana wieku pacjentek włączonych do badania wyniosła 63 lata (zakres 36-90). U większości pacjentek (87,0%) przed rozpoczęciem badania występowały przerzuty. U 55% pacjentek występowały przerzuty do narządów miąższowych przed rozpoczęciem badania. Łącznie 17,1% pacjentek otrzymało wcześniej chemioterapię z powodu choroby zaawansowanej, a u 84,2% pacjentek możliwa była ocena wielkości zmian nowotworowych.
W większości predefiniowanych podgrup pacjentek otrzymane wyniki były spójne. W podgrupie pacjentek z chorobą ograniczoną do przerzutów innych niż przerzuty do narządów miąższowych (n=208) wartość HR wyniosła 0,592 (95% CI: 0,419; 0,837) w grupie otrzymującej fulwestrant w porównaniu z grupą leczoną anastrozolem. W podgrupie pacjentek z przerzutami do narządów miąższowych (n=254) HR wyniósł 0,993 (95% CI: 0,740; 1,331) w grupie otrzymującej fulwestrant w porównaniu z grupą leczoną anastrozolem. Wyniki dotyczące skuteczności w badaniu FALCON przedstawiono w tabeli 4. i na rycinie 1.
Tabela 4 Podsumowanie wyników dotyczących pierwszorzędowego punktu końcowego i najważniejszych drugorzędowych punktów końcowych oceny skuteczności (ocena badacza, populacja zgodna z zaplanowanym leczeniem) - badanie FALCON
Fulwestrant 500 mg (N=230) | Anastrozol 1 mg (N=232) | |
Przeżycie wolne od progresji choroby | ||
Liczba zdarzeń PFS (%) | 143 (62,2%) | 166 (71,6%) |
Współczynnik ryzyka (95% CI) i wartość p dla PFS | HR 0,797 (0,637 – 0,999) p = 0,0486 | |
Mediana PFS [miesiące (95% CI)] | 16,6 (13,8, 21,0) | 13,8 (12,0, 16,6) |
Liczba zdarzeń OS* | 67 (29,1%) | 75 (32,3%) |
Współczynnik ryzyka (95% CI) i wartość p dla OS | HR 0,875 (0,629 – 1,217) p = 0,4277 | |
ORR** | 89 (46,1%) | 88 (44,9%) |
Iloraz szans (95% CI) i wartość p dla ORR | OR 1,074 (0,716 – 1,614) p = 0,7290 | |
Mediana DoR (miesiące) | 20,0 | 13,2 |
CBR | 180 (78,3%) | 172 (74,1%) |
Iloraz szans (95% CI) i wartość p dla CBR | OR 1,253 (0,815 – 1,932) p = 0,3045 | |
* (31% dojrzałość danych) - nieostateczna analiza OS
** u pacjentek ze zmianami mierzalnymi
Rycina 1 Wykres Kaplana-Meiera dla przeżycia wolnego od progresji choroby (ocena badacza, populacja zgodna z zaplanowanym leczeniem) - badanie FALCON
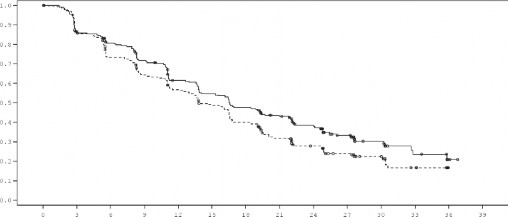
Czas od randomizacji (miesiące)
Leczenie Fulwestrant 500 mg (N=230) Anastrozol 1 mg (N=232)
Liczba pacjentek obarczonych ryzykiem:
FUL500 | 230 | 187 | 171 | 150 | 124 | 110 | 96 | 81 | 63 | 44 | 24 | 11 | 2 | 0 |
ANAS1 | 232 | 194 | 162 | 139 | 120 | 102 | 84 | 60 | 45 | 31 | 22 | 10 | 0 | 0 |
Dwa badania kliniczne III fazy ukończyło 851 kobiet po menopauzie z zaawansowanym rakiem piersi z nawrotem choroby w trakcie lub po zakończeniu hormonalnego leczenia uzupełniającego lub
z progresją choroby podczas leczenia hormonalnego zaawansowanego raka piersi. U 77% pacjentek biorących udział w badaniach wykazano obecność receptora estrogenowego w komórkach raka piersi. W badaniach tych porównywano bezpieczeństwo stosowania i skuteczność fulwestrantu podawanego co miesiąc w dawce 250 mg i anastrozolu (inhibitora aromatazy) podawanego codziennie w dawce
Wchłanianie
Po podaniu fulwestrantu w postaci roztworu do wstrzykiwań domięśniowych o długotrwałym działaniu, fulwestrant jest powoli wchłaniany i osiąga maksymalne stężenie w osoczu (Cmax) po około 5 dniach. Podawanie fulwestrantu w dawce 500 mg powoduje osiąganie stałych lub w przybliżeniu stałych wartości ekspozycji przez pierwszy miesiąc stosowania (średnie wartości odpowiednio: AUC 475 [33,4%] ng.dni/ml, Cmax 25,1 [35,3%] ng/ml, Cmin 16,3 [25,9%] ng/ml). W stanie stacjonarnym
wartości stężeń fulwestrantu w osoczu mieszczą się w stosunkowo wąskim zakresie, dla którego stężenie maksymalne jest do 3 razy większe niż różnica stężeń w stanie stacjonarnym. Po podaniu domięśniowym w zakresie dawek od 50 do 500 mg, ekspozycja jest w przybliżeniu proporcjonalnie zależna od dawki.
Dystrybucja
Fulwestrant ulega rozległej i szybkiej dystrybucji w tkankach. Duża pozorna objętość dystrybucji w stanie stacjonarnym (Vdss) (wynosi około 3 do 5 l/kg), wskazuje, że lek znajduje się głównie w przestrzeni pozanaczyniowej.
Fulwestrant silnie wiąże się z białkami osocza w 99%. Fulwestrant wiąże się przede wszystkim
z frakcjami lipoprotein o bardzo małej (VLDL), małej (LDL) i dużej gęstości (HDL). Nie badano interakcji dotyczących konkurencyjnego mechanizmu wiązania z białkami. Nie określono wiązania z globuliną wiążącą hormony płciowe (SHBG).
Metabolizm
Metabolizm fulwestrantu nie został w pełni poznany i zachodzi na drodze wielu przemian biologicznych, w sposób podobny jak w przypadku endogennych steroidów. Wykryte metabolity (włączając 17-keton, sulfoniany, 3-siarczan, 3- i 17-glukuroniany) mają mniejszą niż fulwestrant lub porównywalną z nim aktywność antyestrogenową. W badaniach z wykorzystaniem ludzkich komórek wątroby i rekombinowanych ludzkich enzymów wątrobowych stwierdzono, że CYP 3A4 jest jedynym izoenzymem cytochromu P-450 biorącym udział w procesie oksydacji fulwestrantu, natomiast w warunkach in vivo główną rolę wydają się pełnić enzymy nienależące do grupy cytochromu P-450. Na podstawie badań in vitro można sądzić, że fulwestrant nie wpływa hamująco na izoenzymy układu cytochromu CYP450.
Eliminacja
Fulwestrant jest wydalany głównie w postaci zmetabolizowanej. Fulwestrant jest wydalany przede wszystkim z kałem, mniej niż 1% jest wydalany w moczu. Fulwestrant ma duży klirens,
11±1,7 ml/min/kg, co może świadczyć o dużym udziale wątroby w wydalaniu leku. Końcowy okres półtrwania (t1/2) po podaniu domięśniowym zależy przede wszystkim od szybkości wchłaniania
i wynosi około 50 dni.
Szczególne grupy pacjentów
Z przeprowadzonych badań III fazy wynika, że nie ma różnic w farmakokinetyce leku stosowanego u pacjentek z różnych grup wiekowych (pacjentki w wieku 33 do 89 lat), o różnej masie ciała (40- 127 kg) lub u pacjentek różnych ras.
Zaburzenia czynności nerek
Łagodne do umiarkowanych zaburzenia czynności nerek nie wpływają na farmakokinetykę fulwestrantu w stopniu istotnym klinicznie.
Zaburzenia czynności wątroby
Farmakokinetykę fulwestrantu oceniono w badaniu klinicznym u kobiet z łagodnymi i umiarkowanymi zaburzeniami czynności wątroby (grupa A i B według skali Child-Pugh) po podaniu pojedynczej dawki. W badaniu stosowano dużą dawkę o krótkim działaniu podawaną domięśniowo. W grupie pacjentek z zaburzeniami czynności wątroby obserwowano mniej więcej 2,5-krotne zwiększenie AUC w porównaniu do wartości uzyskanych w grupie zdrowych uczestników badania. Oczekuje się, że takie zwiększenie ekspozycji u kobiet otrzymujących fulwestrant będzie dobrze tolerowane. Nie badano kobiet z ciężkimi zaburzeniami wątroby (grupa C według skali Child-Pugh).
Dzieci i młodzież
Farmakokinetykę fulwestrantu oceniono w badaniu klinicznym przeprowadzonym wśród 30 dziewcząt w wieku od 1 do 8 lat z przedwczesnym dojrzewaniem płciowym związanym z zespołem McCune Albrighta (patrz punkt 5.1). Otrzymywały one flulwestrant domięśniowo w dawce 4 mg/kg masy ciała co miesiąc. Geometryczna mediana (SD) wartości stężeń fulwestrantu w osoczu w stanie stacjonarnym
(Cmin,ss) i powierzchnia pola pod krzywą AUCss wynosiły odpowiednio: 4,2 (0,9) ng/ml i 3680 (1020) ng*hr/ml. Pomimo, iż zebrane dane są ograniczone, wartości stężeń fulwestrantu w stanie stacjonarnym w osoczu u dzieci wydają się odpowiadać wartościom stwierdzanym u pacjentów dorosłych.
Fulwestrant wykazuje niewielką toksyczność po podaniu jednorazowym (ostrą).
Referencyjny produkt leczniczy oraz fulwestrant w innych postaciach był dobrze tolerowany przez wszystkie gatunki zwierząt, na których wykonano badania po podaniu wielokrotnym. Reakcje obserwowane w miejscu podania, tj. zapalenie mięśni i ziarniniaki wynikały z działania substancji pomocniczych leku. Jednakże w badaniach przeprowadzonych na królikach zaobserwowano, że zapalenie mięśni było bardziej nasilone w miejscu podania fulwestrantu niż w miejscu podania próbki kontrolnej, która zawierała sól fizjologiczną. Badania przeprowadzone na szczurach i psach wykazały, że po podaniu wielokrotnych dawek fulwestrantu w postaci wstrzyknięcia domięśniowego występują objawy związane z działaniem antyestrogenowym leku. Przede wszystkim obserwowano wpływ na żeński układ rozrodczy oraz na te narządy, które są wrażliwe na działanie hormonów u obu płci.
U niektórych psów, które przyjmowały lek długotrwale (12 miesięcy), obserwowano zapalenie tętnic obejmujące różne tkanki.
W badaniach na psach, gdy fulwestrant był podawany doustnie i dożylnie, stwierdzono, że wpływa on na układ sercowo-naczyniowy (niewielkie uniesienie odcinka ST w badaniu EKG [ podanie doustne], zahamowanie zatokowe u jednego psa [podanie dożylne]). Reakcje te wystąpiły, gdy stężenia leku stwierdzane u zwierząt były wielokrotnie większe niż stwierdzone u ludzi (Cmax >15 razy). Należy zatem uznać, że w praktyce klinicznej ma to niewielkie znaczenie dla bezpieczeństwa ludzi.
Fulwestrant nie wykazywał działania genotoksycznego.
Wpływ fulwestrantu na reprodukcję i działanie uszkadzające na zarodek/płód, gdy był on podawany w dawkach zbliżonych do dawek terapeutycznych, wynika z jego działania antyestrogenowego.
Fulwestrant podawany szczurom powodował odwracalne zmniejszenie płodności samic i zmniejszone przeżycie zarodków, a także dystocję i zwiększoną ilość różnych zaburzeń u płodu, w tym patologiczne zgięcie śródstopia. U samic królików, którym podawano fulwestrant występowały poronienia. Obserwowano zwiększoną masę łożyska i obumieranie zarodków po zagnieżdżeniu.
Stwierdzono zwiększoną liczbę nieprawidłowości u płodów (w tym wsteczne przesunięcie obręczy miednicy i 27. kręgu przedkrzyżowego).
W dwuletnim badaniu działania rakotwórczego u szczurów (domięśniowe podawanie fulwestrantu) wykazano zwiększoną częstość występowania łagodnych ziarniszczaków w jajniku u samic szczurów po podaniu dużych dawek, 10 mg/szczur/15 dni oraz zwiększoną częstość występowania nowotworów z komórek Leydiga w jądrach u samców. Podczas dwuletniego badania rakotwórczości u myszy po podaniu dawek 150 mg/kg/dobę i 500 mg/kg/dobę (doustnie, codziennie) stwierdzono zwiększoną częstość występowania nowotworów sznura płciowego i zrębu jajnika (zarówno łagodnych jak i złośliwych). Wyniki ekspozycji układowej (AUC), obserwowane po podaniu największej dawki bez obserwowanego działania (NOEL), były u szczurów w przybliżeniu 1,5-krotnością, oczekiwanej ekspozycji u kobiet i 0,8-krotnością ekspozycji u mężczyzn, a u myszy w przybliżeniu 0,8-krotnością, oczekiwanej ekspozycji zarówno u kobiet jak i u mężczyzn. Indukcja takich nowotworów jest związana z farmakologicznymi zaburzeniami hormonalnego sprzężenia zwrotnego dla stężenia gonadotropin, spowodowanymi przez antyestrogeny u zwierząt czynnych rozrodczo. W związku z tym wyniki tych badań nie są uznane za odpowiednie do zalecenia stosowania fulwestrantu w leczeniu zaawansowanego raka piersi u kobiet po menopauzie.
Badania oceny ryzyka dla środowiska wykazały, że fulwestrant ma potencjał do powodowania działań niepożądanych w środowisku wodnym (patrz punkt 6.6).
Etanol 96% Alkohol benzylowy Benzylu benzoesan
Olej rycynowy oczyszczony
Ponieważ nie wykonywano badań dotyczących zgodności, produktu leczniczego nie wolno mieszać z innymi lekami.
Przechowywać i przewozić w stanie schłodzonym (2˚C - 8˚C).
Należy ograniczyć przechowywanie produktu w temperaturze innej niż 2°C - 8°C. Należy unikać przechowywania w temperaturze wyższej niż 30°C i nie przekraczać okresu 28 dni ze średnią temperaturą przechowywania poniżej 25°C (ale powyżej zakresu 2°C - 8°C). Jeśli zakres temperatur zostanie przekroczony, należy natychmiast zastosować zalecane warunki przechowywania (przechowywać i transportować w lodówce 2°C - 8°C). Przekroczenie właściwej temperatury przechowywania może mieć skumulowany wpływ na jakość produktu, a 28-dniowy okres nie może być przekroczony w ciągu 2 lat ważności fulwestrantu (patrz punkt 6.3). Ekspozycja na temperaturę poniżej 2°C nie powoduje uszkodzenia produktu, jeśli nie jest on przechowywany w temperaturze poniżej -20°C.
Ampułko-strzykawkę przechowywać w oryginalnym opakowaniu w celu ochrony przed światłem.
Zestaw zawierający ampułko-strzykawkę składa się z:
Jednej ampułko-strzykawki wykonanej z bezbarwnego szkła (typ I) z tłokiem z polistyrenu zakończonym korkiem z elastomeru, z końcówką zabezpieczającą w tekturowym pudełku, zawierającej 5 ml roztworu fulwestrantu do wstrzykiwań.
W opakowaniu znajduje się również jedna igła z systemem zabezpieczającym (BD SafetyGlide), do podawania produktu leczniczego.
lub
Dwóch ampułko-strzykawek wykonanych z bezbarwnego szkła (typ I) z tłokami z polistyrenu, zakończonymi korkami z elastomeru, z końcówkami zabezpieczającymi w tekturowym pudełku, zawierających po 5 ml roztworu fulwestrantu do wstrzykiwań, każda.
W opakowaniu znajdują się również dwie igły z systemem zabezpieczającym (BD SafetyGlide), do podawania produktu leczniczego.
lub
2 x dwóch ampułko-strzykawek wykonanych z bezbarwnego szkła (typ I) z tłokami z polistyrenu, zakończonymi korkami z elastomeru, z końcówkami zabezpieczającymi w tekturowych pudełkach, zawierających po 5 ml roztworu fulwestrantu do wstrzykiwań, każda.
W każdym opakowaniu znajdują się również po dwie igły z systemem zabezpieczającym (BD SafetyGlide), do podawania produktu leczniczego.
lub
Czterech ampułko-strzykawek wykonanych z bezbarwnego szkła (typ I) z tłokami z polistyrenu, zakończonymi korkami z elastomeru, z końcówkami zabezpieczającymi w tekturowym pudełku, zawierających po 5 ml roztworu fulwestrantu do wstrzykiwań, każda.
W opakowaniu znajdują się również cztery igły z systemem zabezpieczającym (BD SafetyGlide), do podawania produktu leczniczego.
lub
Sześciu ampułko-strzykawek wykonanych z bezbarwnego szkła (typ I) z tłokami z polistyrenu, zakończonymi korkami z elastomeru, z końcówkami zabezpieczającymi w tekturowym pudełku, zawierających po 5 ml roztworu fulwestrantu do wstrzykiwań, każda.
W opakowaniu znajduje się również sześć igieł z systemem zabezpieczającym (BD SafetyGlide), do podawania produktu leczniczego.
Nie wszystkie wielkości opakowań muszą znajdować się w obrocie.
Instrukcja podawania
Należy podawać wstrzyknięcie zgodnie z lokalnymi wytycznymi dotyczącymi wykonywania wstrzyknięć domięśniowych o dużej objętości.
UWAGA: Należy zachować ostrożność, jeśli fulwestrant jest podawany w górno-boczną okolicę pośladka ze względu na bliskość nerwu kulszowego (patrz punkt 4.4).
Uwaga – nie należy umieszczać w autoklawie igły z systemem osłaniającym (BD SafetyGlide Shielding Hypodermic Needle) przed jej zastosowaniem. Podczas stosowania leku i usuwania pozostałości należy unikać kontaktu rąk z igłą.
Dotyczy obydwu strzykawek:
UWAGA: Postępuj tak, aby zapewnić bezpieczeństwo sobie i innym. Nasłuchuj kliknięcia i wizualnie potwierdź, czy końcówka igły jest całkowicie ukryta.
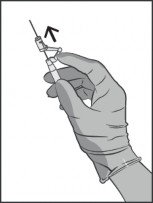
Rysunek 5.
Usuwanie pozostałości
Ampułko-strzykawki są przeznaczone wyłącznie do jednorazowego użycia.
Ten lek może stanowić zagrożenie dla środowiska wodnego. Wszelkie niewykorzystane resztki produktu leczniczego lub jego odpady należy usunąć zgodnie z lokalnymi przepisami (patrz punkt 5.3).
Zentiva, k.s.
U kabelovny 130 Dolní Měcholupy 102 37 Praga 10 Republika Czeska
Pozwolenie nr 26214
Data wydania pierwszego pozwolenia na dopuszczenie do obrotu: 25.01.2021r.
01/2021








